
High-performance aqueous copper-ion batteries based on iron hexacyanoferrate cathodes for enhanced energy storage†
Jinshu
Zhang
 a,
Lexian
Liu
a,
Yuao
Wang
c,
Yantuo
Li
a,
Yang
Yang
a,
Mingyi
Ning
a,
Jianxue
Wu
a,
Bingjie
Ma
a and
Wei
Liu
*ab
a,
Lexian
Liu
a,
Yuao
Wang
c,
Yantuo
Li
a,
Yang
Yang
a,
Mingyi
Ning
a,
Jianxue
Wu
a,
Bingjie
Ma
a and
Wei
Liu
*ab
aSchool of Physics, Key Laboratory of Quantum Materials and Devices of Ministry of Education, Frontiers Science Center for Mobile Information Communication and Security, Southeast University, Nanjing 211189, China. E-mail: 101012931@seu.edu.cn
bPurple Mountain Laboratories, Nanjing 211111, China
cKey Laboratory of Superlight Materials and Surface Technology of Ministry of Education, College of Materials Science and Chemical Engineering, Harbin Engineering University, Harbin 150001, China
First published on 18th March 2025
Abstract
The integration of renewable energy sources, such as solar and wind, requires efficient energy storage systems. Aqueous batteries, with their safety, low cost, and flexibility, have gained attention as promising solutions for energy storage. In this study, we developed an aqueous copper-ion storage device based on an iron hexacyanoferrate (FeHCF) cathode, which offers high capacities of 190 mA h g−1 at 1 A g−1 and 102 mA h g−1 even at 3 A g−1, with a discharge plateau at 0.59 V vs. SHE and a low polarization voltage of 0.2 V. In situ XRD, Raman, and XPS characterization techniques show that copper-ion insertion induces structural changes in FeHCF, leading to a valence state transition between Fe2+ and Fe3+, with a partial conversion of Cu2+ to Cu+. To improve the working voltage, we replaced the Cu2+/Cu0 anode reaction with the lower potential Zn/Zn(OH)42− reaction, achieving an aqueous battery with a voltage range of 1.6–2.5 V. These findings highlight FeHCF-based aqueous batteries’ potential for high-performance and safe energy storage.
 Wei Liu | Dr Wei Liu is a Professor at the School of Physics, Southeast University. He received his Bachelor's and Master's degrees in Materials Physics from Sun Yat-sen University. He obtained his Ph.D in NGS from the Department of Chemistry at the National University of Singapore under the supervision of Prof. Loh Kian Ping. After 4 years as a postdoctoral researcher at the National University of Singapore and RIKEN, Japan, he joined Southeast University in 2021. His research interest focuses on innovative synthesis of functional materials for energy storage. |
1. Introduction
The transition to renewable energy sources, such as solar, wind, and tidal power, is essential for reducing fossil fuel consumption and improving environmental sustainability.1,2 However, integrating these energy sources into the grid effectively requires reliable energy storage systems. Electrochemical energy storage systems have attracted significant attention due to their rapid response, flexibility, and environmental friendliness.3,4 Although lithium-ion batteries currently dominate the energy storage market, their reliance on organic electrolytes raises safety concerns.5Aqueous batteries have emerged as a safer and more cost-effective alternative, attracting interest for their inherent safety and affordability, and they are considered to contribute to the development of green energy and the achievement of carbon neutrality goals.6–10 The performance of aqueous batteries is significantly influenced by the choice of electrode materials. Prussian blue analogues (PBAs), also known as metal hexacyanoferrates (MHCFs), are particularly promising due to their ease of synthesis and low cost.11–13 Initially used in applications such as dyeing and energy conversion, PBAs were first explored as electrode materials by Neff et al. in 1978, who demonstrated the reversible electrochemical insertion and extraction of K+ in K2Fe2+Fe2+(CN)6. This breakthrough paved the way for the development of PBAs in energy storage applications.14 The open framework structure of PBAs, combined with transition metals like Zn, Cu, Ni, and Fe, results in large lattice parameters that facilitate ion diffusion and storage. PBAs are now applied as electrode materials in various ion batteries, including Li+, Na+, K+, Zn2+ and Mg2+ batteries.15,16 In particular, in aqueous multivalent ion batteries, PBAs have demonstrated high capacity and stable cycling performance, making them a key focus in the advancement of aqueous batteries.17–20 Among these, iron hexacyanoferrate (FeHCF) stands out due to its high theoretical capacity, simple synthesis, and abundant elemental resources, positioning it as a promising material for energy storage.21–23
When FeHCF is used as a zinc-ion storage material, it exhibits a high capacity of 120 mA h g−1 at a current density of 0.01 A g−1, with a discharge potential of 0.34 V vs. SHE. The charge–discharge voltage difference is approximately 0.4 V.24 However, at a low current density of 0.06 A g−1, the capacity significantly drops to only 30 mA h g−1. High-voltage scanning can effectively activate the C-coordinated iron in the FeHCF cathode, resulting in higher reaction potentials and improved capacity.23 Under these conditions, the material achieves a reaction voltage of 0.74 V vs. SHE and a capacity of 76 mA h g−1 at a high current of 1 A g−1. However, this improvement comes at the trade-off of a wider charge–discharge voltage range, approximately 2.3 V.
Due to the unique redox properties of copper ions, aqueous copper-ion batteries have the potential to further enhance the energy density of aqueous batteries, and related research has been extensively reported in recent years.25–27 In this study, we report FeHCF nanocrystals as an effective material for copper-ion storage, offering a working voltage of 0.59 V vs. SHE. Within a narrower voltage range of 0.6 V, FeHCF delivers a stable discharge plateau and high capacity, reaching 190 mA h g−1 at a current density of 1 A g−1. Furthermore, FeHCF exhibits minimal voltage polarization during copper-ion storage, with a voltage difference of 0.2 V, which helps reduce energy loss during the storage process. The storage mechanism of Cu2+ in FeHCF was thoroughly investigated using in situ X-ray diffraction (XRD), Raman spectroscopy, X-ray photoelectron spectroscopy (XPS), and other characterization techniques. Finally, by using FeHCF as the cathode and Zn/Zn(OH)42− as the anode, we constructed an aqueous battery that operates within a voltage range of 1.6–2.5 V, significantly enhancing its potential for practical applications.
2. Results and discussion
FeHCF nanocrystals were prepared using the well-established coprecipitation method.23 The structure of FeHCF, as shown in Fig. 1a, exhibits a face-centered cubic structure. The FeC6 and FeN6 octahedra are connected by CN triple bonds, forming an open framework structure with large spatial voids, which can serve as a fast ion transport channel or storage site. The resulting FeHCF powder consists of fine, uniform cubic particles with edge lengths of approximately 100–200 nm (Fig. 1b and S1†). The nanoscale size provides a larger specific surface area, directly exposing more active sites, which is beneficial for fully activating the energy storage capability of the electrode material.28 The XRD diffraction pattern of the obtained sample is shown in Fig. 1c, with diffraction peaks matching the characteristic peaks of face-centered cubic FeHCF (PDF#73-0687). The diffraction peaks at 17.5°, 24.8°, 35.4°, and 39.8° correspond to the (200), (220), (400), and (420) crystal planes, respectively. The chemical structure of FeHCF was characterized by Fourier transform infrared (FTIR) spectroscopy, as shown in Fig. 1d. Peaks at 499 cm−1 and 607 cm−1 correspond to the out-of-plane bending vibration of Fe–O bonds and the in-plane bending vibration of Fe–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N, respectively, while the peak at 2090 cm−1 is attributed to the stretching vibration of the CN bond.29 In the Raman spectrum shown in Fig. 1e, the vibration peaks between 2000 and 2200 cm−1 represent the characteristic spectra of different valence states of Fe coordinated with cyanide, where the peaks at 2100 cm−1 and 2150 cm−1 can be assigned to CN–Fe2+ and CN–Fe3+, respectively. In the XPS spectrum shown in Fig. S2,† the presence of C, N, and Fe elements is clearly observed. A detailed XPS spectrum of the Fe 2p orbitals (Fig. 1f) shows fitting peaks at 708.4/721.4 eV and 712.1/725.54 eV corresponding to Fe3+ and fitting peaks at 710.0/723.6 eV and 714.1/727.3 eV corresponding to Fe2+.23 This indicates the coexistence of Fe2+ and Fe3+ in the synthesized FeHCF powder, consistent with previous reports.30
N, respectively, while the peak at 2090 cm−1 is attributed to the stretching vibration of the CN bond.29 In the Raman spectrum shown in Fig. 1e, the vibration peaks between 2000 and 2200 cm−1 represent the characteristic spectra of different valence states of Fe coordinated with cyanide, where the peaks at 2100 cm−1 and 2150 cm−1 can be assigned to CN–Fe2+ and CN–Fe3+, respectively. In the XPS spectrum shown in Fig. S2,† the presence of C, N, and Fe elements is clearly observed. A detailed XPS spectrum of the Fe 2p orbitals (Fig. 1f) shows fitting peaks at 708.4/721.4 eV and 712.1/725.54 eV corresponding to Fe3+ and fitting peaks at 710.0/723.6 eV and 714.1/727.3 eV corresponding to Fe2+.23 This indicates the coexistence of Fe2+ and Fe3+ in the synthesized FeHCF powder, consistent with previous reports.30
 | ||
| Fig. 1 Morphology, structure and chemical composition of FeHCF. (a) The cubic crystal structure and (b) scanning electron microscopy (SEM) image of FeHCF. (c) XRD patterns, (d) FTIR spectroscopy and (e) Raman spectra of FeHCF. (f) XPS spectra of Fe 2p for FeHCF. | ||
To investigate the copper-ion storage capability of FeHCF, it was used as the working electrode in a coin cell assembled with copper as the counter electrode and 1 M CuSO4 as the electrolyte. Fig. 2a shows the initial cyclic voltammetry (CV) curve of the FeHCF electrode at a scan rate of 0.2 mV s−1. In the first cycle, the first reduction peak appears at 0.39 V vs. SHE, which can be attributed to the initial insertion of Cu2+ into the FeHCF lattice. In subsequent cycles, a new reduction peak at 0.59 V vs. SHE emerges, likely due to the activation of C-coordinated Fe in the FeHCF cathode during higher potential scans.23 Unlike the zinc-ion storage process, the reduction peak at the lower potential for copper-ion storage does not disappear in the subsequent cycles but gradually stabilizes at around 0.44 V vs. SHE. The presence of dual active sites (dual reduction peaks) effectively ensures the copper-ion storage capability of FeHCF. The initial galvanostatic charge/discharge (GCD) curves also reflect this result, as shown in Fig. 2b. During the first discharge cycle, nearly all of the capacity is provided by the discharge plateau at 0.39 V, while in subsequent cycles, the discharge plateau at 0.59 V vs. SHE contributes nearly half of the capacity. The decay of the low-potential plateau capacity leads to a decrease in the specific capacity of the FeHCF electrode, but the activation of new active sites alleviates this issue and provides a higher operating potential, which is beneficial for improving the overall energy density of the battery. Furthermore, compared to the storage of other ions such as Zn2+, Al3+, and K+, FeHCF exhibits smaller voltage polarization (∼0.2 V) and a narrower charge–discharge window (0.6 V) when storing Cu2+, which better meets the requirements for voltage stability and reduced energy loss in energy storage batteries during use.23,31–33
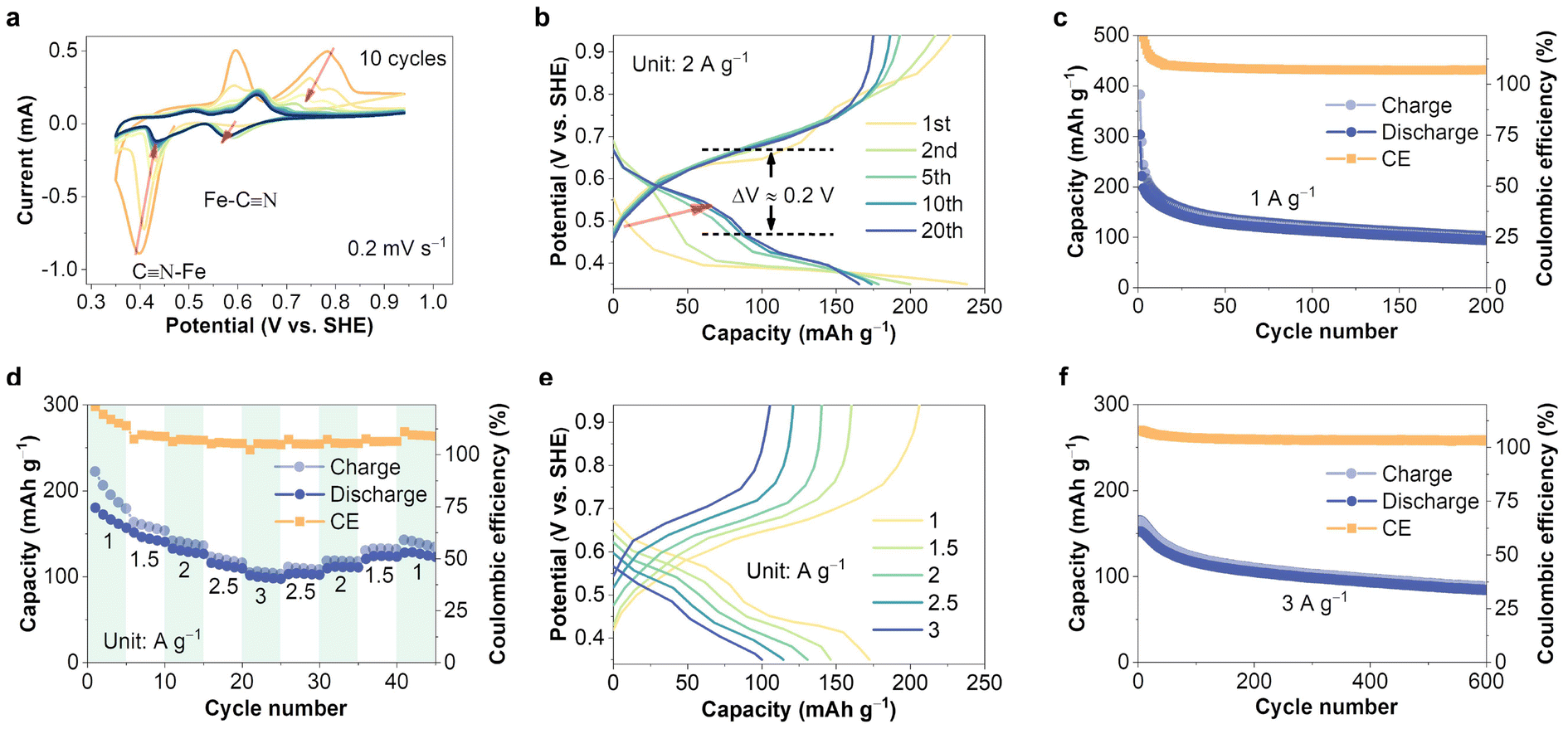 | ||
Fig. 2 Electrochemical copper-ion storage performance of FeHCF. (a) In situ CV curves recorded at 0.2 mV s−1. (b) Change of the GCD curves of FeHCF. (c) Long-term cycling of the FeHCF electrode (FeHCF![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :super P:PVDF = 7:2:1) in 1 M CuSO4 solution at a fixed rate of 1 A g−1. (d) Rate performance and (e) GCD curves of FeHCF at different current densities. (f) Long-term cycling of FeHCF at a fixed rate of 3 A g−1. :super P:PVDF = 7:2:1) in 1 M CuSO4 solution at a fixed rate of 1 A g−1. (d) Rate performance and (e) GCD curves of FeHCF at different current densities. (f) Long-term cycling of FeHCF at a fixed rate of 3 A g−1. | ||
In subsequent long-term cycling, the FeHCF electrode exhibits good stability, as evidenced by the electrochemical impedance spectroscopy (EIS) tests at different cycle numbers (Fig. S3†). In addition, FeHCF is detected in the electrode after 100 cycles, indicating that the active material does not degrade significantly (Fig. S4†). These results contribute to the maintenance of the electrode's electrochemical activity. The copper-ion storage electrochemical performance of the FeHCF electrode in the coin cell was evaluated (Fig. 2c and S5, 6†). As shown in Fig. 2c, at a current density of 1 A g−1, FeHCF (electrochemical tests were conducted under the conditions of FeHCF:super P:PVDF = 7:2:1 and 1 M CuSO4 electrolyte, unless otherwise specified) provides an initial capacity of 304 mA h g−1; after activation at higher potentials (3 cycles), the capacity is 190 mA h g−1. As mentioned earlier, the initial capacity decay may be due to the reduced activity of the low-potential active sites. At the same time, side reactions such as the partial decomposition of Prussian blue due to water attack can also lead to capacity degradation and the phenomenon where the charging capacity is greater than the discharging capacity.34,35 Even at higher current densities, the FeHCF electrode retains a capacity of 102 mA h g−1 at 3 A g−1, as shown in Fig. 2d. The GCD curves in Fig. 2e show voltage plateaus at approximately 0.44 V vs. SHE and 0.59 V vs. SHE, with sloping curves at different current densities, indicating that the excellent rate performance is due to the combined maintenance of both battery-type and capacitor-type capacities. At a high current density of 3 A g−1, the FeHCF electrode maintains stable cycling and retains a capacity of 84 mA h g−1 after 600 cycles (Fig. 2f). Compared to recent reports on FeHCF-based energy storage devices, these performance metrics are highly competitive, as shown in Fig. S7.†
The reason for the excellent rate performance of the FeHCF electrode was investigated through CV tests at different scan rates, as shown in Fig. 3a. As the scan rate increased from 0.2 to 1 mV s−1, the CV curves showed a systematic change, with only a slight shift in the redox peak positions. This demonstrates the fast redox reaction capability of the FeHCF electrode. The relationship between current (i) and scan rate (v) can be expressed by the following equation:36
| i = avb | (1) |
| I(V) = k1v + k2v1/2 | (2) |
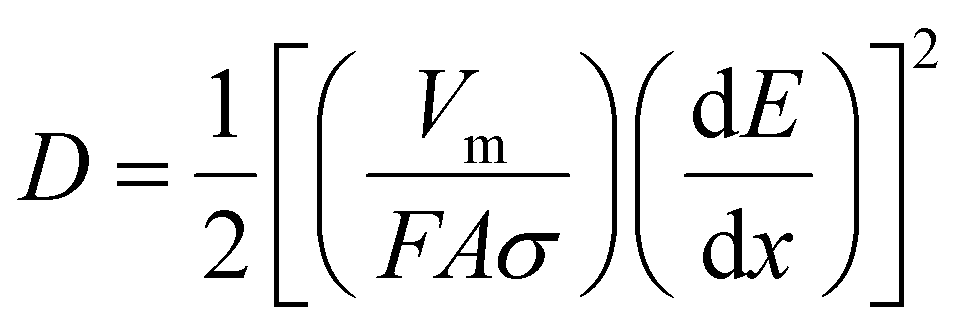 | (3) |
 | (4) |
 | ||
| Fig. 3 (a) CV curves of FeHCF at various scan rates. (b) Plots of log(i) versus log(v) of FeHCF. (c and d) The pseudocapacitive and diffusion-controlled charge storage contributions at different scan rates of FeHCF. (e) Diffusion coefficient as a function of potential derived from the impedance spectra. (f) Diffusion coefficient of copper-ion of FeHCF measured by the GITT. | ||
To further understand the storage mechanism of Cu2+ in FeHCF, in situ XRD, Raman, and XPS were used to analyze the state changes of the FeHCF electrode during charge–discharge cycles. Fig. 4a–e and S9† show the in situ XRD results for the Cu-FeHCF battery. From the full spectrum in Fig. S9,† it is clear that in the initial state, the FeHCF electrode exhibits a diffraction pattern consistent with that of the FeHCF powder described earlier, where the diffraction peaks at 43.2° and 50.4° correspond to the window tape used in the testing cell. To better observe the impact of Cu2+ insertion and extraction on the FeHCF structure, typical diffraction peaks of crystal planes from the earlier cycles (Fig. 4a) were selected for further analysis, as shown in Fig. 4b–e. During the first discharge process, when Cu2+ ions are initially inserted, no significant change in the characteristic peaks of FeHCF is observed. However, as Cu2+ ions are nearly fully inserted, the diffraction peaks corresponding to (200), (220), and (400) disappear, and a new peak appears at 28.5°. During the first charge (Cu2+ extraction), the 28.5° diffraction peak gradually disappears, and the originally disappeared (200), (220), and (400) diffraction peaks reappear, indicating the gradual recovery of the FeHCF structure. Notably, these diffraction peaks show a shift towards higher angles compared to the initial FeHCF electrode. This transition is attributed to the incomplete removal of the inserted Cu ions or their partial substitution for Fe ions, leading to a slight distortion of the FeHCF lattice structure. The emergence of a new high plateau in subsequent cycles may also be associated with changes in the FeHCF framework. This is likely the cause of the initial capacity decay. In subsequent cycles, the diffraction peaks of various crystal planes showed regular changes, indicating that the FeHCF lattice structure undergoes reversible changes during the insertion and extraction of Cu2+ ions.
 | ||
| Fig. 4 Evolution of FeHCF during the storage of copper ions. (a) Charge and discharge curves corresponding to in situ XRD of the FeHCF electrode. (b–e) The in situ XRD curve of FeHCF corresponding to (a). (f) Charge and discharge curves and (g) Raman spectra of FeHCF at the corresponding potentials in (f). | ||
The Raman spectroscopy characterization of the FeHCF electrode at different charge–discharge states is shown in Fig. 4f and g. The vibration peaks in the 2000–2200 cm−1 range are attributed to CN stretching vibrations affected by Fe2+/Fe3+. In the initial state, two vibration peaks appear at 2096 cm−1 and 2153 cm−1, which are consistent with those of the powder sample. After the first cycle, some residual copper ions caused a change in the relative intensity of the FeHCF vibration peaks, shifting them to higher wavenumbers at 2123 cm−1 and 2166 cm−1. Upon further discharge, the extensive insertion of copper ions further affected the CN vibration, causing the peaks at 2123 cm−1 and 2166 cm−1 to shift to 2178 cm−1, corresponding to the main peak. This change was reversed during the subsequent charging (Cu2+ extraction) process.
XPS shows the trend of elemental valence changes during the insertion and extraction of copper ions in FeHCF, as shown in Fig. 5. In the initial FeHCF electrode, no Cu element was detected. After the first cycle, residual Cu was detected in the FeHCF electrode, indicating that not all inserted copper ions were extracted, which is consistent with the in situ XRD and Raman results.40 At this point, the remaining copper ions include both Cu2+ and Cu+, suggesting a partial conversion of Cu2+ to Cu+ during this process. In the subsequent discharge process, the intensity of the characteristic Cu 2p peaks gradually increases, as shown in Fig. 5a and b, indicating the insertion of copper ions. The detailed spectrum of the Cu 2p orbitals shows a change in the ratio of Cu2+ to Cu+ (Fig. 5c and d). Initially, Cu+ accounted for 24.2% of the total copper ion content, but as discharge progressed and Cu2+ was inserted, this value increased to 46.3%. This further confirms the conversion of Cu2+ to Cu+ during the process, as the insertion of only Cu2+ would not explain this change. The discharge process is the reverse of charging, with Cu+ being re-oxidized to Cu2+ and extracted, as indicated by the restoration of Cu+ content to 23.9%, accompanied by a decrease in the intensity of the Cu 2p characteristic peaks in the full spectrum. Corresponding changes are also observed in the Fe 2p spectrum (Fig. 5e). After the first cycle, the content of Fe3+ decreased, while that of Fe2+ increased, which correlates with the phenomenon of some copper ions remaining in the electrode. During subsequent copper ion insertion, more Fe3+ is reduced to Fe2+, and the copper ion extraction process follows the reverse trend. The C 1s and N 1s characteristic peaks also exhibit systematic changes during the same cycling process, as shown in Fig. 5f and g.
 | ||
| Fig. 5 (a) Charge and discharge curves. (b) XPS survey spectrum of the FeHCF electrode at the corresponding stages in panel (a). Core-level XPS spectra of (c) Cu 2p, (e) Fe 2p, (f) C 1s and (g) N 1s collected at the corresponding stages in panel (a), and (d) the change in the Cu+ and Cu2+ content ratio corresponding to the stage in (c). | ||
While FeHCF demonstrates excellent copper-ion storage capability, using Cu as the anode results in a battery voltage of only around 0.25 V, limiting its practical applications. To increase the battery's operating voltage, it is essential to select an anode with a working potential much lower than the Cu2+/Cu0 potential. Previous studies have shown that the Zn metal anode, in alkaline electrolytes, undergoes a Zn/Zn(OH)42− redox reaction, with a potential as low as −1.22 V vs. SHE.41 At the same time, Zn metal electrodes offer a high theoretical capacity and a lower cost, making Zn an ideal candidate for increasing the battery's operating voltage.42–44 By designing a decoupled battery system, the overall operating voltage can be effectively enhanced. The reaction process can be expressed as follows:
| Cathode: FeHCF + xCu2+ + 2xe− ↔ CuxFeHCF | (5) |
| Anode: Zn + 4OH− ↔ Zn(OH)42− + 2e− | (6) |
The battery structure is shown in Fig. 6a, where the anode is a Zn metal electrode, and the cathode is the FeHCF electrode, with the anode side (alkaline electrolyte) and cathode side (acidic electrolyte) consisting of 1 M NaOH and 1 M CuSO4, respectively. The middle section uses 1 M Na2SO4 as a salt bridge to balance the charge, and cation exchange membranes (CEMs) and anion exchange membranes (AEMs) separate the different electrolytes. The CV results of the Zn//FeHCF decoupled battery are shown in Fig. 6b, where two sets of redox peaks confirm its redox activity within the 1.6 V–2.5 V voltage range. Correspondingly, the GCD curves at different current densities also exhibit discharge plateaus at the corresponding potentials, as shown in Fig. 6c, with an average discharge voltage of around 2 V. As the current density increases, the voltage polarization difference gradually increases, which is due to the combined effect of ion transport in both the electrode material and the ion exchange membranes. As shown in Fig. 6d and e, at current densities of 0.5, 1, 1.5, and 2 A g−1, the Zn//FeHCF decoupled battery exhibits capacities of 239, 180, 120 and 70 mA h g−1, respectively. In conclusion, the constructed Zn//FeHCF decoupled battery demonstrates excellent storage capacity and operating voltage, which is undoubtedly advantageous compared to other recently reported aqueous batteries using PBAs as cathode materials, as shown in Fig. 6f.18,23,34,45–50
 | ||
| Fig. 6 Performance of the Zn//FeHCF decoupled battery. (a) Structure and mechanism diagram of the Zn//FeHCF decoupled battery. (b) CV curves recorded at 0.2 mV s−1. (c) GCD curves at different current densities. (d) Rate performance of the Zn//FeHCF decoupled battery. (e) Electrochemical cycling stability at a fixed rate of 0.5 A g−1. (f) Comparison of the Zn//FeHCF decoupled battery with recently reported batteries using PBAs as cathode materials. | ||
3. Conclusions
In conclusion, we have developed an aqueous copper-ion storage device based on FeHCF nanocrystal cathodes. FeHCF proves to be an efficient copper-ion storage material, delivering a high capacity of 190 mA h g−1 within a narrow voltage range of 0.6 V, with a stable discharge plateau at 0.59 V vs. SHE and a minimal polarization voltage of 0.2 V. The FeHCF cathode shows excellent rate performance, maintaining a substantial capacity of 102 mA h g−1 even at high current densities, attributed to the fast copper-ion transport. To further enhance the operating voltage, the Cu2+/Cu0 anode reaction was replaced with the lower potential Zn/Zn(OH)42− reaction. This modification successfully extended the working voltage of the FeHCF-based battery, which operates within a voltage range of 1.6–2.5 V. These findings highlight the potential of FeHCF-based aqueous batteries for high-performance and safe energy storage applications.Author contributions
Jinshu Zhang: writing – review & editing, writing – original draft, investigation, formal analysis, and data curation. Lexian Liu: investigation, formal analysis, and data curation. Yuao Wang: investigation and formal analysis. Yantuo Li: methodology and data curation. Yang Yang: data curation. Mingyi Ning: data curation. Jianxue Wu: data curation. Bingjie Ma: data curation. Wei Liu: writing – review & editing, supervision, investigation, and conceptualization.Data availability
The data have been included in the paper or the ESI.†Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
The authors acknowledge the financial support from the National Key Research and Development Program of China (2022YFB3807200), the National Natural Science Foundations of China (22371041), the Open Fund of Key Laboratory for Intelligent Nano Materials and Devices of the Ministry of Education NJ2023002 (INMD-2023M08) and the SEU Innovation Capability Enhancement Plan for Doctoral Students (CXJH_SEU 24136). The authors thank the Center for Fundamental and Interdisciplinary Sciences of Southeast University for the support of in situ XRD and Raman measurements.References
- M. A. Giovanniello, A. N. Cybulsky, T. Schittekatte and D. S. Mallapragada, Nat. Energy, 2024, 9, 197–207 CrossRef CAS.
- R. Fan, C. Liu, Z. Li, H. Huang, J. Feng, Z. Li and Z. Zou, Nat. Sustain., 2024, 7, 158–167 CrossRef.
- J. Janek and W. G. Zeier, Nat. Energy, 2023, 8, 230–240 CrossRef.
- H. Wan, J. Xu and C. Wang, Nat. Rev. Chem., 2024, 8, 30–44 CrossRef CAS PubMed.
- J. Duan, X. Tang, H. Dai, Y. Yang, W. Wu, X. Wei and Y. Huang, Electrochem. Energy Rev., 2020, 3, 1–42 CrossRef CAS.
- G. Liang, F. Mo, X. Ji and C. Zhi, Nat. Rev. Mater., 2021, 6, 109–123 CrossRef CAS.
- L. Chang, J. Li, Q. Sun, X. Liu, X. Lu and H. Cheng, Small, 2024, 2408124 CrossRef CAS PubMed.
- D. Chao, W. Zhou, F. Xie, C. Ye, H. Li, M. Jaroniec and S.-Z. Qiao, Sci. Adv., 2020, 6, eaba4098 CrossRef CAS PubMed.
- M. Xia, J. Zhou and B. Lu, Adv. Energy Mater., 2024, 2404032 CrossRef.
- Y. Wang, T. Wang, P. Cui, G. Mao, K. Ye, F. Hu, M. Chen, D. Cao and K. Zhu, Adv. Funct. Mater., 2025, 2421363 CrossRef.
- H. Wu, J. Hao, Y. Jiang, Y. Jiao, J. Liu, X. Xu, K. Davey, C. Wang and S.-Z. Qiao, Nat. Commun., 2024, 15, 575 CrossRef CAS PubMed.
- K. Du, Y. Liu, Y. Zhao, H. Li, H. Liu, C. Sun, M. Han, T. Ma and Y. Hu, Adv. Mater., 2024, 36, 2404172 CrossRef CAS PubMed.
- T.-U. Wi, C. Park, S. Ko, T. Kim, A. Choi, V. Muralidharan, M. Choi and H.-W. Lee, Nano Lett., 2024, 24, 7783–7791 CrossRef CAS PubMed.
- V. D. Neff, J. Electrochem. Soc., 1978, 125, 886 CrossRef CAS.
- B. Singh, Y. Arya, G. K. Lahiri and A. Indra, Coord. Chem. Rev., 2025, 523, 216288 CrossRef CAS.
- C. Xu, Z. Yang, X. Zhang, M. Xia, H. Yan, J. Li, H. Yu, L. Zhang and J. Shu, Nano-Micro Lett., 2021, 13, 166 CrossRef CAS PubMed.
- X. Zhang, J. Zhang, H. Yu, T. L. Madanu, M. Xia, P. Xing, A. Vlad, J. Shu and B.-L. Su, Angew. Chem., Int. Ed., 2024, e202414479 Search PubMed.
- L. Ma, S. Chen, C. Long, X. Li, Y. Zhao, Z. Liu, Z. Huang, B. Dong, J. A. Zapien and C. Zhi, Adv. Energy Mater., 2019, 9, 1902446 CrossRef CAS.
- W. Chang, J. Peng, W. Mao, Q. Wang, Y. Zhu and N. Peng, Chem. Eng. J., 2024, 500, 157204 CrossRef CAS.
- L. Wang, N. Liu, Q. Li, X. Wang, J. Liu, Y. Xu, Z. Luo, N. Zhang and F. Li, Angew. Chem., Int. Ed., 2024, e202416392 Search PubMed.
- X. Bai, L. Zhang, D. Zhang, K. Li, T. Li, Y. Ren, H. He, B. Xie, G. Sun, J. Xu and X. Yin, Chem. Eng. J., 2024, 497, 154902 CrossRef CAS.
- J. Guo, F. Feng, S. Zhao, Z. Shi, R. Wang, M. Yang, F. Chen, S. Chen, Z.-F. Ma and T. Liu, Carbon Energy, 2023, 5, e314 CrossRef CAS.
- Q. Yang, F. Mo, Z. Liu, L. Ma, X. Li, D. Fang, S. Chen, S. Zhang and C. Zhi, Adv. Mater., 2019, 31, 1901521 CrossRef PubMed.
- Z. Liu, G. Pulletikurthi and F. Endres, ACS Appl. Mater. Interfaces, 2016, 8, 12158–12164 CrossRef CAS PubMed.
- X. Wu, A. Markir, L. Ma, Y. Xu, H. Jiang, D. P. Leonard, W. Shin, T. Wu, J. Lu and X. Ji, Angew. Chem., Int. Ed., 2019, 58, 12640–12645 CrossRef CAS PubMed.
- H. Sun, M. Li, J. Zhu, J. Ni and L. Li, ACS Nano, 2024, 18, 21472–21479 CrossRef CAS PubMed.
- C. Dai, L. Hu, H. Chen, X. Jin, Y. Han, Y. Wang, X. Li, X. Zhang, L. Song, M. Xu, H. Cheng, Y. Zhao, Z. Zhang, F. Liu and L. Qu, Nat. Commun., 2022, 13, 1863 CrossRef CAS PubMed.
- J. Lin, R. Chenna Krishna Reddy, C. Zeng, X. Lin, A. Zeb and C.-Y. Su, Coord. Chem. Rev., 2021, 446, 214118 CrossRef CAS.
- P. K. Pathak, K. P. Agrim, C. Park, H. Ahn and R. R. Salunkhe, ACS Appl. Energy Mater., 2024, 7, 11352–11360 CrossRef CAS.
- L.-P. Wang, P.-F. Wang, T.-S. Wang, Y.-X. Yin, Y.-G. Guo and C.-R. Wang, J. Power Sources, 2017, 355, 18–22 CrossRef.
- D. Du, Z. Zhu, K.-Y. Chan, F. Li and J. Chen, Chem. Soc. Rev., 2022, 51, 1846–1860 RSC.
- S. Kumar, T. Salim, V. Verma, W. Manalastas and M. Srinivasan, Chem. Eng. J., 2022, 435, 134742 CrossRef CAS.
- M. Wang, H. Wang, H. Zhang and X. Li, J. Energy Chem., 2020, 48, 14–20 CrossRef.
- L. Jiang, Y. Lu, C. Zhao, L. Liu, J. Zhang, Q. Zhang, X. Shen, J. Zhao, X. Yu, H. Li, X. Huang, L. Chen and Y.-S. Hu, Nat. Energy, 2019, 4, 495–503 CrossRef CAS.
- W. Shu, J. Li, G. Zhang, J. Meng, X. Wang and L. Mai, Nano-Micro Lett., 2024, 16, 128 CrossRef CAS PubMed.
- T. Shang, J. Zhang, Y. Li, Y. Yang, J. Wu, M. Ning and W. Liu, Batteries Supercaps, 2024, 7, e202300613 CrossRef CAS.
- Y. Wang, B. Wang, J. Zhang, D. Chao, J. Ni and L. Li, Carbon Energy, 2023, 5, e261 CrossRef CAS.
- J. Zhang, Y. Wang, M. Yu, J. Ni and L. Li, ACS Energy Lett., 2022, 7, 1835–1841 CrossRef CAS.
- Y. Zhu, Y. Xu, Y. Liu, C. Luo and C. Wang, Nanoscale, 2013, 5, 780–787 RSC.
- Z. Zhang, Y. Li, G. Zhao, L. Zhu, Y. Sun, F. Besenbacher and M. Yu, ACS Appl. Mater. Interfaces, 2021, 13, 40451–40459 CrossRef CAS PubMed.
- X. Zhang, B. Zhang, J.-L. Yang, J. Wu, H. Jiang, F. Du and H. J. Fan, Adv. Mater., 2024, 36, 2307298 CrossRef CAS PubMed.
- Y. Huang, Y. Wang, X. Peng, T. Lin, X. Huang, N. S. Alghamdi, M. Rana, P. Chen, C. Zhang, A. K. Whittaker, L. Wang and B. Luo, Mater. Futures, 2024, 3, 035102 Search PubMed.
- C. Fan, W. Meng and J. Ye, J. Energy Chem., 2024, 93, 79–110 CrossRef CAS.
- Y. Wang, T. Wang, Y. Mao, Z. Li, H. Yu, M. Su, K. Ye, D. Cao and K. Zhu, Adv. Energy Mater., 2024, 14, 2400353 CrossRef CAS.
- Z.-L. Xie, Y. Zhu, J.-Y. Du, D.-Y. Yang, N. Zhang, Q.-Q. Sun, G. Huang and X.-B. Zhang, Angew. Chem., Int. Ed., 2024, 63, e202400916 Search PubMed.
- X. Wu, Y. Xu, H. Jiang, Z. Wei, J. J. Hong, A. S. Hernandez, F. Du and X. Ji, ACS Appl. Energy Mater., 2018, 1, 3077–3083 Search PubMed.
- X. Wu, J. J. Hong, W. Shin, L. Ma, T. Liu, X. Bi, Y. Yuan, Y. Qi, T. W. Surta, W. Huang, J. Neuefeind, T. Wu, P. A. Greaney, J. Lu and X. Ji, Nat. Energy, 2019, 4, 123–130 Search PubMed.
- W. Deng, Z. Li, Y. Ye, Z. Zhou, Y. Li, M. Zhang, X. Yuan, J. Hu, W. Zhao, Z. Huang, C. Li, H. Chen, J. Zheng and R. Li, Adv. Energy Mater., 2021, 11, 2003639 CrossRef CAS.
- Z. Wang, Y. Xu, J. Peng, M. Ou, P. Wei, C. Fang, Q. Li, J. Huang, J. Han and Y. Huang, Small, 2021, 17, 2101650 CrossRef CAS PubMed.
- Y. Zeng, X. F. Lu, S. L. Zhang, D. Luan, S. Li and X. W. Lou, Angew. Chem., Int. Ed., 2021, 60, 22189–22194 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr05203j |
| This journal is © The Royal Society of Chemistry 2025 |