
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The fungal natural product class of the sorbicillinoids: structures, bioactivities, biosynthesis, and synthesis†
Tobias M.
Milzarek
 *a and
Tobias A. M.
Gulder
*ab
*a and
Tobias A. M.
Gulder
*ab
aDepartment of Natural Product Biotechnology, Helmholtz Institute for Pharmaceutical Research Saarland (HIPS), Helmholtz Centre for Infection Research (HZI), Department of Pharmacy at Saarland University, PharmaScienceHub (PSH), 66123 Saarbrücken, Germany. E-mail: tobias.milzarek@helmholtz-hips.de; tobias.gulder@helmholtz-hips.de
bChair of Technical Biochemistry, Technische Universität Dresden, Bergstraße 66, 01069 Dresden, Germany. E-mail: tobias.gulder@tu-dresden.de
First published on 27th January 2025
Abstract
Covering 1948 up to October 2024
Sorbicillinoids are a growing class of natural products (NPs) that stem from a variety of fungi including members of the orders Hypocreales and Eurotiales. This compound class is unique in its combination of structural complexity and pharmaceutically relevant biological activities. The majority of the sorbicillinoids, which are named after the common hexaketide precursor sorbicillin, exhibit anti-inflammatory, antimicrobial, cytotoxic, phytotoxic, and other selective enzyme inhibitory activities. Over the last eight decades, more than 170 sorbicillinoids, many with strong pharmaceutical potential, have been isolated and described in the literature. This review aims to provide an overview of the structural diversity, biosynthetic pathways, and synthetic studies of this exceptional NP class.
 Tobias M. Milzarek | Tobias M. Milzarek received his BSc (2015) and MSc (2018) in Chemistry from the Technical University of Munich. He received his PhD (2021) in the field of bio- and chemo-catalytic total synthesis from the Technical University of Dresden under the supervision of Prof. Gulder. During this time, he received a doctoral scholarship from the Stiftung der Deutschen Wirtschaft (sdw). Following his graduate work, he spent further time at the TU Dresden (2021) and at the Laboratory of Catalysis and Organic Synthesis (LCSO, Prof. Waser) at the Ecole Polytechnique Fédérale de Lausanne (2022–2023) as a Post-Doc. He is currently a Senior Scientist at the Department of Natural Product Biotechnology (Prof. Gulder) at the Helmholtz Institute for Pharmaceutical Research Saarland (HIPS). His projects concern the development of new chemo-enzymatic strategies towards bioactive compounds. |
 Tobias A. M. Gulder | Tobias A. M. Gulder holds degrees in Chemistry from the University of Würzburg (Diploma 2004, PhD 2008). After postdoctoral training at the Scripps Institution of Oceanography with Bradley Moore, he started his independent work in 2010 as a Liebig and Emmy Noether fellow at the University of Bonn under the mentorship of Jörn Piel. In 2014, he joined the Technical University of Munich as Professor of Biosystems Chemistry at the Department of Chemistry and the Center for Integrated Protein Science Munich (CIPSM). In 2019, he became Chair of Technical Biochemistry at the Technical University of Dresden. Since 2023, he is Chair of Natural Product Biotechnology at Saarland University and the Head of the Department of Natural Product Biotechnology at the Helmholtz Institute for Pharmaceutical Research Saarland (HIPS). His research interests focus on the discovery and (bio-) synthesis of microbial natural products. |
1. Introduction
The first representative of this class of natural products (NPs), sorbicillin (1), was initially isolated and characterized in 1948 by Cram from Penicillium notatum.1,2 Three decades later, more complex bicyclic compounds were found in the culture medium of Verticillium intertextum and Trichoderma longibrachiatum.3–5 Since then, a large number of new analogues were isolated, and structurally elucidated from marine and terrestrial sources.6–8 It is important to note that the taxonomy of the fungi has been reclassified in the literature since initial sorbicillinoid isolation publications (e.g. Clonostachys rosea instead of Verticillium intertextum). Throughout this article, the original references are used to avoid confusion. One major characteristic of this class of NPs is their broad range of biological activities. Due to their anti-inflammatory, antimicrobial, cytotoxic, phytotoxic, and enzyme inhibitory properties, sorbicillinoids are potential candidates for pharmaceutical and agrochemical applications. The combination of stereochemically complex three-dimensional architectures and highly promising biological properties makes sorbicillinoids a special class of NPs.Since the last comprehensive review of Harned in 2011, multiple further sorbicillinoid NPs were isolated and significant progress has been made in understanding sorbicillinoid biosynthesis.6 In this review, we first focus on the full biosynthetic pathway accessing the sorbicillinoids NP class. Second, an overview of the structural diversity ranging from monomeric to polymeric and hybrid sorbicillinoids will be given. Finally, synthetic approaches and chemo-enzymatic strategies to assemble these NPs will be presented.
2. Biosynthesis
In the course of the isolation of the initial monomeric and dimeric sorbicillinoids, the first biosynthetic hypotheses were suggested by Dreiding and Rast.4,9 They determined that sorbicillin (1) is the key biosynthetic precursor of all compounds isolated up to that point. Further hypotheses10–12 led to biosynthetic investigations using stable-isotope labelling experiments employing [1-13C] and [1,2-13C2] labeled sodium acetate.13 This work firmly established the hexaketide origin of the sorbicillinoids and the exact labelling pattern, pointing at the polyketide origin of this NP family.14,15 The exact mechanism of assembly of sorbicillin (1) was finally uncovered by in-depth biosynthetic investigation and functional analysis of the sorbicillinoid biosynthetic gene cluster from Penicillium chrysogenum by the Cox lab,16 including the mechanism of the crucial oxidative dearomatization of 1 to sorbicillinol (2). The BGC (Fig. 1A) contains two iterative polyketide synthases (iPKS), SorbA and SorbB, that build up sorbicillin (1). The non-reducing (NR) iPKS SorbA (orf4) consists of a starter-unit acyl transferase (SAT), β-ketoacylsynthase (KS), acyl transferase (AT), product template (PT), acyl carrier protein (ACP), C-methyl transferase (CMeT), and reductive release (Red) domain. Open reading frame 5 (orf5) encodes the highly reducing (HR) iPKS SorbB comprising KS, AT, dehydratase (DH), CMeT, β-ketoacylreductase (KR), enoylreductase (ER), and ACP domains. SorbA fuses three C2 units with reduction by the KR and DH domains after each elongation step to provide the SorbA-bound sorbyl side chain (Fig. 1B). This is further extended with three additional C2 units accompanied with two C-methylation events to deliver a SorbB-bound hexaketide precursor. The latter is released by action of the reductive domain (Red) as an intermediate aldehyde that undergoes a Knoevenagel cyclization to give sorbicillin (1). | ||
| Fig. 1 (A) Sorbicillinoid biosynthetic gene cluster in P. chrysogenum. (B) Biosynthesis of monomeric sorbicillinoids. | ||
In addition to the PKS machinery, there are two FAD-dependent monooxygenases (FMO) located in the BGC, which fulfill different tasks. Oxidative dearomatisation of sorbicillin (1) to sorbicillinol (2) is carried out by the FMO SorbC, which uses FAD and NADH or NADPH as cofactors.16 The conversion into sorbicillinol (2) occurs with perfect regio- and stereocontrol. For the production of higher oxidised monomeric sorbicillinoids, such as epoxysorbicillinol (3) and oxosorbicillinol (4), it already became apparent at an early stage that a further oxidising enzyme is required.17 Two decades later, it was confirmed by the Cox lab that the FMO Sor(b)D catalyses exactly these reactions.15,18,19 Furthermore, the flavin-dependent monooxygenase Sor(b)D is supposed to additionally be involved in the intermolecular reactions to sorbicillinoid dimers.18,19 Oxosorbicillinol (4) can also be considered as precursor of the recently isolated nitrogen-containing aminosorbicillinol (5).20 Their interconversion could be catalysed by an aminotransferase. However, this hypothesis has not yet been experimentally validated.
The diversity of sorbicillinoids is further extended by two derivatisations of the sorbyl side chain: reduction to 2′-3′-dihydro analogues and hydroxylation to 6′-hydroxy derivates. In rare cases, a combination of both functionalisations was observed.21 Biosynthetically, the formation of the corresponding 2′-3′-dihydro species is likely caused by a reduction step during sorbyl side chain biosynthesis by the ER domain within the SorbA PKS machinery. The exact timing of this transformation and the enzyme involved in the hydroxylation of the sorbyl chain has not yet been described in the literature. The isolation of hydroxysorbicillin (1c) and the fact that heterologously expressed monooxygenase SorbC is able to oxidise 1c to hydroxysorbicillinol suggest that the hydroxylation might take place at the level of sorbicillin (1a).21–23 However, a hydroxylation of the sorbicillinoids at the end of the biosynthetic pathway leading to a specific compound cannot be excluded.
The biosynthesis of sorbicillinols (2) provides the key building blocks of all known sorbicillinoids. In addition to the monomeric representatives, there is a high tendency within the sorbicillinoid NP class to form dimeric derivatives. The reactivity-based mechanisms for the generation of these dimers will be explained in detail in Section 3.2.
3. Structural overview
In general, only those compounds that contain the, in part heavily modified, carbon skeleton of sorbicillin (1a) are designated as sorbicillinoids. The NP class can be divided into monomeric, dimeric, polymeric (so far only trimeric), and hybrid sorbicillinoids. Monomeric to trimeric representatives are exclusively composed of sorbicillin (1a) or closely related precursors, whereas hybrid ones contain additional structural elements. In the following sections, the different subclasses of sorbicillinoids are described and special aspects of these compounds, such as structural scaffolds, biological activities, and biosynthetic mechanisms, are highlighted.In order to provide a comprehensive knowledge base to the field, the ESI† contains a fact sheet for each currently known sorbicillinoid that includes: full structure, fungal producer, known bioactivities, biosynthesis, previous total syntheses, and full analytical data (e.g., melting point, thin layer chromatography, specific optical rotation, NMR, IR, and HRMS data, as far as available).
3.1. Monomeric sorbicillinoids
The monomeric sorbicillinoids include three main groups: sorbicillin (1a) and its derivatives with alterations at the sorbyl side chain (Fig. 2A), sorbicillinol (2a) and congeners including higher oxidised analogues (Fig. 2B), and the vertinolides (Fig. 3). In addition, there are many other monomeric compounds that can be classified as sorbicillinoids in a broader context due to similar structural characteristics and/or co-isolation in sorbicillinoid-forming fungi. These types of compounds are described in Section 3.4. | ||
| Fig. 2 (A) Structures of sorbicillins 1 and definition of different sorbyl side chain variations (shown in green). (B) Overview of structures of sorbicillinols 2 and derivatives 3–5. | ||
 | ||
| Fig. 3 Structures of the monomeric subclass of vertinolides. | ||
Oxidative dearomatization of the sorbicillins 1a–d at C5 by SorbC gives rise to the sorbicillinols 2–5 (Fig. 2B), thereby installing the required reactivity for downstream formation of all complex sorbicillinoids (for more details, see Section 3.2). Sorbicillinol (2a) was first proposed as a starting material for dimeric sorbicillinoids by Dreiding and co-workers in 1983.3 Two decades later, the structure of the unstable sorbicillinol (2a) was elucidated by trapping experiments.10 In the same manner as sorbicillin (1a), sorbicillinol (2a) was also identified in its reduced form, dehydrosorbicillinol (2b), in Penicillium chrysogenum.16 In addition to sorbicillinol (2a), even higher oxidised sorbicillinols 3–5 were isolated. This includes the epoxysorbicillinols 3a–b,17,31 oxosorbicillinols 4a–b,32–34 and aminosorbicillinol (5a).20 Besides aminosorbicillinol (5a), further nitrogen-containing monomers, sorbicillinoides E–F (5b–d) were isolated from the deep-sea derived fungus Penicillium rubens F54.35 Retrobiosynthetically, these compounds 5b–d can be formed by a Michael addition of the amino group to the Δ2′,3′ double bond of the sorbyl side chain (dark blue bond). This would require the hypothetical iso-aminosorbicillinol (5e) as a precursor for sorbicillinoids E–F (5b–d). Among the sorbicillinols, only the oxosorbicillinols 4a–b have significant reported biological activities. Oxosorbicillinol (4a) and hydroxyoxosorbicillinol (4b) both exhibit antioxidative properties due to 2,2-diphenyl-1-picrylhydrazyl (DPPH) radical scavenging activity for 4a and the inhibition of lipoxygenases and cyclooxygenases for 4b.32,34
3.2. Oligomeric sorbicillinoids
The formation of oligomeric sorbicillinoids is based on the high reactivity of sorbicillinol (2a). On the one hand, its cyclic dienone functionality (s-cis conformation) together with the double bonds within the sorbyl side are prone to Diels–Alder reactions, carrying both, a dienophile and an enophile. On the other hand, the cyclic dienone displays a Michael acceptor and donor, allowing classical Michael-addition chemistry. Diels–Alder reactions generate the typical bicyclo[2.2.2]octane scaffold of sorbicillinoids, whereas the Michael reactivity leads to cage-like sorbicillinoids. This inherent reactivity of sorbicillinols explains the formation of all complex sorbicillinoids and hybrid sorbicillinoids (Section 3.3) known to date, which will be discussed in detail in the following chapters.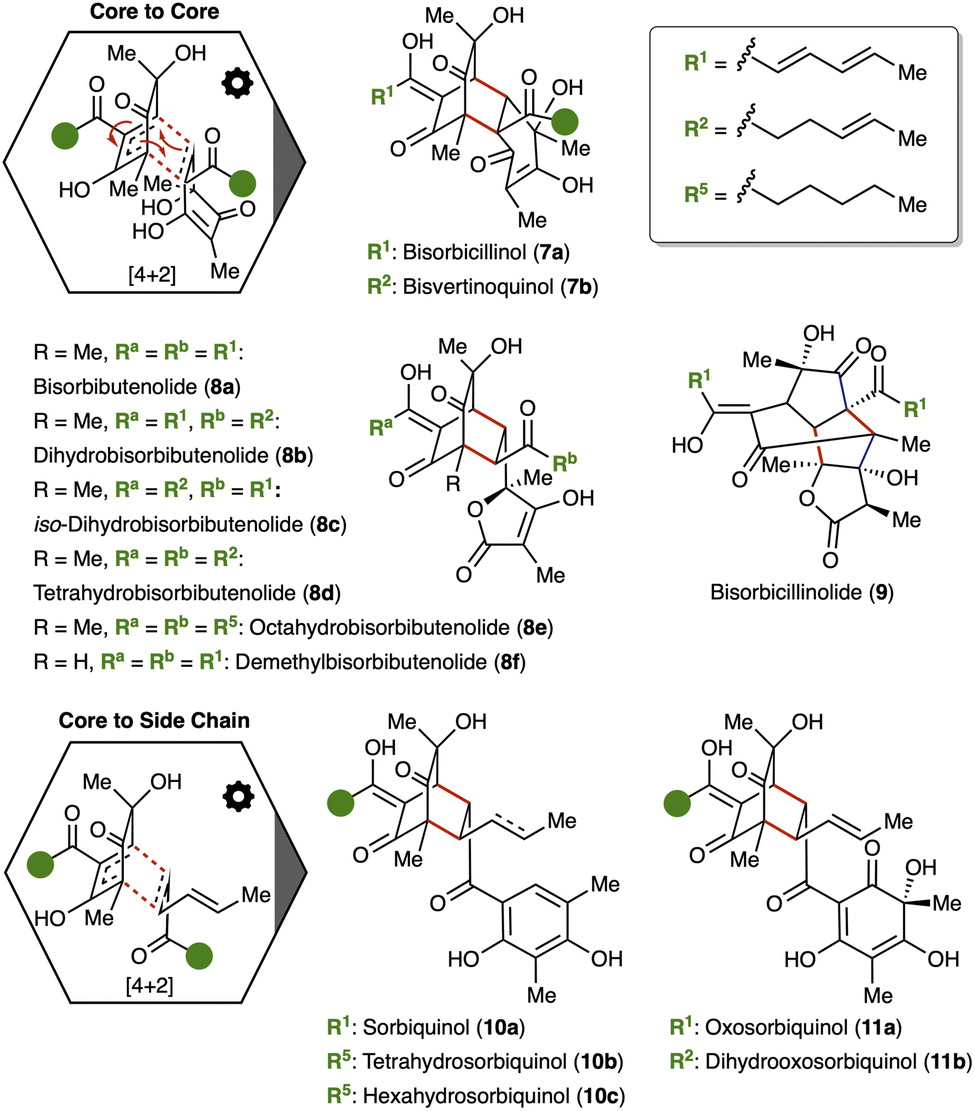 | ||
| Fig. 4 Dimeric sorbicillinoids based on Diels–Alder reactions (shown in red). | ||
Second, a Diels–Alder reaction using the sorbyl side chain as dienophile (Fig. 4, lower hexagon) leads to the sorbiquinols (10a–c) and oxosorbiquinols (11a, 11b). Tetra- and hexahydrosorbiquinol (10b, 10c) have never been isolated from nature. Instead, these compounds were synthesised by reduction of 10a to verify the structure of the sorbiquinols.53 Carrying out the same Diels–Alder reaction with oxosorbicillinol (4a) rather than sorbicillinol (2a) yields the oxosorbiquinols 11a, 11b. Similar to bisorbutenolide 8a, the dimers 11a, 11b have cytotoxic activities against diverse cancer cells (leukemic P388 and HL60 cells, lung cancer cell line A549, carcinoma cells BEL7402).54
 | ||
| Fig. 5 Dimeric sorbicillinoids based on a Michael additions (shown in dark blue). | ||
The structurally most sophisticated representatives of the sorbicillinoids are the trichodimerols (15). Their cage-like core is derived of two Michael reactions and ketalizations between two sorbicillinol units (2). Trichodimerol (15a) was isolated from Trichoderma longibrachiatum5 and further fungal species, like Penicillium chrysogenum57,61–63 and Trichoderma.47–49,64,65 Compound 15a is known for the inhibition of bacterial endotoxin-induced production of tumor necrosis factor (TNF-α), which plays diverse roles in cellular mechanisms like cell survival, proliferation, differentiation, and death.62 Concerning bioactivities, trichodimerols (15a–f) are typical examples among the sorbicillinoids. Their broad bioactive spectra is ranging from cytotoxicity (15b: IC50 = 2.10–33.0 μM, 15c: IC50 = 4.30–8.80 μM, 15d: IC50 = 5.10–13.7 μM; against A549, HCT116, Hs683, OE21, U373 cell lines)50,56,66 over antibacterial (15c: MIC = 128 μg mL−1 against S. aureus sp.)29 and antifungal properties (15e: IC50 = 69.1 μM against C. neoformans)67 to 2,2-diphenyl-1-picrylhydrazyl (DPPH) radical scavenging activity (15f: ED50 = 42.4 μM).43 Bisorbibetanone (16), which was found in Trichoderma sp. USF-2690 by Abe and co-workers, features a slightly different Michael-addition pattern (see Fig. 5) compared to the trichodimerols (15: C1′–4, 16: C2′–4) and displays DPPH-radical scavenging activity (ED50 = 62.5 μM).33,68 Comparable to the trichobisvertinols D (13), the trichodimerols can undergo a further Michael addition within the sorbyl side chain leading to the ustisorbicillinol (17a, 17b) and trichodimer ethers (17c–e).57,58,69
The sorbicillamines B–D (18, 19a, 19b) represent one of the first nitrogen-containing sorbicillinoids (see Fig. 6).70 The retrobiosynthetic basis of these compounds lies in an isomeric precursor 5e of the recently isolated aminosorbicillinol (5a).20 Sorbicillamine D (18) is the amine analogue of the bisvertinolones (14) using the aminated precursor instead of oxosorbicillinol (4a) in the cyclization reactions. Subsequent Michael addition of the amine to the sorbyl chain results in sorbicillamines B (19a) and C (19a), as isolated from Penicillium sp. F23-2. The sorbicillamines (18, 19a, 19b) show cytotoxic properties against various cancer cells (BEL7402, HCT 116, HEK293, HeLa; IC50 > 10.0 μM).70
 | ||
| Fig. 6 Nitrogen-containing Michael-type dimers. | ||
 | ||
| Fig. 7 Examples of trimeric sorbicillinoids. | ||
The trimeric sorbicillinoids are another notable example of sorbicillinoids with cytotoxicity against various cancer cell lines (20–23: HL60, K562, P388; IC50 = 3.14–88.2 μM).70,71,74 Furthermore, tetrahydrotrisorbicillinone C (22b) displays high affinities in targeting diabetes and cancer related proteins (GLP-1R: Kd = 16.2 nM, EF2K: Kd = 74.6 nM).72
3.3. Hybrid sorbicillinoids
Sorbicillinoids derived of reactions between monomeric sorbicillinoids (Section 3.1) and an additional non-sorbicillinoid building block are categorised as hybrid sorbicillinoids. Like the dimeric sorbicillinoids (Section 3.2), hybrid sorbicillinoids can be divided into Diels–Alder-type (Section 3.3.1) and Michael-type sorbicillinoids (Section 3.3.2).An example of the incorporation of cyclic olefins is sorbicillamine A (26), in which a pyrrole species acts as a dienophile (see Fig. 8).70 Other nitrogen-containing dienophiles such as lactams and cyclic ureas are leading to trichsorbicillin A (27) and sorbicillinoid urea 28a, respectively.21,75 In 2023, the first sorbicillinoid glycoside, paeciureallin (28b), was isolated from the rhizospheric soil-derived fungus Paecilomyces sp. KMU21009.76 Paeciureallin (28b) contains a N-ribofuranose functionality, which is based on the N-glycosidation of urea 28a with β-D-ribose (β-D-Rib). The glycoside 28b exhibits cytotoxicity against colorectal cancer (SW480, IC50 = 32.0 μM) and adenocarcinomic cell lines (A549, IC50 = 34.4 μM).76 Nature also integrates oxygenated cyclic olefins, such as lactones and furanes. One of the simplest representatives is ustisorbicillinol E (29), which was isolated from the causal pathogen of rice false smut, fungus Ustilaginoidea virens UV8b, by Zhou and co-workers.58 Together with the antibacterial rezishanone A (30), both compounds are derived of an unsaturated lactone.44,77 A tetrahydrofuran derivative provides the dienophilic precursor for the sorbicillfurans A (31) and B (32).78 The saturated five-membered ring in 31 is obtained upon epoxidation (and epoxide opening) of the Diels–Alder product between sorbicillinol (2a) and furan-2-ylmethanol (see ESI, Section 2†). Sorbicillfuran A (31) was evaluated for cytotoxicity against human renal cancer (ACHN, OS-RC2, 786O) and leukemia cell lines (HL60, K562, MOLT4). Notably, hybrid sorbicillinoid 31 only showed a cytotoxic effect against HL60 cells (IC50 = 9.60 μM) after further derivatisation via an oxo-Diels–Alder reaction with citrinin leading to sorbicillfuran B (32).78 This might reveal that the citrinin moiety contributes significantly to the cytotoxicity. In recent years, other complex, furane-based, hybrid sorbicillinoids such as bisorbicillchaetones A–C (33a–c) and tanshisorbicin (34) were isolated from Trichoderma and Penicillium species.20,79,80 The anti-inflammatory bisorbicillchaetones (33a: IC50 = 80.3 μM, 33b: IC50 = 38.4 μM), which inhibit nitric oxide production, differ in their sorbyl chain reduction and carboxylic acid protection (methyl ester for 33a, 33b; free acid for 33c). Tanshisorbicin (34) displays antibacterial properties against Bacillus species and Staphylococcus aureus (IC50 = 16.0–125 μM). Structurally, it is derived of the diterpenoid tanshinone IIA.79
 | ||
| Fig. 8 Hybrid Diels–Alder-type sorbicillinoids based on cyclic alkenes as dienophiles. | ||
There are also multiple sorbicillinoids that appear to be derived of a Diels–Alder reaction in which a terminal olefin served as dienophile (see Fig. 9). This includes, for example, the remaining representatives of the rezishanones.81,82 The biosynthesis of the rezishanones B–D (35a–c) would be based on vinyl ethers as dienophiles. Since vinyl ether have never been isolated from natural sources, an artificial origin (like contaminations in the organic solvent used for extraction) cannot be excluded. In bioactivity screens, rezishanones B–D (35a–c) displayed weak antibacterial activity against S. aureus and B. subtilis as well as cytotoxicity against murine leukemic lymphoblasts L5178y (35b: IC50 > 10.0 mg mL−1).24,44 The saturnispols,40 acresorbicillinol B,67 and sorbicatechols83,84 (36a–c, 37a–d) are structurally related to rezishanones. Instead of vinyl ethers, various substituted styrenes now act as dienophiles. Presumably due to the increased size of the denophiles, the exo-Diels–Alder product (cf. Section 3.2.3) is also obtained, here in the form of sorbicatechol B (37b). In addition to antibacterial properties similar to those of rezishanones (35a–c), this group of sorbicillinoids (36a–c, 37a–d) exhibits antiviral (H1N1, 37a: IC50 = 85.0 μM, 37b: IC50 = 113 μM)83 and cytotoxic effects (37d: HT29 cells).84 It should be emphasized that further evaluation of the antiviral activity against influenza A virus has revealed cytotoxicity against the host cell system, rather than true antiviral effects.85 The spirosorbicillinols A–D (38a–d) were first isolated from Trichoderma sp. USF-4860 by Hirota and co-workers.86 Their uniqueness lies in the fact that they are composed of two NPs: sorbicillinol (2a) and scytolide. The spirosorbicillinols only differ in their Diels–Alder selectivity (exo: 38a, 38d; endo: 38b, 38c) and the double bond isomer of scytolide (Δ12,13: 38a, 38b; Δ13,14: 38c, 38d) used.31,77,86 A structurally and biomedically highly interesting subgroup of the hybrid sorbicillinoids are the chloctanspirones (39a, 39b), which are among the only halogen-containing sorbicillinoids. Chloctanespirones A (39a) and B (39b) possess cytotoxic activities against leukemic (HL60, 39a: IC50 = 9.20 μM, 39b: IC50 = 37.8 μM) and adenocarcinoma cell lines (A549, 39a: IC50 = 39.7 μM).87 Acresorbicillinol A (40) is named after the fungal source: Acremonium chrysogenum C10.67 This compound was isolated in recent years together with citrisorbicillinol (41).88 Similarly, the name of compound 41 is again based on the fungal source (Penicillium citrinum ZY-2) and furthermore on the employed dienophile, a citrinin derivative.
 | ||
| Fig. 9 Hybrid Diels–Alder-type sorbicillinoids based on terminal alkenes as dienophiles. | ||
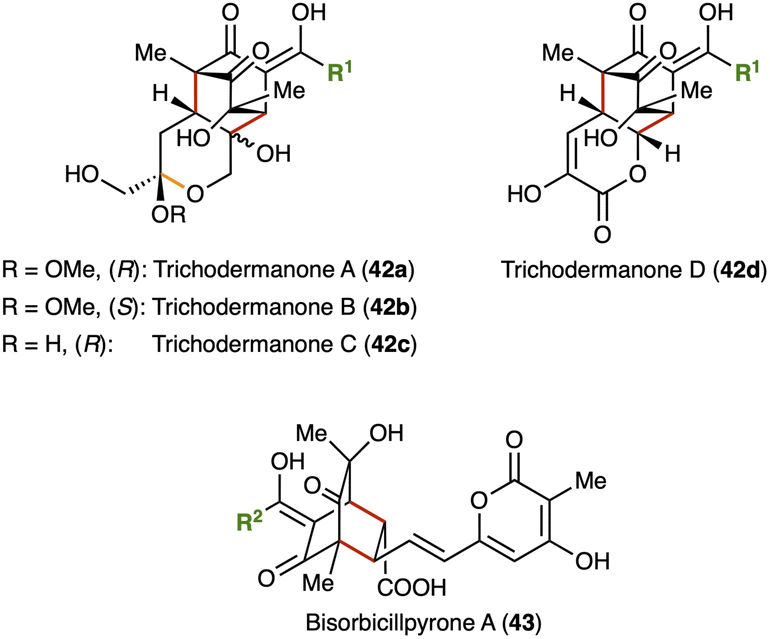
Some hybrid Diels–Alder-type sorbicillinoids are not derived of cyclic or terminal alkenes (see Fig. 10). These comprise the trichodermanones A–D (42a–d) and bisorbicillpyrone A (43). At first glance, trichodermanones 42a–d are expected to incorporate cyclic dienophiles. However, the six-membered ring with ether/ester functionality is generated after [4+2] cycloaddition by ketalisation (orange bond) or lactonisation. Accordingly, (Z)-5-oxohex-2-enoic acid serves as the dienophile for the trichodermanones A–D (42a–d).48 Further biosynthetic steps, like hydroxy-lation, methylation, and redox processes, lead to the final compounds (ESI, Fig. S5†). The trichodermanones 42a–c are another example of sorbicillinoids showing DPPH-radical scavenging activities.48 Besides, bisorbicillpyrone A (43) isolated from Penicillium species, inhibits α-glycosidase (IC50 = 156 μM) making it a potential antihyperglycemic agent.41,80 Bisorbicillpyrone A (43) is derived of sorbicillinol (2a) and a further PKS-derived metabolite, trichopyrone.
 | ||
| Fig. 10 Hybrid Diels–Alder-type sorbicillinoids based on cis-/trans-disubstituted, acyclic alkenes as dienophiles. | ||
 | ||
| Fig. 11 Hybrid Michael-type sorbicillinoids based on previously introduced dimers (A) and new scaffolds (B). | ||
3.4. Other related structures
In addition to the approximately 120 sorbicillinoids, there are also numerous compounds with similar structural characteristics (e.g. sorbyl chain) related to this NP class. Trifonov and co-workers reported the isolation of the first trichodermolide 49a along with sorbiquinol (10a) in 1996.53 The trichodermolides (49a–d, 50) differ in the saturation and oxidation level of the sorbyl side chains (Fig. 12).41,56,93 Dihydrotrichodermolide (49b) is the only representative with cytotoxicity against leukemic P388 (IC50 = 11.5 μM) and K562 cells (IC50 = 22.9 μM).93 The biosynthesis of the trichodermolides is still unknown. However, the presence of two sorbyl side chains suggests a dimerization of two sorbicillin units (see Section 3.2). Retrobiosynthetic analyses indicate that the trichodermolides are based on a single sorbicillinol unit. The second sorbyl side chain is probably introduced by incorporation of another PKS-based metabolite. The same applies to acremonilactone (51), which was isolated from Acremonium sp. AN-13.29 | ||
| Fig. 12 Pseudo-dimeric sorbicillinoids with unknown biosynthesis. | ||
Structural homologues to the vertinolides 6a–h (Fig. 3) were found in the recently isolated subfamily of anti-neuroinflammatory sorbicillinolides 52, 53 (Fig. 13A).35 Sorbicillinolides A–D (52, 53) are based on an oxidative dearomatization of sorbicillins 1a and 1b at position C3 instead of C5 (for sorbicillinol, 2a). This oxidation pattern is reminiscent of the azaphilones and tropolones, which will be briefly discussed later in Section 4.2.2.94,95 Sorbicillinolide J (54) is derived of 2′,3′-dihydroepoxysorbicillinol (3b) performing the same lactonization as seen for the vertinolides 6a–h (ESI, Fig. S7W†).
 | ||
| Fig. 13 (A) Monomeric derivatives related to vertinolides. (B) Nitrogen-containing monomeric analogues. | ||
Nitrogen-containing related compounds (see Fig. 13B) include sorbicillinoid-type pyridine 55 and sorbicillasins (56a, 56b).57,96 The latter were isolated from Phialocephala sp. FL30r and could be based on oxidated 2′,3′-dihydrosorbicillin (1b) and L-asparagine. The tricyclic system is built by via subsequent imination.
The largest group of monomeric related structures are phenols (Fig. 14). These differ in the number of substituents, substitution patterns (catechols, resorcinols), sorbyl chain variations (R1 to R12) and comprise the subfamilies of scalbucillins,97,98 sohirnones,26,44,65 and trichosorbicillin.21 The phenols 57–66 possess many different types of bioactivities, including anti-inflammatory activity due to inhibition of nitric oxide production (mainly for trichosorbicillins: 57c, 57g, 60b, 61a, 61b),21 antibacterial activity against various S. aureus strains (57b, 60a, 62, 64),29,41,44,99 and cytotoxicity against leukemic cell lines (57e, 57f, 58b, 62).65,93,100
 | ||
| Fig. 14 Monomeric phenols related to sorbicillin. | ||
Derived from phenols, other metabolites, such as quinones (67–71), pyranones (72, 73), and chromanones (74, 75), are found in sorbicillinoid-forming fungi (Fig. 15, detailed structures are given in the ESI†). Information on these structural classes and further details on the phenols can be found in the ESI (Section 1.4).†
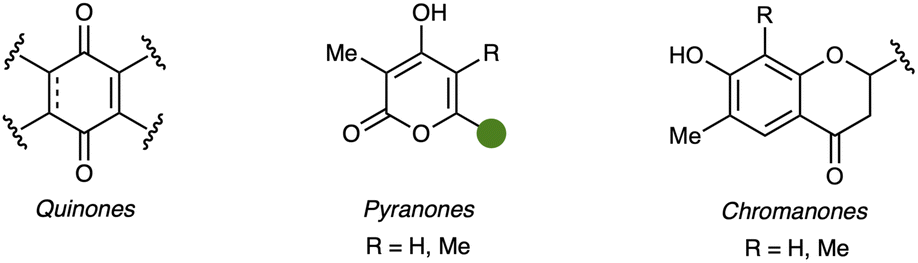 | ||
| Fig. 15 Overview of core scaffolds and functionalities of further related compounds 67–75 (detailed structures are given in the ESI†). | ||
4. Synthesis of the sorbicillinoids
The unique biological activities and three-dimensional architectures of sorbicillinoids have lead to high interest in their synthetic access. Over the years, K. C. Nicolaou and E. J. Corey independently reported purely chemical approaches to generate dimeric sorbicillinoids.101–103 Since 2014, chemo-enzymatic strategies applying the heterologously expressed monooxygenase SorbC have been established.16 In the upcoming sections, we aim to provide a brief overview on previous synthetic work (Section 4.1) followed by in-depth discussion of the straightforward chemo-enzymatic approaches using the biosynthetic key enzyme SorbC (Section 4.2). Detailed information on all total syntheses can be found in the ESI (Section 3).†4.1. Chemical total synthesis
Efficient synthetic access to sorbicillin (1a) is an important basis of total syntheses of sorbicillinoids. In 1954, Kuhn and Staab reported the first synthesis of sorbicillin (1a).104 After the synthesis of dimethylresorcinol (77), sorbicillin (1a) can be obtained upon formation of a chlorinated acetophenone followed by addition of crotonaldehyde. Due to the low overall yield of less than 1%, further strategies were developed. The key step in all recent approaches is a Friedel–Crafts acylation using different Lewis acids (Grignard,105 boron trifluoride,103 aluminium chloride59) to introduce the sorbyl side chain (Fig. 16A). Aluminium chloride in combination with sorbyl chloride yields 1a in up to 61% yield.59 | ||
| Fig. 16 Overview of different total syntheses accessing sorbicillin (A) as well as sorbicillinol and dimers (B). | ||
The first total syntheses of dimeric sorbicillinoids (Fig. 16B) were independently reported by the groups of Corey and Nicolaou.101,103 Both groups used a biomimetic approach (Fig. 16B, reaction 1) in which the oxidative dearomatisation of sorbicillin (1a) with Pb(OAc)4 was the key step in the formation of acetate rac-79. Acetyl deprotection under basic conditions directly led to the formation of bisorbicillinol (7a) and trichodimerol (15a). However, the overall yields were low due to the lack of regio- (see 78) and stereocontrol in the oxidative dearomatization reaction. Improvements were introduced by Pettus et al. using a chiral pyrrolidine tether (80) and a hypervalent iodine species in the oxidative dearomatisation (Fig. 16B, reaction 2).106 Bisorbicillinol (7a) was obtained upon hydrolysis of intermediate 81 in 51% enantiomeric excess (ee). Further progress was achieved by Deng et al. synthesizing sorbicillinol synthon 84 in 92% ee starting with an enantioselective cyanosilylation of 82 to give 83 (Fig. 16B, reaction 3).107 Compared to the previous strategies, this represents a fundamentally different approach, in which the stereocenter is selectively introduced in the first step. The required sorbicillinol core structure is assembled in eight further steps to deliver 84, and upon PMB-removal, bisorbicillinol (7a) is formed. Access to Diels–Alder-type dimer 7a readily enabled the formation of bisorbibutenolide (8a) and bisorbicillinoide (9) after treatment of 7a with KHMDS or MeOH.103,107 Overall, all these previous total syntheses either lack stereoselectivity or require long linear synthetic sequences.
Another strategy that uses hypervalent iodine reagents for oxidation is found in the biomimetic synthesis of sorbiterrin A (46a).108 After Michael addition of 4-hydroxypyrone to acetoxy sorbicillinol 79, the [3.3.1] ring system was generated by a silver nanoparticle catalyzed bridged aldol condensation (ESI, Fig. S13†).
Further total syntheses were reported for monomeric sorbicillinoids epoxysorbicillinol (3a) and vertinolide (6a). Epoxysorbicillinol (3a) was synthesized with a similar pyrrolidine tether as for bisorbicillinol (7a).106 Notably, the use of twice the amount of (bis(trifluoroacetoxy)iodo)benzene (PIFA) achieved the introduction of the characteristic epoxide functionality. Another strategy based on diethyl methylmalonate used a novel 1,3-dipolar cycloaddition between an α-diazo ketone and a propiolate ester to generate monomer 3a over 13 steps.109 To date, a total of six approaches leading to the monomeric sorbicillinoid 6a were introduced (ESI, Section 3).† The main difference is the implementation of stereo information using asymmetric reactions,110 cinchonine resolution,111 or enantio-pure building blocks ((R)-phenylethanamine,112 (R)-lactic acid113). In analogy to Corey's short synthetic route, Takabe and co-workers developed a four step, racemic synthesis of vertinolide (6a) representing the fastest access to monomer 6a.114
In summary, the synthesis of monomeric sorbicillinoids is significantly more efficient in terms of stereocontrol and overall yield compared to more complex, dimeric NPs of this class. Thereby, the key problem is the regio- and stereoselective formation of the reactive intermediate, sorbicillinol (2a), through oxidative dearomatization of sorbicillin (1a). The optimization of this step would be the basis for a better access to complex sorbicillinoids.
4.2. Chemo-enzymatic approaches
As outlined in Section 4.1, the regio- and stereoselective oxidative dearomatisation of sorbicillin (1a) into sorbicillinol (2a) represents a challenging key step in the synthesis of sorbicillinoids. Pioneering research by Cox and co-workers revealed that this transformation is biosynthetically catalysed by the flavin-dependent monooxygenase SorbC in exceptional regio- and stereoselectivity.16 The successful cloning of the encoding gene SorbC from Penicillium chrysogenum and its heterologous expression in Escherichia coli provided an excellent basis for the development of chemo-enzymatic approaches towards sorbicillinoids. In general, chemo-enzymatic strategies constitute a beneficial alternative to classical total synthesis by combining traditional organic-synthetic methods with biocatalysis. In this section, we introduce examples of applied chemo-enzymatic syntheses and display how this approach can be used for the formation of artificial sorbicillinoids. | ||
| Fig. 17 Examples of the chemo-enzymatic synthesis of sorbicillinoids ((A and B) monomers, (C and D) dimers and hybrid sorbicillinoids) cs: co-solvent. | ||
The straightforward access to sorbicillinol (2a) can be used to generate further monomeric sorbicillinoids. Direct addition of potassium hydroxide and tert-butylhydroperoxide to the sorbicillinol-containing reaction batch enabled the stereoselective formation of epoxysorbicillinol (3a) in 23% yield (Fig. 17A).119 Furthermore, the same enzymatic system can be used together with other substrates (see Fig. 17B), such as 6-hydroxysorbicillin (85) and 2,5-dimethylsorbicillin (86). Slight adjustment of the catalytic loading and reaction time thereby leads to oxosorbicillinol (4a) and sorrentanone (67a) in yields of 21% and 29%, respectively.119 Biosynthetically, the generation of these monomers might indicate that the FMO, SorbC, accepts a broad range of substrates, which provides further insight into the biosynthesis of the NP class.
Despite the reactivity towards dimerization described in Section 4.1, sorbicillinol (2a) is relatively stable in aqueous solution. Only after quenching of the aqueous reaction with larger quantities of organic solvent (usually dichloromethane), the formation of dimeric sorbicillinoids is observed.59,119 The tendency to form Diels–Alder or Michael addition type dimers can be controlled via the co-solvent employed (see Fig. 17C). Acetone as a co-solvent favours the formation of bisorbicillinol (7a, 27%), whereas dimethylformamide (DMF) promotes the slower generation of alternative dimerization products, such as trichodimerol (15a, 27%; along 7a, 20%). The addition of pyridine to the enzymatic set up together with DMF and heat resulted in the synthesis of sorbiquinol (10a, 7%). Bisvertinolone (14a) can be obtained in a DMF-promoted Michael addition (20% yield) upon combination of sorbicillinol (2a) and epoxysorbicillinol (3a).
In addition to dimeric sorbicillinoids, hybrid sorbicillinoids can also be synthesised by incorporating external dienophiles and Michael donors (Fig. 17C and D). The use of unsaturated pyrroles and imidazoles allows the synthesis of trichosorbicillin A (27, 14%) and sorbicillinoid urea (28a, 21%).23,117 Rezishanones B (35a, 18%) and C (35b, 29%), saturnispol C (36a, 47%) and D (36b, 62%), as well as sorbicatechol A (37a, 30%) are formed by reaction with vinyl-type dienophiles.23,85,119 The synthesis of saturnispol D (36b) should be emphasised, as 6′-hydroxysorbicillin (1c) was used as a substrate instead of sorbicillin (1a). The direct conversion of 6′-hydroxysorbicillin (1c) by SorbC once again illustrates the broad substrate tolerance of the monooxygenase. The more complex examples within the hybrid sorbicillinoids include the spirosorbicillinols A–C (38a–c) and sorbicillactone A (48a). The spirosorbicillinols A–C (38a–c) require the synthetically challenging (epi-)scytolide as dienophile reaction partner, which is itself a NP. In our previous work, we presented four different scytolide isomers, which were produced over 7–11 synthetic steps in yields between 2% and 49%.120 Spirosorbicillinol A (38a) and its endo-analogue, spirosorbicillinol B (38b) were synthesized in one pot from scytolide. Notably, the [4+2] cycloaddition led to endo-38b in higher yield (52%) compared to the exo-product 38a (9%). Spirosorbicillinol C (38c) was generated analogously using epi-scytolide (45%). The chemo-enzymatic synthesis of the Michael-type hybrid sorbicillinoid, sorbicillactone A (48a), is based on a fumarylazlactone building block 89 serving as Michael donor (Fig. 18A).121 The synthesis of the fumarylazlactone was initiated by an amide coupling between alanine precursor 87 and ethyl fumarate to give 88. Azlactone 89 was finally obtained after saponification of intermediate 88 by ring closure. Convergent application of 89 in the chemo-enzymatic one-pot formation of sorbicillinol (2a) directly furnished sorbicillactone A (48a, R![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Me) in 26% yield with perfect stereocontrol at all three established, contiguous stereocenters. From the mechanistic side, a coordination of the C5 alcohol to the enolate form of 89 was proposed as starting point (see 90). Thus, Micheal addition and subsequent lactonization (see 91) led to the final NP, sorbicillactone A (48a). The strategy was further extended to introduce different C9 analogs (of 48a) by applying derivatives of the fumarylazlactone. Compared to the also reported chemical synthesis of 48a using PIFA, several advantages of the chemo-enzymatic strategy are immediately apparent.121 When comparing the relative performances of both approaches from the same starting point (2-methylresorcinol), the number of reaction steps is reduced (chemical: 12, chemo-enzymatic: 4) in addition to improved yield (chemical: 0.13%, chemo-enzymatic: 29%) and stereoselectivity (chemical: none, chemo-enzymatic: ee > 99%).122 This observation applies not only to sorbicillactone A (48a) but to all chemo-enzymatically produced sorbicillinoids, making this strategy the currently best approach for the synthesis of sorbicillinoids.
Me) in 26% yield with perfect stereocontrol at all three established, contiguous stereocenters. From the mechanistic side, a coordination of the C5 alcohol to the enolate form of 89 was proposed as starting point (see 90). Thus, Micheal addition and subsequent lactonization (see 91) led to the final NP, sorbicillactone A (48a). The strategy was further extended to introduce different C9 analogs (of 48a) by applying derivatives of the fumarylazlactone. Compared to the also reported chemical synthesis of 48a using PIFA, several advantages of the chemo-enzymatic strategy are immediately apparent.121 When comparing the relative performances of both approaches from the same starting point (2-methylresorcinol), the number of reaction steps is reduced (chemical: 12, chemo-enzymatic: 4) in addition to improved yield (chemical: 0.13%, chemo-enzymatic: 29%) and stereoselectivity (chemical: none, chemo-enzymatic: ee > 99%).122 This observation applies not only to sorbicillactone A (48a) but to all chemo-enzymatically produced sorbicillinoids, making this strategy the currently best approach for the synthesis of sorbicillinoids.
 | ||
| Fig. 18 (A) Synthesis of azlactones. (B) Chemo-enzymatic synthesis of sorbicillactones. | ||
 | ||
| Fig. 19 (A) Promiscuity profile of the monooxygenase SorbC. Reaction conditions by Narayan:117 2.5 mM substrate, 2.5 μM SorbC, 1.0 mM NADP+, 5.0 mM glucose-6-phosphate (G6P), 1.0 U ml−1 glucose-6-phosphate dehydrogenase (G6PDH), 50 mM potassium phosphate buffer, pH 8.0, 30 °C, 1 h. TTN: total turnover numbers (complete conversion: TTN = 1000, dark green). Reaction conditions by Gulder:23 2.0 mM substrate, 20 μM SorbC, 4.0 mM NADH, 50 mM potassium phosphate buffer, pH 8.0, 17% co-solvent v/v (acetone or DMF), 25 °C, 1.5 h. Yield of 90% and higher are shown in dark green. nd: not determined. (B) Formation of artificial new-to-science sorbicillinoids. brsm: based on reisolated starting material. | ||
The above-mentioned substrates were used in the one pot chemo-enzymatic synthesis of unknown sorbicillinoids (Fig. 19B). Various bisorbicillinols 99, 100a–e and saturnispol C analogues 101a–e were obtained using acetone as co-solvent.23 The use of hydroxysorbicillin (1c) gave access to dihydroxybisorbicillinol (99) in 41% yield. In this case, DMF was used as cosolvent instead due to the higher polarity of the substrate. Particularly noteworthy is the [4+2] cycloaddition using 3-ethyl derivative 88b, enabling the formation of the corresponding bisorbicillinol 100c in increased yield (61%, natural 7a: 27%). A possible explanation might be the stronger donor activity of the ethyl group compared to the methyl group in sorbicillinol (2a). Switching to DMF as a co-solvent allowed the formation of trichobisvertinol D analogue 102 in 27% yield.
In addition to the diversification of the substrate, the additional structural elements (dienophile, Michael donor) can also be exchanged within the hybrid sorbicillinoids. One example of this is the demethylated species of trichosorbicillin A (103, Fig. 19B), which was synthesized in analogous fashion to trichosorbicillin A (27) using 1,5-dihydro-2H-pyrrol-2-one (10% yield).23 Sorbicatechols can be derivatised in the same manner using comparatively simpler, and in some cases commercially available, dienophiles. In previous work of our group, a library of sorbicatechol structural analogues was established by using diverse substituted vinyls, including phenyls, heterocycles, ethers, and carbonyls (37a, 104a–l).85 The sorbicatechol derivatives 104a–l were obtained in yields between 21 and 31% and were submitted to anti-HIV assays based on the reported antiviral activity of natural sorbicatchol A (37a, IC50 = 85 μM).83 However, in-depth analysis of their bioactivity against the influenza A virus displayed cytotoxicity against the host cell system instead of having a true antiviral effect. Nevertheless, this work illustrates the potential of the one-pot chemo-enzymatic approach for the synthesis of focussed compound libraries to obtain structure activity relationship data.
Another approach for broadening the substrate spectrum of the monooxygenase SorbC lies in the introduction of dummy groups. This strategy is based on mimicking the sorbyl structure within the natural substrate 1a. The big advantage here is the flexibility for downstream late stage modification after removal of the dummy group. This principle was developed in parallel by our group (Fig. 20A) as well as Narayan and co-workers (Fig. 20B).123,124 Starting with the same dimethylated resorcinol 77, which was used in the total synthesis of sorbicillin (1a, Fig. 16A), carboxylic acid 105 can be obtained by treatment with potassium bicarbonate. Esterification with multiple alcohols enabled access to the sorbyl mimicking ester analogues 106a–g in yields of 59–93%. HPLC analysis of their enzymatic conversion to sorbicillinols 107a–g revealed the requirement for an alkyl chain length of at least three carbon atoms. The optimum is reached with n-pentyl (107e, 83%) and n-butyl species (107d, 79%). The latter has already been successfully dimerised into the corresponding bisorbicillinol 108 (15%, brsm: 33%). In contrast, the work of Narayan and co-workers124 focused on the use of crotyl esters (106g, TTN of 827) and amides (109, TTN of 909), which showed higher activities in their studies (Fig. 20B). The exploration of the tolerance of modifications in position R3 (112a–d) and R5 (114a–c) indicated the same trends as observed for the sorbyl analogues 94a–e and 95a–c (see Fig. 19A). Deprotection of the acetylated crotyl ester 115 to the free acid 116 can be performed under reductive conditions in presence of a palladium catalyst. Furthermore, crotyl ester bearing sorbicillinol 107g can be transformed into a hybrid sorbicillinoid by addition of external dienophiles, such as pyrrole-2,5-dione. This allowed the synthesis of pyrrolidine 117 in 23% yield. Subsequent deprotection of the crotyl ester 117 or basic saponification of bisorbicillinol 108 would enable the generation of artificial sorbicillinoids bearing a directly modifiable free acid moiety. Further linkage with suitable probes combined with target docking studies would allow the in-depth analysis of the mode of action of all current members of the sorbicillinoid family.
 | ||
| Fig. 20 Ester mimicking strategies by our group (A) and Narayan (B). Pie chart(s) in (A) shows enzymatic conversion in percentage yield, whereas in (B), the total turnover numbers are given. | ||
Besides derivatization of the substrates, the flavoprotein monooxygenase used can also be exchanged.125 Biocatalytic transformations by Narayan et al. using the FMOs TropB or AzaH lead to the structurally related compound classes of tropolones and azaphilones (see Fig. 21A, 118–120).117,126,127 Their biosyntheses are based on oxidative dearomatisation reactions comparable to those of the sorbicillinoids, but with a different site and stereo selectivity (C3/C5). Similar to SorbC, the enzymes were successfully used in chemo-enzymatic approaches (Fig. 21B), for example in the synthesis of stipitatic aldehyde 123 or trichoflectin (126). Screening experiments by Cox and co-workers revealed a broad substrate scope of the monooxygenase TropC, similar to SorbC.128 In another interesting study by Narayan et al., identification of the AzaH FMO homologe AfoD enabled the formation the corresponding (S)-trichoflectin (126) with excellent enantioselectivity.129 In addition to the identification of natural enzyme homologues, selective protein engineering would enable the generation of highly specific enzymes for the targeted control of regio- and stereoselectivity. This represents an exciting strategy and its use within the sorbicillinoids would open up further derivatisation possibilities.
 | ||
| Fig. 21 (A) Representatives of the structurally related tropolones and azaphilones. (B) FMO diversity in chemo-enzymatic syntheses. | ||
Overall, the biocatalytic transformation with ester substrate mimics and the variation of the flavin-dependant monooxygenase demonstrates the impact of substrate and protein alteration in the expansion of the utility of enzymes in synthetic procedures.
5. Conclusions and outlook
Over the last eight decades, the NP class of the sorbicillinoids has grown steadily and up to now includes around 120 natural representatives and over 60 related and new-to-science analogs. Their molecular architectures and manifold biological activities have attracted the interest of NP researchers including synthetic organic chemists, biochemists, and molecular biologists. Since the last comprehensive review of Harned in 2011,6 significant progress was made in understanding sorbicillinoid biosynthetic assembly by the Cox laboratory and also concerning efficient synthetic access towards this NP family. Only few open questions remain, including the enzymatic process behind sorbyl side-chain hydroxylation or the installation of the amino groups in some sorbicillinoids. The chemo-enzymatic approaches presently offer the shortest access combined with the highest overall synthetic yields and nearly perfect stereocontrol. For the first time, sufficient amounts of synthetic natural and artificially modified sorbicillinoids are thus available to enable structure–activity studies and in-depth investigations into molecular modes of action leading to the diverse bioactivities within the sorbicillinoid NP class. However, there is still enormous potential to improve chemo-enzymatic access, in particular concerning the yields in the reactions capturing the reactive sorbicillinol intermediate (2), which are currently in most cases around 30% of the desired product, owing to the high reactivity of 2 and hence the occurrence of undesired side-reactions. Strategies to tame this inherent reactivity of 2 and to guide downstream functionalization reactions to exclusively furnish the desired products are thus needed. Furthermore, approaches that allow enzyme recovery and regeneration would be highly beneficial. In addition to the identification of further natural SorbC homologues with different product selectivity or substrate scope, the development of SorbC homologues with altered properties by protein engineering has the potential to significantly expand the accessible structural and functional chemical space.On the purely chemical side, the Holy Grail of sorbicillinoid total synthesis lies in the development of an oxidising agent that would allow for a regio- and stereoselective dearomatisation of sorbicillin (1a) to sorbicillinol (2a), with similar overall efficiency when compared to the SorbC-catalyzed enzymatic transformation. However, previous strategies using, e.g., asymmetric hypervalent iodine reagent have highlighted the many challenges in developing such a reagent.130 All in all, the combination of new developments with the already existing synthetic and biocatalytic toolkit will offer enormous potential for the assembly of sorbicillinoid structural libraries with high potential in diverse medical applications.
6. Author contributions
The review was conceptualised by TMM and TAMG. TMM prepared the initial draft and figures. Both authors contributed to the reviewing and editing process of the manuscript.7. Conflicts of interest
There are no conflicts to declare.8. Acknowledgements
We thank the DFG for the generous financial support of the work in our laboratory (GU 1233/7-1, INST 269/971-1). All current and former team members working with sorbicillinoids, including Dr A. Sib, Dr M. Einsiedler, Dr J. I. Müller, Dr A. Matura, J. Schuricht, F. Gattwinkel, M. Gehmlich, E. Schulze, B. Kirmizigül, and T. Bernauer are acknowledged for their scientific contribution. We thank Prof. Russell Cox for fruitful discussions on the manuscript draft.9. References
- D. J. Cram and M. Tishler, J. Am. Chem. Soc., 1948, 70, 4238 CrossRef CAS PubMed.
- D. J. Cram, J. Am. Chem. Soc., 1948, 70, 4240 CrossRef CAS PubMed.
- L. Trifonov, J. Bieri, R. Prewo and A. Dreiding, Tetrahedron, 1983, 39, 4243–4256 CrossRef CAS.
- L. Trifonov, H. Hilpert, P. Floersheim, A. Dreiding, D. Rast, R. Skrivanova and L. Hoesch, Tetrahedron, 1986, 42, 3157 CrossRef CAS.
- R. Andrade, W. A. Ayer and P. P. Mebe, Can. J. Chem., 1992, 70, 2526 CrossRef CAS.
- A. M. Harned and K. A. Volp, Nat. Prod. Rep., 2011, 28, 1790 RSC.
- J. Meng, X. Wang, D. Xu, X. Fu, X. Zhang, D. Lai, L. Zhou and G. Zhang, Molecules, 2016, 21, 715 CrossRef PubMed.
- X. Hou, X. Zhang, M. Xue, Z. Zhao, H. Zhang, D. Xu, D. Lai and L. Zhou, J. Fungi, 2022, 8, 62 CrossRef CAS.
- L. S. Trifonov, A. S. Dreiding, L. Hoesch and D. M. Rast, Helv. Chim. Acta, 1981, 64, 1843 CrossRef CAS.
- N. Abe, O. Sugimoto, K. ichi Tanji and A. Hirota, J. Am. Chem. Soc., 2000, 122, 12606 CrossRef CAS.
- N. Abe, K. Yamamoto, T. Arakawa and A. Hirota, Chem. Commun., 2001, 23 RSC.
- K. Sugaya, H. Koshino, Y. Hongo, K. Yasunaga, J. ichi Onose, K. Yoshikawa and N. Abe, Tetrahedron Lett., 2008, 49, 654 CrossRef CAS.
- N. Abe, T. Arakawa, K. Yamamoto and A. Hirota, Biosci. Biotechnol. Biochem., 2002, 66, 2090 CrossRef CAS PubMed.
- I. S. Druzhinina, E. M. Kubicek and C. P. Kubicek, BMC Evol. Biol., 2016, 16, 269 CrossRef PubMed.
- F. Guzmán-Chávez, O. Salo, Y. Nygård, P. P. Lankhorst, R. A. L. Bovenberg and A. J. M. Driessen, Microb. Biotechnol., 2017, 10, 958 CrossRef PubMed.
- A. Al Fahad, A. Abood, K. M. Fisch, A. Osipow, J. Davison, M. Avramović, C. P. Butts, J. Piel, T. J. Simpson and R. J. Cox, Chem. Sci., 2014, 5, 523 RSC.
- S. Sperry, G. J. Samuels and P. Crews, J. Org. Chem., 1998, 63, 10011 CrossRef CAS.
- L. Kahlert, R. J. Cox and E. Skellam, Chem. Commun., 2020, 56, 10934 RSC.
- L. Kahlert, E. F. Bassiony, R. J. Cox and E. J. Skellam, Angew. Chem., Int. Ed., 2020, 59, 5816 CrossRef CAS PubMed.
- A. Yang, Y. Hong, F. Zhou, L. Zhang, Y. Zhu, C. Wang, Y. Hu, L. Yu, L. Chen and X. Wang, Molecules, 2023, 28, 6301 CrossRef CAS PubMed.
- P. Zhang, Y. Deng, X. Lin, B. Chen, J. Li, H. Liu, S. Chen and L. Liu, J. Nat. Prod., 2019, 82, 947 CrossRef CAS PubMed.
- S.-Z. Liu, G.-X. Xu, F.-M. He, W.-B. Zhang, Z. Wu, M.-Y. Li, X.-X. Tang and Y.-K. Qiu, Mar. Drugs, 2022, 20, 213 CrossRef CAS PubMed.
- T. M. Milzarek, S. Schuler, A. Matura and T. A. Gulder, ACS Catal., 2022, 12, 1898 CrossRef CAS.
- G. Bringmann, G. Lang, T. A. M. Gulder, H. Tsuruta, J. Mühlbacher, K. Maksimenka, S. Steffens, K. Schaumann, R. Stöhr, J. Wiese, J. F. Imhoff, S. Perović-Ottstadt, O. Boreiko and W. E. G. Müller, Tetrahedron, 2005, 61, 7252 CrossRef CAS.
- N. Abe, T. Murata and A. Hirota, Biosci. Biotechnol. Biochem., 1998, 62, 2120 CrossRef CAS PubMed.
- L. Du, T. Zhu, L. Li, S. Cai, B. Zhao and Q. Gu, Chem. Pharm. Bull., 2009, 57, 220 CrossRef CAS.
- Y. M. Ying, Z. J. Zhan, Z. S. Ding and W. G. Shan, Chem. Nat. Compd., 2011, 47, 541 CrossRef CAS.
- Y. Yao, J. Li, C. S. Jiang, X. X. Zhao, Z. H. Miao, H. T. Liu, P. Zheng, W. X. Yao and W. Q. Li, Pharmazie, 2015, 70, 394 CAS.
- C.-L. An, Q.-Y. Ma, Q.-Y. Xie, L. Yang, J.-Z. Yuan, H.-F. Dai and Y.-X. Zhao, J. Asian Nat. Prod. Res., 2023, 1, 489 Search PubMed.
- W.-J. Lan, Y. Zhao, Z.-L. Xie, L.-Z. Liang, W.-Y. Shao, L.-P. Zhu, D.-P. Yang, X.-F. Zhu and H.-J. Li, Nat. Prod. Commun., 2012, 7, 1337 CAS.
- M. T. Ngo, M. V. Nguyen, J. W. Han, M. S. Park, H. Kim and G. J. Choi, J. Fungi, 2021, 7, 428 CrossRef CAS PubMed.
- N. Abe, K. Yamamoto and A. Hirota, Biosci. Biotechnol. Biochem., 2000, 64, 620 CrossRef CAS PubMed.
- N. Abe, O. Sugimoto, T. Arakawa, K. ichi Tanji and A. Hirota, Biosci. Biotechnol. Biochem., 2001, 65, 2271 CrossRef CAS PubMed.
- T. Komoda and M. Nishikawa, Biosci. Biotechnol. Biochem., 2012, 76, 1404 CrossRef CAS PubMed.
- J. Wu, Q. Meng, D. Liu, A. Fan, J. Huang and W. Lin, Phytochem., 2024, 219, 113976 CrossRef CAS PubMed.
- L. Trifonov, J. Bieri, R. Prewo, A. Dreiding, D. Rast and L. Hoesch, Tetrahedron, 1982, 38, 397 CrossRef CAS.
- D. R. McMullin, J. B. Renaud, T. Barasubiye, M. W. Sumarah and J. D. Miller, Can. J. Microbiol., 2017, 63, 621 CrossRef CAS PubMed.
- U. Supratman, T. Suzuki, T. Nakamura, Y. Yokoyama, D. Harneti, R. Maharani, S. Salam, F. F. Abdullah, T. Koseki and Y. Shiono, Nat. Prod. Res., 2019, 35, 1525 CrossRef PubMed.
- P. Han, X. Zhang, D. Xu, B. Zhang, D. Lai and L. Zhou, J. Fungi, 2020, 6, 229 CrossRef CAS.
- J. Meng, W. Cheng, H. Heydari, B. Wang, K. Zhu, B. Konuklugil and W. Lin, Mar. Drugs, 2018, 16, 226 CrossRef PubMed.
- X. Pang, X. Zhou, X. Lin, B. Yang, X. Tian, J. Wang, S. Xu and Y. Liu, Bioorg. Chem., 2021, 107, 104600 CrossRef CAS.
- A. Takaiwa and K. Yamashita, Agric. Biol. Chem., 1983, 47, 429 CAS.
- N. Abe, T. Murata and A. Hirota, Biosci. Biotechnol. Biochem., 1998, 62, 661 CrossRef CAS PubMed.
- R. P. Maskey, I. Grün-Wollny and H. Laatsch, J. Nat. Prod., 2005, 68, 865 CrossRef CAS.
- K. Sugaya, T. Terajima, A. Takahashi, J. ichi Onose and N. Abe, Bioorg. Med. Chem. Lett., 2019, 29, 832 CrossRef CAS PubMed.
- R. Andrade, W. A. Ayer and L. S. Trifonov, Aust. J. Chem., 1997, 50, 255 CrossRef CAS.
- O. Shirota, V. Pathak, C. F. Hossain, S. Sekita, K. Takatori and M. Satake, J. Chem. Soc., Perkin Trans. 1, 1997, 2961 RSC.
- K. Neumann, A. Abdel-Lateff, A. D. Wright, S. Kehraus, A. Krick and G. M. König, Eur. J. Org. Chem., 2007, 2007, 2268 CrossRef.
- A. Evidente, A. Andolfi, A. Cimmino, S. Ganassi, C. Altomare, M. Favilla, A. D. Cristofaro, S. Vitagliano and M. A. Sabatini, J. Chem. Ecol., 2009, 35, 533 CrossRef CAS PubMed.
- E. S. Balde, A. Andolfi, C. Bruyère, A. Cimmino, D. Lamoral-Theys, M. Vurro, M. V. Damme, C. Altomare, V. Mathieu, R. Kiss and A. Evidente, J. Nat. Prod., 2010, 73, 969 CrossRef CAS PubMed.
- J. ya Ueda, J. Hashimoto, S. Inaba, M. Takagi and K. Shin-ya, J. Antibiot., 2010, 63, 203 CrossRef.
- J. Yu, H. Han, X. Zhang, C. Ma, C. Sun, Q. Che, Q. Gu, T. Zhu, G. Zhang and D. Li, Mar. Drugs, 2019, 17, 446 CrossRef CAS PubMed.
- R. Andrade, W. A. Ayer and L. S. Trifonov, Can. J. Chem., 1996, 74, 371 CrossRef CAS.
- D. Li, F. Wang, S. Cai, X. Zeng, X. Xiao, Q. Gu and W. Zhu, J. Antibiot., 2007, 60, 317 CrossRef CAS PubMed.
- N. Koyama, T. Ohshiro, H. Tomoda and S. Ōmura, Org. Lett., 2007, 9, 425 CrossRef CAS PubMed.
- S. U. Rehman, L.-J. Yang, Y.-H. Zhang, J.-S. Wu, T. Shi, W. Haider, C.-L. Shao and C.-Y. Wang, Front. Microbiol., 2020, 11, 1334 CrossRef PubMed.
- W. Ding, F. Wang, Q. Li, Y. Xue, Z. Dong, D. Tian, M. Chen, Y. Zhang, K. Hong and J. Tang, Chem. Biodivers., 2021, 18, e2100229 CrossRef CAS.
- J. Meng, G. Gu, P. Dang, X. Zhang, W. Wang, J. Dai, Y. Liu, D. Lai and L. Zhou, Front. Chem., 2019, 7, 435 CrossRef CAS PubMed.
- A. Sib and T. A. M. Gulder, Angew. Chem., Int. Ed., 2017, 56, 12888 CrossRef CAS PubMed.
- W. Liu, Q. Gu, W. Zhu, C. Cui and G. Fan, J. Antibiot., 2005, 58, 441 CrossRef CAS PubMed.
- Q. Gao, J. E. Leet, S. T. Thomas, J. A. Matson and D. P. Bancroft, J. Nat. Prod., 1995, 58, 1817 CrossRef CAS.
- G. A. Warr, J. A. Veitch, A. W. Walsh, G. A. Hesler, D. M. Pirnik, J. E. Leet, P. F. M. Lin, I. A. Medina, K. D. Mcbrien, S. Forenza, J. M. Clark and K. S. Lam, J. Antibiot., 1996, 49, 234 CrossRef CAS PubMed.
- O. Salo, F. Guzmán-Chávez, M. I. Ries, P. P. Lankhorst, R. A. L. Bovenberg, R. J. Vreeken and A. J. M. Driessen, Appl. Environ. Microbiol., 2016, 82, 3971 CrossRef CAS PubMed.
- A. Abdel-Lateff, K. Fisch and A. D. Wright, Z. Naturforsch., 2009, 64, 186 CrossRef CAS PubMed.
- L. Ma, W. Liu, Y. Huang and X. Rong, J. Antibiot., 2011, 64, 645 CrossRef CAS PubMed.
- W. Liu, Q. Gu, W. Zhu, C. Cui and G. Fan, J. Antibiot., 2005, 58, 621 CrossRef CAS PubMed.
- C. Duan, S. Wang, R. Huo, E. Li, M. Wang, J. Ren, Y. Pan, L. Liu and G. Liu, J. Fungi, 2022, 8, 530 CrossRef CAS PubMed.
- N. Abe, T. Murata, K. Yamamoto and A. Hirota, Tetrahedron Lett., 1999, 40, 5203 CrossRef CAS.
- M.-M. Zhai, F.-M. Qi, J. Li, C.-X. Jiang, Y. Hou, Y.-P. Shi, D.-L. Di, J.-W. Zhang and Q.-X. Wu, J. Agric. Food Chem., 2016, 64, 2298 CrossRef CAS PubMed.
- W. Guo, J. Peng, T. Zhu, Q. Gu, R. A. Keyzers and D. Li, J. Nat. Prod., 2013, 76, 2106 CrossRef CAS PubMed.
- D. Li, F. Wang, X. Xiao, Y. Fang, T. Zhu, Q. Gu and W. Zhu, Tetrahedron Lett., 2007, 48, 5235 CrossRef CAS.
- M.-J. Cao, T. Zhu, J.-T. Liu, L. Ouyang, F. Yang and H.-W. Lin, Nat. Prod. Res., 2020, 34, 2880 CrossRef CAS PubMed.
- X.-P. Peng, G. Li, L.-M. Wang, Q. Wang, C. Wang, L.-X. Ji, C.-X. Cao, G.-F. Lin, Z.-Y. Jiang, Z. qian He, P. Wang and H.-X. Lou, Front. Microbiol., 2022, 13, 800626 CrossRef PubMed.
- D. Li, S. Cai, T. Zhu, F. Wang, X. Xiao and Q. Gu, Tetrahedron, 2010, 66, 5101 CrossRef CAS.
- G. M. Cabrera, M. Butler, M. A. Rodriguez, A. Godeas, R. Haddad and M. N. Eberlin, J. Nat. Prod., 2006, 69, 1806 CrossRef CAS PubMed.
- X.-Y. Guo, H.-T. Li, Y.-T. Shao, C.-Y. Li, W.-Y. Huang and W. Li, Fitoterapia, 2023, 166, 105443 CrossRef CAS PubMed.
- R. Marra, R. Nicoletti, E. Pagano, M. DellaGreca, M. M. Salvatore, F. Borrelli, N. Lombardi, F. Vinale, S. L. Woo and A. Andolfi, Nat. Prod. Res., 2019, 33, 3389 CrossRef CAS PubMed.
- J. Wang, K. Li, X. Luo, Z. Wu, T. Gu, S. Liao, X. Lin, B. Yang, Y. Liu, W. Fang and X. Zhou, Org. Biomol. Chem., 2019, 17, 8721 RSC.
- W. He, M. Liu, X. Li, X. Zhang, W. M. Abdel-Mageed, L. Li, W. Wang, J. Zhang, J. Han, H. Dai, R. J. Quinn, H. wen Liu, H. Luo, L. Zhang and X. Liu, Appl. Microbiol. Biotechnol., 2016, 100, 8349 CrossRef CAS PubMed.
- X. Pang, P. Wang, S. Liao, X. Zhou, X. Lin, B. Yang, X. Tian, J. Wang and Y. Liu, Phytochem., 2022, 202, 113311 CrossRef CAS PubMed.
- D. Lee, J. H. Lee, X. F. Cai, J. C. Shin, K. Lee, Y.-S. Hong and J. J. Lee, J. Antibiot., 2005, 58, 615 CrossRef CAS PubMed.
- R. T. M. el dien, B. K. Mahmoud, M. F. Abdelwahab, A. I. M. Khedr, M. S. Kamel, R. Yahia, N. M. Mohamed, A. E. Zawily, E. S. Kamel, A. K. Salem, U. R. Abdelmohsen and M. A. Fouad, BMC Microbiol., 2023, 23, 308 CrossRef PubMed.
- J. Peng, X. Zhang, L. Du, W. Wang, T. Zhu, Q. Gu and D. Li, J. Nat. Prod., 2014, 77, 424 CrossRef CAS PubMed.
- C.-L. Xie, D. Zhang, T. Lin, Z.-H. He, Q.-X. Yan, Q. Cai, X.-K. Zhang, X.-W. Yang and H.-F. Chen, Front. Microbiol., 2021, 11, 636948 CrossRef PubMed.
- A. Sib, T. M. Milzarek, A. Herrmann, L. Oubraham, J. I. Müller, A. Pichlmair, R. Brack-Werner and T. A. M. Gulder, ChemBioChem, 2020, 21, 492 CrossRef CAS PubMed.
- K. Washida, N. Abe, Y. Sguiyma and A. Hirota, Biosci. Biotechnol. Biochem., 2009, 73, 1355 CrossRef CAS PubMed.
- D. Li, L. Chen, T. Zhu, T. Kurtán, A. Mándi, Z. Zhao, J. Li and Q. Gu, Tetrahedron, 2011, 67, 7913 CrossRef CAS.
- Y. Zhang, J. Zhang, Q. Du, X. M. Wu, Y. Chen and R. X. Tan, Fitoterapia, 2024, 173, 105836 CrossRef CAS PubMed.
- T. Kawahara, M. Takagi and K. Shin-ya, J. Antibiot., 2012, 65, 45 CrossRef CAS PubMed.
- D. Lai, J. Meng, X. Zhang, D. Xu, J. Dai and L. Zhou, Org. Lett., 2019, 21, 1311 CrossRef CAS PubMed.
- L. Chen, T. Zhu, Y. Ding, I. A. Khan, Q. Gu and D. Li, Tetrahedron Lett., 2012, 53, 325 CrossRef CAS.
- G. Bringmann, G. Lang, T. Bruhn, K. Schäffler, S. Steffens, R. Schmaljohann, J. Wiese and J. F. Imhoff, Tetrahedron, 2010, 66, 9894 CrossRef CAS.
- D. Li, S. Cai, T. Zhu, F. Wang, X. Xiao and Q. Gu, Chem. Biodivers., 2011, 8, 895 CrossRef CAS PubMed.
- J.-M. Gao, S.-X. Yang and J.-C. Qin, Chem. Rev., 2013, 113, 4755 CrossRef CAS PubMed.
- H. Guo, D. Roman and C. Beemelmanns, Nat. Prod. Rep., 2019, 36, 1137 RSC.
- Z. Zhang, X. He, Q. Che, G. Zhang, T. Zhu, Q. Gu and D. Li, Mar. Drugs, 2018, 16, 245 CrossRef PubMed.
- R. F. Reátegui, D. T. Wicklow and J. B. Gloer, J. Nat. Prod., 2006, 69, 113 CrossRef PubMed.
- T. El-Elimat, H. A. Raja, M. Figueroa, S. M. Swanson, J. O. F. III, D. M. Lucas, M. R. Grever, M. C. Wani, C. J. Pearce and N. H. Oberlies, J. Antibiot., 2015, 68, 191 CrossRef CAS PubMed.
- C. S. Jiang, Z. F. Zhou, X. H. Yang, L. F. Lan, Y. C. Gu, B. P. Ye and Y. W. Guo, Chin. J. Nat. Med., 2018, 16, 358 CAS.
- S. Liu, X. Yan, M. Yu, J. Chen and L. Zhang, Chem. Nat. Compd., 2010, 46, 116 CrossRef CAS.
- D. Barnes-Seeman and E. J. Corey, Org. Lett., 1999, 1, 1503 CrossRef CAS PubMed.
- K. C. Nicolaou, K. B. Simonsen, G. Vassilikogiannakis, P. S. Baran, V. P. Vidali, E. N. Pitsinos and E. A. Couladouros, Angew. Chem., Int. Ed., 1999, 38, 3555 CrossRef CAS PubMed.
- K. C. Nicolaou, G. Vassilikogiannakis, K. B. Simonsen, P. S. Baran, Y.-L. Zhong, V. P. Vidali, E. N. Pitsinos and E. A. Couladouros, J. Am. Chem. Soc., 2000, 122, 3071 CrossRef CAS.
- R. Kuhn and H. A. Staab, Chem. Ber., 1954, 87, 266 CrossRef CAS.
- F. Bigi, G. Casiraghi, G. Casnati, S. Marchesi, G. Sartori and C. Vignali, Tetrahedron, 1984, 40, 4081 CrossRef CAS.
- L. H. Pettus, R. W. V. D. Water and T. R. R. Pettus, Org. Lett., 2001, 3, 905 CrossRef CAS PubMed.
- R. Hong, Y. Chen and L. Deng, Angew. Chem., Int. Ed., 2005, 44, 3478 CrossRef CAS PubMed.
- C. Qi, T. Qin, D. Suzuki and J. A. Porco, J. Am. Chem. Soc., 2014, 136, 3374 CrossRef CAS PubMed.
- J. L. Wood, B. D. Thompson, N. Yusuff, D. A. Pflum and M. S. P. Matthäus, J. Am. Chem. Soc., 2001, 123, 2097 CrossRef CAS PubMed.
- J. E. Wrobel and B. Ganem, J. Org. Chem., 1983, 48, 3761 CrossRef CAS.
- A. Takaiwa and K. Yamashita, Agric. Biol. Chem., 1984, 48, 961 CAS.
- D. Desmaële, Tetrahedron, 1992, 48, 2925 CrossRef.
- K. Matsuo and Y. Sakaguchi, Chem. Pharm. Bull., 1997, 45, 1620 CrossRef CAS.
- K. Takabe, N. Mase, M. Nomoto, M. Daicho, T. Tauchi and H. Yoda, J. Chem. Soc., Perkin Trans. 1, 2002, 2, 500 RSC.
- M. M. Huijbers, S. Montersino, A. H. Westphal, D. Tischler and W. J. van Berkel, Arch. Biochem. Biophys., 2014, 544, 2 CrossRef CAS PubMed.
- H. K. Chenault and G. M. Whitesides, Appl. Biochem. Biotechnol., 1987, 14, 147 CrossRef CAS PubMed.
- S. A. B. Dockrey, A. L. Lukowski, M. R. Becker and A. R. H. Narayan, Nat. Chem., 2018, 10, 119 CrossRef PubMed.
- S. Visitsatthawong, P. Chenprakhon, P. Chaiyen and P. Surawatanawong, J. Am. Chem. Soc., 2015, 137, 9363 CrossRef CAS PubMed.
- A. Sib and T. A. M. Gulder, Angew. Chem., Int. Ed., 2018, 57, 14650 CrossRef CAS PubMed.
- T. M. Milzarek and T. A. M. Gulder, Commun. Chem., 2023, 6, 187 CrossRef CAS PubMed.
- J. I. Müller and T. A. M. Gulder, Commun. Chem., 2024, 7, 39 CrossRef PubMed.
- K. A. Volp, D. M. Johnson and A. M. Harned, Org. Lett., 2011, 13, 4486 CrossRef CAS PubMed.
- T. M. Milzarek, M. Einsiedler, H. Aldemir, P. M. D'Agostino, J. K. Evers, G. Hertrampf, K. Lamm, M. Malay, A. Matura, J. I. Müller and T. A. M. Gulder, Org. Lett., 2019, 21, 4520 CrossRef CAS PubMed.
- S. A. B. Dockrey, C. E. Suh, A. R. Benítez, T. Wymore, C. L. Brooks and A. R. H. Narayan, ACS Cent. Sci., 2019, 5, 1010 CrossRef CAS PubMed.
- W. van Berkel, N. Kamerbeek and M. Fraaije, J. Biotechnol., 2006, 124, 670 CrossRef CAS PubMed.
- J. Davison, A. Al Fahad, M. Cai, Z. Song, S. Y. Yehia, C. M. Lazarus, A. M. Bailey, T. J. Simpson and R. J. Cox, Proc. Natl. Acad. Sci., 2012, 109, 7642 CrossRef CAS PubMed.
- K. Matsuzaki, H. Tahara, J. Inokoshi, H. Tanaka, R. Masuma and S. Omura, J. Antibiot., 1998, 51, 1004 CrossRef CAS PubMed.
- A. Abood, A. Al-Fahad, A. Scott, A. E.-D. M. S. Hosny, A. M. Hashem, A. M. A. Fattah, P. R. Race, T. J. Simpson and R. J. Cox, RSC Adv., 2015, 5, 49987 RSC.
- J. B. Pyser, S. A. B. Dockrey, A. R. Benítez, L. A. Joyce, R. A. Wiscons, J. L. Smith and A. R. H. Narayan, J. Am. Chem. Soc., 2019, 141, 18551 CrossRef CAS PubMed.
- A. M. Harned, Tetrahedron Lett., 2014, 55, 4681 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Fact sheets of sorbicillinoids, structure determination guide. See DOI: https://doi.org/10.1039/d4np00059e |
| This journal is © The Royal Society of Chemistry 2025 |