DOI:
10.1039/D4NR03492A
(Paper)
Nanoscale, 2025,
17, 965-971
Exfoliated MoS2 nanosheets immobilized in porous microbeads as recoverable photocatalysts†
Received
26th August 2024
, Accepted 14th November 2024
First published on 14th November 2024
Abstract
Molybdenum disulfide (MoS2) is a highly effective visible light photocatalyst when used as well-exfoliated 2D nanosheets. The ability to make effective use of these properties is significantly compromised by the challenge of preventing nanosheet aggregation or restacking in fluid suspensions. We report a strategy for immobilizing chemically exfoliated MoS2 as single- and few-layer nanosheets in porous crosslinked polymers prepared as microbeads. The polymeric support prevents aggregation of the nanosheets while allowing access to the nanomaterial for model organic compounds present in the surrounding fluid. Exposure to visible light results in high degradation yields (>99%) of these organic species in aqueous media, and the MoS2 nanosheets maintain their photocatalytic efficacy through multiple cycles of use. The recoverability of the porous beads and the persistent photocatalytic activity of the polymer-supported MoS2 offer the potential of realizing an effective, environmentally sustainable platform for photocatalytic degradation of dissolved solutes.
Introduction
Semiconducting nanomaterials have gained widespread utilization for the photocatalytic degradation of contaminants in aqueous fluids, including dyes,1–3 antibiotics,4,5 and bacteria.6,7 Typically, these semiconducting photocatalysts harness light energy to generate electron–hole pairs, which subsequently lead to the formation of reactive radicals that drive catalytic processes.8–10 The potential to use natural light as an energy source is compelling in terms of cost-effectiveness and environmental sustainability. Titanium dioxide (TiO2) has been extensively explored for its photocatalytic potential,11–13 but its wide bandgap (∼3.2 eV) limits its photocatalytic efficiency given the limited UV component of sunlight (∼4%).14 Two-dimensional molybdenum disulfide (MoS2) has attracted attention as a potential alternative due to its narrow direct bandgap of ∼1.9 eV, which is well-suited for visible light operation.15,16 In contrast to its optically inactive bulk form, MoS2 in the form of single-layer nanosheets exhibits a high density of exposed active edge sites, which enhances its photocatalytic capabilities.17,18 In addition, its high specific surface area and fast charge carrier dynamics contribute to high catalytic efficiency, thereby enhancing its suitability as a novel photocatalyst with broadened application potential.19–22 To date, however, it remains difficult to use MoS2 readily as a photocatalyst. This is primarily due to its limited stability when dispersed in aqueous or organic media, caused by strong attractive van der Waals interactions between nanosheets.23 Surface chemical modification and the use of select surface active agents can produce relatively stable dispersions under controlled conditions,24–26 but these systems are generally not robust against the presence of other species, and are therefore not amenable for general purpose photocatalysis.
Immobilization of MoS2 nanosheets on suitable supports presents an alternative approach for presenting single- and few-layer MoS2 active sites to advance the chemical transformations of interest using photocatalysis. Nanocomposites of MoS2 nanosheets have been successfully prepared in different contexts, including using polymeric,27,28 liquid crystal,29 and hydrogel matrices.30,31 For the purpose of advancing photocatalysis in solution, we desire a system in which MoS2 nanosheets can be immobilized in discrete yet porous solid materials. The porous structure would facilitate access of species in solution to the catalyst particles, allowing catalysis to occur effectively.32,33 Moreover, the ability to prepare porous solid materials on colloidal length scales, and potentially with magnetic inclusions, offers the possibility of facile catalyst recovery by centrifugation, filtration, or the use of magnetic methods.34 Taking advantage of these characteristics, various studies have described the immobilization of UV photocatalysts, such as TiO2,35,36 copper oxide (CuO),37,38 zinc oxide (ZnO),39,40 and graphitic carbon nitride (g-CN),41,42 on porous supports. Although there are several previous studies related to MoS2-based photocatalysts (Table S1†),43–51 there is a notable scarcity of studies directly addressing the immobilization of MoS2 nanosheets and the visible light photocatalysis thereby enabled.
Here, we demonstrate a successful immobilization strategy for MoS2 nanosheets. We leverage the concept of microporous supports that effectively mitigate the agglomeration of MoS2 through physical anchoring. We conducted a comprehensive analysis of the dispersion state of MoS2 within porous beads through electron microscopy observations and Raman analysis. We demonstrate that the porous bead-supported MoS2 catalysts exhibit excellent activity for visible light photodegradation of model compounds, while allowing readily retrieval and reuse.
Results and discussion
Design of the MoS2-immobilized porous bead photocatalyst
MoS2 has a crystal structure consisting of weakly coupled layers of S–Mo–S, where a Mo atom layer is sandwiched between two layers of S atoms (Fig. S1a†). These layers are held together by weak van der Waals interactions that drive stacking.52 To exploit MoS2 as a photocatalyst, chemically active edge sites must be exposed by exfoliation into nanosheets. Maintaining high catalytic activity requires that the nanosheet morphology is preserved, which necessitates suppressing random aggregation and/or restacking of the nanosheets. We propose a microbead photocatalyst system in which MoS2 nanosheets are immobilized in alginate-based porous microbeads, as illustrated in Fig. 1. First, MoS2 nanosheets were obtained by chemical exfoliation using n-butyllithium using previously reported methods.53 Briefly, bulk MoS2 powder was subjected to a reaction with n-butyllithium in hexane, yielding Li-intercalated MoS2. Subsequently, this compound was hydrolyzed in water, resulting in the formation of a stable suspension comprising discrete single- and few-layer nanosheets. The obtained MoS2 nanosheets were dispersed in sodium alginate solution through ultrasonic dispersion to prepare a precursor solution. The precursor solution was extruded through a syringe needle into a CaCl2 solution, leading to the formation of MoS2-immobilized porous beads (MPBs) (Fig. 1a). When this precursor solution comes into contact with the CaCl2 medium, ionic crosslinking between the G units of alginate and Ca2+ ions is initiated at the surface and gradually progresses toward the center of the bead, eventually solidifying the entire construct. These MPBs are used for photodegradation of a model organic compound, MO, under visible light, as schematically illustrated in Fig. 1b.
 |
| | Fig. 1 Schematic illustration for (a) the fabrication of MoS2-immobilized porous microbeads (MPBs) and (b) the visible-light-induced photocatalytic performance of MPBs. | |
Characterization of MoS2 nanosheets inside microbeads
We first investigated the morphology of the chemically exfoliated MoS2 nanosheets. The MoS2 nanosheets exhibited an average width of ∼158 nm and an average thickness of ∼1.2 nm (Fig. 2a and S2†); the average thickness is consistent with a combination of single- (0.65 nm thickness) and few-layer nanosheets. Chemical exfoliation leads to a mixture of metallic (1T) and semiconducting (2H) nanosheets.54 We used the optoelectronic properties of the semiconducting fraction for diagnostic purposes. Photoluminescence (PL) spectra confirm the presence of the 2H semiconducting phase contained in these chemically exfoliated MoS2 nanosheets. As shown in Fig. 2b, the MoS2 aqueous dispersion showed a sharp peak at 1.9 eV, which is consistent with the bandgap energy of single-layer MoS2. The PL emission was maintained when dispersed in alginate solution. Using Raman spectroscopy, we further confirmed that the MoS2 exists as nanosheets within the Ca2+–alginate composite (Fig. 2c) by analyzing the wavenumber difference (Δk) between the E12g and A1g vibrational modes.55 MoS2 nanosheets dispersed in the polymer beads had a Δk value of 22.3 ± 0.3 cm−1, which is notably smaller than the value of 25.9 ± 0.6 cm−1 observed for bulk MoS2 powder (Fig. 2c) and consistent with the preservation of exfoliated nanosheet structures in the polymer beads.
 |
| | Fig. 2 (a) TEM image of chemically exfoliated MoS2 nanosheets. (b) Photoluminescence spectra of MoS2 dispersion. (c) Raman spectra of the alginate composite, bulk MoS2 and MPBs. (d) Photographs of (i) alginate porous beads (PBs) and MPBs with varying concentrations of incorporated MoS2: (ii) 0.1, (iii) 0.2, and (iv) 0.5 mg g−1, respectively (scale bar = 10 mm). (e) Cross-section SEM image of the MPBs. (f) Magnified internal morphology of the MPBs and EDX mapping of the MoS2 nanosheets inside the microbeads (scale bar = 100 μm). | |
MPBs created via a conventional extrusion dripping method displayed a spherical morphology (Fig. 2d). Employing a syringe pump facilitated the production of uniform beads with an average diameter of approximately 2.7 mm (Fig. S3†). The size of the beads was adjustable based on the inner diameter of the needle (Fig. S4†). We prepared MPBs using different concentrations of MoS2, denoted as MPBn, where n represents the MoS2 concentration (mg g−1). The concentration of added MoS2 affected the color of the beads, from dark brown to black, but had no noticeable effect on their size. However, it is noteworthy that the Δk value for beads with the largest MoS2 concentration, MPB0.5, exhibited a minor increase compared to that of MPB0.2 (Fig. S5†). This suggests that the propensity for nanosheet restacking increases at elevated mass loadings. Fig. 2e shows the internal morphology of the MPBs, whose structure is characterized by pores formed by a dense alginate–calcium layer. The surface of the alginate–Ca layer without MoS2 is smooth, whereas the surface containing MoS2 is relatively rough (Fig. S6a–c†). For MPB0.1 and MPB0.2, MoS2 nanosheets are well dispersed within the alginate–Ca layer. Elemental dispersive X-ray spectroscopy (EDS) mapping indicates visually that the distribution of MoS2 nanosheets within MPB0.2 is spatially uniform (Fig. 2f). However, in MPB0.5, some nanosheets remain well anchored, while others exhibit restacking and aggregation (Fig. S6d†). These observations are consistent with the Raman spectroscopy data and are expected to have a significant impact on photocatalytic performance.
Photocatalytic performance of MPBs under visible light
We assessed the efficacy of MPBs as a “green” photocatalytic system for the photodegradation of molecular species. Methyl orange (MO) was chosen as a model compound, given its well-known role as an environmental pollutant.56 The photocatalytic degradation of MO was initiated by the introduction of MPBs into an aqueous MO solution, followed by stirring under visible light generated by a custom-made cylindrical photoreactor enveloped by an LED strip (Fig. S7†). To avoid the sedimentation of MPBs, which may reduce the effective surface area for light absorption, mild stirring is required. MO exhibits distinctive colors depending on the pH of its environment: red under acidic conditions and yellow in neutral and basic environments. The investigation of MO photodegradation involved UV-visible spectroscopy, with absorbance intensity monitored at a wavelength of 500 nm in the acidic solution and 464 nm under neutral and basic conditions, respectively (Fig. S8†). The adsorption–desorption equilibrium of MO was reached in 30 minutes in a dark environment (Fig. S9a†). The amount of MO adsorbed onto the beads decreased with increasing pH. This is attributed to the fact that MO exhibits a negative charge above pH 7,57 rendering it less amenable to adsorption by the alginate beads, which also possess a negative charge. Photolysis of MO was negligible when subjected to visible light in the absence of the beads (Fig. S9b†). In the presence of the beads, as shown in Fig. 3a–c, there was no noticeable photodegradation of MO under neutral and basic conditions, whereas under acidic conditions, a significant decrease in absorbance was observed as a function of irradiation time. This means that an active photocatalytic reaction occurs, which is further evidenced by the loss of color of the MO solution.
 |
| | Fig. 3 UV–vis spectra of methyl orange (MO) during photodegradation with the MPB catalyst at (a) pH 3, (b) pH 7, and (c) pH 10, respectively. The insets show the photographs of MO solution before (left) and after (right) photodegradation. Plots of C/C0versus time depending on the reaction conditions: (d) pH and (e) MoS2 concentration. Ci is the initial concentration of MO and C is the concentration of MO at reaction time t. The inset of (e) shows the photograph of MO solution after the photodegradation reaction (from left to right: PB, MPB0.1, MPB0.2, and MPB0.5). (f) Plots of ln(C0/C) versus time for the photodegradation of MO using different catalysts at pH 3. | |
This acidic condition-favorable photodegradation of MO can be understood through the photocatalytic mechanism of MoS2.46 Semiconducting MoS2 nanosheets generate electron–hole pairs when exposed to visible light (1). These photogenerated electrons participate in reactions with dissolved oxygen to form superoxide radicals (˙O2−) (2). Subsequently, ˙O2− can further react with protons (H+) to generate hydrogen peroxide (H2O2) species (3). The ensuing decomposition of H2O2 results in the formation of hydroxyl radicals (˙OH) (4). These hydroxyl radicals play a pivotal role in the degradation of organic species (5), with ˙O2− also contributing, though to a lesser extent.48,58,59
| |  | (1) |
| | 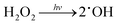 | (4) |
| | | MO dye + ˙OH → degraded products | (5) |
Within this sequence of processes, the presence of H+ serves to facilitate the generation of hydroxyl radicals. The observed pH dependence (Fig. 3d) in the photodegradation of MO by the bead-supported MoS2 nanosheets is consistent with this proton facilitated mechanism.
The photocatalytic efficiency of MPBs is affected by several parameters, including bead size, the degree of crosslinking, and the concentration of MoS2 in the beads. Larger bead sizes and lower degrees of crosslinking tend to yield slightly reduced degradation rates under visible light of 92.7% and 82.3% after 4 h, respectively (Fig. S10†). In general, the smaller the bead, the larger the specific surface area, leading to better photocatalytic efficiency. With respect to the degree of crosslinking, we speculate that higher levels of crosslinking result in better preservation of nanosheet dispersion during bead formation. Independent of photocatalytic degradation, the incorporation of MoS2 nanosheets increases the adsorption capacity of the beads for MO, as revealed by experiments conducted in the dark (Fig. 3e). The quantity of MO adsorbed by the beads (represented by the initial decline in the MO concentration in the dark) increased steadily with the concentration of the MoS2 suspension used to prepare the beads. The effectiveness of the photodegradation increased as the concentration of MoS2 used to prepare the beads increased from 0.1 to 0.2 mg g−1. At 0.5 mg g−1, however, the rate of photodegradation decreased. Although the difference in absolute values is minor, the observed increase in Δk values at this higher concentration is likely attributed to the restacking of the nanosheets, as corroborated by the Raman spectroscopy data (Fig. S5†). We presume that the photodegradation efficacy is a function of the quantity of semiconducting vs. metallic MoS2 nanosheets present in the system. The fraction of semiconducting nanosheets is unknown, but their prevalence can be increased through thermal annealing,60 which we conducted here to investigate its effect on photodegradation. As seen from the UV–Vis data (Fig. S11a†), this enables the characteristic excitonic absorption peaks at 605 and 648 nm to be readily distinguished from the background absorption. However, the increased tendency of semiconducting sheets to restack limits the use of higher concentrations in bead preparation (Fig. S11b†). Regarding the reaction kinetics, the MPB catalyst exhibits behavior consistent with pseudo-first-order kinetics (Fig. 3f). The degradation rate constant (k) can be deduced from the equation ln(C0/C) = kt, where C0 represents the initial concentration of MO after adsorption, C signifies the concentration of MO, and t denotes the reaction time. The calculated degradation rate constants for MO in conjunction with MPBs were determined to be 0.21 h−1 for MPB0.1, 0.80 h−1 for MPB0.2, and 0.28 h−1 for MPB0.5, respectively.
Recoverable and reusable microbead photocatalyst
We assessed the reusability of the MPB catalyst. Following its initial use, the beads were collected by centrifugation. After a gentle rinse with DI water, the catalyst was prepared for the subsequent reaction. For each reaction cycle, the establishment of an adsorption–desorption equilibrium for MO was initiated in the dark for 30 minutes before the operation of the visible light photoreactor. The adsorption amount, which was close to 60% in the first cycle, decreased to about 25% in the next cycle, probably due to residual MO degraded products adsorbed within the beads from the last batch (Fig. 4a). Nevertheless, the degradation efficiency remained consistently high at 99% even after four cycles. In the context of the C/Ci plot, a relatively modest slope was evident within the first hour following visible light irradiation during the first cycle, primarily attributable to the preferential degradation of MO adsorbed within the beads (Fig. 4b). Thereafter, similar slopes were consistently observed throughout the subsequent cycles, reflecting the kinetic trends observed in all four reuse cases. The MPB catalyst demonstrated consistent performance without significant discernible physical deterioration (Fig. 4c). This excellent performance is comparable to existing MoS2-based catalysts and is especially noteworthy in terms of the economical catalyst amount, mild light intensity, and easy reuse (Table S1†). These findings imply that our MPB catalysts have great potential as high-performance green catalyst platforms.
 |
| | Fig. 4 Recycling properties of the photocatalytic degradation of MO over the MPB catalyst. (a) Adsorption and photodegradation efficiency for each cycle. The remaining MO is calculated as C/Ci × 100 (%), where Ci is the initial MO concentration and C is the concentration of MO after adsorption and photodegradation, respectively. (b) Plots of C/Civersus time for each cycle. (c) TGA curves of MPBs after each cycle. | |
Experimental
Materials
Sodium alginate (SA, from brown algae), calcium chloride (CaCl2, anhydrous, >97%), molybdenum(IV) sulfide (MoS2, powder, 99%, particle size <2 μm), n-butyllithium solution (n-BuLi, 2.5 M in hexane), and hexane (anhydrous, 95%) were purchased from Sigma-Aldrich (USA). Methyl orange (MO), sodium hydroxide solution (1N), and hydrochloric acid solution (1N) were purchased from Fisher Chemical (USA). Deionized double-distilled (DI) water was used in all experiments.
Preparation of MoS2 nanosheets
2D MoS2 nanosheets were prepared via a typical chemical exfoliation procedure. Initially, bulk MoS2 powder was introduced into an n-BuLi solution at a molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 in an N2-filled glovebox. This mixture was allowed to stir for 48 hours under an N2 atmosphere. Subsequently, it underwent several cycles of washing with hexane and centrifugation at 6000 rpm for 30 minutes to separate excess n-BuLi from the Li-intercalated MoS2. This washing process was repeated three times. Then, the residual hexane was evaporated under vacuum. Degassed water was then added to the resulting pellet. Subsequently, the solution was bath sonicated for 90 minutes to promote the exfoliation of the Li-intercalated 2D nanosheets. Following sonication, the unexfoliated material was isolated through centrifugation at 2000 rpm for 10 minutes, a step that was repeated twice. The dark supernatant obtained was subsequently transferred into dialysis bags with a molecular weight cutoff of 20 kDa (Sigma) for dialysis in DI water over a period of 3 days. The concentration of the final MoS2 suspension was determined by weighing the restacked nanosheets on a glass coverslip after the water had evaporated.
:3 in an N2-filled glovebox. This mixture was allowed to stir for 48 hours under an N2 atmosphere. Subsequently, it underwent several cycles of washing with hexane and centrifugation at 6000 rpm for 30 minutes to separate excess n-BuLi from the Li-intercalated MoS2. This washing process was repeated three times. Then, the residual hexane was evaporated under vacuum. Degassed water was then added to the resulting pellet. Subsequently, the solution was bath sonicated for 90 minutes to promote the exfoliation of the Li-intercalated 2D nanosheets. Following sonication, the unexfoliated material was isolated through centrifugation at 2000 rpm for 10 minutes, a step that was repeated twice. The dark supernatant obtained was subsequently transferred into dialysis bags with a molecular weight cutoff of 20 kDa (Sigma) for dialysis in DI water over a period of 3 days. The concentration of the final MoS2 suspension was determined by weighing the restacked nanosheets on a glass coverslip after the water had evaporated.
Fabrication of MPBs
We fabricated MPBs using an extrusion dripping method. The initial step involved the preparation of a precursor solution, in which MoS2 nanosheets were dispersed in SA aqueous solution. To achieve this, SA was completely dissolved in DI water. Subsequently, the MoS2 nanosheet solution was introduced into the prepared SA solution, followed by probe sonication (Q700, QSonica, USA) at 60% amplitude to ensure homogeneity. The concentration of SA was set to 5 wt%, while the concentration of MoS2 varied at 0.1, 0.2, and 0.5 mg g−1 within the entire solution, denoted as MPB0.1, MPB0.2, and MPB0.5, respectively. The resulting precursor solutions were extruded into CaCl2 solution (5 wt%) dropwise using a syringe equipped with a needle (25 G) under mild stirring. For uniform bead fabrication, a syringe pump (KDS 210, KD Scientific, USA) was employed, and the flow rate was set to 0.2 ml min−1. The Ca2+ ion crosslinking proceeded for 12 hours at room temperature. Finally, MPBs were successfully obtained. The MPBs were rinsed using DI water to wash the excess Ca2+ ions.
Characterization of MPBs
The morphology of the MoS2 nanosheets was observed with a transmission electron microscope (TEM, JEM-F200 F2, JEOL, USA) operating at 200 kV. The thickness of the MoS2 nanosheets was measured from an atomic force microscopy (AFM, Dimension Icon, Bruker, USA) image. PL spectra were collected using an Edinburgh Instruments FLS1000 fluorometer with an excitation light of 532 nm. Raman spectra were obtained using a Horiba LabRam Evolution confocal Raman microscope with 633 nm light excitation and 300 s exposure under a 100× objective. The average diameter of MPBs was determined using ImageJ software. The morphology of freeze-dried MPBs and the distribution of MoS2 nanosheets were analyzed by environmental scanning electron microscopy (SEM, FEI Quanta 600, FEI, USA) coupled with energy-dispersive X-ray spectroscopy (EDX). Thermogravimetric analysis (TGA) was carried out using an SDT Q650 (TA Instruments, USA) at a scan rate of 10 °C min−1.
Photocatalytic performance test
To confirm the photocatalytic performance of the MPB catalyst, MO was employed as a representative reaction model. In a typical experimental setup, 10 mL of MO aqueous solution was prepared in a glass vial at a concentration of 20 ppm (20 mg L−1). Subsequently, the pH of the solution was adjusted to 3, 7, and 10, respectively, utilizing HCl and NaOH solutions. MPBs were then introduced into each of these solutions. As a visible light source, a 24 W m−1 LED strip was utilized. The vial containing the MPB and MO solutions was placed inside a custom-made cylindrical reactor enveloped by a 5-meter LED strip (Fig. S7†). To mitigate temperature increases resulting from LED illumination, a USB-powered fan was positioned above the reactor, and an additional fan was placed adjacent to the reactor to facilitate continuous cooling. Before the light irradiation, the vial was shielded with foil and stirred for 30 minutes in the dark to establish an adsorption–desorption equilibrium of MO. The photodegradation process was subsequently initiated with continuous stirring. The degradation progress was tracked by recording UV–visible spectra at designated time intervals using a Varian Cary 100 Bio UV-visible spectrometer.
Conclusions
In summary, we described here a method to support MoS2 nanosheets within porous polymeric microbeads, ensuring their high dispersion stability. The porous network of the beads serves as a physical scaffold that prevents aggregation and restacking of the MoS2 nanosheets. As a result, the photocatalytic activity of the nanosheets is preserved through several cycles of use. The ability to use visible light to induce photocatalytic degradation, or other carrier-driven processes, and the ease with which the beads can be retrieved and reused stand out as attractive properties. These results highlight the potential of these polymeric microbeads for the development of innovative green and sustainable technologies for water treatment or other carrier-driven chemical processes.
Author contributions
This manuscript was written through the contributions of all authors. D. Park: conceptualization, methodology, formal analysis, and writing. J. W. Kim: writing and funding acquisition. C. O. Osuji: conceptualization, writing, supervision, and project administration.
Data availability
The data supporting this article have been included as part of the ESI.†
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgements
This work was supported by the Postdoctoral Research Program of Sungkyunkwan University (2022). The authors acknowledge financial support from the Center for Engineering MechanoBiology (CEMB) under the National Science Foundation (NSF) grant number CMMI: 1548571 at the University of Pennsylvania.
References
- A. Rafiq, M. Ikram, S. Ali, F. Niaz, M. Khan, Q. Khan and M. Maqbool, J. Ind. Eng. Chem., 2021, 97, 111–128 CrossRef CAS.
- M. Saeed, M. Muneer, A. U. Haq and N. Akram, Environ. Sci. Pollut. Res. Int., 2022, 29, 293–311 CrossRef CAS PubMed.
- P. Sun, Z. Xing, Z. Li and W. Zhou, Chem. Eng. J., 2023, 458, 141399 CrossRef CAS.
- C. Feng, Z. Lu, Y. Zhang, Q. Liang, M. Zhou, X. Li, C. Yao, Z. Li and S. Xu, Chem. Eng. J., 2022, 435, 134833 CrossRef CAS.
- E. M. Bayan, L. E. Pustovaya and M. G. Volkova, Environ. Technol. Innovation, 2021, 24, 101822 CrossRef CAS.
- J. Zeng, Z. Li, H. Jiang and X. Wang, Mater. Horiz., 2021, 8, 2964–3008 RSC.
- Z. Zhou, B. Li, X. Liu, Z. Li, S. Zhu, Y. Liang, Z. Cui and S. Wu, ACS Appl. Bio Mater., 2021, 4, 3909–3936 Search PubMed.
- X. Xiao, S. Tu, M. Lu, H. Zhong, C. Zheng, X. Zuo and J. Nan, Appl. Catal., B, 2016, 198, 124–132 CrossRef CAS.
- N. Serpone and A. V. Emeline, J. Phys. Chem. Lett., 2012, 3, 673–677 Search PubMed.
- M.-H. Wu, L. Li, N. Liu, D.-J. Wang, Y.-C. Xue and L. Tang, Process Saf. Environ. Prot., 2018, 118, 40–58 CrossRef CAS.
- Q. Guo, C. Zhou, Z. Ma and X. Yang, Adv. Mater., 2019, 31, e1901997 CrossRef PubMed.
- M. Ni, M. K. H. Leung, D. Y. C. Leung and K. Sumathy, Renewable Sustainable Energy Rev., 2007, 11, 401–425 CrossRef CAS.
- C. B. Anucha, I. Altin, E. Bacaksiz and V. N. Stathopoulos, Chem. Eng. J. Adv., 2022, 10, 100262 CrossRef CAS.
- T. Larbi, M. A. Amara, B. Ouni and M. Amlouk, Mater. Res. Bull., 2017, 95, 152–162 Search PubMed.
- Z. Li, X. Meng and Z. Zhang, J. Photochem. Photobiol., C, 2018, 35, 39–55 Search PubMed.
- E. Parzinger, B. Miller, B. Blaschke, J. A. Garrido, J. W. Ager, A. Holleitner and U. Wurstbauer, ACS Nano, 2015, 9, 11302–11309 Search PubMed.
- M.-H. Wu, L. Li, Y.-C. Xue, G. Xu, L. Tang, N. Liu and W.-Y. Huang, Appl. Catal., B, 2018, 228, 103–112 Search PubMed.
- W.-Q. Chen, L.-Y. Li, L. Li, W.-H. Qiu, L. Tang, L. Xu, K.-J. Xu and M.-H. Wu, Engineering, 2019, 5, 755–767 Search PubMed.
- A. Rahman, J. R. Jennings, A. L. Tan and M. M. Khan, ACS Omega, 2022, 7, 22089–22110 Search PubMed.
- S. Huang, C. Chen, H. Tsai, J. Shaya and C. Lu, Sep. Purif. Technol., 2018, 197, 147–155 Search PubMed.
- H. Shen, S. Liao, C. Jiang, J. Zhang, Q. Wei, R. A. Ghiladi and Q. Wang, Carbohydr. Polym., 2022, 277, 118853 Search PubMed.
- Y. Yu, L. Lu, Q. Yang, A. Zupanic, Q. Xu and L. Jiang, ACS Appl. Nano Mater., 2021, 4, 7523–7537 CAS.
- M. Chhowalla, H. S. Shin, G. Eda, L. J. Li, K. P. Loh and H. Zhang, Nat. Chem., 2013, 5, 263–275 CrossRef.
- W. Osim, A. Stojanovic, J. Akbarzadeh, H. Peterlik and W. H. Binder, Chem. Commun., 2013, 49, 9311–9313 RSC.
- D. M. Sim, H. J. Han, S. Yim, M. J. Choi, J. Jeon and Y. S. Jung, ACS Omega, 2017, 2, 4678–4687 CrossRef CAS PubMed.
- K. Zhou, J. Liu, P. Wen, Y. Hu and Z. Gui, Appl. Surf. Sci., 2014, 316, 237–244 CrossRef CAS.
- D. Xuan, Y. Zhou, W. Nie and P. Chen, Carbohydr. Polym., 2017, 155, 40–48 CrossRef CAS PubMed.
- X. Feng, X. Wang, W. Xing, K. Zhou, L. Song and Y. Hu, Compos. Sci. Technol., 2014, 93, 76–82 CrossRef CAS.
- U. R. Gabinet, C. Lee, R. Poling-Skutvik, D. Keane, N. K. Kim, R. Dong, Z. Vicars, Y. Cai, A. U. Thosar, A. Grun, S. M. Thompson, A. J. Patel, C. R. Kagan, R. J. Composto and C. O. Osuji, ACS Mater. Lett., 2021, 3, 704–712 CrossRef CAS.
- H. P. Lee, G. Lokhande, K. A. Singh, M. K. Jaiswal, S. Rajput and A. K. Gaharwar, Adv. Mater., 2021, 33, e2101238 CrossRef.
- W. Xu, W. Wang, S. Chen, R. Zhang, Y. Wang, Q. Zhang, L. Yuwen, W. J. Yang and L. Wang, J. Colloid Interface Sci., 2021, 586, 601–612 CrossRef CAS PubMed.
- M. Xia, S.-M. Kang, G.-W. Lee, Y. S. Huh and B. J. Park, J. Ind. Eng. Chem., 2019, 73, 306–315 CrossRef CAS.
- L. Ai, H. Yue and J. Jiang, J. Mater. Chem., 2012, 22, 23447–23454 RSC.
- D. Park, C. O. Osuji and J. W. Kim, Small Methods, 2023, 7, e2201195 CrossRef.
- N. X. D. Mai, J. Bae, I. T. Kim, S. H. Park, G.-W. Lee, J. H. Kim, D. Lee, H. B. Son, Y.-C. Lee and J. Hur, Environ. Sci.: Nano, 2017, 4, 955–966 RSC.
- S. Ullah, E. P. Ferreira-Neto, A. A. Pasa, C. C. J. Alcântara, J. J. S. Acuña, S. A. Bilmes, M. L. Martínez Ricci, R. Landers, T. Z. Fermino and U. P. Rodrigues-Filho, Appl. Catal., B, 2015, 179, 333–343 CrossRef CAS.
- S. Sharma and S. Basu, Sep. Purif. Technol., 2021, 279, 119759 CrossRef CAS.
- M. Diab, K. Shreteh, M. Volokh and T. Mokari, Molecules, 2021, 26, 6067 CrossRef CAS.
- K. O. Kassem, M. A. T. Hussein, M. M. Motawea, H. Gomaa, Z. A. Alrowaili and M. Ezzeldien, J. Cleaner Prod., 2021, 326, 129416 CrossRef CAS.
- M. Hasanpour, S. Motahari, D. Jing and M. Hatami, Top. Catal., 2024, 67, 1334–1347 CrossRef.
- Q. Cao, J. Barrio, M. Antonietti, B. Kumru, M. Shalom and B. V. K. J. Schmidt, ACS Appl. Polym. Mater., 2020, 2, 3346–3354 CrossRef CAS.
- X. Zhang, H. Zhang, P. Chen, M. Liu, P. Wu, C. Liu and W. Jiang, Colloids Surf., A, 2022, 644, 128814 CrossRef CAS.
- T.-M. Tien, C.-H. Chen, C.-T. Huang and E. L. Chen, Catalysts, 2022, 12, 1474 CAS.
- J. Kisała, R. Wojnarowska-Nowak and Y. Bobitski, Sci. Rep., 2023, 13, 14148 CrossRef.
- X. Lin, X. Wang, Q. Zhou, C. Wen, S. Su, J. Xiang, P. Cheng, X. Hu, Y. Li, X. Wang, X. Gao, R. Nözel, G. Zhou, Z. Zhang and J. Liu, ACS Sustainable Chem. Eng., 2018, 7, 1673–1682 Search PubMed.
- W. Liu, Q. Hu, F. Mo, J. Hu, Y. Feng, H. Tang, H. Ye and S. Miao, J. Mol. Catal. A: Chem., 2014, 395, 322–328 CrossRef CAS.
- W. Zhang, X. Xiao, L. Zheng and C. Wan, Appl. Surf. Sci., 2015, 358, 468–478 CrossRef CAS.
- H. K. Sadhanala, S. Senapati, K. V. Harika, K. K. Nanda and A. Gedanken, New J. Chem., 2018, 42, 14318–14324 RSC.
- D. Sahoo, J. Shakya, N. Ali, W. J. Yoo and B. Kaviraj, Langmuir, 2022, 38, 1578–1588 CrossRef CAS PubMed.
- D. Sahoo, B. Kumar, J. Sinha, S. Ghosh, S. S. Roy and B. Kaviraj, Sci. Rep., 2020, 10, 10759 CrossRef CAS.
- Z. Xia, Y. Tao, Z. Pan and X. Shen, Results Phys., 2019, 12, 2218–2224 CrossRef.
- N. Thomas, S. Mathew, K. M. Nair, K. O’Dowd, P. Forouzandeh, A. Goswami, G. McGranaghan and S. C. Pillai, Mater. Today Sustainability, 2021, 13, 100073 CrossRef.
- X. Lu, U. R. Gabinet, C. L. Ritt, X. Feng, A. Deshmukh, K. Kawabata, M. Kaneda, S. M. Hashmi, C. O. Osuji and M. Elimelech, Environ. Sci. Technol., 2020, 54, 9640–9651 CrossRef CAS.
- S. Jayabal, J. Wu, J. Chen, D. Geng and X. Meng, Mater. Today Energy, 2018, 10, 264–279 CrossRef.
- H. Li, Q. Zhang, C. C. R. Yap, B. K. Tay, T. H. T. Edwin, A. Olivier and D. Baillargeat, Adv. Funct. Mater., 2012, 22, 1385–1390 CAS.
- M. Farhan Hanafi and N. Sapawe, Mater. Today: Proc., 2020, 31, A141–A150 CAS.
- E. Akama, A. J. Tong, M. Ito and S. Tanaka, Talanta, 1999, 48, 1133–1137 CrossRef.
- Z. Zhang, G. Wang, W. Li, L. Zhang, T. Chen and L. Ding, Colloids Surf., A, 2020, 601, 125034 CrossRef CAS.
- A. P. Nagvenkar and A. Gedanken, ACS Appl. Mater. Interfaces, 2016, 8, 22301–22308 CrossRef CAS PubMed.
- Z. Wang, Y. J. Zhang, M. Liu, A. Peterson and R. H. Hurt, Nanoscale, 2017, 9, 5398–5403 RSC.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence abc,
Jin Woong
Kim
abc,
Jin Woong
Kim





