
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The role and mechanisms of canonical and non-canonical tailoring enzymes in bacterial terpenoid biosynthesis
Yuya
Kakumu
 ab,
Ayesha Ahmed
Chaudhri
ab and
Eric J. N.
Helfrich
*abc
ab,
Ayesha Ahmed
Chaudhri
ab and
Eric J. N.
Helfrich
*abc
aInstitute for Molecular Bio Science, Goethe University Frankfurt, Max-von-Laue Strasse 9, 60438 Frankfurt am Main, Germany. E-mail: eric.helfrich@bio.uni-frankfurt.de
bLOEWE Center for Translational Biodiversity Genomics (TBG), Senckenberganlage 25, 60325 Frankfurt am Main, Germany
cSenckenberg Gesellschaft für Naturforschung, Senckenberganlage 25, 60325 Frankfurt am Main, Germany
First published on 3rd February 2025
Abstract
Covering: up to April 2024
Terpenoids represent the largest and structurally most diverse class of natural products. According to textbook knowledge, this diversity arises from a two-step biosynthetic process: first, terpene cyclases generate a vast array of mono- and polycyclic hydrocarbon scaffolds with multiple stereocenters from a limited set of achiral precursors, a process extensively studied over the past two decades. Subsequently, tailoring enzymes further modify these complex scaffolds through regio- and stereocontrolled oxidation and other functionalization reactions, a topic of increasing interest in recent years. The resulting highly functionalized terpenoids exhibit a broad spectrum of unique biological activities, making them promising candidates for drug development. Recent advances in genome sequencing technologies along with the development and application of sophisticated genome mining tools have revealed bacteria as a largely untapped resource for the discovery of complex terpenoids. Functional characterization of a limited number of bacterial terpenoid biosynthetic pathways, combined with in-depth mechanistic studies of key enzymes, has begun to reveal the versatility of bacterial enzymatic processes involved in terpenoid modification. In this review, we examine the various tailoring reactions leading to complex bacterial terpenoids. We first discuss canonical terpene-modifying enzymes, that catalyze the functionalization of unactivated C–H bonds, incorporation of diverse functional groups, and oxidative and non-oxidative rearrangements. We then explore non-canonical terpene-modifying enzymes that facilitate oxidative rearrangement, cyclization, isomerization, and dimerization reactions. The increasing number of characterized tailoring enzymes that participate in terpene hydrocarbon scaffold fomation, rather than merely decorating pre-formed scaffolds suggests that a re-evaluation of the traditional two-phase model for terpenoid biosynthesis might be warranted. Finally, we address the potential and challenges of mining bacterial genomes to identify terpene biosynthetic gene clusters and expand the bacterial terpene biosynthetic and chemical space.
 Yuya Kakumu | Yuya Kakumu received his BSc (2018) and MSc (2020) from Gifu University, Japan. Following a short period in industry, he returned to Gifu University as a research associate. Supported by a JSPS research fellowship, he earned his PhD from Gifu University under the tutelage of Prof. Tohru Mitsunaga in 2023. His doctoral studies focused on the metabolomics-based discovery, structural elucidation, and biological activities of plant-derived terpenoids. He is currently a postdoctoral researcher at Goethe University Frankfurt, Germany, working with Prof. Eric J. N. Helfrich. His research is centered around the genome mining-based identification and characterization of bacterial terpenoid biosynthetic pathways. |
 Ayesha Ahmed Chaudhri | Ayesha Ahmed Chaudhri received her BSc from Punjab University and M.Phil. (2020) from Quaid-i-Azam University, Pakistan. She is currently pursuing her PhD at Goethe University Frankfurt, Germany, in the research group of Prof. Eric J. N. Helfrich, supported by an HEC/DAAD fellowship. Her research involves the genome mining-based discovery and functional characterization of non-canonical bacterial terpenoid biosynthetic gene clusters, with an emphasis on tailoring reactions involved in the biosynthesis of complex terpenoids. |
 Eric J. N. Helfrich | Eric Helfrich earned his BSc (2010) from the University of Bonn and his MSc (2013) from the University of Jena, Germany. He pursued his PhD (2017) at ETH Zurich, Switzerland, under the supervision of Prof. Jörn Piel. He received an SNSF Fellowship to conduct postdoctoral research in the lab of Prof. Jon Clardy at Harvard Medical School, Boston, USA. In 2020, he was appointed Professor of Natural Product Genomics at Goethe University Frankfurt, Germany. His research group develops genome mining tools to uncover overlooked biosynthetic gene clusters and investigates non-canonical biosynthetic pathways and enzymatic transformations in terpenoid, peptide and alkaloid biosynthesis. |
1. Introduction
Terpenoids are ubiquitously biosynthesized in nature as primary or secondary metabolites. Their physiological roles range from coenzymes (e.g., Q10) and vitamins (A, D, and K) to hormones (e.g., steroids), pigments (e.g., carotenoids), and defensive metabolites (e.g., momilactones). Terpenoids, with about 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 characterized compounds (Dictionary of Natural Products),1 represent the largest and structurally most diverse class of natural products. Terpenoids can be categorized into more than 400 structural families.1 When taking diastereomers and natural product hybrids (i.e., meroterpenoids) consisting of a terpenoid component and a substructure that is derived from other natural product classes (e.g., polyketides and alkaloids) into consideration, the number of identified compounds rises to 180000 that have been reported from all domains of life including plants, marine invertebrates, fungi, bacteria, and archaea.2 Many terpenoids feature highly oxygenated sp3-rich carbon scaffolds with multiple stereocenters. These three-dimensionally distinct terpenoids frequently show potent biological activities and selectively interact with their molecular target.3,4 In these interactions, hydrogen bonds between highly oxygenated terpenoids and their targeted molecules play a crucial role. Several complex terpenoids have been exploited as drugs in human and veterinary medicine, agrochemicals, and chemical probes for biological studies. Notable examples include the anti-cancer agent taxol,5 the antibiotic pleuromutilin,6 the insecticide azadirachtin,7 and the phosphoinositide 3-kinase inhibitor wortmannin.8 Consequently, the biosyntheses of these complex terpenoids has garnered attention which resulted in the meticulous characterization of a variety of tailoring enzymes that catalyze unprecedented biosynthetic transformations. Moreover, complex terpenoids have served as a source of inspiration for the development of multi-step organic synthesis. Most notable is the recent progress in the development of efficient strategies for the rigorous stereocontrolled functionalization of seemingly undistinguishable aliphatic C–H bonds.9
000 characterized compounds (Dictionary of Natural Products),1 represent the largest and structurally most diverse class of natural products. Terpenoids can be categorized into more than 400 structural families.1 When taking diastereomers and natural product hybrids (i.e., meroterpenoids) consisting of a terpenoid component and a substructure that is derived from other natural product classes (e.g., polyketides and alkaloids) into consideration, the number of identified compounds rises to 180000 that have been reported from all domains of life including plants, marine invertebrates, fungi, bacteria, and archaea.2 Many terpenoids feature highly oxygenated sp3-rich carbon scaffolds with multiple stereocenters. These three-dimensionally distinct terpenoids frequently show potent biological activities and selectively interact with their molecular target.3,4 In these interactions, hydrogen bonds between highly oxygenated terpenoids and their targeted molecules play a crucial role. Several complex terpenoids have been exploited as drugs in human and veterinary medicine, agrochemicals, and chemical probes for biological studies. Notable examples include the anti-cancer agent taxol,5 the antibiotic pleuromutilin,6 the insecticide azadirachtin,7 and the phosphoinositide 3-kinase inhibitor wortmannin.8 Consequently, the biosyntheses of these complex terpenoids has garnered attention which resulted in the meticulous characterization of a variety of tailoring enzymes that catalyze unprecedented biosynthetic transformations. Moreover, complex terpenoids have served as a source of inspiration for the development of multi-step organic synthesis. Most notable is the recent progress in the development of efficient strategies for the rigorous stereocontrolled functionalization of seemingly undistinguishable aliphatic C–H bonds.9
Despite their vast structural diversity, all terpenoids share a common biosynthetic origin.10,11 The biosynthesis of terpenoids begins with the formation of simple C5 isoprene units, isopentenyl pyrophosphate (IPP) and dimethylallyl pyrophosphate (DMAPP), which are synthesized via two distinct pathways: the mevalonate (MVA) pathway12 or the methylerythritol phosphate (MEP) pathway.13 Textbook knowledge describes terpenoid biosynthesis as a “two-step” process comprising a backbone assembly and a hydrocarbon scaffold modification phase (Fig. 1).14
 | ||
| Fig. 1 Schematic overview of the two-phase terpenoid biosynthetic process. Phase 1: terpene hydrocarbon backbone assembly, phase 2: extensive modification of terpene backbone by tailoring enzymes. | ||
In the first phase, the isoprene units are condensed in a head-to-tail fashion by oligoprenyl synthases to produce the methyl-branched acyclic and achiral polyene pyrophosphates, geranyl (GPP, C10), farnesyl (FPP, C15), geranylgeranyl (GGPP, C20), geranylfarnesyl (GFPP, C25), hexaprenyl (HexPP, C30), and heptaprenyl pyrophosphates (HepPP, C35).15,16 Additionally, FPP and GGPP are condensed in a head-to-head fashion to yield squalene (C30) and phytoene (C40), respectively.17 The linear polyenes then undergo a series of carbocation-driven cyclization and rearrangement reactions catalyzed by two canonical types of terpene cyclases (TCs)10,18–20 or non-canonical TCs.21,22 Notably, this enzymatic process results in the formation of numerous mono- or polycyclic hydrocarbon scaffolds with multiple stereocenters.23 For the biosynthesis of meroterpenes, the oligoprenyl pyrophosphates are transferred by prenyltransferases (PTs) onto nonterpenyl molecules such as polyketides and amino acid-derived compounds. The resulting hybrid compounds often undergo cyclization catalyzed by TCs.24
In the second phase, (mero)terpenes undergo extensive modifications by oxidation of double bonds and unactivated C–H bonds, oxidative rearrangements, and incorporation of various functional groups.14,25 These modification reactions result in a myriad of structurally distinct terpenoids. Enzymes responsible for the decoration of terpene hydrocarbon scaffolds are primarily oxidoreductases and transferases, such as cytochrome P450 monooxygenases (P450s), nonheme iron α-ketoglutarate (Fe/αKG)-dependent oxygenases, flavin-dependent monooxygenases (FMOs), NAD-dependent short-chain dehydrogenases/reductases (SDRs), methyltransferases (MTs), acetyltransferases (ATs), and glycosyltransferases (GTs). While most tailoring enzymes are chemo-, regio-, and stereoselective, a subset of specialized tailoring enzymes are multifunctional or promiscuous, leading to the production of a range of structurally similar terpenoids from the same pathway.26–30 Among these enzymes, P450s are particularly noteworthy for their ability to introduce various modifications to the terpene scaffold, such as hydroxylations, multi-electron oxidations, epoxidations, and oxidative structural rearrangements.31
Complex terpenoids have predominantly been isolated from eukaryotes.2 Bacteria, on the other hand, have for the longest time been regarded as incapable of producing complex terpenoids.11 However, with the advancements in analytical techniques, highly complex terpenoids have been discovered from bacterial origins, albeit fewer in number compared to those from eukaryotic origin.32 These bacterial terpenoids often exhibit remarkable biological activities and have the potential to serve as lead compounds for therapeutic applications. Examples of complex terpenoids with promising bioactivities include the immunosuppressant brasilicardin A (1),33 the selective fatty acid synthase inhibitor platensimycin (2),34 the glyceraldehyde-3-phosphate dehydrogenase (GAPDH) inhibitor pentalenolactone (3),35 and persicamidine C (4) which shows anti-SARS-CoV-2 activity (Fig. 2).36 The ever increasing number of publicly available genome sequences in the post-genomic era, along with the development of sophisticated genome mining tools, have revealed that terpenoid biosynthetic gene clusters (BGCs) are widely distributed in bacterial genomes.37,38 Genes encoding TCs in bacterial genomes are frequently co-localized with multiple genes encoding tailoring enzymes, particularly P450s.37,38 As a result, bacteria are increasingly regarded as an almost untapped resource for the discovery of novel complex terpenoids.32 Despite the immense biosynthetic potential to produce complex terpenoids as indicated by genome mining studies, the small number of characterized bacterial terpenoids suggests that most bacterial terpenoid BGCs are silent under standard laboratory culture conditions.
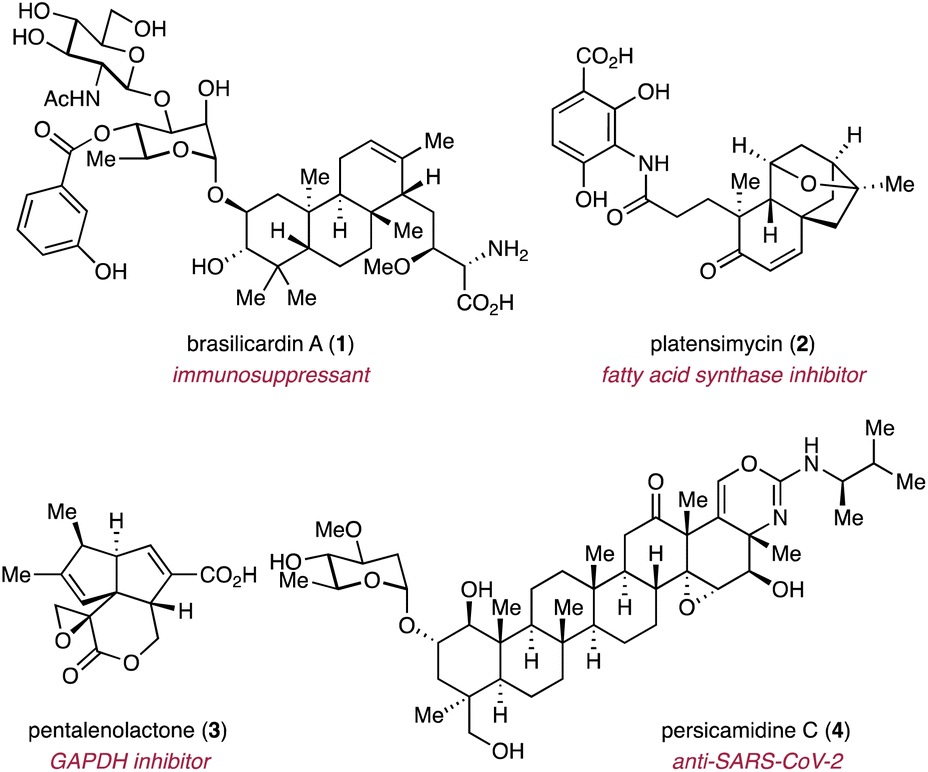 | ||
| Fig. 2 Representative examples of complex bacterial terpenoids with promising biological activities. | ||
Over the past two decades, molecular biology techniques, such as heterologous expression, have emerged as powerful tools for the production of bacterial terpenoids and the characterization of the corresponding biosynthetic pathways. These techniques have not only enabled the identification of numerous highly functionalized terpenoids but also showcased the diverse enzymatic repertoire of tailoring enzymes involved in their biosynthesis. The vast majority of terpene hydrocarbon scaffold modifications involves functionalization of unactivated C–H bonds. However, a subset of tailoring enzymes acts as catalysts for unprecedented transformations, significantly modifying terpene frameworks and resulting in distinct terpenoid scaffolds. This growing body of knowledge on bacterial terpenoid biosynthesis prompts us to review the broad spectrum of characterized tailoring reactions that we divided into canonical and non-canonical modifications.
Canonical modifications involve archetypical functionalizations catalyzed by oxidoreductases and transferases that operate via well-characterized catalytic mechanisms, such as oxygen rebound of iron-dependent enzymes and the methyl transfer reactions to nucleophiles by S-adenosyl-L-methionine (SAM)-dependent MTs. The reaction outcomes are highly conserved within enzyme families, as evidenced by numerous characterized examples. Canonical modifications of the terpene hydrocarbon scaffolds can encompass, but are not limited to, hydroxylations, epoxidations, four or six electron oxidations, dehydrogenations, methylations, glycosylations, benzoylations, and Baeyer–Villiger (BV) oxidations.
Non-canonical modifications, on the other hand, result from tailoring enzymes that employ previously uncharacterized reaction mechanisms to catalyze unprecedented biochemical transformations. These enzymatic activities often arise from neofunctionalization, enabling enzymes to drive key biosynthetic steps in shaping unique skeletal architectures of terpenoids. Examples of non-canonical modifications include oxidative rearrangements, cyclization, dimerization, and nonoxidative cyclization reactions. Furthermore, non-canonical modifications can also encompass the functionalization by members of known enzyme families with unprecedented catalytic activities and enzymes annotated as hypothetical proteins. The catalytic mechanisms underlying non-canonical tailoring enzymes are typically absent from textbooks, as these reactions have only recently been described with a limited number of characterized examples. To date, the majority of terpene biosynthetic studies have focused on the TC-catalyzed cyclization reactions. These canonical and non-canonical cyclizations have been covered in excellent review articles and are hence not addressed in this review.10,18–22
In this review, we will begin with a brief description of a selection of hydrocarbon scaffolds that are biosynthesized across kingdom borders, albeit with different modification patterns and as a result of convergent evolution. The following sections will delve into recent advancements in our understanding of canonical and non-canonical modifications of the terpene hydrocarbon backbone catalyzed by tailoring enzymes in bacterial terpenoid biosynthetic pathways. Each section will focus on a family of tailoring enzymes and will be further subdivided based on individual reaction types catalyzed by members of the enzyme family. Rather than reviewing the biosynthetic models of each bacterial terpenoid family separately, we have structured our review to focus on specific families of tailoring enzymes. This approach allows us to highlight similarities and differences between tailoring reactions and pathways, providing a more cohesive and informative narrative. We provide a brief overview of the biosynthetic model for each terpenoid family upon its first mention before delving into detailed descriptions of the respective tailoring reaction. We believe this structure offers a more insightful perspective compared to the traditional approach of reviewing terpenoid families individually. Our review does not include proteins with previously unknown functions involved in the cyclization phase, e.g., Stig cyclases participating in cyanobacterial hapalindole biosynthesis39 or Pyr4-family transmembrane cyclases involved in meroterpenoid production.40,41 Furthermore, we will discuss the potential and limitation of genome mining strategies for the identification of highly oxidized bacterial terpenoids to further expand bacterial terpene biosynthetic and chemical space in the future.
2. Structural similarity and divergence of bacterial terpenoids and terpenoids of other origin
Terpenoids produced by bacteria often fall into the same structural families as those found in other kingdom of life.32 Since the number of oligoprenyl precursors is limited, it is not surprising that the same hydrocarbon scaffolds are produced across kingdom borders. Despite the commonality of terpene hydrocarbon backbones, most bacterial terpenoids exhibit distinct oxidation patterns when compared to eukaryote-derived terpenoids. However, in certain cases (e.g., the gibberellins), the same modification patterns originate from different pathways as a result of convergent evolution. This section will introduce examples of bioactive terpenoids where the distinct modification patterns observed for bacterial and non-bacterial terpenoids are responsible for, in many cases, non-overlapping bioactivities. These drastic changes in bioactivity between members of a scaffold family highlight the importance of tailoring reactions during the biosynthesis of complex terpenoids.2.1. Gibberellins
Gibberellins are norditerpenoid phytohormones that are widely found in plants, plant-associated fungi, and bacteria.42 In case of plant-associated microorganisms, gibberellins are utilized for the suppression of the plant host's immune response or the promotion of nodule formation.42 Gibberellins that function as phytohormones—GA1, GA3, GA4, and GA7 (5–8)—share the 6/5/6/5-tetracyclic ent-gibberellane scaffold with a C-3β hydroxy group, C-6 carboxylic acid, and γ-lactone bridge (Fig. 3A).43,44 The C-3β hydroxy functionality is crucial for the promotion of plant growth and development.42,43 Plant pathogenetic bacteria such as Xanthomonas oryzae pv. oryzicola produce 7.45–47 In contrast, most nodule-forming symbionts, such as Rhizobium spp., do not produce plant growth-promoting gibberellins and instead produce GA9 (9) that does not act as a phytohormone,46–48 although some rhizobia produce 7.49 The difference in gibberellin production between pathogenic and symbiotic bacteria is hypothesized to be related to their respective types of relationships with plants.42 Plant pathogenetic bacteria might strategically produce 7 to compromise the plant host's immune system to facilitate infection.45,46 Symbiotic bacteria, on the other hand, delegate the C-3β hydroxylation step to their plant hosts so that plants can balance the production of gibberellins.47 This strategy allows symbionts to support plant growth without suppressing the plant's immune system.50 | ||
| Fig. 3 Structural similarity and difference of diterpenoids from bacterial and eukaryotic origins. (A) Gibberellin norditerpenoids that are active or inactive as phytohormones from different taxonomic origins. (B) Selected fusicoccane diterpenoids from bacteria and fungi. (C) Selected clerodane diterpenoids from plants, fungi, and bacteria. (D) Selected eunicellane diterpenoids from soft corals and bacteria. | ||
2.2. Fusicoccane diterpenoids
Fusicoccane diterpenoids are produced by fungi, bacteria, and plants.51 The family features a 5/8/5-tricyclic dicyclopenta[a,d]cyclooctane ring system with characteristic oxidation and modification patterns that are dependent on the producing organism.51,52 Phytopathogenic fungus-derived fusicoccin A (10) and cotylenin A (11) feature a tetra-oxygenated terpene scaffold with a reverse-isoprenylated glycosyl group (Fig. 3B).53–56 Both compounds act as molecular glues that stabilize protein–protein interactions in the eukaryotic 14-3-3 protein family.57 On the other hand, cyclooctatin (12),58,59 found in various Streptomyces spp.,60–63 exhibits a different oxidation pattern compared to the fungus-derived fusicoccane-type diterpenoids such as cotylenol (13) which is an aglycone of 11 (Fig. 3B).64,65 Interestingly, 12 is a potent lysophospholipase inhibitor58 and hybrids of 12 and a polyketide from Streptomyces violascens, e.g., fusicomycin B (14), suppress migration and invasion of human hepatocarcinoma cells through the inhibition of matrix metalloproteases.60 The biological activities of fusicoccane diterpenoids largely rely on the oxidation patterns and the incorporation of other building blocks.2.3. Clerodane diterpenoids
Clerodane diterpenoids are mainly found in plants, where they typically feature a branched carbon chain connected to a decalin core, often forming heterocycles such as furan and furanone rings.66 Furthermore, the stereochemical configuration of the decalin ring is divided into eight types, namely four neo-clerodane types and four ent-neo-clerodane types, providing a wide range of structural diversity.66 Representative examples from plants include the insecticide clerodin (15)67–70 and the potent selective κ-opioid receptor agonist salvinorin A (16).71,72 In contrast, actinobacteria are capable of producing a small family of clerodane-type diterpenoids, such as terpentecin (17)73,74 and UCT4B (18) (Fig. 3C).75,76 Bacterial clerodanes feature a highly oxygenated alkyl chain that exists in equilibrium between the α-ketoformyl aldehyde, hemiacetal, and dioxane dimer forms in solution.77 Interestingly, fungi produce the highly similar diterpenoid clerocidin (19) that features a slightly different oxidation pattern at the decalin ring.77,78 These microbial diterpenoids act as topoisomerase II inhibitors that prevent DNA religation and result in DNA cleavage.75,79–81 This activity is significantly influenced by the presence of the unique oxygenated alkyl chain, wherein a strained epoxide adjacent to a ketolactol residue alkylates guanine bases.82,832.4. Eunicellane diterpenoids
Historically, eunicellane diterpenoids have been almost exclusively identified from soft corals.84–86 Extensive isolation and structure elucidation efforts have shown that most of the coral-derived eunicellane diterpenoids feature a 6/10-cis-bicyclic ring system, which typically contains a transannular ether bridge.84–86 These diterpenoids often exhibit remarkable biological activities, such as the taxol-like microtuble stabilization activity of eleutherobin (20)87,88 and the anti-migration and anti-invasion activities of (+)-polyanthelin A (21).89 Only recently, bacteria have been shown to produce various eunicellane diterpenoids with either cis- or trans-fused bicyclic scaffolds.90 The oxidation patterns of bacterial eunicellanes are distinct from their coral-derived counterparts. It is noteworthy that all previously identified bacterial terpenoids lack the ether bridge on the 10-membered ring, although the number of identified compounds remains relatively small.90 Instead, almost all bacterial eunicellanes feature fully oxidized allyl methyls. Notable examples include the antibacterial benditerpenoic acid (22),91 the antiproliferative microeunicellol A (23),92 and aridacin A (24) with weak cytotoxicity against cancer cells (Fig. 3D).932.5. Other terpenoids
In addition to the above-described examples, plant-like oxygenated eudesmane sesquiterpenoids have been identified from several Streptomyces spp.94–99 Furthermore, among structurally diverse bacterial carotenoids (>300 from ProCarDB),100 some compounds such as β-carotene and astaxanthin are also produced by eukaryotes.101 Actinomycetes produce (iso)pimarane-type norditerpenoids that lack one of the gem-methyls, which is a feature almost exclusively found in (iso)pimaranes of bacterial origin.94,102–104The structural diversity of bacterial terpenoids and their unique oxidation patterns reflect the versatile biosynthetic capabilities of bacteria, demonstrating their potential to produce novel bioactive compounds albeit retaining the common frameworks observed across kingdom borders. Moreover, examples like the gibberellins showcase that some biosynthetic pathways have convergently evolved to produce structurally identical terpenoids using different strategies across the tree of life.
3. Modification of terpene hydrocarbon scaffolds by canonical tailoring enzymes in bacteria
3.1. Oxidation by cytochrome P450s
P450s are heme-thiolate-containing monooxygenases that play a crucial role in the oxidative transformation of members of various natural product classes.105 They are predominantly known to catalyze hydroxylations of unactivated C–H bonds, epoxidations of alkenes, and four- and six-electron oxidations, the formation of aldehydes, ketones, and carboxylic acids, as well as biaryl couplings.106–108 Most P450s require redox partners, e.g., a ferredoxin (Fd) and a ferredoxin reductase (FdR) in bacteria, to obtain single electrons for the reduction of the heme iron species during the catalytic cycle.105 Although the catalytic mechanism of P450s has been extensively reviewed,106–109 we briefly describe the most crucial steps of its archetypical catalytic cycle using C–H hydroxylation reactions through the classical oxygen rebound mechanism as an example. The purpose of this brief introduction is to compare the catalytic cycle of canonical modifications to that of non-canonical modifications, such as skeletal rearrangements (vide infra). In the oxygen rebound mechanism (Fig. 4), a highly electrophilic oxoiron(IV) porphyrin cation radical (Compound I) abstracts a hydrogen atom from the substrate to generate a substrate radical. At the same time, Compound I is converted to a hydroxoiron(IV) intermediate (Compound II). The rebound of the hydroxyl radical from Compound II onto the substrate radical yields the hydroxylated substrate, returning Compound II to the incipient ferric state of the catalytic cycle. The formation of aldehydes, ketones, and carboxylic acids can be explained by multiple rounds of C–H hydroxylation to form diols coupled with H2O elimination. The epoxidation of olefinic bonds is also realized via the radical rebound mechanism (Fig. 4).106 This almost concerted reaction begins with coupling of an olefin and Compound I to yield the heme iron-substrate radical complex, with the components connected by a C–O bond. The resulting iron alkoxy radical intermediate immediately forms a second C–O bond through the addition of an iron(IV) oxyl radical to the alkyl radical, leading to the formation of an epoxide.106 | ||
| Fig. 4 Consensus oxygen rebound mechanism of the P450-catalyzed hydroxylation of unactivated C–H bonds and epoxidation of alkenes. | ||
P450s serve as the archetypical biocatalyst for oxidative functionalization in terpenoid biosynthetic pathways.31 In bacterial genomes, most core biosynthetic genes encoding TCs and oligoprenyl synthases are clustered with one or multiple genes encoding P450s that are likely involved in the decoration of the terpene hydrocarbon scaffold.37,38 The vast majority of these P450s encoded in bacterial terpenoid BGCs have not yet been functionally characterized. However, several studies have demonstrated the diverse catalytic activities of P450s involved in bacterial terpenoid biosynthesis.
3.1.1.1. Cyclooctatin. Cyclooctatin (12) was isolated from Streptomyces melanosporofaceins MI614-43F2.58,59 The corresponding BGC (cot) is comprised of four genes encoding a GGPP synthase (GGPPS), a TC, and two P450s (CotB3, CotB4).114 Heterologous expression of the cot BGC-encoded genes revealed that CotB3 installs a hydroxy group at C-5 of cyclooctat-9-en-7-ol (25) to produce cyclooctat-9-ene-5,7-diol (26). Subsequently, CotB4 hydroxylates C-18 of 26 to yield 12 (Fig. 5).114 These two P450s catalyze regio- and stereospecific reactions with the assistance of an endogenous redox system of the heterologous host. Further studies revealed that the catalytic efficiencies of CotB3 and CotB4 are influenced by the redox system.115 Specifically, the AfR/Afx redox system identified from Streptomyces afghaniensis shows increased compatibility with CotB3 and CotB4 than the Pseudomonas putida-derived PdR/Pdx system. Heterologous expression of CotB3 and CotB4 combined with AfR/Afx results in higher production titers of 12 (15 mg L−1) compared to the native producer (0.35 mg mL−1).115 Additionally, substrate scopes of CotB3 and CotB4 were explored using an Escherichia coli heterologous host harboring genes encoding diterpene TCs and AfR/Afx.115 These combinatorial biosynthetic studies revealed that CotB3 is also capable of hydroxylating a casbane-type diterpene backbone produced by the plant-derived TC JcCSH, leading to the formation of sinulacasbane D (27) (Fig. 5).115 Homologous BGCs of the cot BGC have been identified in the genomes of at least 72 actinomycetes.116In vitro mechanistic investigations of CotB2 homologs revealed that ScCotB2 from Streptomyces collinus Tü365 converts GGPP to collinodiene (cyclooct-5,7-diene) as the main product, along with 25.116 Collinodiene may also serve as a substrate for the P450 homologs of CotB3 and CotB4 in S. collinus.
 | ||
| Fig. 5 Sequential hydroxylations in the cyclooctatin biosynthetic pathway by P450s and substrate promiscuous activity of the P450 CotB3 in dependence of redox partners. | ||
3.1.1.2. Phenalinolactones. Phenalinolactone A (28) is an anti–anti–syn-fused tricyclic perhydrophenanthrene diterpenoid which is decorated with a methyl pyrrolate, a dihydroxy furanone, and a methyl L-amicetose moiety.117 The pla BGC identified in the genome of Streptomyces sp. Tü6071 spans 42 kbp and harbors four P450 genes (plaO2, plaO3, plaO4, plaO5).118 Targeted inactivation of phenalinolactone biosynthetic genes and structure determination of the resulting intermediates indicated the functions of PlaO3, PLaO4, and PlaO5 (Fig. 6).118,119 PlaO3 first catalyzes hydroxylation of the terminal methyl C-19 at the A-ring of the dihydroxyfuranone-bearing PL HS6 (29) to form 30. The installed hydroxy group is further decorated by the acyltransferase PlaP2 to yield 31. Subsequently, PlaO4 converts 31 to 32 by installing a hydroxy group at C-20. The resulting OH-20 is used for the condensation with L-amicetose which is transferred by the glycosyltransferase PlaA6, leading to the formation of 33. Afterwards, the acyltransferase PlaV acetylates at OH-3 to produce phenalinolactone D (34). Finally, PlaO5 hydroxylates C-1 of 34 to yield 28. The remaining PlaO2 is not essential for the biosynthesis of 28.119
 | ||
| Fig. 6 Proposed tailoring steps catalyzed by the three P450s, PlaO3, PlaO4, and PlaO5 in the biosynthesis of phenalinolactone A. | ||
3.1.1.3. Brasilicardins. Brasilicardin A (1), an anti–syn–anti-fused tricyclic diterpenoid glycoside, is a promising immunosuppressive drug candidate.33,120,121 The mechanism of action of 1 is distinct from that of currently used immunosuppressants such as FK506, cyclosporin, and phingolimod.122 Structure–activity relationship studies revealed that the functional groups at C-2, composed of L-rhamnose, N-acetylglucosamine, and 3-hydroxybenzoate, are vital for the immunosuppressive activity of 1.121,123 Functional analyses of the bra gene products from Nocardia strains showed that the P450 Bra6 catalyzes the C-2β hydroxylation of the perhydrophenanthrene intermediate 35 (Fig. 7).124,125 The concomitant transamination of the α-ketoacid catalyzed by the aminotransferase Bra1 results in the production of brasilicardin E (36). The aglycone 36 undergoes methoxylation at C-16, catalyzed by the Fe/αKG-dependent oxygenase Bra0 and the methyltransferase Bra11, followed by extensive functionalization at OH-2 by transferases derived from the bra BGC, ultimately yielding 1.124,125
 | ||
| Fig. 7 Proposed tailoring steps during brasilicardin A biosynthesis. | ||
3.1.1.4. Other terpenoids. In addition to the above mentioned examples, C–H hydroxylation by P450s are found in biosynthetic pathways of various bacterial terpenoids, e.g., 37–43 (Fig. 8).112,113,126 Notably, CryP derived from Crossiella cryophila CGMCC 4.1710 installs a hydroxy group on a bridgehead carbon to yield cryophilain (39). Although hydroxy bridgeheads are frequently found in complex terpenoids, CryP stands out as one of the handful examples that is biochemically validated to oxidate a bridgehead carbon in a stereospecific manner.113
 | ||
| Fig. 8 Selected bacterial terpenoids decorated by P450s. Red oxygen atoms are installed by P450 hydroxylases. | ||
3.1.2.1. Platensimycin. Platensimycin (2) is an antibiotic that selectively inhibits the β-ketoacyl-(acyl-carrier-protein) synthase I (FabF) in bacterial fatty acid biosynthesis.34 It also selectively inhibits the mammalian fatty acid synthase and acts as a potent suppressor of hepatic de novo lipogenesis.127 In comparison to the structurally related ent–seco-atisane diterpenoid platencin (44), an inhibitor of both FabF and β-ketoacyl-(acyl-carrier-protein) synthase III (FabH),1282 harbors an ether bond between the C and D rings of the ent-kaurane derived carbon skeleton, which likely contributes to its selective inhibition of FabF.129 Inactivation of candidate biosynthetic genes and in vitro biochemical characterization experiments revealed the biosynthetic origin of the tetrahydrofuran ring in 2.130,131 The P450 PtmO5 stereoselectively hydroxylates C-11β of 16α-hydroxy-ent-kauranoic acid 45 (Fig. 9).130 The resulting diol 46 undergoes nonenzymatic dehydroxylation of the tertiary alcohol OH-16, forming the tertiary cation IM1. The cation IM1 is subsequently quenched by the C-11 hydroxy group which results in the formation of 11,16-epoxy-ent-kauranoic acid 47.130,131 The C-11 hydroxy group installed by PtmO5 is positioned in close proximity to the generated carbocation, leading to a spontaneous intramolecular SN1 reaction.130 The intramolecular ether formation of 46 is only observed under strong acidic conditions in vitro. Therefore, the possibility that PtmO5 or other enzymes encoded in the ptm BGC are involved in the ether formation in vivo cannot be excluded. After the formation of the ether bridge, 47 undergoes various modifications, including B-ring oxidation (Chapter 3.2.3), A-ring cleavage (Chapter 4.2.5), β-oxidation, and non-terpenyl building block incorporation (Chapter 3.3.4), to yield 2.132
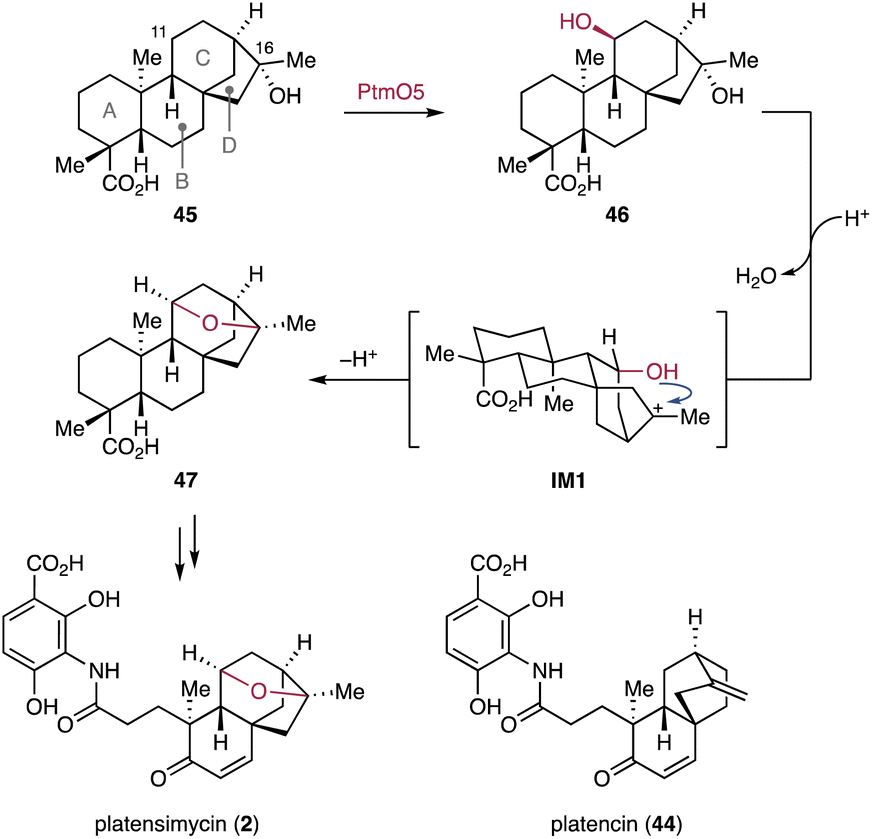 | ||
| Fig. 9 P450-mediated hydroxylation and nonenzymatic ether bond formation in platensimycin biosynthesis. | ||
3.1.3.1. Pentalenolactone. The GAPDH inhibitor pentalenolactone (3) is a sesquiterpenoid lactone that features a densely oxidized 5/5/6/3-tetracyclic ring flamework.133 The ptl gene cluster for the biosynthesis of pentalenolactone-type sesquiterpenoids was identified in the genome of Streptomyces avermitilis MA-4680.134 The hydrocarbon backbone of 3, namely pentalenene (48), is biosynthesized by the TC PtlA from FPP.134 Decoration of the scaffold 48 begins with a multi-electron oxidation of the C-13 methyl by the P450 PtlI (Fig. 10A).135In vitro experiments showed that PtlI catalyzes the oxidation of 48 to yield the alcohol 49, the aldehyde 50, and trace amounts of the carboxylic acid 51.135 Consequently, PtlI was proposed to catalyze a two-step oxidation that forms the aldehyde from the allylic methyl at C-13. Given the low efficient conversion of 48 into 51, it remains unclear whether PtlI or another oxygenase, with appropriate redox partners, catalyzes the oxidation of the aldehyde in 50 to the carboxylic acid in 51.135
 | ||
| Fig. 10 Examples of P450-catalyzed sequential oxidations in bacterial terpenoid biosynthetic pathways. (A) PtlI-catalyzed carboxylation of pentalenenolactone biosynthetic pathways. (B) Ketone formation in albaflavone catalyzed by CYP170A1. (C) Other examples of terpenoids with ketones or carboxylic acids installed by P450s. | ||
3.1.3.2. Albaflavenone. Another prominent example of P450-catalyzed multi-electron oxidations is the CYP170A1-mediated ketone formation during albaflavenone (52) biosynthesis in Streptomyces coelicolor A3(2) (Fig. 10B).136 CYP170A1 performs two sequential allylic oxidations at the C-5 methylene to yield 52. Interestingly, the CYP170A1-catalyzed ketone formation in epi-isozizaene (53) proceeds through the formation of the epimeric albaflavenols (54), both of which are converted to 52.136
3.1.3.3. Venezuelaene B. Venezuelaene B (55) is a fragrant diterpenoid. The ven BGC has been identified in the genome of Streptomyces venezuelae ATCC 15439 (Fig. 10C).137 This compound features an unprecedented 6/5/5/7-tetracyclic ring system with a ketone as the lone oxygenated functionality. Among ven gene products, the P450 VenC catalyzes the formation of the ketone by four-electron oxidation. Interestingly, another BGC-encoded P450, VenB, with 65% sequence homology to VenC shows no enzymatic activity using either 55 or the unmodified hydrocarbon scaffold as a substrate.137
3.1.3.4. Gibberellins. In the biosynthetic pathway of bacterial gibberellins, CYP117 catalyzes a six-electron oxidation of the C-19 methyl in ent-kaurene to yield ent-kaurenoic acid (56) (Fig. 10C).48,138
3.1.3.5. Pimarane-type diterpenoids. The asp BGC that encodes type I and type II TCs (aspT1, aspT2) and two P450s (aspP1, aspP2) was identified from the genome of Actinomadura sp. NAK0032.112 Heterologous expression studies in the S. albus J1074M chassis revealed that the terpene scaffold pimara-8(14),15-diene is first hydroxylated by Asp2 to give a terpene alcohol. The methyl C-18 of the resulting alcohol is then sequentially oxidized by Asp1 to the carboxylic acid (57).112
3.1.4.1. Cyslabdans. Cyslabdans are hybrids of a labdane diterpenoid and an acyl cysteine linked via a thioether bond.141–143 These compounds have been isolated from Streptomyces cyslabdanicus K04-0144. They enhance the activity of imipenem against methicillin-resistant Staphyrococcus aureus.144 Genome mining identified the cld BGC comprised of four genes encoding a GGPPS, type I and II TCs, and a P450.145 Subsequent heterologous expression of the cld genes in Streptomyces avermitilis SUKA22 indicated that the P450 CldC catalyzes hydroxylation at C-7 and epoxidation at the C-8/C-17 olefin of labda-8(17)-12,14-triene (58) to yield 59 (Fig. 11A). The epoxide of 59 is opened by nucleophilic addition of mycothiol. The resulting mycothiol derivative is then hydrolyzed by a mycothiol-S-conjugate amidase to yield cyslabdan A (60). The rmn and lab BGCs were discovered as homologous BGCs of the cld BGC in the genomes of Streptomyces anulatus GM95 and Streptomyces sp. KIB 015, respectively.145,146 Interestingly, the P450s RmnC and LabC, homologs of the multifunctional CldC, are monofunctional and only install a hydroxy group at C-7β of 58 to form raimonol (61) (Fig. 11A).145,146
 | ||
| Fig. 11 Representative examples of bacteria terpenoid functionalization by multifunctional P450s. Proposed biosynthesis of cyslabdan A and raimonol (A), ent-sandarapimaradiene derived diterpenoids (B), and aridacins (C). | ||
3.1.4.2. Nor-ent-sandaracopimaradiene diterpenoids. The nor-ent-sandaracopimaradiene diterpenoids have been identified through heterologous expression of the vsp BGC encoded in the genome of Verrucosispora sp. NA02020.112 Functional characterization of the vsp BGC revealed that the P450 VspP is a versatile oxygenase involved in all oxidative modifications of the ent-sandaracopimaradiene scaffold (62).112 Heterologous expression of the type I and II TC genes (vspT1 and vspT2) of the vsp BGC in S. albus J1074M afforded 62.112 The expression of vspP alongside vspT1 and vspT2 in S. albus led to the production of several oxidized diterpenoids, including 63–66 (Fig. 11B).112 Based on the characterized products, VspP was proposed to be a mutifunctional oxygenase that catalyzes ketone formation at C-2 and six electron oxidations of the methyl C-19 in 62, resulting in the formation of 64.112 Furthermore, VspP decarboxylates C-19 of 64 and subsequently hydroxylates C-18 to form 66.
3.1.4.3. Aridacins. The recent discovery and biosynthetic studies of aridacins, eunicellane-type diterpenoids from Amycolaptosis arida CGMCC 4.5579, led to the identification of one of the highest oxidizing P450s, AriD, to date.93 AriD forms a glyceric acid moiety from an isopropenyl residue of the 6/7/5-tricyclic arida-3,6,15-triene scaffold (67) to produce aridacin A (24) (Fig. 11C). The formation of the glyceric acid moiety was proposed to result from carboxylation of the C-17 methyl and epoxidation of the C-15 alkene, followed by epoxide ring opening through the addition of H2O.93 Additionally, AriD is also capable of hydroxylating the C-19 methyl in 24 to form aridacin B (68). Another P450, AriG, catalyzes the hydroxylation of the methylene C-12 in 24, yielding aridacin C (69).93 Although the catalytic activity of AriD likely follows the classic oxygen rebound mechanism, the incorporation of at least four oxygens in the terpene hydrocarbon backbone is quite rare.
3.2. Oxidation by other oxidoreductases
In bacterial terpenoid biosynthesis, the vast majority of enzymes that decorate the hydrocarbon scaffold during the modification phase are P450s. However, many large bacterial terpenoid BGCs also harbor genes encoding other oxidoreductases, such as FMOs, Fe/αKG-dependent oxygenases, and SDRs. These oxidoreductases are frequently utilized in the biosynthesis of highly oxidized terpenoids.3.2.1.1. Sesquisabinene-type sesquiterpenoids. The recently identified net BGC in Streptomyces netropsis DSM 40259 harbors genes encoding a type I TC (netT), a P450 (netP), an acetyltransferase (netA), and a FMO (netO).112 Based on the functional characterization of the net BGC by heterologous expression, NetO epoxidizes the terminal olefin C-10/C-11 of the hydrocarbon scaffold 7-epi-cis-sesquisabinene hydrate (70) generated by NetT (Fig. 12).112 The stereoselectivity of the NetO-catalyzed epoxidation remains undetermined, as the products isolated from the co-expression of netT and netO are diastereomeric diols resulting from epoxide hydrolysis of 71 in the heterologous host.112 The epoxidation of the dimethyl allyl group by NetO resembles the epoxidation of FMOs acting on oligoprenyl pyrophosphates and squalene to set up type II TC-catalyzed cyclization reactions. In contrast, NetO is a tailoring enzyme that epoxidizes the cyclized terpene scaffold 70. The P450 NetP is proposed to convert 71 into the ketone-containing intermediate 72. Alternatively, it has been suggested that the enzyme order can be reversed: 70 might be first converted to the ketone-containing intermediate 73 by NetP, followed by the NetO catalyzed epoxidation to yield 72 (Fig. 12).112
 | ||
| Fig. 12 Oxidative functionalization of sesquisabinene hydrate by the FMO NetO and the P450 NetP. | ||
3.2.2.1. Pentalenolactones. Pentalenolactone (3) and neopentalenoketolactone (74) are representatives of highly oxygenated sesquiterpenoids isolated from various Streptomyces sp.133,150 The structure of 3 bears an epoxidized δ-lactone moiety that is likely derived from the methyl cyclopentane ring of 1-deoxypentalenoic acid (51) through ketone formation followed by BV oxidation. The biosynthetic transformations from 51 to pentalenolactone F (75) and 74 were thoroughly characterized using a combination of in vitro biochemical assays of tailoring enzymes, gene inactivation, and gene overexpression studies as well as single X-ray crystallography of the Fe/αKG-dependent oxygenase PtlH.150–154 BGCs, named ptl, pnt, and pen, were characterized from the genomes of S. avermitilis, Streptomyces arenae Tü 469, and Streptomyces exfoliatus UC5319, respectively.134,151 After the production of 51 through a six electron oxidation (Fig. 10A), PtlH (PntH, PenH) hydroxylates the methylene C-11 to form 1-deoxy-11β-hydroxypentalenic acid (76). The alcohol 76 is subsequently converted to 1-deoxy-11-oxopentalenic acid (77) by the SDR PtlF (PntF, PenF) (Fig. 13).152–154 The BVMO PntE (PenE) then catalyzes the BV oxidation of 77 to afford pentalenolactone D (78).151 The orthologous BVMO PtlE unexpectedly shows the opposite regioselectivity of BV oxidation on the ketone of 77, resulting in the production of neopentalenolactone D (79) (Fig. 13).150 Since the BVMOs involved in pentalenolactone and neopentalenoketolactone biosynthesis share high sequence identity, crucial amino acid residues that dictate the distinct regioselectivity of PntE (PenE) and PtlE remain unknown.151 Nevertheless, it is the distinct regioselectivity of these BVs that is the bases for the diverging pathways of the pentalenolactones and neopentalenoketolactones.151
 | ||
| Fig. 13 Biosynthetic pathways of pentalenolactone F and neopentalenoketolactone branched by BVMO-mediated lactone formation. | ||
3.2.3.1. Pentalenolactone. In the biosynthetic pathways of the pentalenolactone family of sesquiterpenoids, pentalenolactone D (78) and neopentalenolactone D (79) are structurally diversified by multi-step oxidizations catalyzed by the multifunctional Fe/αKG-dependent oxygenases PntD (PenD) and PtlD, respectively (Fig. 13).151 In these pathways, PntD (PenD) desaturates 78 to form pentalenolactone E (80) and epoxidizes the generated exo-methylene to produce 75.151In vitro biochemical studies using H218O or 18O2 and site-directed mutagenesis of PtlD confirmed its remarkable multifunctionality in the structural diversification of neopentaketolenolactones.155,156 PtlD converts 79 into either neopentalenolactone E (81) by dehydrogenation or the ketoacid product 82 by hydroxylation-mediated δ-lactone cleavage. Notably, in the context of the PtlD-catalyzed desaturation reaction, a hydrogen at C-9 is abstracted by the iron(IV)-oxo species and the subsequent electron transfer yields a carbocation intermediate, which is stabilized by the π–cation interaction with the active site residue Tyr113 in PtlD.156 The carbocation intermediate is then converted to the oxocarbenium ion, facilitating the deprotonation of H-10 by the basic nature of Lys288 in PtlD to give 81.156 The exo-methylene 81 is epoxidized by PtlD to yield the epoxyketal IM2, which immediately undergoes a spontaneous rearrangement reaction to form the ketoester 74 or a hydrolytic reaction to yield the δ-lactone-opened product 83.155 The remarkable functionality of PtlD in catalyzing different reactions with various substrates gives rise to the complex and branched biosynthetic pathways of neopentalactone-related sesquiterpenoids.
3.2.3.2. Hapalindoles. In bacterial terpenoid biosynthesis, a representative example of Fe/αKG-dependent oxygenases is the enantioselective halogenation of hapalindoles. The family of hapalindoles is composed of structurally diverse terpenoid indole alkaloids produced from stigonematalean cyanobacteria Fischerella spp. and Hapalosiphon spp.39 The widespread occurrence of C-13 chlorinated hapalindoles suggests that chlorination is likely an early functionalization step in the oxidative maturation of hapalindole alkaloids (Fig. 14). The wel and amb BGCs for welwitindolinones and ambiguines have been identified in the genomes of Hapalosiphon welwitschii UTEX B1830 and Fischerella ambigua UTEX1903, respectively. Each BGC harbors genes encoding five nonheme iron-dependent oxygenases including a Fe/αKG-dependent oxygenase (welO5 and ambO5).157,158In vitro enzymatic assays demonstrated that WelO5 stereospecifically converts 12-epi-fischerindole U (84) and 12-epi-hapalindole C (85) to the chlorinated products, 12-epi-fischerindole G (86) and 12-epi-hapalindole E (87), respectively (Fig. 14).157,159 In addition to the chlorination, in vitro enzymatic assays showed that WelO5 is also capable of incorporating bromine in 84.160 The apo and holo structures of WelO5 provide insights into the catalytic mechanism of the stereoselective halogenation that requires ligand rearrangement.161 Mutating active site residues in WelO5 indicated that a single residue (Ser189) plays a key role in determining the selectivity of the halogenation reaction through regulating the position of an oxygenic group in the Fe(IV)-oxo intermediate by hydrogen bonding.161 In contrast to WelO5, the homologous AmbO5 (79% id) chlorinates the C-13 position of various premature hapalindole scaffolds.162 AmbO5 converts structurally distinct hapalindoles 84, 85, and 88–90 into the C-13 chlorinated molecules 86, 87, and 91–93, respectively (Fig. 14). Sequence comparison of WelO5 and AmbO5 and mutagenesis of WelO5's C-terminal residues revealed that the distinct C-terminal 18 amino acids determine the substrate scope.162
 | ||
| Fig. 14 Site-selective halogenation by Fe/αKG-dependent halogenases in the biosynthesis of hapalindoles. | ||
3.2.4.1. Platensimycin and platencin. In the biosynthesis of platensimycin (2) and platencin (44), stereospecific hydroxylation through cryptic carbonylation is orchestrated by the redundant αKG-dependent oxygenase pair PtmO3/PtmO6 and SDR family enzymes PtmO8 and PtmO1, a pair of NAD+/NADPH-dependent dehydrogenases.167 Targeted gene inactivation and in vitro enzyme assays revealed that the C-7 oxidation and epimerization are initiated by the Fe/αKG-dependent oxygenases PtmO3/PtmO6 that convert 47 and 94 to 95 and 96, respectively, that each harbor a C-7β hydroxy group (Fig. 15A).167 The C-7β hydroxy groups of 95 and 96 are oxidized by the SDR PtmO8 to the C-7 carbonyls 97 and 98. Subsequently, 97 and 98 are reduced by PtmO1 to afford the C-7α hydroxy 99 and 100, respectively, resulting in a net epimerization of the C-7 hydroxy group.167 The discovery of a three-enzyme cascade responsible for stereospecific hydroxylation, oxidation, and reduction at C-7 explains the origin of the C-7 oxidation patterns commonly found in ent-kaurane and ent-atisane-derived diterpenoids.167
 | ||
| Fig. 15 B-ring functionalization of ent-kaurane and ent-atisane diterpenoids by oxidoreductases. (A) Cryptic carbonylation and hydroxy epimerization in the platensimycin and platencin biosynthetic pathways. (B) SDR-assisted conversion of an aldehyde into a carboxylic acid moiety during bacterial gibberellin biosynthesis. | ||
3.2.4.2. Gibberellins. The characteristic carbon backbone of the gibberellins is derived from an ent-kaurane scaffold through contraction of the B-ring (vide infra). In the bacterial gibberellin biosynthetic pathway, the P450 CYP114 from Sinorhizobium fredii NGR234 converts 56 to GA12-aldehyde (101) along with a low amount of GA12 (102) (Fig. 15B).48 Heterologous expression and gene inactivation experiments have demonstrated that the SDRGA efficiently catalyzes the conversion of the aldehyde 101 to the carboxylic acid 102 (Fig. 15B).48 The unique tandem enzymatic conversion in bacterial gibberellin biosynthesis differs from that in plants and fungi, where P450s catalyze both the ring contraction and the complete conversion of the extruded C-7 methylene into a carboxylic acid moiety.48
3.3. Functionalization by transferases
Transferase-encoding genes are mostly found in BGCs associated with highly functionalized terpenoids where they further decorate terpene scaffolds by installing functional groups including methyls, sugars, and aromatic compounds onto functional groups installed during the decoration phase. In most cases, these BGCs are relatively large (>10 kb) because the genes encoding enzymes for the biosynthesis of the building blocks to be transferred typically cluster with genes encoding terpene synthases, transferases, and oxygenases that install the handles for building block attachment. | ||
| Fig. 16 Selected bacterial terpenoids that are modified by the incorporation of non-terpenyl building blocks. | ||
3.4. Modification of terpene precursors
Modifications introduced by tailoring enzymes do not always occur after cyclization of the oligoprenyl precursors. In some cases, MTs and P450s act as modifying enzymes involved in either the assembly of unusual oligoprenyl pyrophosphates or the construction of novel hydrocarbon backbones through cyclization reactions (see Chapters 4.1.5 and 4.2.7).93,176–179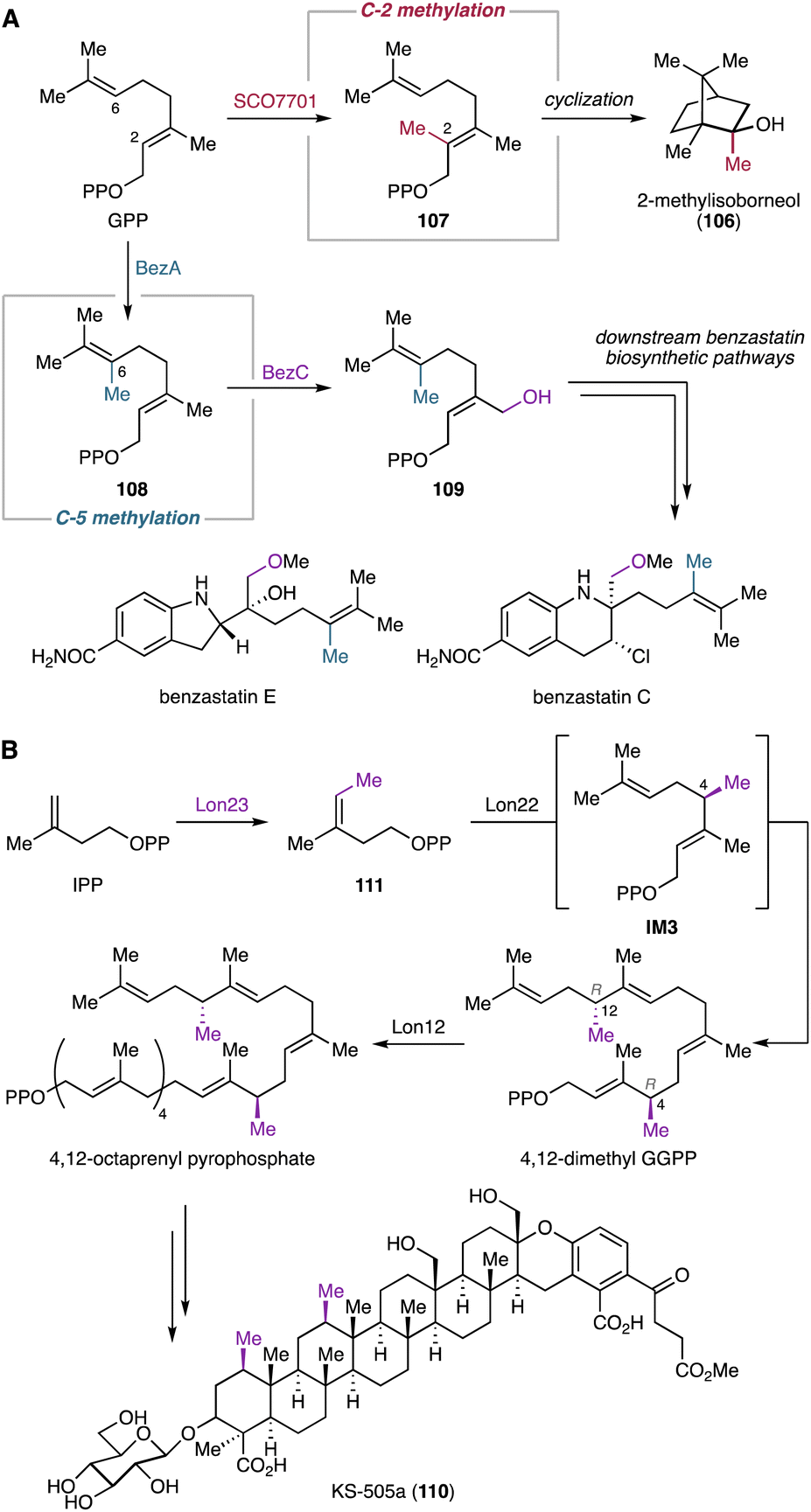 | ||
| Fig. 17 Early-stage modification of oligoprenyl pyrophosphates by MTs before the formation of cyclic terpene hydrocarbon backbones. (A) Biosynthetic models of 2-methylisoborneol and benzastatins. (B) Biosynthetic pathway of KS-505a. | ||
4. Modification of terpene hydrocarbon scaffolds by non-canonical tailoring enzymes in bacteria
4.1. Non-canonical modifications by P450s
4.1.1.1. Pentalenolactone. Biosynthetic studies on pentalenolactone (3) revealed an oxidative rearrangement from 75 to 3 catalyzed by the P450 PntM (PenM).192,193 Typically, neopentyl radicals generated through P450 catalysis do not cause skeletal rearrangements.194 Given the structures of shunt products biosynthesized in trace amounts from pentalenolactone-producing Streptomyces spp.,195,196 the PntM-catalyzed oxidative methyl migration was proposed to proceed through the formation of a neopentyl cation (Fig. 18).192 Specifically, the neopentyl radical IM4 generated by H-1 Si hydrogen abstraction undergoes a rapid electron transfer from IM4 to the highly oxidizing heme FeIII–OH radical cation to produce the neopentyl cation IM5. This cation facilitates the syn-1,2-methyl shift and the antarafacial deprotonation of H-3α, resulting in the formation of the final product 3 that features the vic-dimethylcyclopentene ring (Fig. 18).192
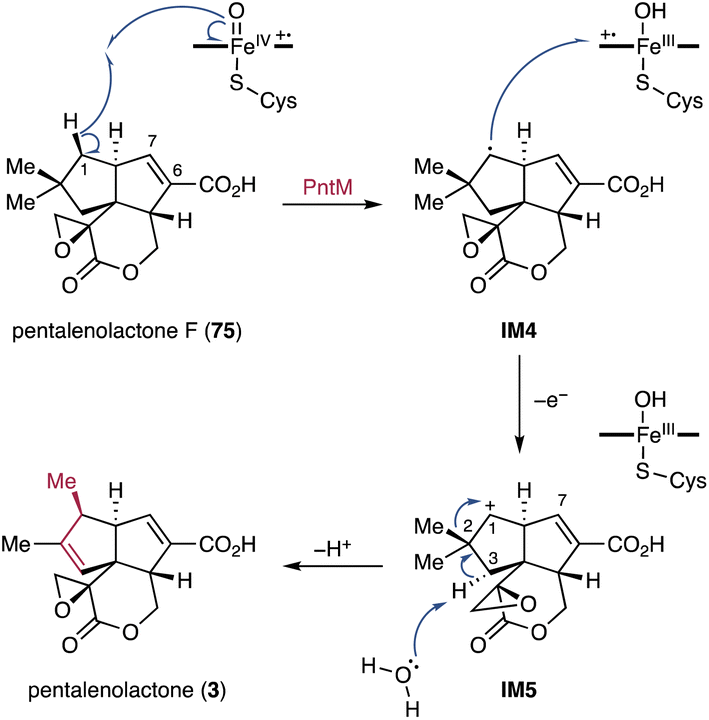 | ||
| Fig. 18 PntM-catalyzed oxidative methyl migration in the pentalenolactone biosynthetic pathway. | ||
X-ray structure analyses of the wild-type and single amino acid mutated PntM variants imply that a complex network of noncovalent interactions tightly regulates the positioning of both substrates and products so that the Si face of C-1 is located close to the heme iron species.193 The appropriate orientation of 75 is further retained by steric hindrance caused by the C-2 methyl and C-7 olefin adjacent to the C-1 Si face. The typical oxygen rebound of P450s requires the rapid rotation of the Fe-bound OH group formed after hydrogen abstraction, and the rate constant for oxygen rebound is >1010–1011 s−1.197–199 In contrast, the thermodynamically unfavorable electron transfer to the heme FeIII–OH radical species has a significantly slower rate (<108–109 s−1).200 However, in the case of a radical center with β-branched alkyl groups such as the neopentyl radical, oxygen rebound is extremely slow because of the steric hindrance and thus the electron transfer is compatible with oxygen rebound.200,201 The naturally occurring steric barrier in 75 due to the presence of the C-2 gem-dimethyls and H-7 olefinic proton makes the kinetically insignificant electron transfer favorable, resulting in the dominant production of 3.193 In addition, quantum mechanical/molecular mechanics (QM/MM) calculations support that the electron transfer process for the formation of the C-1 cation IM5 from IM4 is favorable compared to the oxygen rebound mechanism because the overlap between the π* orbital of Fe–OH and the σz2 orbital of the C-1 radical in IM4 is blocked by the presence of H-2.202 The migration reaction is likely terminated by a H-3α abstraction, wherein a water molecule that shows hydrogen bonding with the carboxylic acid of porphyrin acts as Brønsted base.193,202
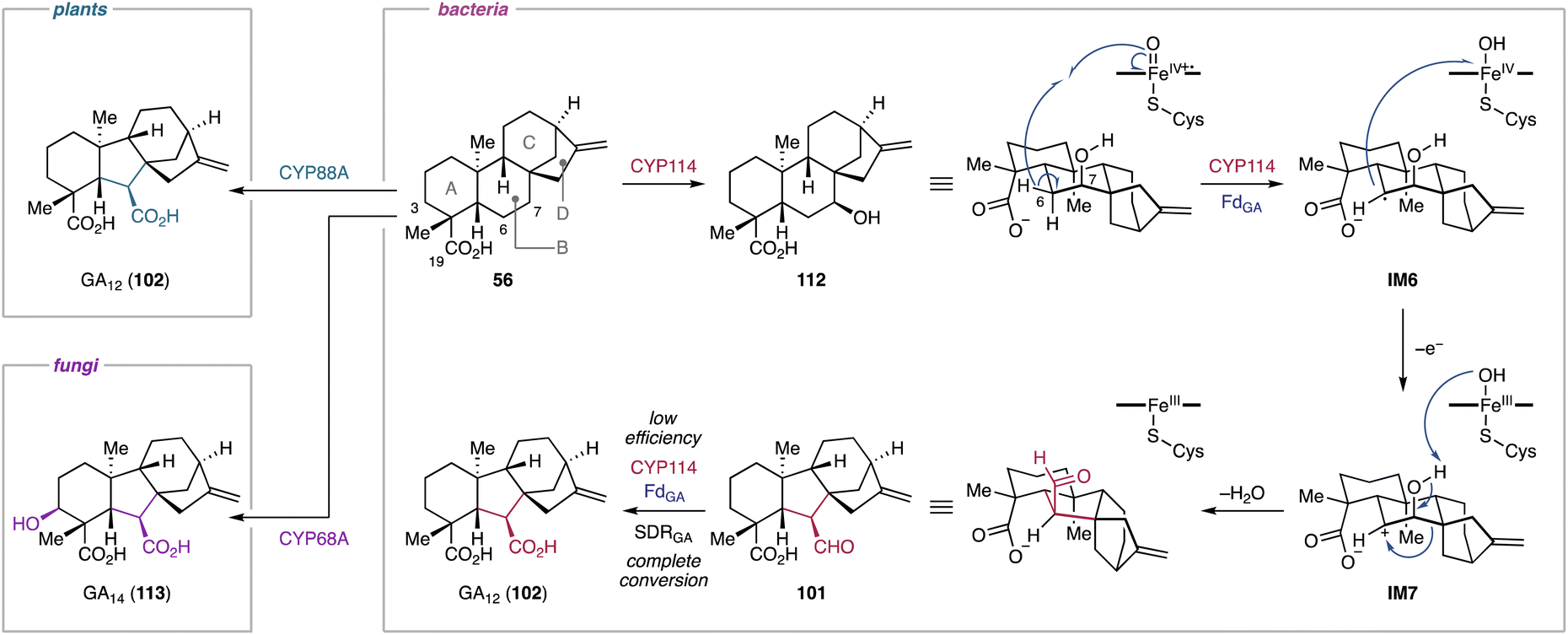
4.1.2.1. Gibberellins. A prominent example of a semipinacol rearrangement can be found in the biosynthesis of gibberellins. Its characteristic 6/5/6/5-tetracyclic carbon framework is derived from the contraction of the B-ring of the ent-kaurane-type hydrocarbon skeleton.212 Isotope-labeling studies of fungal and plant gibberellin biosynthesis revealed that the semipinacol rearrangement-based B-ring contraction proceeds through the abstraction of H-6β in ent-7β-hydroxykaurenoic acid (112), forming the radical intermediate IM6.213 A subsequent single electron transfer from IM6 to the hydroxoiron(IV) species generates the secondary cation IM7.213 This intermediate undergoes an 1,2-alkyl migration onto the C-6 carbocation, ultimately leading to the formation of GA12-aldehyde (101) (Fig. 19).213–217 The P450s responsible for this rearrangement reaction have been identified from both plants and fungi.42 Furthermore, recent studies on the bacterial gibberellin biosynthetic pathways provided insights into the enzymatic structure–function relationships underlying the semipinacol rearrangement-based ring contraction reaction.218
 | ||
| Fig. 19 Semipinacol rearrangement-based ring contraction in the gibberellin biosynthetic pathways in bacteria, plants, and fungi. | ||
Functional characterization of biosynthetic enzymes encoded in the gibberellin BGC in S. fredii revealed that CYP114 not only catalyzes the hydroxylation at C-7 of ent-kaurenoic acid (56), but also the B-ring contraction of 56 to produce 101 and trace amounts of GA12 (102).48 The ring contraction reaction catalyzed by CYP114 requires the redox partner FdGA which is encoded in the gibberellin BGC.48 In the absence of FdGA, the enzymatic activity of CYP114 is limited to C-7β hydroxylation with the help of an endogenous Fd from the heterologous host.48 The relationships between CYP114 and FdGA are unique compared to their counterparts from plants and fungi, where the P450s do not require a dedicated redox partner and rather use ubiquitous cytochrome P450 reductases for their ring contraction activity.219–221 Furthermore, the enzymatic capability of plant- and fungi-derived P450s (CYP88A/CYP68A) encompass the full oxidation of the extruded C-7 into a carboxylic acid moiety. The fungal P450, CYP68A, exhibits an additional hydroxylation activity at C-3β of 102 to yield GA14 (113) (Fig. 19).42 Nevertheless, the substrate and the unique rearrangement reaction of the P450s are conserved beyond kingdom borders. In light of the structural rearrangements observed in plant and fungal gibberellin biosynthetic pathways, the contraction reaction of the B-ring was hypothesized to be a semipinacol rearrangement.48 Feeding experiments of ent-kaurenal and ent-kaurenoic acid methyl ester revealed that the C-19 carboxylic acid anchimerically assists in stabilizing the C-6 carbocation of IM7 that is presumably generated during the semipinacol-type rearrangement.48 Further isotope labeling studies supported the CYP114-catalyzed semipinacol rearrangement extruding C-7 from the B-ring of 112via H-6β abstraction and 1,2-alkyl migration (Fig. 19).222
Recently, the reaction mechanism of the CYP114-catalyzed ring contraction has been elucidated.218In vitro enzymatic assays and in vivo feeding experiments indicated that EtCYP114 from Erwinia tracheiphila converts 56 to 101 along with 112 and 102 in the presence of the redox partner EtFdGA from E. tracheiphila or BjFdGA from Bradyrhizobium japonicum. However, EtCYP114 loses its ability to contract the B-ring when incubated with spinach Fd, which results exclusively in the formation of 112. While EtCYP114 is able to accommodate both 56 and 112, its ring contraction activity is limited to using 56 as a substrate.47,218,222 Structural analysis of EtCYP114 bound to 56 showed the positioning of H-7β (3.7 Å) and H-6β (3.9 Å) in close proximity to the heme iron. The crystal structure of EtCYP114 shows a notable lack of ionic interactions at the C-18 carboxylic acid, which provides the anchimeric effect for the C-6 carbocation of IM7. This interaction strongly supports the carbocation-mediated semipinacol rearrangement reaction (Fig. 19), although an unusual radical mediated rearrangement cannot be ruled out.218 Furthermore, EtCYP114 lacks the highly conserved acid–alcohol motif (e.g., Asp–Thr) that facilitates sequential protonation of the heme iron species in P450s.218,223 When the acidic amino acid residue is introduced into EtCYP114, the enzyme completely loses its catalytic ability to contract the ring.218 These results along with the apo structure of EtCYP114:A261D suggested that complementation of the acid–alcohol motif in EtCYP114 may alter the hydrogen-bonding network within the active site, potentially disrupting its interaction with the dedicated redox partner FdGA.218 Consequently, the ring contraction reaction catalyzed by CYP114 requires both the characteristic redox partner and the absence of the acidic amino acid residue typically conserved in other P450s.48,218
4.1.3.1. Gibberellins. Many gibberellin family norditerpenoids possess a γ-lactone bridge at the A-ring.212 The formation of the lactone bridge with the concurrent loss of C-20 in 102 or 113 is catalyzed by the Fe/αKG-dependent oxygenase GA20ox in plants or the P450 CYP68B in fungi.42 In a similar manner, CYP112 in bacteria is capable of converting 102 into GA9 (9).48
In vitro enzymatic assays and 18O2 labeling studies were conducted to gain insights into the CYP112-catalyzed C-20 dissociation and γ-lactone formation.230EtCYP112 converts 102 into 9 by decarboxylation of C-20 in the presence of spinach Fd, E. tracheiphila derived EtFdR, and NADPH. The mixed origin of the γ-lactone oxygen from molecular oxygen and the C-18 carboxylic acid indicates that CYP112 is a multifunctional enzyme that catalyzes demethylation and γ-lactone formation via two proposed diverging routes (Fig. 20).230 Initially, CYP112 hydroxylates the terminal methyl C-20 of 102 to form 20-hydroxy-GA12 (114), followed by dehydration to yield the δ-lactone-harboring GA15 (115). Subsequently, 114 and 115 each follow two sequential oxidation reactions along separate routes (routes A and B) (Fig. 20). In route A, CYP112 first converts 114 into the gem-diol form (116) of GA24 through the oxygen rebound mechanism. Furthermore, H-20 of 116 is abstracted by Compound I of CYP112, generating the radical intermediate IM8. Compound II then abstracts the hydrogen of the C-18 carboxylic acid in IM8. The resulting diradical undergoes intramolecular radical coupling, yielding the gem-diol cyclic anhydride 117 of GA25. In route B, 115 undergoes two rounds of hydroxylation at C-20 to form 117 through the formation of the lactol form (118) of GA24. The key intermediate 117, convergently produced from both proposed routes, is further oxidized by CYP112 to obtain 9. Specifically, the hydrogens of the C-20 gem-diol are sequentially abstracted by the heme iron species, which induces a radical-based rearrangement of IM9 to form the carbonate diradical IM10. Subsequent intramolecular radical coupling results in the formation of 9 through decarboxylation at C-20 (Fig. 20).230 The unusual diradical-mediated C–C bond scission and lactonization in bacterial gibberellin biosynthesis are remarkable for their similarity to the corresponding reactions in plant and fungal-derived gibberellin biosynthetic pathways.213,231 Despite the involvement of evolutionally independent oxygenase families (P450s vs. αKG-dependent oxygenases) or P450 classes (cytosolic vs. membrane-bound), these enzymes showcase that tailoring enzymes for the biosynthesis of terpenoids have converged to utilize similar mechanistic routes across different branches of life.230
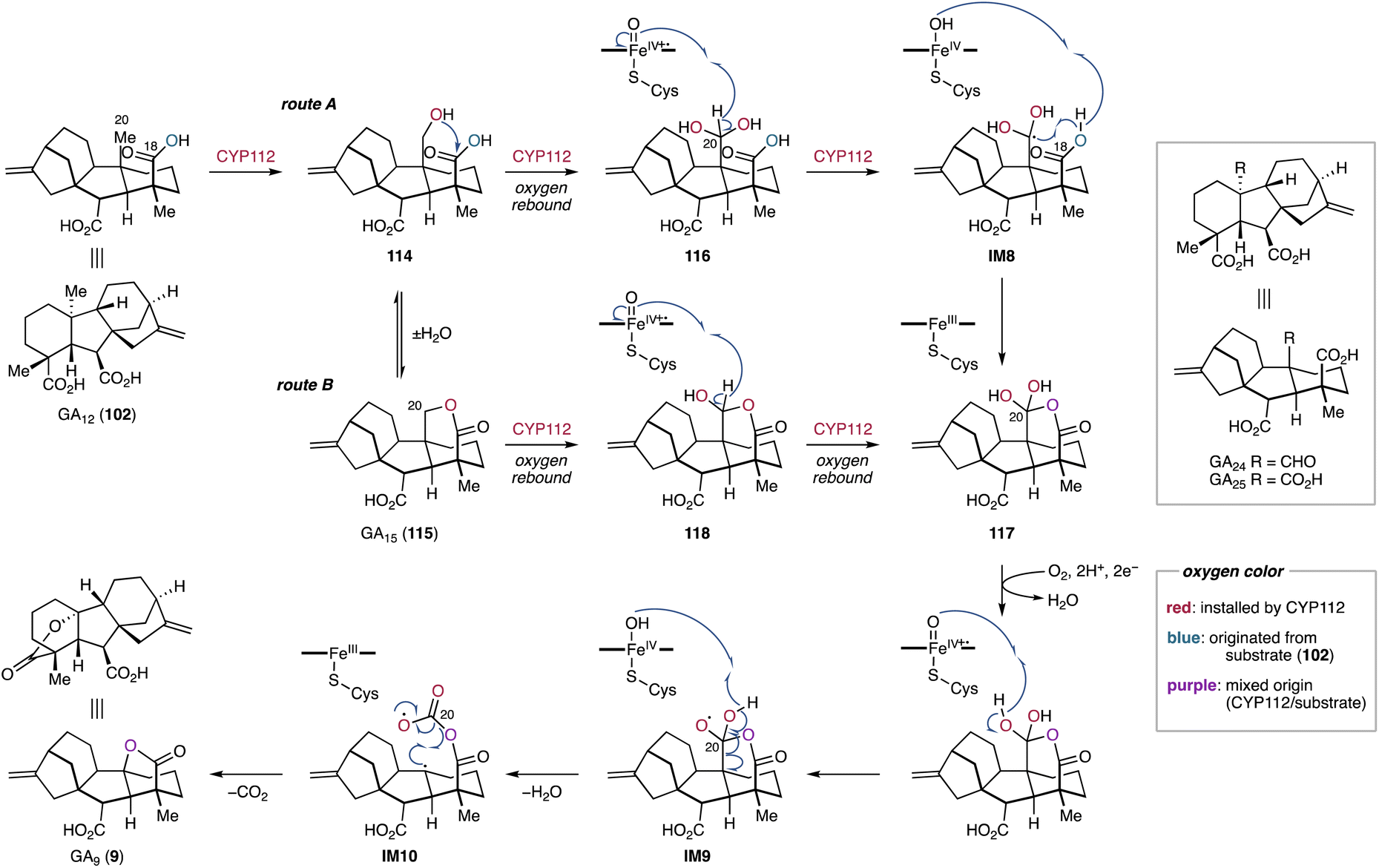 | ||
| Fig. 20 CYP112-catalyzed C-20 elimination and γ-lactone formation in the biosynthetic pathway of bacterial gibberellins. | ||
4.1.4.1. Benzastatins. Benzastatins are p-aminobenzoic acid-derived meroterpenoids with neural cell protective and antiviral activities.234–237 Most of benzastatins bear tetrahydroquinoline or indoline scaffolds which result from the cyclization of a geranyl moiety. The benzastatin BGC and its homologs have been identified in the genomes of Streptomyces sp. RI18 and several other actinobacteria.179 Heterologous expression of bez genes in Streptomyces lividans and in vitro enzymatic assays have led to the characterization of the benzastatin biosynthetic pathway.179 The pathway is initiated by the N-hydroxylase BezJ and the AT BezG, which sequentially catalyze N-hydroxylation and O-acetoxylation of p-aminobenzoic acid (119), respectively, to form p-N-acetoxybenzoic acid (120). The PT BezF then incorporates modified GPPs 108 or 109 into 120, resulting in the formation of geranyl p-N-acetoxybenzoic acid (121), a bona fide intermediate of the cyclized benzastatins.179 The highly unstable 121 is nonenzymatically converted into the indoline 122 and the tetrahydroquinoline 123 with low efficiency, as well as the dihydrobenzo[b]azepine shunt product 124 (Fig. 21).179 The production of 122–124 likely involves the nonenzymatic generation of the arylnitrenium ion IM11 through hydrolysis of the N-acetoxy moiety. This highly electrophilic ion reacts with the C-9/C-10 olefin to generate an azirinium ion, which is hydrolyzed to yield 122 and 123. Alternatively, the arylnitrenium ion IM11 reacts with the C-17 methyl of the geranyl moiety to form 124.179 More importantly, the P450 BezE has been identified as the heterocyclization catalyst that mediates the formation of the benzastatins (122, 125). During the cyclization reaction, BezE does not require a redox partner for its catalytic cycle, indicating a nonoxidative reaction mechanism.179
 | ||
| Fig. 21 Nonoxidative heterocyclization reaction catalyzed by the P450 nitrene transferase BezF in the benzastatin biosynthetic pathway. | ||
Mechanistically, the BezE-catalyzed nonoxidative heterocyclization reaction is proposed to employ a nitrene transfer mechanism (Fig. 21).179 In case of azirination catalyzed by P450 variants engineered from P450BM3, the nitrene transfer begins with the acquisition of N2 from an azide-containing compound to form an iron nitrenoid intermediate, which further reacts with olefins to yield aziridines.238 Instead of using an azide to form the iron nitrenoid,238 BezE utilizes the N-acetoxy group as nitrene source.179,239 The heme-iron nitrenoid (IM12) of BezE reacts with an olefin of the geranyl moiety through nitrene transfer, resulting in aziridine formation (IM13, IM14). Subsequently, the highly strained aziridine ring is opened by nucleophilic attack of a hydroxide or a chloride to form indoline 122 or tetrahydroquinoline 125, respectively.179 The chlorine in 125 is nonenzymatically substituted to afford 123 and its C-9 diastereomer. The higher yield of 123 compared to its diastereomer suggested that the nonenzymatic conversion proceeds via an SN1 reaction rather than an SN2 reaction.179 This preference is likely due to the steric hindrance of the dimethylpentenyl moiety at C-10.179 BezE is the first example of a native P450 that catalyzes nitrene transfer. The N-acetoxylation likely facilitates bypassing of the conventional P450 catalytic cycle, which typically involves oxene transfer.240 This unusual catalytic mechanism enables BezE to act without the need for electron supplementation from redox partners.179 Consequently, BezE is classified as a P450 nitrene transferase.179
4.1.5.1. Aridacins. Aridacins A–C (24, 68, 69) are cis-eunicellane diterpenoids that feature a densely oxidized 6/7/5-tricyclic scaffold.93 Heterologous expression of the ari BGC-encoding genes in S. lividans TK64 and an GGPP-overproducing E. coli strain and in vitro enzymatic experiments revealed that the P450 AriF is capable of constructing a 6/7/5 tricyclic scaffold 67 from a 6/10-bicyclic framework 126. However, an earlier report showed that heterologous expression of the ari BGC in S. albus J1074M only produced the 6/10-bicyclic eunicellane 127 decorated with a C-19 hydroxy group and a C-20 carboxylic acid.112 Further feeding experiments using S. lividans and S. albus and in vitro assays showed that AriF oxidizes 126 to produce 6/10-bicyclic eunicellanes 127–130 as shunt products (Fig. 22). The production of the shunt metabolites 127–130 in low titers might originate from the limited activity of AriF, potentially due to the use of incompatible redox partners.93
 | ||
| Fig. 22 AriF-mediated oxidative C–C bond formation in the aridacin biosynthetic pathway and canonical oxygen rebound-based shunt pathway. | ||
The reaction mechanism of the AriF-catalyzed C–C bond formation has been proposed to proceed through either a radical rearrangement or a carbocation-based reaction. Density functional theory (DFT) calculations suggested that the terpenyl radical species IM15 generated by H-20 abstraction can undergo a barrierless and exothermic single electron transfer with a concomitant carbocation-based cyclization reaction (Fig. 22).93 In contrast, the radical-mediated rearrangement requires a ca. 15 kcal mol−1 barrier to reach the transition state toward the tricyclic ring system in the doublet state of the heme iron species. The thermodynamically favorable electron transfer is likely due to the coupled reaction with the energetically barrierless cyclization of the bicyclic carbocation IM16 to form the tricyclic carbocation IM17. Consequently, the AriF-catalyzed cyclization is most likely a carbocation-driven reaction, although the radical rearrangement remains a possibility albeit with some energetic challenges. Further DFT calculations revealed that the canonical oxygen rebound for the formation of the C-20 hydroxy eunicellane IM18 is energetically feasible with a barrier of ca. 4 kcal mol−1 (Fig. 22).93 These calculations explain the formation of 127–130, which are produced via the AriF-catalyzed oxygen rebound route as shunt products.
4.2. Non-canonical modification by other oxidoreductases
4.2.1.1. Aurachins. Aurachins are sesquiterpenoid quinolines that have been isolated from the myxobacterium Stigmatella aurantiaca Sg a15.246 They inhibit mitochondrial respiration and photosynthesis.246,247 Structurally, aurachins are grouped into A- and C-types based on the location of a farnesyl moiety at the quinolone.248 Feeding experiments using 13C- and 18O-labeled anthranilic acid, 13C-acetate, and 18O2 indicate that A-type aurachins are derived from C-type aurachins through farnesyl migration.249 Furthermore, the oxygen atoms at C-3 in aurachins A and B (131, 132) are derived from molecular oxygen. Mining the genome of S. aurantiaca Sg a15 revealed that the biosynthetic genes for aurachins are split into three genetic loci.248,250 Based on gene inactivation experiments and in vitro enzymatic assays using synthetic variants of aurachin C (133), the FMO AuaG catalyzes 1,2-farnesyl migration in 133 through either of two possible reaction routes (Fig. 23, routes A and B).243,244,248 In route A, AuaG initially epoxidizes the quinolone double bond of 133, yielding IM19. The subsequent deprotonation of N-OH induces epoxide ring opening and 1,2-farnesyl migration through an α-ketol rearrangement to give the highly unstable ketone oxime 134. Alternatively, the 1,2-farnesyl migration in route B involves tandem sigmatropic rearrangements. Following epoxidation and base-mediated ring opening, the intermediate IM20 undergoes a concerted retro-[2,3]-Wittig rearrangement to yield the O-nerolidyl quinoline N-oxide IM21. A following Claisen rearrangement results in the formation of 134. The NAD-dependent oxidoreductase AuaH then reduces the C-3 ketone and eliminates OH-4 to form 132. Bioinformatic analysis of aua genes and 18O2 feeding experiments suggest that another FMO AuaJ likely epoxidizes an olefin proximal to the quinoline core in 132, resulting in the formation of IM22 (Fig. 23).248,249 This oxidation reaction follows epoxide hydrolysis and heterocylization catalyzed by the epoxide hydrolase AuaI to afford 131.
 | ||
| Fig. 23 FMO-catalyzed 1,2-farnesyl migration and heterocyclization in the late-stage aurachin biosynthetic pathways. | ||
4.2.2.1. Xiamycins. Xiamycin A (135), sespenine (136), and their congeners have been isolated from Streptomyces spp. from various origins such as soil, mangrove trees, and sea sediments.256–260 Most of them exhibit antimicrobial and antiviral activities.256–261 The xia BGCs have simultaneously been discovered by genome mining of Streptomyces sp. HKI0576 and Streptomyces sp. SCSIO 02999.262,263 Bioinformatic analyses, single gene disruption, and heterologous expression of the xia BGC demonstrated that two unparalleled cyclization steps construct the characteristic pentacyclic scaffolds that feature the carbazole or 2-azabicyclo[3.3.1]nonane in 135 and 136, respectively.262–264 The first cyclization forming a trans-decalin ring is catalyzed by XiaE, a homolog of the Pyr4-type transmembrane TC.41 After a six electron oxidation catalyzed by the P450 XiaJ,265 the latter cyclization step forming the central ring of 135 and 136 is catalyzed by a new member of the group D FMOs, XiaF. Phylogenic analysis of XiaF and its homologs revealed that XiaF evolved from xenobiotic degrading oxygenases and groups into the class of indigo-forming oxygenases.262,266 Based on feeding experiments and in vitro enzymatic assays, XiaF converts indosespene (137) into the dihydrocarbazole-bearing prexiamycin (138) that spontaneously oxidizes to form 135.263 The cyclization activity of XiaF depends on flavin reduction catalyzed by a flavin reductase partner. In fact, the dedicated flavin reductase encoding gene xiaP is located in the flanking region of the xia BGC.266 Furthermore, XiaF also hydroxylates C-3 of the indole ring as a shunt reaction, leading to the autooxidation of the resulting hydroxyindole to form indigo. Based on the catalytic potential of XiaF, cyclization and rearrangement reactions for the formation of 135 and 136 have been proposed (Fig. 24A).262,263,266 Initially, the 3-hydroxy iminium species IM23 is formed from 137via nucleophilic attack by the C-2/C-3 double bond to the hydroperoxide group of the activated FAD. This cation formation triggers ring closure through the addition of the exo-methylene (C-21 of IM23) to the iminium, yielding IM24. Subsequent dehydration and deprotonation lead to the formation of 138 that is readily converted into 135. For the construction of the 2-azabicyclo[3.3.1]nonane framework in 136, the carbocation intermediate IM24 produced through C–C bond formation between C-2 and C-21 undergoes phenyl migration. Based on the proposed rearrangement mechanism to form 136 and its stereochemical outcomes, the C-3 hydroxylation catalyzed by XiaF likely occurs from the Si face. Intramolecular addition of C-21 to the imine species follows to form the (2R,3S)-cis-fused dihydroindole moiety in IM24, which is the configuration required for the subsequent phenyl migration. However, the stereoselectivity of the XiaF-catalyzed hydroxylation has not yet been experimentally confirmed. The divergent pathways for the biosynthesis of 135 and 136 may be explained by the degree of conformational freedom in the rearrangement reaction of the carbocation species.262
 | ||
| Fig. 24 Non-canonical modifications in the biosynthetic pathways of xiamycins and dixiamycins. (A) Proposed XiaF-mediated cyclization for the formation of xiamycin and sespenine. (B) Proposed dimerization reaction of the dixiamycins and sulfadixiamycins biosynthesis initiated by the XiaH-mediated N-hydroxylation. | ||
X-ray crystallography analysis of XiaF's apo structure and docking studies with indole revealed that the regiospecific hydroxylation of indole is regulated by precisely coordinating the C-3 position close to the hydroperoxide group of the activated FAD.266 Broad-range xenobiotic oxygenases generally have a small active site cavity lined with bulky amino acid residues.267 In contrast, XiaF harbors a relatively large substrate binding channel to facilitate the acceptance of the bulky substrate 137. Considering that XiaF is promiscuous enough to also hydroxylate indole but no other typical xenobiotic substrates, XiaF is a specialized tailoring enzyme that has evolved from xenobiotic detoxification enzymes to catalyze a non-canonical cyclization reaction of indolosesquiterpenoids.266
4.2.3.1. Dixiamycins. Dixiamycins are dimers of xiamycin (135) fused via C–N, N–N, or sulfonyl linkages.259,262,270–272 Structurally, most C2 or C1 symmetric dixiamycins with aryl C–N or N–N bonds show atropoisomerism and both atropodiastereomers are found in nature.259,270,272
Targeted gene deletion, heterologous expression, and feeding experiments revealed that the FMO XiaH is responsible for the production of bisindolosesquiterpenoids with aryl and sulfonyl bridges, e.g., 139–142 (Fig. 24B).270,271In vitro studies, however, show that XiaH only hydroxylates 135 to provide N-hydroxyxiamycin (143).273 NMR analysis and electron paramagnetic resonances of 143 revealed that the nitroxyl radical species IM25 is also presented in solution. Furthermore, time-course biochemical experiments showed conversion of 143 into 135, suggesting that the aminyl radical species IM26 can be formed via homocoupling of IM25.273 Subsequent deoxygenation results in the formation of structurally diverse xiamycin monomer analogs.273
The formation of dimeric xiamycins may be explained by a radical-based coupling mechanism (Fig. 24B).270,271,273 XiaH initially N-hydroxylates 135 to yield 143. The N-hydroxide 143 is in equilibrium with its nitroxyl radical species IM25. The radical dimerizes and the resulting endoperoxide IM27 is deoxygenated to form the aminyl radical IM26. This intermediate readily undergoes radical migration on the π-conjugated carbazole moiety (e.g., IM28). Afterwards, an intermolecular coupling reaction leads to the formation of axially chiral aryl C–N or N–N bond bearing dixiamycins 139–141. Alternatively, the aminyl radical IM26 reacts with sulfur dioxide to form the sulfonyl radical IM29, which leads to the formation of sulfadixiamycins, e.g., 142.271
4.2.4.1. Phenalinolactones. The pla BGC harbors a gene encoding a Fe/αKG-dependent oxygenase (plaO1).118 Inactivation of plaO1 in the native producer resulted in the accumulation of PL CD6 (144) that bears an acyclic pyruvate-derived C4 side chain at C-14, instead of a dihydroxyfuranone moiety.118,119In vitro enzymatic assays confirmed the function of PlaO1 that converts 144 into the dihydroxyfuranone-containing 29.119 A mechanism for the formation of the γ-butyrolactone moiety by PlaO1 has been proposed (Fig. 25).118 PlaO1 putatively initially hydroxylates C-16 of 144 to form the enol IM30, which is converted into the cyclopropanone IM31 through enolization and intramolecular nucleophilic addition of C-15 to C-17. The proposed subsequent cyclopropanone ring opening results in the formation of the formyl α-ketoacid IM32, which undergoes ring closure to yield the hemiacetal IM33. Tautomerization of the C-17 ketone follows to yield 29. Since PlaO1 is a stand-alone enzyme that produces 29 from 144,119 the keto–enol tautomerization of C-17 likely occurs spontaneously or in a PlaO1-dependent fashion. Although further studies are needed to verify the putative reaction mechanism, the PlaO1-catalyzed ketolactol formation is highly unusual for Fe/αKG-dependent oxygenases.119
 | ||
| Fig. 25 Proposed ketolactol formation catalyzed by the Fe/αKG-dependent oxygenase PlaO1 during phenalinolactone biosynthesis. | ||
4.2.5.1. Platensimycin and platencin. Platensimycin (2) and platencin (44) are examples of 4,5-seco-ent-kaurane and seco-ent-atiserene diterpenoids, respectively.129,277 The formation of the 4,5-seco-diterpene skeleton is proposed to proceed through C-5 hydroxylation with a concomitant retro-aldol reaction.278–280 In-depth biosynthetic studies of 2 and 44 led to the discovery of a non-canonical nonheme diiron enzyme that installs a hydroxy group at C-5 to set the stage of the intricate opening reaction.280
The gene encoding the C-5 hydroxylase was initially elusive as all oxygenases encoded in the ptm BGC had been functionally characterized to be involved in other biosynthetic steps leading up to the formation of 2 and 44.130–132,167 Therefore, deletion of genes with unknown function was performed using S. platensis SB12029, a dual-producer of 2 and 44.279,280 Comparative metabolomic studies of extracts derived from mutant strains suggest that PtmU3 is likely responsible for the C-5 hydroxylation of the C-19 CoA thioesters 145 and 146.280In vitro enzymatic assays imply that PtmU3 is a nonheme iron-dependent oxygenase that converts 145 and 146 into the C-5β hydroxylated CoA thioesters 147 and 148, respectively (Fig. 26A).280 The generated tertiary alcohols can facilitate a retro-aldol reaction to form 4,5-seco-terpenyl CoAs 149 and 150.
 | ||
| Fig. 26 PtmU3-catalyzed oxidation in the biosynthesis of platensimycin and platensin. (A) PtmU3-mediated hydroxylation to set up the retro-aldol ring cleavage reaction. (B) Proposed unusual catalytic mode of PtmU3. | ||
X-ray crystallographic studies reveal that PtmU3 forms a trioseophosphate isomerase (TIM)-like barrel structure, one of the most common tertiary structures found in nature.281 While TIM-barrel structures are present in aldolases, glycosidases, enolases, and metal-dependent hydrolases,281,282 no TIM-barrel-containing metal-dependent oxygenase had been reported.280 Furthermore, PtmU3 harbors two iron atoms in its active site bridged by Glu241 and Asp308. Their coordination is distinct from canonical nonheme diiron enzymes that typically feature μ-oxo or μ-peroxo bridging ligands between the two metal centers.283 Mutagenesis studies of PtmU3 and QM/MM calculations of the C-5 hydroxylation reaction reveal that the two iron-bridging amino acid residues are vital for the enzymatic activity and a saturated octahedral iron plays a structural but likely not a catalytic role.280,284 The unprecedented diiron coordination of PtmU3 follows neither canonical diiron monooxygenases that usually use one or both iron atoms for catalysis283 nor other diiron oxygenases, e.g., myo-inositol oxygenase MIOX285 and organophosphonate oxidase PhnZ,286 where one iron binds to the respective substrate. In addition to the distinct structural features, QM/MM calculations suggest an unusual catalytic cycle for PtmU3 (Fig. 26B).284 The unsaturated iron in the active site of PtmU3 readily binds molecular oxygen to form a ferric-superoxide species. Spin density calculations for all possible states of the superperoxo-diiron species revealed electron transfers from the iron center to dioxygen and the diiron-coordinating amino acid residues, indicating that these amino acid residues play a role in the electron reorganization of the iron center.284 The generated FeIII-superoxo species abstracts H-5β of 145 or 146 and the resulting hydroperoxide-iron(III) intermediates bind to the respective substrate radicals (IM34) to give 147 or 148 with the formation of an oxoiron(IV). The oxoiron(IV) species then react with the same substrates via proton abstraction and oxygen rebound to yield the hydroxylated products. Oxygen rebound in both states of ferric-superoxo and oxoiron(IV) species are feasible based on the energy barrier of the rate-limiting proton abstraction. Therefore, the proposed unusual catalytic mode of PtmU3 successively uses molecular oxygen for the efficient formation of C-5β hydroxy CoA thioesters.284
Sequence similarity network analysis revealed that homologs of PtmU3 are widely distributed in actinobacteria, with conserved key residues for diiron coordination and binding of CoA-activated substrates.280 Noteworthily, half of the genes encoding PtmU3 homologs are located in proximity to genes encoding CoA ligases. This observation suggests that these non-canonical nonheme diiron oxygenases may prefer CoA-activated substrates.280
4.2.6.1. Napyradiomycins. The napyradiomycins are a family of chlorine-bearing naphthoquinone meroterpenoids that are widely found in terrestrial and marine Streptomyces spp.300–302 Based on the nucleophilicity of the 1,3,6,8-tetrahydroxynaphthalene (THN) nucleus, prenylation patterns are expected at the nucleophilic C-2 or C-4 positions. However, some napyradiomycins feature prenyl groups at the non-nucleophilic C-3 position. As a result, the napyradiomycins are classified into class I (at C-2 or C-4 positions) and class II (at C-3 position).245 Some napyradiomycins feature a chlorinated gem-dimethyl tetrahydropyran ring fused with dihydronaphthoquinone and a chlorinated gem-dimethyl cyclohexane ring, both of which originate from the terpene components.
Earlier biosynthetic studies using 13C-labeled acetic acid suggest that napyradiomycin B1 (151) is biosynthesized from a symmetric THN and two isoprene units, i.e., DMAPP and FPP.303 Investigation of the genes encoding a THN-forming type III polyketide synthase (PKS) and a PT led to the identification of nap BGCs in the genomes of Streptomyces sp. CNQ-525 and Streptomyces aculeolatus NRRL 18422 with lengths of 36 kbp and 43 kbp, respectively.291 Both nap BGCs include three genes, namely napH1, napH3, and napH4, encoding VHPOs.291 Based on heterologous expression studies in S. albus, in vitro enzymatic assays, mutagenesis of VHPO genes, and biomimetic syntheses, the biosynthetic pathway of napyradiomycins was characterized (Fig. 27A).245,291,292,297,299 All VHPOs play key roles in the maturation of napyradiomycins. Following the biosynthesis of the THN core by NapB1, the PT NapT9 installs a geranyl group at C-4 to yield 4-geranyl THN (152). NapH1 then catalyzes dichlorination at C-2 and OH-3 of 152. The resulting dichloro THN (IM35) is then dearomatized through the loss of a chlorine atom at OCl-3, followed by quenching of the benzylic cation IM36 with H2O to yield α′-hydroxyenone 153. NapH4 also converts 152 into 153, albeit with a low catalytic efficiency.297 With strict enzymatic stereocontrol, another PT NapT8 performs the asymmetric prenylation at C-2 of 153, resulting in the formation of the α-hydroxyketone 154.245 Strikingly, NapH3, a homolog of NapH1 and NapH4, is highly substrate-specific and catalyzes α-ketol rearrangement of 154 to form naphthomevalin (155) independent of Na3VO4 and H2O2.245 DFT calculations indicate that the 1,2-suprafacial-geranyl migration is thermodynamically favorable and requires a gem-substitution at C-2 to disrupt the π-conjugated system, which contributes to the planar structure of the naphthalene-derived bicyclic compound.245 NapH1 is also capable of catalyzing heterocyclization to afford napyradiomycin A1 (156) via the enantiospecific formation of the chloronium IM37 using 155 as a substrate.292 Therefore, NapH1 is a dual-functional enzyme that performs chlorination-induced dearomatization and heterocyclization in a substrate-dependent manner. In analogy to the heterocyclization reaction of NapH1, NapH4 chlorinates the olefin C-21/C-22 of 156.297 The resulting intermediate IM38 undergoes chloronium-induced cyclization, forming the tertiary cyclohexyl cation IM39. Subsequent deprotonation yields 151 that features an exo-olefin-containing cyclohexane ring.297
 | ||
| Fig. 27 VHPO-mediated rearrangement and cyclization in THN-based meroterpenoid biosynthetic pathways. (A) Biosynthetic pathway of napyradiomycin B1. (B) Biosynthetic pathway of merochlorins A–D. | ||
Despite the relatively high homology between NapH1, NapH3, and NapH4, these enzymes catalyze different reactions. Phylogenetic analysis of functionally validated VHPOs demonstrated that VHPOs derived from Streptomyces spp., including NapH1, NapH3, and NapH4, group separately from other microbial and eukaryotic VHPOs.299 Within the Streptomyces VHPOs clade, NapH3 is phylogenetically distant from NapH1 and NapH4, suggesting it has evolved to exclusively catalyze the α-ketol rearrangement by losing its halogenation capability.245,299 The apo structure of NapH1 and mutagenesis of its active site residues revealed the presence of two key amino acid residues that are crucial for the enantioselective chlorofunctionalization and halide oxidation, respectively.299 Notably, these amino acid residues are not conserved in other VHPOs that show substrate promiscuous activities.299 Insights from the biomimetic synthesis of merochlorins using chloramine suggest Lys324 likely forms a chloramine with hypohalous acid generated from the vanadate and regulates the stereospecific transportation of chlorine to the substrate.295,299 By contrast, the apo structure of NapH3 revealed that His445, which is corresponding to His494 of NapH1, is post-translationally phosphorylated to form a τ-phosphohistidine.299 Instead of the vanadate, the phosphate group noncovalently interacts with active site residues that are essential for vanadate stabilization. This post-translational modification of His445 explains the loss of the haloperoxidase activity in NapH3 by preventing vanadate binding.245,299 Mutagenesis studies show that His445 does not play any role in the NapH3-catalyzed α-ketol rearrangement reaction, indicating that substrate binding is independent of His445.299
4.2.6.2. Merochlorins. Merochlorins are another type of THN-based meroterpenoids with potent antibiotic activity.302 Structurally, these compounds are derived from the TNH core and a rare isosesquilavandulyl moiety with various modifications.293,302 The mcl BGC (58 kbp) was identified by mining the genome of Streptomyces sp. CNH-189, which produces merochlorins A–D (157–160).293 Like the nap BGC, the mcl BGC harbors two genes (mcl24, mcl40) encoding VHPOs with high homology to napH1, napH3, and napH4 respectively. Heterologous expression of the mcl genes in S. coelicolor M1152 indicated that Mcl40 is responsible for macrocyclization to produce the 15-membered cyclic ether-bearing 159 from 160 (Fig. 27B), presumably through chloronium ion formation (IM40) as demonstrated for NapH1.293 Further in vitro biochemical studies suggest that Mcl24 is a multifunctional VHPO that converts pre-merochlorin (161) into the structurally diverse products 157, 158, and 160.245,295 Mcl24 precisely controls the site-selectivity of the arene and alkene halofunctionalization, as well as the timing and stereospecificity of the oxidative dearomatization and polycyclization (Fig. 27B).245,295 Similar to the catalytic activity of NapH1, Mcl24 initially chlorinates both C-2 and OH-3 of 161 and subsequently dearomatizes the THN core by chloride elimination. The resulting benzylic cation IM41 is converted to the spirocyclic carbocation IM42via addition of the olefin C-14/C-15. Subsequent cation quenching by nucleophilic addition of the C-2/C-3 olefin or OH-3 yields the polycyclic products 157 and 158, respectively. Alternatively, quenching of the benzylic cation by H2O gives rise to the enone IM43, which is subsequently converted to the C-2 gem-dichloride IM44 by Mcl24. The intermediate IM44 undergoes a thermodynamically favored α-ketol rearrangement to produce 162.245 This rearrangement reaction is promoted under basic conditions (pH 8.0), where cation quenching by hydration is preferred over alkene addition and deprotonation.245 Compared to the gem-dichlorinated product 162 identified from the in vitro catalytic reaction of Mcl24,245 the C-2 position in both 159 and 160 is substituted with methyl and chlorine groups.293 Therefore, the pathway for the biosynthesis of 160 may proceed through C-2 methylation of the hydrated product IM43, presumably catalyzed by the MT Mcl21,293 followed by the Mcl24-catalyzed α-ketol rearrangement.295,302
4.2.7.1. Sodorifen. Sodorifen (163), a unique homosesquiterpene that features the polymethyl bicyclo[3.2.1]octene system, is the major component of the volatile bouquet of organic compounds produced by Serratia plymuthica.310 Comparative transcriptomic analysis of 163-producing and non-producing strains have revealed that the biosynthesis of 163 is dependent on the catalytic activity of a SAM-dependent C-MT (SodMT) and the sodorifen terpene cyclase (SodS).311 The biosynthesis of 163 has been thoroughly investigated through a combination of in vitro and in vivo experiments and complemented with DFT calculations.307,312,313 The complex scaffold of 163 is formed from the sequential cyclization of FPP catalyzed by SodMT and SodS (Fig. 28 and 29). SodMT methylates FPP at C-10 resulting in carbocation formation at C-11. Subsequent ring formation yields the cyclohexyl cation IM45. This cationic intermediate undergoes a base-induced cyclopropanation by H-9 deprotonation. In turn, cyclopropane ring opening of the resulting intermediate IM46 by reprotonation leads to the formation of a C-8-extruded cyclopentyl cation IM47. The successive 1,2-hydride and 1,2-methyl shifts to IM48, followed by deprotonation of H-10 yield 164.312,313
 | ||
| Fig. 28 Non-canonical C-MT-mediated cyclization in the biosynthesis of the homo- and bishomosesquiterpenes sodorifen and chlororaphens. | ||
 | ||
| Fig. 29 Fragmentation–recombination mechanism of the SodS-catalyzed cyclization in the biosynthesis of sodorifen. | ||
SodS catalyzes the unusual cyclization reaction of 164 to produce 163 (Fig. 29).307,313 The reaction begins with substrate ionization by pyrophosphate abstraction in 164, generating the allylic carbocation IM50. Sequential 1,4- and 1,2-hydride shifts convert IM50 to the cyclopentyl cation IM51. After fragmentation of IM51 into a cyclopentene and a pentadienyl cation (IM52), hydride transfer from the cyclopentene to the pentadienyl cation and rotation of the resulting pentadiene fragment take place to yield IM53. Subsequently, proton transfer from C-7 to C-5 results in the formation of a pentenyl cation, which rotates to align for recombination with the cyclopentadiene fragment (IM54). The recombination of the two generated fragments (IM54) proceeds through an asynchronous [4 + 3] cycloaddition to form the bicyclo[3.2.1]octene framework IM55. The cationic intermediate IM55 then undergoes Wagner–Meerwein rearrangement, followed by deprotonation of H-16, to yield 163.
Mining of the NCBI RefSeq genome database using protein sequences of SodMT and SodS as query revealed BGCs similar to the sodorifen BGC in more than 180 different bacterial species, mainly belonging to the phylum Proteobacteria.314 The identified BGCs were classified into four types based on the presence of genes encoding MTs and TCs. Heterologous expression of selected BGCs in a yeast host led to the identification of various C16 terpenes that feature a polymethylated and highly strained cage-like hydrocarbon scaffold.314 These C16 terpenes are derived from 164 as a common intermediate that undergoes TC-catalyzed cyclization. More recently, in-depth mechanistic studies of TCs that produce non-canonical homoterpenes revealed that the fragmentation–recombination mechanism is a common theme in their biosynthetic pathways,315 as exemplified in the biosynthetic pathway of 163.313 The diverse skeletal architectures of these TC-catalyzed reaction products arise from varying modes of recombination of the two components generated by the ionization-induced fragmentation of 164.315
4.2.7.2. Chlororaphens. A similar MT-mediated cyclization has been observed in the biosynthesis of the bishomosesquiterpenes, chlororaphens A and B (165, 166).308,316 These unusual C17 terpenes have recently been identified from Pseudomonas chlororaphis O6, which harbors a BGC similar to the sodorifen BGCs.308 In addition to genes encoding the non-canonical C-MT (Pc-FPP-MT) and the type I TC (Pc-ChloS), a gene encoding another C-MT (Pc-γ-PSPP-MT) is contained in the chlororaphen BGC. Heterologous expression of the chlororaphen BGC and in vitro enzymatic assays using isotope-labeled FPP revealed that the catalytic activity of Pc-FPP-MT follows a similar cyclization mechanism to that of SodMT (Fig. 28).308,316 However, instead of cation quenching by H-10 deprotonation, an additional 1,2-hydride shift and subsequent deprotonation of H-16 follow to form γ-PSPP (167) with a methylidenecyclopentane moiety. Subsequently, Pc-γ-PSPP-MT methylates the exocyclic olefin, producing the non-canonical bishomofarnesyl pyrophosphate, α-PCPP (168). Finally, α-PCPP is cyclized by Pc-ChloS to yield 165 and 166, as well as the premature bicyclic prechlororaphen (169) as a minor product.308,316
Mechanistic characterization of Pc-ChloS revealed an unusual cyclization pathway that proceeds through the generation of the neutral compound 169en route to 165 and 166 (Fig. 30).316 The pyrophosphate of the C17 intermediate 168 is abstracted by Pc-ChloS to generate the allylic cation IM56. The intermediate IM56 undergoes C–C bond formation between C-3 and C-11, followed by 1,2-hydride shift to form the cis-fused bicyclo[3.3.0]octane intermediate IM57. Deprotonation of IM57 yields the premature bicyclic product 169. Afterwards, reprotonation on C-1 of 169 facilitates the second C–C bond formation between C-2 and C-7, generating the tricyclic cationic intermediate IM58. Subsequent 1,3-hydride shift results in the formation of IM59, which is further converted to IM60 through a Wagner–Meerwein-type 1,2-alkyl shift. Finally, cation quenching of IM60 by deprotonation of H-14 yields 165. Alternatively, two additional Wagner–Meerwein rearrangements (IM61, IM62), followed by deprotonation of H-12, lead to the formation of 166. The cyclization mechanism catalyzed by Pc-ChloS is distinct from the fragmentation–recombination mechanism commonly observed in the biosynthesis of C16 terpenes like 163.316
 | ||
| Fig. 30 Pc-ChloS-catalyzed cyclization mechanism in the biosynthesis of chlororaphens. | ||
4.2.7.3. Teleocidins. Teleocidins and their derivatives are terpenoid indole alkaloids that are potent protein kinase activators, produced by actinobacteria and cyanobacteria.317–320 Among these, teleocidin Bs feature an indole framework fused with a nine-membered lactam and a C11 terpene moiety. The BGC for teleocidin Bs was identified in the genome of Streptomyces blastmyceticus NBRC 12747 by mining for genes homologous to those responsible for the production of lyngbyatoxin A (170), a teleocidin analog bearing a linear C10 terpene moiety.309,321 The tle BGC contains genes encoding a non-ribosomal peptide synthetase (NRPS, tleA), a P450 (tleB), and a PT (tleC). Notably, the tle BGC that lacks genes encoding a TC or a C-MT is solely responsible for the production of 170 that has a linalyl group.309,321 Investigation of candidate MT genes located outside of the tle BGC led to the identification of tleD encoding a SAM-dependent C-MT involved in the biosynthesis of teleocidins B-1, B-4, and des-O-methyl-olivoretin C (171–173).309 Further in vitro enzymatic assays with isotope-labeled 170 indicate that TleD catalyzes methylation-induced cyclization of the linalyl group through a cationic cascade reaction (Fig. 31).309 Initially, TleD installs a methyl group at C-25 to generate the tertiary cation IM63. After 1,2-hydride migration to IM64, aromatic electrophilic substitution at C-7 results in the formation of the spiro-fused cyclohexadienyl iminium intermediates (IM65, IM66). Subsequently, the intermediates undergo iminium cation-driven cascade reactions including 1,2-alkyl shift at the spiro-cyclopentane ring, followed by deprotonation to yield 171–173. The nucleophilic addition of C-7 to the C-25 carbocation from the Re face is favored over the Si face, as indicated by the predominant production of 172 and 173.309
 | ||
| Fig. 31 Non-canonical C-MT-mediated cyclization in the biosynthesis of teleocidins. | ||
X-ray crystallographic analysis of TleD complexed with S-adenosylhomocysteine and 170 revealed that the active site residues provide an ideal hydrophobic environment to accommodate 170.322 This hydrophobicity excludes water molecules and thus prevents the premature quenching of the cation species during methyl transfer-induced cyclization. The linalyl group of 170 appears to be flexible in the crystal structure of TleD. Molecular dynamics simulations of TleD suggest that the conformation of the linalyl group dictates the facial selectivity of the nucleophilic attack to C-25.322 The predominant conformation of the linalyl group supports the favored formation of 172 and 173.
4.2.8.1. Albireticulones. The first trans-eunicellane synthase (AlbS) was recently discovered in Streptomyces albireticuli NRRL B-1670.331 In the genome of S. albireticuli, albS is clustered with genes encoding a GGPPS (albG), two P450s (albP1, albP2), and a protein with unknown function (albU).112,329 Heterologous expression of the alb BGC in S. albus J1074M resulted in the production of albireticulones A and B (174, 175).112 Overexpression of alb genes in the native producer and in vitro enzymatic assays revealed that AlbU catalyzes the irreversible isomerization of albireticulene (176) to form iso-albireticulene (177).329 Since AlbU exhibits its enzymatic activity in the absence of cofactors, an acid–base catalytic mechanism, involving the generation of IM67, has been proposed for the isomerization reaction (Fig. 32A).329 Although the suggested mechanism is similar to that of type 1 isopentenyl diphosphate isomerases,332 AlbU has neither a detectable Pfam domain nor an InterPro designation, indicating that it is the first member of a novel family of isomerases. Moreover, AlbU may isomerize the allyl alcohol IM68 derived from 177 through the AlbP1-catalyzed hydroxylation at C-6.329 Specifically, protonation at C-19 of IM68 generates the tertiary cation IM69, which undergoes deprotonation to form the enol intermediate IM70. Subsequent tautomerization of IM70 results in the formation of 174.329 Further mechanistic studies of AlbU are needed to ascertain the proposed dual functions in isomerizing both 176 and the C-6 hydroxylated intermediate of 177.
 | ||
| Fig. 32 Scaffold functionalization of trans-eunicellane diterpenoids by enzymes previously annotated as hypothetical proteins. (A) Proposed isomerization mechanism of AlbU and biosynthetic pathway of albireticulones. (B) Proposed isomerization and oxidation activity of EutD and biosynthetic pathway of euthailols. | ||
4.2.8.2. Euthailols. More recently, the eut BGC from Streptomyces euthainensis N2458 was identified to be responsible for the biosynthesis of trans-fused eunicellane diterpenoids.330 Although the eut BGC shows high similarity to the alb BGC, it harbors unique genes encoding a TC fused with a truncated (E)-4-hydroxy-3-methyl-but-2-enyl pyrophosphate reductase (eutB) and a P450 that is split into three fragments (eutEFG).330 Heterologous expression of the eut BGC in S. avermitilis SUKA22 led to the identification of eutailols A–C (178–180).330In vivo functional characterization of the eut genes revealed partial functional redundancy between the P450 EutD and the hypothetical protein EutC, both of which hydroxylate C-17 of 176 (Fig. 32B).330 While EutD hydroxylates C-5 and C-17 of 176 to produce euthailol D (181), EutC oxidates 176 to yield 174 and 175, as well as the epoxide diastereomers, euthailols F and G (182, 183). Additionally, EutC is also capable of isomerizing 176 to generate 177.330 This observation suggests that EutC is a bifunctional enzyme, acting as an isomerase and an oxygenase. Despite the high sequence similarity of EutC to AlbU, which solely acts as an isomerase,329 the dual functionality of EutC suggests a functional divergence between two enzymes.330 The requirement of cofactors for the oxygenation activity of EutC remains unknown,330 leaving its catalytic mechanism to be elucidated in future studies.
5. Potential and limitations of genome mining for the targeted discovery of terpenoids
Natural product biosynthesis, much like textbook terpene biosynthesis, is a two-step process consisting of scaffold formation and decoration. The importance of the decoration phase varies among different natural product classes, with terpenoids relying more heavily on this phase to achieve structural diversity compared to other classes. Natural product biosynthesis can be classified into two fundamentally different strategies:333(1) Complex polyketides and non-ribosomal peptides (NRPs) are biosynthesized by large multi-enzyme complexes in an assembly line-like fashion. Each assembly line is composed of individual modules, which can be further subdivided into multiple enzymatic domains that work together to add or modify a building block to the nascent natural product backbone.334–336 Diversity in these systems is achieved through the incorporation of a wide array of building blocks recognized by specific domains, as well as facultative enzymatic domains integrated into the modules of the assembly line.335,337 Consequently, assembly line-like systems generate structural diversity using distinct architectures composed of vectorial arrangements of modules with varying domain compositions. Post-assembly line tailoring reactions play a subordinate role, as a large number of functional groups are already present in the building blocks or are generated during the assembly process. The biosynthesis of these systems is well understood, and seemingly universal biosynthetic principles have been deciphered to predict the core structure of the assembly line product from genome sequence information.335,338 The biosynthetic logic of assembly line-like pathways, along with a set of conserved enzymatic domains that exhibit a high degree of sequence homology, facilitates the mapping of the full biosynthetic diversity of these pathways. It is assumed that the majority of assembly line-like pathways can be identified and annotated by current genome mining tools. Assembly line biosynthetic gene clusters are identified using profile Hidden Markov Models (pHMMs) trained to recognize each commonly occurring enzymatic domain in assembly line modules.339 Module composition, number, and order, as well as the substrate specificity of individual enzymatic domains within each module, are used to predict the core structure of the assembly line product.338,340,341
(2) In contrast, complex terpenes are produced by a linear arrangement of discrete, typically monofunctional enzymes.333 Terpene hydrocarbon scaffolds are composed of multiple isoprene units condensed into a small subset of achiral oligoprenyl precursors. Terpene BGCs do not encode large proteins composed of highly conserved enzymatic domains. Instead, TC genes are typically the only genes consistently present in all canonical terpene BGCs.339 In many cases, oligoprenyl synthases are encoded within these clusters, indicating the specific subclass of terpenes produced by a pathway.342 The structural diversity of terpenes arises partly from cyclization and rearrangement processes catalyzed by TCs. Unlike the domains in assembly line-like pathways, the primary sequence of TCs is not highly conserved, with conservation typically limited to a set of catalytically important motifs.37 Nevertheless, their tertiary structures are relatively conserved. State-of-the-art genome mining algorithms are capable of identifying the most common types of TCs.340 There have been multiple reports, however, of TCs lacking characteristic motifs343 or entirely different enzyme families acting as TCs.21,22,39,344 This non-canonical TC subpopulation currently evades detection due to the limited number of characterized examples available to train pHMMs to map the full biosynthetic diversity of TCs. Consequently, it is difficult to estimate the proportion of TCs and their associated BGCs that remain unrecognized. TCs merely act as chaperones that guide cationic intermediates through a cascade of cyclization and rearrangement reactions.11 During the cyclization process, the achiral precursors are transformed into stereochemically intricate polycyclic structures. This process, however, currently cannot be predicted. The challenge is further compounded by the fact that a single TC can yield multiple scaffolds.10,345 Moreover, some TCs are only a few mutations away from producing an almost non-overlapping spectrum of products.268 Due to the limited number of oligoprenyl precursors, the diversity achievable through TC-mediated cyclization reactions is inherently constrained. As a result, the structural diversity of complex terpenoids is further enhanced through a wide range of tailoring reactions that decorate the terpene scaffolds. Genes encoding these biosynthetic enzymes can be annotated by genome mining algorithms in the vicinity of TC-encoding genes using pHMMs. Predicting the regio- and stereochemistry of the catalyzed reactions, let alone the type of reaction catalyzed, however, is currently impossible.
Several dozen different enzymatic transformations involved in terpene maturation have been described, yet the number of characterized enzymes per enzyme family and type of reaction catalyzed is too small to serve as training datasets for either hard-coded or artificial intelligence-based prediction tools. In summary, the principles of terpene biosynthesis, along with the small number of characterized BGCs, hinder the development of genome mining algorithms that are truly capable of charting the full biosynthetic diversity of terpene BGCs beyond the canonical terpene BGCs identified by state-of-the-art tools. Recently, a protein 3D structure-model-based genome mining approach has been developed to identify non-canonical TCs that lack conserved motifs typically used for the identification of canonical TCs.343 BGCs were selected by focusing on the presence of genes encoding oligoprenyl synthases and hypothetical proteins upstream and downstream of oligoprenyl synthase encoding genes. The authors speculated that these hypothetical proteins serve as non-canonical TCs.343 AlphaFold predictions suggested that the identified hypothetical proteins exhibit high structural similarity to canonical type I TCs indicating their potential role as TCs.343 Sequence homology searches of the identified non-canonical TCs revealed such TSs to be encoded either within a BGCs or as standalone genes in a wide range of bacterial genera and in eukaryotes.343 The outlined strategy is one of many approaches to expanding terpene biosynthetic space, and to bring us closer to the goal of charting this space as comprehensively as we have for assembly line-like pathways.
The development of genome mining tools to comprehensively chart terpene biosynthetic space and predict terpenoid structures associated with a BGC of interest require a larger number of characterized terpene BGCs. Artificial intelligence-based approaches could help us predict both the type of scaffold formed by any given TC and the regio- and stereospecificity of tailoring enzymes, along with the types of reactions they catalyze.
6. Conclusion
Tailoring reactions of terpene hydrocarbon scaffolds are crucial biosynthetic steps en route to complex terpenoids. For the longest time, bacteria had not been considered as a rich source for the discovery of heavily modified terpenoids. However, as outlined in this review, bacteria undoubtedly produce complex bioactive terpenoids using dedicated sets of tailoring enzymes, such as P450s, FMOs, and MTs. These versatile modifications range from the regio- and stereospecific functionalization of unactivated C–H bonds that are chemically largely undistinguishable to unprecedented oxidative or nonoxidative structural rearrangements, executed in either promiscuous or enantiospecific fashion. This remarkable enzymatic repertoire is the bases for the extraordinary structural diversity of bacterial terpenoids. Particularly, P450s play a central role in the oxidative functionalization of terpene scaffolds, with most bacterial terpenoid BGCs including multiple P450 genes alongside TC encoding genes. The functional characterization of the full biosynthetic repertoire of P450s and other oxidoreductases will fundamentally change our understanding of how terpenoids strategically gain their unique chemical and biological features.Ongoing efforts to characterize bacterial terpenoid biosynthetic pathways have uncovered unusual functions of tailoring enzymes. Unlike the canonical enzymes that follow well-established mechanistic routes, such as the oxygen rebound mechanism of iron-dependent oxygenases, the characterized non-canonical tailoring enzymes catalyze fascinating reactions mostly involved in skeletal transformations through unusual enzymatic mechanisms. Notable examples discussed in this review include oxidative alkyl migration (PntM, AuaG, NapH3), ring contraction (CYP114), and cyclization (BezE, XiaE, PlaO1, TleD) reactions. These non-canonical tailoring enzyme-catalyzed transformations frequently occur in the later stage of biosynthetic pathways, contributing to the structural complexity of terpenoids. Intriguingly, some enzymes act earlier to forge unusual terpene scaffolds rather than merely decorating hydrocarbon flameworks, as shown in the biosynthesis of aridacins (AriF) and sodorifen (SodMT). The catalytic diversity of non-canonical tailoring enzymes underscores the immense potential for the characterization of novel enzymes with unique and distinct activities from bacterial terpenoid biosynthetic pathways.
Motivated by the sophisticated catalytic function of bacterial terpenoid tailoring enzymes, both native and engineered variants have been applied for the efficient synthesis of biologically important terpenoids.9 Hence, the discovery of new enzymology opens up opportunities to develop elaborate biocatalysts for (chemo)enzymatic late-stage functionalization and inspires biomimetic syntheses of naturally occurring complex terpenoids and even new-to-nature molecules.
Although almost all bacterial terpenoid BGCs are silent or expressed at low levels under standard laboratory culture conditions, recent advancements in molecular biology, such as the efficient heterologous expression of terpene BGCs in heterologous hosts, optimized for terpene production,112,346 have paved the way for the discovery of novel terpenoids and the identification of new enzymatic functions. Coupled with the ever-increasing availability of genome data, state-of-the-art genome mining techniques readily identify canonical terpenoid BGCs. To date, the antiSMASH database, an online repository of BGCs predicted by antiSMASH, lists more than 28800 BGCs from publicly available genomes of bacteria.347 However, the targeted identification of non-canonical bacterial terpenoid BGCs still remains challenging. Many terpenoid BGCs evade detection by current genome mining platforms due to the relatively low sequence homology between TCs and the frequent absence of detectable Pfam domains in non-canonical TCs responsible for terpene scaffold formation.22,38 Moreover, unlike thiotemplate assembly line-based BGCs, structure predictions of terpenoids from BGC information remains challenging. The difficulty of structural predictions hinders the prioritization of orphan BGCs for the discovery of novel terpenoids. A deeper understanding of the principles that govern bacterial terpenoid biosynthesis may enable the construction of training datasets for the development of artificial intelligence-based structure prediction tools specific to modifications introduced by tailoring enzymes. These tools can be integrated into the genome mining workflow to thoroughly analyze BGCs and facilitate the targeted discovery of novel terpenoid biosynthetic pathways.
For the majority of the past two decades, research on bacterial terpenoid biosynthesis has predominantly concentrated on the cyclization phase mediated by TCs. During this period, various tailoring enzymes encoded in the BGCs studied for their TC have been discovered. As the mechanistic characterization of terpenoid tailoring enzymes rapidly advances, it is evident that multidisciplinary approaches—combining bioinformatics, synthetic biology, organic chemistry, enzymology, and computational chemistry—are crucial for deepening our understanding of bacterial terpenoid biosynthesis. Emerging evidence now suggests that more and more characterized tailoring enzymes are involved in the formation of terpene backbones, rather than merely decorating pre-formed hydrocarbon scaffolds. These insights might necessitate a re-evaluation of the traditional ‘two-phase’ model of terpenoid biosynthesis, which strictly separates scaffold formation from decoration processes.
7. Conflicts of interest
There are no conflicts to declare.8. Acknowledgements
EJNH gratefully acknowledges funding by the LOEWE Center for Translational Biodiversity Genomics (LOEWE TBG), AAC is supported by a fellowship from the Higher Education Commission, Pakistan (HEC PAK)/Deutscher Akademischer Austauschdienst (DAAD Germany).9. References
- Dictionary of Natural Products 32.2, http://dnp.chemnetbase.com/, accessed May 2024.
- T. Zeng, Y. Chen, Y. Jian, F. Zhang and R. Wu, New Phytol., 2022, 235, 662–673 CrossRef CAS PubMed.
- J. Clardy and C. Walsh, Nature, 2004, 432, 829–837 CrossRef CAS PubMed.
- B. C. Doak, B. Over, F. Giordanetto and J. Kihlberg, Chem. Biol., 2014, 21, 1115–1142 CrossRef CAS PubMed.
- K. C. Nicolaou, W.-M. Dai and R. K. Guy, Angew. Chem., Int. Ed., 1994, 33, 15–44 CrossRef.
- O. Goethe, A. Heuer, X. Ma, Z. Wang and S. B. Herzon, Nat. Prod. Rep., 2019, 36, 220–247 RSC.
- E. D. Morgan, Bioorg. Med. Chem., 2009, 17, 4096–4105 CrossRef CAS PubMed.
- M. Ui, T. Okada, K. Hazeki and O. Hazeki, Trends Biochem. Sci., 1995, 20, 303–307 CrossRef CAS PubMed.
- C. N. Stout and H. Renata, Acc. Chem. Res., 2021, 54, 1143–1156 CrossRef CAS PubMed.
- D. W. Christianson, Chem. Rev., 2017, 117, 11570–11648 CrossRef CAS PubMed.
- E. J. N. Helfrich, G.-M. Lin, C. A. Voigt and J. Clardy, Beilstein J. Org. Chem., 2019, 15, 2889–2906 CrossRef CAS PubMed.
- J. L. Goldstein and M. S. Brown, Nature, 1990, 343, 425–430 CrossRef CAS PubMed.
- A. Frank and M. Groll, Chem. Rev., 2017, 117, 5675–5703 CrossRef CAS PubMed.
- K. Chen and P. S. Baran, Nature, 2009, 459, 824–828 CrossRef CAS PubMed.
- P.-H. Liang, T.-P. Ko and A. H. J. Wang, Eur. J. Biochem., 2002, 269, 3339–3354 CrossRef CAS PubMed.
- D. Sasaki, M. Fujihashi, N. Okuyama, Y. Kobayashi, M. Noike, T. Koyama and K. Miki, J. Biol. Chem., 2011, 286, 3729–3740 CrossRef CAS PubMed.
- C. D. Poulter, Acc. Chem. Res., 1990, 23, 70–77 CrossRef CAS.
- E. Oldfield and F.-Y. Lin, Angew. Chem., Int. Ed., 2012, 51, 1124–1137 CrossRef CAS PubMed.
- X. Pan, J. D. Rudolf and L.-B. Dong, Nat. Prod. Rep., 2024, 41, 402–433 RSC.
- J. S. Dickschat, Nat. Prod. Rep., 2016, 33, 87–110 RSC.
- M. Baunach, J. Franke and C. Hertweck, Angew. Chem., Int. Ed., 2015, 54, 2604–2626 CrossRef CAS PubMed.
- J. D. Rudolf and C.-Y. Chang, Nat. Prod. Rep., 2020, 37, 425–463 RSC.
- Y. Gao, R. B. Honzatko and R. J. Peters, Nat. Prod. Rep., 2012, 29, 1153–1175 RSC.
- Y. Matsuda and I. Abe, Nat. Prod. Rep., 2016, 33, 26–53 RSC.
- C. T. Walsh and M. A. Fischbach, J. Am. Chem. Soc., 2010, 132, 2469–2493 CrossRef CAS PubMed.
- D.-K. Ro, G.-I. Arimura, S. Y. W. Lau, E. Piers and J. Bohlmann, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 8060–8065 CrossRef CAS PubMed.
- S.-S. Gao, N. Naowarojna, R. Cheng, X. Liu and P. Liu, Nat. Prod. Rep., 2018, 35, 792–837 RSC.
- H.-C. Lin, Y. Tsunematsu, S. Dhingra, W. Xu, M. Fukutomi, Y.-H. Chooi, D. E. Cane, A. M. Calvo, K. Watanabe and Y. Tang, J. Am. Chem. Soc., 2014, 136, 4426–4436 CrossRef CAS PubMed.
- J. Feng, F. Surup, M. Hauser, A. Miller, J.-P. Wennrich, M. Stadler, R. J. Cox and E. Kuhnert, Chem. Commun., 2020, 56, 12419–12422 RSC.
- E. Yang, Y. Yao, H. Su, Z. Sun, S.-S. Gao, S. Sureram, P. Kittakoop, K. Fan, Y. Pan, X. Xu, Z.-H. Sun, G. Ma and G. Liu, J. Am. Chem. Soc., 2024, 146, 11457–11464 CAS.
- H. Xiao, Y. Zhang and M. Wang, Trends Biotechnol., 2019, 37, 618–631 CrossRef CAS PubMed.
- J. D. Rudolf, T. A. Alsup, B. Xu and Z. Li, Nat. Prod. Rep., 2021, 38, 905–980 RSC.
- H. Shigemori, H. Komaki, K. Yazawa, Y. Mikami, A. Nemoto, Y. Tanaka, T. Sasaki, Y. In, T. Ishida and J. i. Kobayashi, J. Org. Chem., 1998, 63, 6900–6904 CrossRef CAS PubMed.
- J. Wang, S. M. Soisson, K. Young, W. Shoop, S. Kodali, A. Galgoci, R. Painter, G. Parthasarathy, Y. S. Tang, R. Cummings, S. Ha, K. Dorso, M. Motyl, H. Jayasuriya, J. Ondeyka, K. Herath, C. Zhang, L. Hernandez, J. Allocco, Á. Basilio, J. R. Tormo, O. Genilloud, F. Vicente, F. Pelaez, L. Colwell, S. H. Lee, B. Michael, T. Felcetto, C. Gill, L. L. Silver, J. D. Hermes, K. Bartizal, J. Barrett, D. Schmatz, J. W. Becker, D. Cully and S. B. Singh, Nature, 2006, 441, 358–361 CrossRef CAS PubMed.
- D. E. Cane and J.-K. Sohng, Arch. Biochem. Biophys., 1989, 270, 50–61 CrossRef CAS PubMed.
- L. Keller, E. Oueis, A. Kaur, N. Safaei, S. H. Kirsch, A. P. Gunesch, S. Haid, U. Rand, L. Čičin-Šain, C. Fu, J. Wink, T. Pietschmann and R. Müller, Angew. Chem., Int. Ed., 2023, 62, e202214595 CrossRef CAS PubMed.
- D. E. Cane and H. Ikeda, Acc. Chem. Res., 2012, 45, 463–472 CrossRef CAS PubMed.
- Y. Yamada, T. Kuzuyama, M. Komatsu, K. Shin-ya, S. Omura, D. E. Cane and H. Ikeda, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 857–862 CrossRef CAS PubMed.
- R. M. Hohlman and D. H. Sherman, Nat. Prod. Rep., 2021, 38, 1567–1588 RSC.
- T. Yao, J. Liu, Z. Liu, T. Li, H. Li, Q. Che, T. Zhu, D. Li, Q. Gu and W. Li, Nat. Commun., 2018, 9, 4091 CrossRef PubMed.
- T. Itoh, K. Tokunaga, Y. Matsuda, I. Fujii, I. Abe, Y. Ebizuka and T. Kushiro, Nat. Chem., 2010, 2, 858–864 CrossRef CAS PubMed.
- P. Hedden, Plant Cell Physiol., 2020, 61, 1832–1849 CrossRef CAS PubMed.
- P. Hedden and S. G. Thomas, Biochem. J., 2012, 444, 11–25 CrossRef CAS PubMed.
- P. Hedden and V. Sponsel, J. Plant Growth Regul., 2015, 34, 740–760 CrossRef CAS PubMed.
- X. Lu, D. M. Hershey, L. Wang, A. J. Bogdanove and R. J. Peters, New Phytol., 2015, 206, 295–302 CrossRef CAS PubMed.
- R. Nagel, P. C. G. Turrini, R. S. Nett, J. E. Leach, V. Verdier, M.-A. Van Sluys and R. J. Peters, New Phytol., 2017, 214, 1260–1266 CrossRef CAS PubMed.
- R. Nagel and R. J. Peters, Mol. Plant-Microbe Interact., 2017, 30, 343–349 CrossRef CAS PubMed.
- R. S. Nett, M. Montanares, A. Marcassa, X. Lu, R. Nagel, T. C. Charles, P. Hedden, M. C. Rojas and R. J. Peters, Nat. Chem. Biol., 2017, 13, 69–74 CrossRef CAS PubMed.
- R. S. Nett, T. Contreras and R. J. Peters, ACS Chem. Biol., 2017, 12, 912–917 CrossRef CAS PubMed.
- L. De Bruyne, M. Höfte and D. De Vleesschauwer, Mol. Plant, 2014, 7, 943–959 CrossRef CAS PubMed.
- A. H. de Boer and I. J. d. V.-v. Leeuwen, Trends Plant Sci., 2012, 17, 360–368 CrossRef CAS PubMed.
- J.-Y. Liu, F.-L. Lin, J.-M. Lv, D. Hu and H. Gao, Tetrahedron Lett., 2022, 112, 154224 CrossRef CAS.
- A. Ballio, E. B. Chain, P. De Leo, B. F. Erlanger, M. Mauri and A. Tonolo, Nature, 1964, 203, 297 CrossRef CAS.
- A. Ballio, M. Brufani, C. G. Casinovi, S. Cerrini, W. Fedeli, R. Pellicciari, B. Santurbano and A. Vaciago, Experientia, 1968, 24, 631–635 CrossRef CAS PubMed.
- T. Sassa, T. Tojyo and K. Munakata, Nature, 1970, 227, 379 CrossRef CAS PubMed.
- T. Sassa, M. Togashi and T. Kitaguchi, Agric. Biol. Chem., 1975, 39, 1735–1744 CAS.
- L. M. Stevers, E. Sijbesma, M. Botta, C. MacKintosh, T. Obsil, I. Landrieu, Y. Cau, A. J. Wilson, A. Karawajczyk, J. Eickhoff, J. Davis, M. Hann, G. O'Mahony, R. G. Doveston, L. Brunsveld and C. Ottmann, J. Med. Chem., 2018, 61, 3755–3778 CrossRef CAS PubMed.
- T. Aoyagi, T. Aoyama, F. Kojima, S. Hattori, Y. Honma, M. Hamada and T. Takeuchi, J. Antibiot., 1992, 45, 1587–1591 CrossRef CAS PubMed.
- T. Aoyama, H. Naganawa, Y. Muraoka, T. Aoyagi and T. Takeuchi, J. Antibiot., 1992, 45, 1703–1704 CrossRef CAS PubMed.
- D. Zheng, L. Han, X. Qu, X. Chen, J. Zhong, X. Bi, J. Liu, Y. Jiang, C. Jiang and X. Huang, J. Nat. Prod., 2017, 80, 837–844 CrossRef CAS PubMed.
- W. Yi, Q. Li, T. Song, L. Chen, X.-C. Li, Z. Zhang and X.-Y. Lian, Tetrahedron, 2019, 75, 1186–1193 CrossRef CAS.
- K. Supong, P. Sripreechasak, S. Tanasupawat, K. Danwisetkanjana, P. Rachtawee and P. Pittayakhajonwut, Appl. Microbiol. Biotechnol., 2017, 101, 533–543 CrossRef CAS PubMed.
- J.-P. Jang, M. Jang, G. J. Hwang, M. H. Kim, J. S. Ahn, S.-K. Ko and J.-H. Jang, Bioorg. Med. Chem. Lett., 2022, 57, 128504 CrossRef CAS PubMed.
- T. Sassa, Agric. Biol. Chem., 1972, 36, 2037–2039 CrossRef CAS.
- T. Sassa, A. Takahama and T. Shindo, Agric. Biol. Chem., 1975, 39, 1729–1734 CrossRef CAS.
- R. Li, S. L. Morris-Natschke and K.-H. Lee, Nat. Prod. Rep., 2016, 33, 1166–1226 RSC.
- H. N. Banerjee, Sci. Cult., 1936, 2, 163 CAS.
- G. A. Sim, T. A. Hamor, I. C. Paul and J. M. Robertson, Proc. Chem. Soc., 1961, 75–76 CAS.
- D. H. R. Barton, H. T. Cheung, A. D. Cross, L. M. Jackman and M. Martin-Smith, J. Chem. Soc., 1961, 5061–5073 RSC.
- N. Kato, M. Takahashi, M. Shibayama and K. Munakata, Agric. Biol. Chem., 1972, 36, 2579–2582 CAS.
- A. Ortega, J. F. Blount and P. S. Manchand, J. Chem. Soc., Perkin Trans. 1, 1982, 1, 2505–2508 RSC.
- B. L. Roth, K. Baner, R. Westkaemper, D. Siebert, K. C. Rice, S. Steinberg, P. Ernsberger and R. B. Rothman, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 11934–11939 CrossRef CAS PubMed.
- T. Tamamura, T. Sawa, K. Isshiki, T. Masuda, Y. Homma, H. Inuma, H. Naganawa, M. Hamada, T. Takeuchi and H. Umezawa, J. Antibiot., 1985, 38, 1664–1669 CrossRef CAS PubMed.
- K. Isshiki, T. Tamamura, Y. Takahashi, T. Sawa, H. Naganawa, T. Takeuchi and H. Umezawa, J. Antibiot., 1985, 38, 1819–1821 CrossRef CAS PubMed.
- S. Z. Kawada, Y. Yamashita, Y. Uosaki, K. Gomi, T. Iwasaki, T. Takiguchi and H. Nakano, J. Antibiot., 1992, 45, 1182–1184 CrossRef CAS PubMed.
- Y. Uosaki, S. Kawada, H. Nakano, Y. Saitoh and H. Sano, J. Antibiot., 1993, 46, 235–240 CrossRef CAS PubMed.
- N. R. Andersen and P. R. Rasmussen, Tetrahedron Lett., 1984, 25, 465–468 CrossRef CAS.
- N. R. Andersen, H. O. Lorck and P. R. Rasmussen, J. Antibiot., 1983, 36, 753–760 CrossRef CAS PubMed.
- T. Tamamura, M. Tsuchiya, K. Isshiki, T. Sawa, T. Takeuchi, M. Hori and N. Sakata, J. Antibiot., 1988, 41, 648–654 CrossRef CAS PubMed.
- S.-z. Kawada, Y. Yamashita, N. Fujii and H. Nakano, Cancer Res., 1991, 51, 2922–2925 CAS.
- J. E. McCullough, M. T. Muller, A. J. Howells, A. Maxwell, J. O'Sullivan, R. S. Summerill, W. L. Parker, J. S. Wells, D. P. Bonner and P. B. Fernandes, J. Antibiot., 1993, 46, 526–530 CrossRef CAS PubMed.
- B. Gatto, S. Richter, S. Moro, G. Capranico and M. Palumbo, Nucleic Acids Res., 2001, 29, 4224–4230 CrossRef CAS PubMed.
- S. Richter, B. Gatto, D. Fabris, K. i. Takao, S. Kobayashi and M. Palumbo, Nucleic Acids Res., 2003, 31, 5149–5156 CrossRef CAS PubMed.
- F. Berrue and R. G. Kerr, Nat. Prod. Rep., 2009, 26, 681–710 RSC.
- A. J. Welford and I. Collins, J. Nat. Prod., 2011, 74, 2318–2328 CrossRef CAS PubMed.
- G. Li, J. S. Dickschat and Y.-W. Guo, Nat. Prod. Rep., 2020, 37, 1367–1383 RSC.
- T. Lindel, P. R. Jensen, W. Fenical, B. H. Long, A. M. Casazza, J. Carboni and C. R. Fairchild, J. Am. Chem. Soc., 1997, 119, 8744–8745 CrossRef CAS.
- B. H. Long, J. M. Carboni, A. J. Wasserman, L. A. Cornell, A. M. Casazza, P. R. Jensen, T. Lindel, W. Fenical and C. R. Fairchild, Cancer Res., 1998, 58, 1111–1115 CAS.
- H. M. Hassan, M. A. Khanfar, A. Y. Elnagar, R. Mohammed, L. A. Shaala, D. T. A. Youssef, M. S. Hifnawy and K. A. El Sayed, J. Nat. Prod., 2010, 73, 848–853 CrossRef CAS PubMed.
- Z. Li and J. D. Rudolf, J. Ind. Microbiol. Biotechnol., 2023, 50, kuad027 CrossRef CAS PubMed.
- C. Zhu, B. Xu, D. A. Adpressa, J. D. Rudolf and S. Loesgen, Angew. Chem., Int. Ed., 2021, 60, 14163–14170 CrossRef CAS PubMed.
- L.-F. Ma, M.-J. Chen, D.-E. Liang, L.-M. Shi, Y.-M. Ying, W.-G. Shan, G.-Q. Li and Z.-J. Zhan, J. Nat. Prod., 2020, 83, 1641–1645 CrossRef CAS PubMed.
- Z. Wang, Q. Yang, J. He, H. Li, X. Pan, Z. Li, H.-M. Xu, J. D. Rudolf, D. J. Tantillo and L.-B. Dong, Angew. Chem., Int. Ed., 2023, 62, e202312490 CrossRef CAS PubMed.
- M. Shirai, M. Okuda, K. Motohashi, M. Imoto, K. Furihata, Y. Matsuo, A. Katsuta, Y. Shizuri and H. Seto, J. Antibiot., 2010, 63, 245–250 CrossRef CAS PubMed.
- Z. Yang, Y. Yang, X. Yang, Y. Zhang, L. Zhao, L. Xu and Z. Ding, Chem. Pharm. Bull., 2011, 59, 1430–1433 CrossRef CAS PubMed.
- L. Ding, A. Maier, H.-H. Fiebig, W.-H. Lin, G. Peschel and C. Hertweck, J. Nat. Prod., 2012, 75, 2223–2227 CrossRef CAS PubMed.
- L. Yu, H.-f. Dai, Y.-x. Zhao, W.-j. Zuo, W.-h. Dong, W.-l. Mei and H.-c. Zeng, Phytochem. Lett., 2013, 6, 110–112 CrossRef CAS.
- N. Ding, Y. Jiang, J. Liu, Q. Li, X. Wang, Y. Mu, L. Han and X. Huang, Magn. Reson. Chem., 2016, 54, 930–932 CrossRef CAS PubMed.
- L. Ding and C. Hertweck, J. Nat. Prod., 2020, 83, 2207–2211 CrossRef CAS PubMed.
- L. N. U. Nupur, A. Vats, S. K. Dhanda, G. P. S. Raghava, A. K. Pinnaka and A. Kumar, BMC Microbiol., 2016, 16, 96 CrossRef CAS PubMed.
- M. H. Walter and D. Strack, Nat. Prod. Rep., 2011, 28, 663–692 RSC.
- M. Takagi, K. Motohashi, S. T. Khan, J. Hashimoto and K. Shin-ya, J. Antibiot., 2010, 63, 401–403 CrossRef CAS PubMed.
- B. Shin, B.-Y. Kim, E. Cho, K.-B. Oh, J. Shin, M. Goodfellow and D.-C. Oh, J. Nat. Prod., 2016, 79, 1886–1890 CrossRef CAS PubMed.
- H. Akiyama, N. Oku, E. Harunari, W. Panbangred and Y. Igarashi, J. Antibiot., 2020, 73, 60–65 CrossRef CAS PubMed.
- A. Greule, J. E. Stok, J. J. De Voss and M. J. Cryle, Nat. Prod. Rep., 2018, 35, 757–791 RSC.
- B. Meunier, S. P. de Visser and S. Shaik, Chem. Rev., 2004, 104, 3947–3980 CrossRef CAS PubMed.
- I. G. Denisov, T. M. Makris, S. G. Sligar and I. Schlichting, Chem. Rev., 2005, 105, 2253–2278 CrossRef CAS PubMed.
- X. Huang and J. T. Groves, J. Biol. Inorg. Chem., 2017, 22, 185–207 CrossRef CAS PubMed.
- M.-C. Tang, Y. Zou, K. Watanabe, C. T. Walsh and Y. Tang, Chem. Rev., 2017, 117, 5226–5333 CrossRef CAS PubMed.
- S. Janocha, D. Schmitz and R. Bernhardt, in Biotechnology of Isoprenoids, ed. J. Schrader and J. Bohlmann, Springer, Cham, 2015, vol. 148, ch. 296, pp. 215–250 Search PubMed.
- C. R. Zwick and H. Renata, Nat. Prod. Rep., 2020, 37, 1065–1079 RSC.
- Y. L. Hu, Q. Zhang, S. H. Liu, J. L. Sun, F. Z. Yin, Z. R. Wang, J. Shi, R. H. Jiao and H. M. Ge, Chem. Sci., 2023, 14, 3661–3667 RSC.
- Q. Yang, J. Tian, S. Chen, Z. Yang, Z. Wang, H.-M. Xu and L.-B. Dong, Bioorg. Chem., 2024, 146, 107308 CrossRef CAS PubMed.
- S.-Y. Kim, P. Zhao, M. Igarashi, R. Sawa, T. Tomita, M. Nishiyama and T. Kuzuyama, Chem. Biol., 2009, 16, 736–743 CrossRef CAS PubMed.
- C. Gorner, P. Schrepfer, V. Redai, F. Wallrapp, B. Loll, W. Eisenreich, M. Haslbeck and T. Bruck, Microb. Cell Fact., 2016, 15, 86 CrossRef PubMed.
- K. A. Taizoumbe, S. T. Steiner and J. S. Dickschat, Chem. Eur J., 2023, 29, e202302469 CrossRef CAS PubMed.
- K. Gebhardt, S. W. Meyer, J. Schinko, G. Bringmann, A. Zeeck and H.-P. Fiedler, J. Antibiot., 2011, 64, 229–232 CrossRef CAS PubMed.
- C. Dürr, H.-J. Schnell, A. Luzhetskyy, R. Murillo, M. Weber, K. Welzel, A. Vente and A. Bechthold, Chem. Biol., 2006, 13, 365–377 CrossRef PubMed.
- M. Daum, H.-J. Schnell, S. Herrmann, A. Günther, R. Murillo, R. Müller, P. Bisel, M. Müller and A. Bechthold, ChemBioChem, 2010, 11, 1383–1391 CrossRef CAS PubMed.
- H. Komaki, A. Nemoto, Y. Tanaka, H. Takagi, K. Yazawa, Y. Mikami, H. Shigemori, J. Kobayashi, A. Ando and Y. Nagata, J. Antibiot., 1999, 52, 13–19 CrossRef CAS PubMed.
- K. Komatsu, M. Tsuda, Y. Tanaka, Y. Mikami and J. i. Kobayashi, Bioorg. Med. Chem., 2005, 13, 1507–1513 CrossRef CAS PubMed.
- T. Usui, Y. Nagumo, A. Watanabe, T. Kubota, K. Komatsu, J. i. Kobayashi and H. Osada, Chem. Biol., 2006, 13, 1153–1160 CrossRef CAS PubMed.
- K. Komatsu, M. Tsuda, M. Shiro, Y. Tanaka, Y. Mikami and J. i. Kobayashi, Bioorg. Med. Chem., 2004, 12, 5545–5551 CrossRef CAS PubMed.
- Y. Hayashi, N. Matsuura, H. Toshima, N. Itoh, J. Ishikawa, Y. Mikami and T. Dairi, J. Antibiot., 2008, 61, 164–174 CrossRef CAS PubMed.
- P. N. Schwarz, A. Buchmann, L. Roller, A. Kulik, H. Gross, W. Wohlleben and E. Stegmann, Biotechnol. J., 2018, 13, 1700527 CrossRef PubMed.
- D. Zhang, W. Du, X. Pan, X. Lin, F.-R. Li, Q. Wang, Q. Yang, H.-M. Xu and L.-B. Dong, Beilstein J. Org. Chem., 2024, 20, 815–822 CrossRef CAS PubMed.
- M. Wu, S. B. Singh, J. Wang, C. C. Chung, G. Salituro, B. V. Karanam, S. H. Lee, M. Powles, K. P. Ellsworth, M. E. Lassman, C. Miller, R. W. Myers, M. R. Tota, B. B. Zhang and C. Li, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 5378–5383 CrossRef CAS PubMed.
- J. Wang, S. Kodali, S. H. Lee, A. Galgoci, R. Painter, K. Dorso, F. Racine, M. Motyl, L. Hernandez, E. Tinney, S. L. Colletti, K. Herath, R. Cummings, O. Salazar, I. González, A. Basilio, F. Vicente, O. Genilloud, F. Pelaez, H. Jayasuriya, K. Young, D. F. Cully and S. B. Singh, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 7612–7616 CrossRef CAS PubMed.
- H. Jayasuriya, K. B. Herath, C. Zhang, D. L. Zink, A. Basilio, O. Genilloud, M. T. Diez, F. Vicente, I. Gonzalez, O. Salazar, F. Pelaez, R. Cummings, S. Ha, J. Wang and S. B. Singh, Angew. Chem., Int. Ed., 2007, 46, 4684–4688 CrossRef CAS PubMed.
- J. D. Rudolf, L.-B. Dong, X. Zhang, H. Renata and B. Shen, J. Am. Chem. Soc., 2018, 140, 12349–12353 CrossRef CAS PubMed.
- J. D. Rudolf, L.-B. Dong, K. Manoogian and B. Shen, J. Am. Chem. Soc., 2016, 138, 16711–16721 CrossRef CAS PubMed.
- J. D. Rudolf, L.-B. Dong and B. Shen, Biochem. Pharmacol., 2017, 133, 139–151 CrossRef CAS PubMed.
- D. G. Martin, G. Slomp, S. Mizsak, D. J. Duchamp and C. G. Chidester, Tetrahedron Lett., 1970, 11, 4901–4904 CrossRef PubMed.
- C. N. Tetzlaff, Z. You, D. E. Cane, S. Takamatsu, S. Omura and H. Ikeda, Biochemistry, 2006, 45, 6179–6186 CrossRef CAS PubMed.
- R. Quaderer, S. Omura, H. Ikeda and D. E. Cane, J. Am. Chem. Soc., 2006, 128, 13036–13037 CrossRef CAS PubMed.
- B. Zhao, X. Lin, L. Lei, D. C. Lamb, S. L. Kelly, M. R. Waterman and D. E. Cane, J. Biol. Chem., 2008, 283, 8183–8189 CrossRef CAS PubMed.
- Z. Li, Y. Jiang, X. Zhang, Y. Chang, S. Li, X. Zhang, S. Zheng, C. Geng, P. Men, L. Ma, Y. Yang, Z. Gao, Y.-J. Tang and S. Li, ACS Catal., 2020, 10, 5846–5851 CrossRef CAS.
- R. Nagel and R. J. Peters, Org. Biomol. Chem., 2017, 15, 7566–7571 RSC.
- R. V. K. Cochrane and J. C. Vederas, Acc. Chem. Res., 2014, 47, 3148–3161 CrossRef CAS PubMed.
- U. Bathe and A. Tissier, Phytochemistry, 2019, 161, 149–162 CrossRef CAS PubMed.
- A. Fukumoto, Y.-P. Kim, A. Matsumoto, Y. Takahashi, K. Shiomi, H. Tomoda and S. Ōmura, J. Antibiot., 2008, 61, 1–6 CrossRef CAS PubMed.
- N. Koyama, Y. Tokura, Y. Takahashi and H. Tomoda, Acta Pharm. Sin. B, 2011, 1, 236–239 CrossRef CAS.
- M. Ohtawa, Y. Hishinuma, E. Takagi, T. Yamada, F. Ito, S. Arima, R. Uchida, Y.-P. Kim, S. Ōmura, H. Tomoda and T. Nagamitsu, Chem. Pharm. Bull., 2016, 64, 1370–1377 CrossRef CAS PubMed.
- A. Fukumoto, Y.-P. Kim, H. Hanaki, K. Shiomi, H. Tomoda and S. Ōmura, J. Antibiot., 2008, 61, 7–10 CrossRef CAS PubMed.
- H. Ikeda, K. Shin-ya, T. Nagamitsu and H. Tomoda, J. Ind. Microbiol. Biotechnol., 2016, 43, 325–342 CrossRef CAS PubMed.
- Z.-J. Xiong, J. Huang, Y. Yan, L. Wang, Z. Wang, J. Yang, J. Luo, J. Li and S.-X. Huang, Org. Chem. Front., 2018, 5, 1272–1279 RSC.
- G. J. ten Brink, I. W. C. E. Arends and R. A. Sheldon, Chem. Rev., 2004, 104, 4105–4124 CrossRef PubMed.
- G. R. Krow, Org. React., 2004, 43, 251–798 Search PubMed.
- C. Tolmie, M. S. Smit and D. J. Opperman, Nat. Prod. Rep., 2019, 36, 326–353 RSC.
- J. Jiang, C. N. Tetzlaff, S. Takamatsu, M. Iwatsuki, M. Komatsu, H. Ikeda and D. E. Cane, Biochemistry, 2009, 48, 6431–6440 CrossRef CAS PubMed.
- M.-J. Seo, D. Zhu, S. Endo, H. Ikeda and D. E. Cane, Biochemistry, 2011, 50, 1739–1754 CrossRef CAS PubMed.
- Z. You, S. Omura, H. Ikeda and D. E. Cane, J. Am. Chem. Soc., 2006, 128, 6566–6567 CrossRef CAS PubMed.
- Z. You, S. Omura, H. Ikeda and D. E. Cane, Arch. Biochem. Biophys., 2007, 459, 233–240 CrossRef CAS PubMed.
- Z. You, S. Omura, H. Ikeda, D. E. Cane and G. Jogl, J. Biol. Chem., 2007, 282, 36552–36560 CrossRef CAS PubMed.
- Q. Deng, Y. Liu, L. Chen, M. Xu, N. Naowarojna, N. Lee, L. Chen, D. Zhu, X. Hong, Z. Deng, P. Liu and C. Zhao, Org. Lett., 2019, 21, 7592–7596 CrossRef CAS PubMed.
- L. Chen, Q. Deng, T. Ma, J. Gu, J. Yang, X. Zhang, Y.-Q. Zou, Z. Deng, L. Chen and C. Zhao, ACS Catal., 2024, 14, 7389–7401 CrossRef CAS.
- M. L. Hillwig, H. A. Fuhrman, K. Ittiamornkul, T. J. Sevco, D. H. Kwak and X. Liu, ChemBioChem, 2014, 15, 665–669 CrossRef CAS PubMed.
- M. L. Hillwig, Q. Zhu and X. Liu, ACS Chem. Biol., 2014, 9, 372–377 CrossRef CAS PubMed.
- M. L. Hillwig and X. Liu, Nat. Chem. Biol., 2014, 10, 921–923 CrossRef CAS PubMed.
- Q. Zhu, M. L. Hillwig, Y. Doi and X. Liu, ChemBioChem, 2016, 17, 466–470 CrossRef CAS PubMed.
- A. J. Mitchell, Q. Zhu, A. O. Maggiolo, N. R. Ananth, M. L. Hillwig, X. Liu and A. K. Boal, Nat. Chem. Biol., 2016, 12, 636–640 CrossRef CAS PubMed.
- M. L. Hillwig, Q. Zhu, K. Ittiamornkul and X. Liu, Angew. Chem., Int. Ed., 2016, 55, 5780–5784 CrossRef CAS PubMed.
- H. Jörnvall, B. Persson, M. Krook, S. Atrian, R. Gonzalez-Duarte, J. Jeffery and D. Ghosh, Biochemistry, 1995, 34, 6003–6013 CrossRef PubMed.
- Y. Kallberg, U. Oppermann, H. Jörnvall and B. Persson, Eur. J. Biochem., 2002, 269, 4409–4417 CrossRef CAS PubMed.
- A. Atawong, M. Hasegawa and O. Kodama, Biosci. Biotechnol. Biochem., 2002, 66, 566–570 CrossRef CAS PubMed.
- Z. Wang, D. R. Nelson, J. Zhang, X. Wan and R. J. Peters, Nat. Prod. Rep., 2023, 40, 452–469 RSC.
- L.-B. Dong, X. Zhang, J. D. Rudolf, M.-R. Deng, E. Kalkreuter, A. J. Cepeda, H. Renata and B. Shen, J. Am. Chem. Soc., 2019, 141, 4043–4050 CrossRef CAS PubMed.
- L.-B. Dong, J. D. Rudolf, M.-R. Deng, X. Yan and B. Shen, ChemBioChem, 2018, 19, 1727–1733 CrossRef CAS PubMed.
- L.-B. Dong, J. D. Rudolf and B. Shen, Bioorg. Med. Chem., 2016, 24, 6348–6353 CrossRef CAS PubMed.
- M. J. Smanski, Z. Yu, J. Casper, S. Lin, R. M. Peterson, Y. Chen, E. Wendt-Pienkowski, S. R. Rajski and B. Shen, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 13498–13503 CrossRef CAS PubMed.
- L.-B. Dong, J. D. Rudolf, D. Kang, N. Wang, C. Q. He, Y. Deng, Y. Huang, K. N. Houk, Y. Duan and B. Shen, Nat. Commun., 2018, 9, 2362 CrossRef PubMed.
- L.-B. Dong, J. D. Rudolf and B. Shen, Org. Lett., 2016, 18, 4606–4609 CrossRef CAS PubMed.
- C.-J. Zheng, E. Kalkreuter, B.-Y. Fan, Y.-C. Liu, L.-B. Dong and B. Shen, ACS Chem. Biol., 2021, 16, 96–105 CrossRef CAS PubMed.
- M. R. Prinsep, R. A. Thomson, M. L. West and B. L. Wylie, J. Nat. Prod., 1996, 59, 786–788 CrossRef CAS PubMed.
- D. Back, T. J. O'Donnell, K. K. Axt, J. R. Gurr, J. M. Vanegas, P. G. Williams and B. Philmus, ACS Chem. Biol., 2023, 18, 1797–1807 CrossRef CAS PubMed.
- M. Komatsu, M. Tsuda, S. Ōmura, H. Oikawa and H. Ikeda, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 7422–7427 CrossRef CAS PubMed.
- C.-M. Wang and D. E. Cane, J. Am. Chem. Soc., 2008, 130, 8908–8909 CrossRef CAS PubMed.
- T. Ozaki, S. S. Shinde, L. Gao, R. Okuizumi, C. Liu, Y. Ogasawara, X. Lei, T. Dairi, A. Minami and H. Oikawa, Angew. Chem., Int. Ed., 2018, 57, 6629–6632 CrossRef CAS PubMed.
- H. Tsutsumi, Y. Katsuyama, M. Izumikawa, M. Takagi, M. Fujie, N. Satoh, K. Shin-ya and Y. Ohnishi, J. Am. Chem. Soc., 2018, 140, 6631–6639 CrossRef CAS PubMed.
- N. N. Gerber, CRC Crit. Rev. Microbiol., 1979, 7, 191–214 CrossRef CAS PubMed.
- G. Izaguirre, J. Hwang Cordelia, W. Krasner Stuart and J. McGuire Michael, Appl. Environ. Microbiol., 1982, 43, 708–714 CrossRef CAS PubMed.
- J. S. Dickschat, T. Nawrath, V. Thiel, B. Kunze, R. Müller and S. Schulz, Angew. Chem., Int. Ed., 2007, 46, 8287–8290 CrossRef CAS PubMed.
- L. L. Medsker, D. Jenkins, J. F. Thomas and C. Koch, Environ. Sci. Technol., 1969, 3, 476–477 CrossRef CAS.
- N. N. Gerber, J. Antibiot., 1969, 22, 508–509 CrossRef CAS PubMed.
- R. Bentley and R. Meganathan, FEBS Lett., 1981, 125, 220–222 CrossRef CAS PubMed.
- Y. Saitoh, Y. Ikuina, T. Yasuzawa, S. Kakita, S. Nakanishi, M. Yoshida and H. Sano, Symposium on the Chemistry of Natural Products, symposium papers, 1991, vol. 33, pp. 707–714 Search PubMed.
- H. Seto, H. Watanabe, N. Orihara and K. Furihata, Symposium on the Chemistry of Natural Products, symposium papers, 1996, vol. 38, pp. 19–24 Search PubMed.
- S. Nakanishi, K. Osawa, Y. Saito, I. Kawamoto, K. Kuroda and H. Kase, J. Antibiot., 1992, 45, 341–347 CrossRef CAS PubMed.
- K. Nagashima, S. Toki, S. Nakanishi, H. Kase and Y. Matsuda, J. Antibiot., 1993, 46, 1481–1483 CrossRef CAS PubMed.
- M. Ichimura, R. Eiki, K. Osawa, S. Nakanishi and H. Kase, Biochem. J., 1996, 316, 311–316 CrossRef CAS PubMed.
- Y. Hayashi, H. Onaka, N. Itoh, H. Seto and T. Dairi, Biosci. Biotechnol. Biochem., 2007, 71, 3072–3081 CrossRef CAS PubMed.
- D. Zhu, M.-J. Seo, H. Ikeda and D. E. Cane, J. Am. Chem. Soc., 2011, 133, 2128–2131 CrossRef CAS PubMed.
- L. Duan, G. Jogl and D. E. Cane, J. Am. Chem. Soc., 2016, 138, 12678–12689 CrossRef CAS PubMed.
- J. W. Wilt, in Free Radicals, ed. J. K. Kochi, Wiley-Interscience, New York, 1973, vol. 1, pp. 333–501 Search PubMed.
- D. E. Cane, J. K. Sohng and P. G. Williard, J. Org. Chem., 1992, 57, 844–851 CrossRef CAS.
- H. Seto, T. Sasaki, H. Yonehara, S. Takahashi, M. Takeuchi, H. Kuwano and M. Arai, J. Antibiot., 1984, 37, 1076–1078 CrossRef CAS PubMed.
- P. R. Ortiz de Montellano and R. A. Stearns, J. Am. Chem. Soc., 1987, 109, 3415–3420 CrossRef CAS.
- M. J. Cryle, P. R. Ortiz de Montellano and J. J. De Voss, J. Org. Chem., 2005, 70, 2455–2469 CrossRef CAS PubMed.
- Y. Jiang, X. He and P. R. Ortiz de Montellano, Biochemistry, 2006, 45, 533–542 CrossRef CAS PubMed.
- K. L. Rollick and J. K. Kochi, J. Am. Chem. Soc., 1982, 104, 1319–1330 CrossRef CAS.
- T. H. Lowry and K. S. Richardson, Mechanism and Theory in Organic Chemistry, 1987 Search PubMed.
- X. Wang, J. Shi and Y. Liu, Inorg. Chem., 2018, 57, 8933–8941 CrossRef CAS PubMed.
- R. Fittig, Adv. Cycloaddit., 1860, 114, 54–63 Search PubMed.
- Z.-L. Song, C.-A. Fan and Y.-Q. Tu, Chem. Rev., 2011, 111, 7523–7556 CrossRef CAS PubMed.
- W.-H. Lin, J.-M. Fang and Y.-S. Cheng, Phytochemistry, 1995, 40, 871–873 CrossRef CAS.
- F. Marion, D. E. Williams, B. O. Patrick, I. Hollander, R. Mallon, S. C. Kim, D. M. Roll, L. Feldberg, R. Van Soest and R. J. Andersen, Org. Lett., 2006, 8, 321–324 CrossRef CAS PubMed.
- Q.-Q. Zhao, Q.-Y. Song, K. Jiang, G.-D. Li, W.-J. Wei, Y. Li and K. Gao, Org. Lett., 2015, 17, 2760–2763 CrossRef CAS PubMed.
- L. Jørgensen, S. J. McKerrall, C. A. Kuttruff, F. Ungeheuer, J. Felding and P. S. Baran, Science, 2013, 341, 878–882 CrossRef PubMed.
- C. Tode, Y. Yamano and M. Ito, J. Chem. Soc., Perkin Trans. 1, 2002, 1581–1587 RSC.
- J. H. George, J. E. Baldwin and R. M. Adlington, Org. Lett., 2010, 12, 2394–2397 CrossRef CAS PubMed.
- A. Cernijenko, R. Risgaard and P. S. Baran, J. Am. Chem. Soc., 2016, 138, 9425–9428 CrossRef CAS PubMed.
- L. N. Mander, Chem. Rev., 1992, 92, 573–612 CrossRef CAS.
- J. MacMillan, Nat. Prod. Rep., 1997, 14, 221–243 RSC.
- A. J. Birch, R. W. Richards, H. Smith, A. Harris and W. B. Whalley, Tetrahedron, 1959, 7, 241–251 CrossRef CAS.
- B. E. Cross, R. H. B. Galt and K. Norton, Tetrahedron, 1968, 24, 231–237 CrossRef CAS.
- J. R. Hanson, J. Hawker and A. F. White, J. Chem. Soc., Perkin Trans. 1, 1972, 1892–1895 RSC.
- J. E. Graebe, P. Hedden and J. MacMillan, J. Chem. Soc., Chem. Commun., 1975, 161–162 RSC.
- R. Nagel, L. E. Alexander, C. E. Stewart and R. J. Peters, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2221549120 CrossRef PubMed.
- C. A. Helliwell, P. M. Chandler, A. Poole, E. S. Dennis and W. J. Peacock, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 2065–2070 CrossRef CAS PubMed.
- S. Malonek, M. C. Rojas, P. Hedden, P. Gaskin, P. Hopkins and B. Tudzynski, J. Biol. Chem., 2004, 279, 25075–25084 CrossRef CAS PubMed.
- C. Troncoso, J. Cárcamo, P. Hedden, B. Tudzynski and M. Cecilia Rojas, Phytochemistry, 2008, 69, 672–683 CrossRef CAS PubMed.
- R. S. Nett, J. S. Dickschat and R. J. Peters, Org. Lett., 2016, 18, 5974–5977 CrossRef CAS PubMed.
- K. Hiroya, Y. Murakami, T. Shimizu, M. Hatano and P. R. O. Demontellano, Arch. Biochem. Biophys., 1994, 310, 397–401 CrossRef CAS PubMed.
- Y. Ren and A. D. Kinghorn, J. Med. Chem., 2020, 63, 15410–15448 CrossRef CAS PubMed.
- M. Jiang, Z. Wu, L. Liu and S. Chen, Org. Biomol. Chem., 2021, 19, 1644–1704 RSC.
- T. Nomura, T. Kushiro, T. Yokota, Y. Kamiya, G. J. Bishop and S. Yamaguchi, J. Biol. Chem., 2005, 280, 17873–17879 CrossRef CAS PubMed.
- G. Hansen Bjarne, E. Mnich, F. Nielsen Kristian, B. Nielsen Jakob, T. Nielsen Morten, H. Mortensen Uffe, O. Larsen Thomas and R. Patil Kiran, Appl. Environ. Microbiol., 2012, 78, 4908–4913 CrossRef PubMed.
- Y. Matsuda, T. Wakimoto, T. Mori, T. Awakawa and I. Abe, J. Am. Chem. Soc., 2014, 136, 15326–15336 CrossRef CAS PubMed.
- H. Zeng, G. Yin, Q. Wei, D. Li, Y. Wang, Y. Hu, C. Hu and Y. Zou, Angew. Chem., Int. Ed., 2019, 58, 6569–6573 CrossRef CAS PubMed.
- R. Nagel and R. J. Peters, Angew. Chem., Int. Ed., 2018, 57, 6082–6085 CrossRef CAS PubMed.
- B. Dockerill and J. R. Hanson, Phytochemistry, 1978, 17, 701–704 CrossRef CAS.
- J. Rittle and M. T. Green, Science, 2010, 330, 933–937 CrossRef CAS PubMed.
- Y. Yang and F. H. Arnold, Acc. Chem. Res., 2021, 54, 1209–1225 CrossRef CAS PubMed.
- W. G. Kim, J. P. Kim, C. J. Kim, K. H. Lee and I. D. Yoo, J. Antibiot., 1996, 49, 20–25 CrossRef CAS PubMed.
- W. G. Kim, J. P. Kim and I. D. Yoo, J. Antibiot., 1996, 49, 26–30 CrossRef CAS PubMed.
- W.-G. Kim, J.-P. Kim, H. Koshino, K. Shin-Ya, H. Seto and I.-D. Yoo, Tetrahedron, 1997, 53, 4309–4316 CrossRef CAS.
- J.-G. Lee, I.-D. Yoo and W.-G. Kim, Biol. Pharm. Bull., 2007, 30, 795–797 CrossRef CAS PubMed.
- C. C. Farwell, R. K. Zhang, J. A. McIntosh, T. K. Hyster and F. H. Arnold, ACS Cent. Sci., 2015, 1, 89–93 CrossRef CAS PubMed.
- R. Singh, J. N. Kolev, P. A. Sutera and R. Fasan, ACS Catal., 2015, 5, 1685–1691 CrossRef CAS PubMed.
- P. S. Coelho, E. M. Brustad, A. Kannan and F. H. Arnold, Science, 2013, 339, 307–310 CrossRef CAS PubMed.
- Y.-H. Chooi, Y. J. Hong, R. A. Cacho, D. J. Tantillo and Y. Tang, J. Am. Chem. Soc., 2013, 135, 16805–16808 CrossRef CAS PubMed.
- A. J. King, G. D. Brown, A. D. Gilday, E. Forestier, T. R. Larson and I. A. Graham, ChemBioChem, 2016, 17, 1593–1597 CrossRef CAS PubMed.
- Y. Katsuyama, K. Harmrolfs, D. Pistorius, Y. Li and R. Müller, Angew. Chem., Int. Ed., 2012, 51, 9437–9440 CrossRef CAS PubMed.
- Y. Katsuyama, X.-W. Li, R. Müller and B. Nay, ChemBioChem, 2014, 15, 2349–2352 CrossRef CAS PubMed.
- Z. D. Miles, S. Diethelm, H. P. Pepper, D. M. Huang, J. H. George and B. S. Moore, Nat. Chem., 2017, 9, 1235–1242 CrossRef CAS PubMed.
- B. Kunze, G. Hofle and H. Reichenbach, J. Antibiot., 1987, 40, 258–265 CrossRef CAS PubMed.
- W. Oettmeier, R. Dostatni, C. Majewski, G. Höfle, T. Fecker, B. Kunze and H. Reichenbach, Z. Naturforsch., C: J. Biosci., 1990, 45, 322–328 CrossRef CAS.
- D. Pistorius, Y. Li, A. Sandmann and R. Müller, Mol. Biosyst., 2011, 7, 3308–3315 RSC.
- G. Höfle and B. Kunze, J. Nat. Prod., 2008, 71, 1843–1849 CrossRef PubMed.
- A. Sandmann, J. Dickschat, H. Jenke-Kodama, B. Kunze, E. Dittmann and R. Müller, Angew. Chem., Int. Ed., 2007, 46, 2712–2716 CrossRef CAS PubMed.
- D. Jakubczyk, J. Z. Cheng and S. E. O'Connor, Nat. Prod. Rep., 2014, 31, 1328–1338 RSC.
- S. E. O'Connor and J. J. Maresh, Nat. Prod. Rep., 2006, 23, 532–547 RSC.
- I. S. Marcos, R. F. Moro, I. Costales, P. Basabe and D. Díez, Nat. Prod. Rep., 2013, 30, 1509–1526 RSC.
- T. Ozaki, A. Minami and H. Oikawa, Nat. Prod. Rep., 2023, 40, 202–213 RSC.
- G. M. Cragg and D. J. Newman, J. Ethnopharmacol., 2005, 100, 72–79 CrossRef CAS PubMed.
- K. Takada, H. Kajiwara and N. Imamura, J. Nat. Prod., 2010, 73, 698–701 CrossRef CAS PubMed.
- L. Ding, J. Münch, H. Goerls, A. Maier, H.-H. Fiebig, W.-H. Lin and C. Hertweck, Bioorg. Med. Chem. Lett., 2010, 20, 6685–6687 CrossRef CAS PubMed.
- L. Ding, A. Maier, H.-H. Fiebig, W.-H. Lin and C. Hertweck, Org. Biomol. Chem., 2011, 9, 4029–4031 RSC.
- Q. Zhang, A. Mándi, S. Li, Y. Chen, W. Zhang, X. Tian, H. Zhang, H. Li, W. Zhang, S. Zhang, J. Ju, T. Kurtán and C. Zhang, Eur. J. Org Chem., 2012, 5256–5262 CrossRef CAS.
- S.-H. Kim, T.-K.-Q. Ha, W. K. Oh, J. Shin and D.-C. Oh, J. Nat. Prod., 2016, 79, 51–58 CrossRef CAS PubMed.
- Z. Meng, H. Yu, L. Li, W. Tao, H. Chen, M. Wan, P. Yang, D. J. Edmonds, J. Zhong and A. Li, Nat. Commun., 2015, 6, 6096 CrossRef CAS PubMed.
- Z. Xu, M. Baunach, L. Ding and C. Hertweck, Angew. Chem., Int. Ed., 2012, 51, 10293–10297 CrossRef CAS PubMed.
- H. Li, Q. Zhang, S. Li, Y. Zhu, G. Zhang, H. Zhang, X. Tian, S. Zhang, J. Ju and C. Zhang, J. Am. Chem. Soc., 2012, 134, 8996–9005 CrossRef CAS PubMed.
- H. Li, Y. Sun, Q. Zhang, Y. Zhu, S.-M. Li, A. Li and C. Zhang, Org. Lett., 2015, 17, 306–309 CrossRef CAS PubMed.
- Q. Zhang, H. Li, S. Li, Y. Zhu, G. Zhang, H. Zhang, W. Zhang, R. Shi and C. Zhang, Org. Lett., 2012, 14, 6142–6145 CrossRef CAS PubMed.
- S. Kugel, M. Baunach, P. Baer, M. Ishida-Ito, S. Sundaram, Z. Xu, M. Groll and C. Hertweck, Nat. Commun., 2017, 8, 15804 CrossRef CAS PubMed.
- A. Alfieri, F. Fersini, N. Ruangchan, M. Prongjit, P. Chaiyen and A. Mattevi, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 1177–1182 CrossRef CAS PubMed.
- C. T. Walsh and Y. Tang, Natural Product Biosynthesis, The Royal Society of Chemistry, 2022 Search PubMed.
- Y. Liu, L. Wang, L. Zhao and Y. Zhang, Nat. Prod. Rep., 2022, 39, 1282–1304 RSC.
- M. Baunach, L. Ding, T. Bruhn, G. Bringmann and C. Hertweck, Angew. Chem., Int. Ed., 2013, 52, 9040–9043 CrossRef CAS PubMed.
- M. Baunach, L. Ding, K. Willing and C. Hertweck, Angew. Chem., Int. Ed., 2015, 54, 13279–13283 CrossRef CAS PubMed.
- E. Jin, H. Li, Z. Liu, F. Xiao and W. Li, J. Nat. Prod., 2021, 84, 2606–2611 CrossRef CAS PubMed.
- Q. Zhang, H. Li, L. Yu, Y. Sun, Y. Zhu, H. Zhu, L. Zhang, S.-M. Li, Y. Shen, C. Tian, A. Li, H.-w. Liu and C. Zhang, Chem. Sci., 2017, 8, 5067–5077 RSC.
- W. J. Baas, Phytochemistry, 1985, 24, 1875–1889 CrossRef CAS.
- A. Almeida, L. Dong, G. Appendino and S. Bak, Nat. Prod. Rep., 2020, 37, 1207–1228 RSC.
- P. S. Grant and M. A. Brimble, Chem. Eur J., 2021, 27, 6367–6389 CrossRef CAS PubMed.
- S. B. Singh, H. Jayasuriya, J. G. Ondeyka, K. B. Herath, C. Zhang, D. L. Zink, N. N. Tsou, R. G. Ball, A. Basilio, O. Genilloud, M. T. Diez, F. Vicente, F. Pelaez, K. Young and J. Wang, J. Am. Chem. Soc., 2006, 128, 11916–11920 CrossRef CAS PubMed.
- M. J. Smanski, J. Casper, R. M. Peterson, Z. Yu, S. R. Rajski and B. Shen, J. Nat. Prod., 2012, 75, 2158–2167 CrossRef CAS PubMed.
- J. D. Rudolf, L.-B. Dong, T. Huang and B. Shen, Mol. Biosyst., 2015, 11, 2717–2726 RSC.
- L.-B. Dong, Y.-C. Liu, A. J. Cepeda, E. Kalkreuter, M.-R. Deng, J. D. Rudolf, C. Chang, A. Joachimiak, G. N. Phillips, Jr. and B. Shen, J. Am. Chem. Soc., 2019, 141, 12406–12412 CrossRef CAS PubMed.
- N. Nagano, C. A. Orengo and J. M. Thornton, J. Mol. Biol., 2002, 321, 741–765 CrossRef CAS PubMed.
- A. D. Goldman, J. T. Beatty and L. F. Landweber, J. Mol. Evol., 2016, 82, 17–26 CrossRef CAS PubMed.
- A. J. Jasniewski and L. Que Jr, Chem. Rev., 2018, 118, 2554–2592 CrossRef CAS PubMed.
- S. Zhang, X. Li, Y. Wang, L. Yan, J. Wei and Y. Liu, Inorg. Chem., 2021, 60, 17783–17796 CrossRef CAS PubMed.
- P. M. Brown, T. T. Caradoc-Davies, J. M. J. Dickson, G. J. S. Cooper, K. M. Loomes and E. N. Baker, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15032–15037 CrossRef CAS PubMed.
- L. M. van Staalduinen, F. R. McSorley, K. Schiessl, J. Séguin, P. B. Wyatt, F. Hammerschmidt, D. L. Zechel and Z. Jia, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 5171–5176 CrossRef CAS PubMed.
- K. H. van Pee, C. Dong, S. Flecks, J. Naismith, E. P. Patallo and T. Wage, Adv. Appl. Microbiol., 2006, 59, 127–157 CAS.
- J. M. Winter and B. S. Moore, J. Biol. Chem., 2009, 284, 18577–18581 CrossRef CAS PubMed.
- G. J. Colpas, B. J. Hamstra, J. W. Kampf and V. L. Pecoraro, J. Am. Chem. Soc., 1996, 118, 3469–3478 CrossRef CAS.
- J. N. Carter-Franklin and A. Butler, J. Am. Chem. Soc., 2004, 126, 15060–15066 CrossRef CAS PubMed.
- J. M. Winter, M. C. Moffitt, E. Zazopoulos, J. B. McAlpine, P. C. Dorrestein and B. S. Moore, J. Biol. Chem., 2007, 282, 16362–16368 CrossRef CAS PubMed.
- P. Bernhardt, T. Okino, J. M. Winter, A. Miyanaga and B. S. Moore, J. Am. Chem. Soc., 2011, 133, 4268–4270 CrossRef CAS PubMed.
- L. Kaysser, P. Bernhardt, S.-J. Nam, S. Loesgen, J. G. Ruby, P. Skewes-Cox, P. R. Jensen, W. Fenical and B. S. Moore, J. Am. Chem. Soc., 2012, 134, 11988–11991 CrossRef CAS PubMed.
- R. Teufel, L. Kaysser, M. T. Villaume, S. Diethelm, M. K. Carbullido, P. S. Baran and B. S. Moore, Angew. Chem., Int. Ed., 2014, 53, 11019–11022 CrossRef CAS PubMed.
- S. Diethelm, R. Teufel, L. Kaysser and B. S. Moore, Angew. Chem., Int. Ed., 2014, 53, 11023–11026 CrossRef CAS PubMed.
- L. A. M. Murray, S. M. K. McKinnie, H. P. Pepper, R. Erni, Z. D. Miles, M. C. Cruickshank, B. López-Pérez, B. S. Moore and J. H. George, Angew. Chem., Int. Ed., 2018, 57, 11009–11014 CrossRef CAS PubMed.
- S. M. K. McKinnie, Z. D. Miles, P. A. Jordan, T. Awakawa, H. P. Pepper, L. A. M. Murray, J. H. George and B. S. Moore, J. Am. Chem. Soc., 2018, 140, 17840–17845 CrossRef CAS PubMed.
- S. M. K. McKinnie, Z. D. Miles and B. S. Moore, Methods Enzymol., 2018, 604, 405–424 CAS.
- P. Y.-T. Chen, S. Adak, J. R. Chekan, D. K. Liscombe, A. Miyanaga, P. Bernhardt, S. Diethelm, E. N. Fielding, J. H. George, Z. D. Miles, L. A. M. Murray, T. S. Steele, J. M. Winter, J. P. Noel and B. S. Moore, Biochemistry, 2022, 61, 1844–1852 CrossRef CAS PubMed.
- K. Shiomi, H. Nakamura, H. Iinuma, H. Naganawa, K. Isshiki, T. Takeuchi, H. Umezawa and Y. Iitaka, J. Antibiot., 1986, 39, 494–501 CrossRef CAS PubMed.
- K. Shiomi, H. Iinuma, M. Hamada, H. Naganawa, M. Manabe, C. Matsuki, T. Takeuchi and H. Umezawa, J. Antibiot., 1986, 39, 487–493 CrossRef CAS PubMed.
- L. A. M. Murray, S. M. K. McKinnie, B. S. Moore and J. H. George, Nat. Prod. Rep., 2020, 37, 1334–1366 RSC.
- K. Shiomi, H. Iinuma, H. Naganawa, K. Isshiki, T. Takeuchi and H. Umezawa, J. Antibiot., 1987, 40, 1740–1745 CrossRef CAS PubMed.
- A.-W. Struck, M. L. Thompson, L. S. Wong and J. Micklefield, ChemBioChem, 2012, 13, 2642–2655 CrossRef CAS PubMed.
- Y.-H. Lee, D. Ren, B. Jeon and H.-w. Liu, Nat. Prod. Rep., 2023, 40, 1521–1549 RSC.
- E. Cronan John and T. Luk, Microbiol. Mol. Biol. Rev., 2022, 86, e00013–e00022 Search PubMed.
- S. von Reuss, D. Domik, M. C. Lemfack, N. Magnus, M. Kai, T. Weise and B. Piechulla, J. Am. Chem. Soc., 2018, 140, 11855–11862 CrossRef CAS PubMed.
- N. Magnus, S. H. von Reuss, F. Braack, C. Zhang, K. Baer, A. Koch, P. L. Hampe, S. Sutour, F. Chen and B. Piechulla, Angew. Chem., Int. Ed., 2023, 62, e202303692 CrossRef CAS PubMed.
- T. Awakawa, L. Zhang, T. Wakimoto, S. Hoshino, T. Mori, T. Ito, J. Ishikawa, M. E. Tanner and I. Abe, J. Am. Chem. Soc., 2014, 136, 9910–9913 CrossRef CAS PubMed.
- S. H. von Reuß, M. Kai, B. Piechulla and W. Francke, Angew. Chem., Int. Ed., 2010, 49, 2009–2010 CrossRef PubMed.
- D. Domik, A. Thürmer, T. Weise, W. Brandt, R. Daniel and B. Piechulla, Front. Microbiol., 2016, 7, 737 Search PubMed.
- M. C. Lemfack, W. Brandt, K. Krüger, A. Gurowietz, J. Djifack, J.-P. Jung, M. Hopf, H. Noack, B. Junker, S. von Reuß and B. Piechulla, Sci. Rep., 2021, 11, 3182 CrossRef CAS PubMed.
- H. Xu, L. Lauterbach, B. Goldfuss, G. Schnakenburg and J. S. Dickschat, Nat. Chem., 2023, 15, 1164–1171 CrossRef CAS PubMed.
- Y.-T. Duan, A. Koutsaviti, M. Harizani, C. Ignea, V. Roussis, Y. Zhao, E. Ioannou and S. C. Kampranis, Nat. Chem. Biol., 2023, 19, 1532–1539 CrossRef CAS PubMed.
- H. Xu, B. Goldfuss and J. S. Dickschat, Angew. Chem., Int. Ed., 2024, 63, e202408809 CrossRef CAS PubMed.
- H. Xu, H. Li, B. Goldfuss, G. Schnakenburg and J. S. Dickschat, Angew. Chem., Int. Ed., 2024, 63, e202412040 CAS.
- Y. Hitotsuyanagi, H. Fujiki, M. Suganuma, N. Aimi, S. Sakai, Y. Endo, K. Shudo and T. Sugimura, Chem. Pharm. Bull., 1984, 32, 4233–4236 CrossRef CAS PubMed.
- H. Fujiki, Y. Tanaka, R. Miyake, U. Kikkawa, Y. Nishizuka and T. Sugimura, Biochem. Biophys. Res. Commun., 1984, 120, 339–343 CrossRef CAS PubMed.
- J. H. Cardellina, F.-J. Marner and R. E. Moore, Science, 1979, 204, 193–195 CrossRef CAS PubMed.
- T. Yamashita, M. Imoto, K. Isshiki, T. Sawa, H. Naganawa, S. Kurasawa, B.-Q. Zhu and K. Umezawa, J. Nat. Prod., 1988, 51, 1184–1187 CrossRef CAS.
- D. J. Edwards and W. H. Gerwick, J. Am. Chem. Soc., 2004, 126, 11432–11433 CrossRef CAS PubMed.
- F. Yu, M. Li, C. Xu, B. Sun, H. Zhou, Z. Wang, Q. Xu, M. Xie, G. Zuo, P. Huang, H. Guo, Q. Wang and J. He, Biochem. J., 2016, 473, 4385–4397 CrossRef CAS PubMed.
- J. K. McBee, K. Palczewski, W. Baehr and D. R. Pepperberg, Prog. Retin. Eye Res., 2001, 20, 469–529 CrossRef CAS PubMed.
- M. Lidén and U. Eriksson, J. Biol. Chem., 2006, 281, 13001–13004 CrossRef PubMed.
- J.-M. Zhang, X. Liu, Q. Wei, C. Ma, D. Li and Y. Zou, Nat. Commun., 2022, 13, 225 CrossRef PubMed.
- X. Zhang, X. Pang, L. Zhang, Y. Li, Y. Song, H. Xiao, Y. Liu, J. Wang and Y. Yan, Org. Lett., 2024, 26, 1612–1617 CrossRef CAS PubMed.
- D. Sheehan, G. Meade, V. M. Foley and C. A. Dowd, Biochem. J., 2001, 360, 1–16 CrossRef CAS PubMed.
- J. Yang, T. Mori, X. Wei, Y. Matsuda and I. Abe, Angew. Chem., Int. Ed., 2021, 60, 19458–19465 CrossRef CAS PubMed.
- Z. Li, B. Xu, T. A. Alsup, X. Wei, W. Ning, D. G. Icenhour, M. A. Ehrenberger, I. Ghiviriga, B.-D. Giang and J. D. Rudolf, J. Am. Chem. Soc., 2023, 145, 22361–22365 CrossRef CAS PubMed.
- A. A. Chaudhri, Y. Kakumu, S. Thiengmag, J. C.-T. Liu, G.-M. Lin, S. Durusu, F. Biermann, M. Boeck, C. A. Voigt, J. Clardy, R. Ueoka, A. S. Walker and E. J. N. Helfrich, ACS Chem. Biol., 2024, 19, 2314–2322 CrossRef CAS PubMed.
- Z. Li, B. Xu, V. Kojasoy, T. Ortega, D. A. Adpressa, W. Ning, X. Wei, J. Liu, D. J. Tantillo, S. Loesgen and J. D. Rudolf, Chem, 2023, 9, 698–708 CAS.
- Z. Wu, J. Wouters and C. D. Poulter, J. Am. Chem. Soc., 2005, 127, 17433–17438 CrossRef CAS PubMed.
- F. Biermann, S. L. Wenski and E. J. N. Helfrich, Beilstein J. Org. Chem., 2022, 18, 1656–1671 CrossRef CAS PubMed.
- C. Hertweck, Angew. Chem., Int. Ed., 2009, 48, 4688–4716 CrossRef CAS PubMed.
- R. D. Süssmuth and A. Mainz, Angew. Chem., Int. Ed., 2017, 56, 3770–3821 CrossRef PubMed.
- E. J. N. Helfrich and J. Piel, Nat. Prod. Rep., 2016, 33, 231–316 RSC.
- A. A. Nava, J. Roberts, R. W. Haushalter, Z. Wang and J. D. Keasling, ACS Synth. Biol., 2023, 12, 3148–3155 CrossRef CAS PubMed.
- E. J. N. Helfrich, R. Ueoka, A. Dolev, M. Rust, R. A. Meoded, A. Bhushan, G. Califano, R. Costa, M. Gugger, C. Steinbeck, P. Moreno and J. Piel, Nat. Chem. Biol., 2019, 15, 813–821 CrossRef CAS PubMed.
- M. H. Medema, K. Blin, P. Cimermancic, V. de Jager, P. Zakrzewski, M. A. Fischbach, T. Weber, E. Takano and R. Breitling, Nucleic Acids Res., 2011, 39, W339–W346 CrossRef CAS PubMed.
- K. Blin, S. Shaw, H. E. Augustijn, Z. L. Reitz, F. Biermann, M. Alanjary, A. Fetter, B. R. Terlouw, W. W. Metcalf, E. J. N. Helfrich, G. P. van Wezel, M. H. Medema and T. Weber, Nucleic Acids Res., 2023, 51, W46–W50 CrossRef CAS PubMed.
- M. Röttig, M. H. Medema, K. Blin, T. Weber, C. Rausch and O. Kohlbacher, Nucleic Acids Res., 2011, 39, W362–W367 CrossRef PubMed.
- T. Kawasaki, T. Kuzuyama, K. Furihata, N. Itoh, H. Seto and T. Dairi, J. Antibiot., 2003, 56, 957–966 CrossRef CAS PubMed.
- T. Abe, H. Shiratori, K. Kashiwazaki, K. Hiasa, D. Ueda, T. Taniguchi, H. Sato, T. Abe and T. Sato, Chem. Sci., 2024, 15, 10402–10407 RSC.
- J. Tang and Y. Matsuda, Nat. Commun., 2024, 15, 4312 CrossRef CAS PubMed.
- R. Janke, C. Gorner, M. Hirte, T. Bruck and B. Loll, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2014, 70, 1528–1537 CrossRef CAS PubMed.
- F.-R. Li, X. Lin, Q. Yang, N.-H. Tan and L.-B. Dong, Beilstein J. Org. Chem., 2022, 18, 881–888 CrossRef CAS PubMed.
- K. Blin, S. Shaw, M. H. Medema and T. Weber, Nucleic Acids Res., 2024, 52, D586–D589 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2025 |