
Proteomic and metabolic profiling reveals molecular phenotype associated with chemotrophic growth of Rubrivivax benzoatilyticus JA2 on L-tryptophan†
Shabbir
Ahmad‡
a,
Mujahid
Mohammed§
a,
Lakshmi Prasuna
Mekala¶
a,
Sasikala
Chintalapati
b and
Ramana
Chintalapati
 *a
*a
aDepartment of Plant Sciences, School of Life Sciences, University of Hyderabad, Hyderabad 500046, Telangana, India. E-mail: cvramana449@gmail.com; chvrsl@uohyd.ac.in; Fax: +91 040 23010120&23010145; Tel: +91 040 23134502
bSmart Microbiological Services (SMS), Rashtrapathi Road, Secunderabad 500 003, India
First published on 12th November 2024
Abstract
Rubrivivax benzoatilyticus strain JA2 is an anoxygenic phototrophic bacterium, able to grow under different growth modes. Particularly under chemotrophic conditions, it produces novel Trp-melanin, anthocyanin-like, and pyomelanin pigments. However, the underlying molecular adaptations of strain JA2 that lead to the formation of novel metabolites under chemotrophic conditions remain unexplored. The present study used iTRAQ-based global proteomic and metabolite profiling to unravel the biochemical processes operating under the L-tryptophan-fed chemotrophic state. Exometabolite profiling of L-tryptophan fed chemotrophic cultures revealed production of diverse indolic metabolites, many of which are hydroxyindole derivatives, along with unique pigmented metabolites. Proteomic profiling revealed a global shift in the proteome and detected 2411 proteins, corresponding to 61.8% proteins expressed. Proteins related to signalling, transcription-coupled translation, stress, membrane transport, and metabolism were highly differentially regulated. Extensive rewiring of amino acid, fatty acid, lipid, and energy metabolism was observed under L-tyrptophan fed chemotrophic conditions. Moreover, energy conservation and cell protection strategies such as efflux pumps involved in the efflux of aromatic compounds were activated. The study demonstrated a correlation between some of the detected indole derivatives and the up-regulation of proteins associated with L-tryptophan catabolism, indicating a possible role of aromatic mono/dioxygenases in the formation of hydroxyindole derivatives and pigments under chemotrophic conditions. The overall study revealed metabolic flexibility in utilizing aromatic compounds and molecular adaptations of strain JA2 under the chemotrophic state.
1. Introduction
The genetic and metabolic diversity of microorganisms allows them to produce a wide range of natural compounds; thus they are often referred to as microbial chemical factories.1–3 These diverse chemical compounds of microbial origin have applications in various fields, including the food, cosmetic, textile, and pharmaceutical industries.4–8 There has been renewed interest in discovering new compounds of microbial origin as microorganisms are an invaluable resource of novel chemical compounds.9–11 The increasing availability of bacterial genome sequences has greatly enhanced our understanding of microbial metabolism and the potential novel natural product biosynthesis.5,8 Combining genome sequence analysis with metabolic simulation methods revealed the intricate metabolic pathways responsible for novel biomolecules production.10,12 Though genome-centered approaches are promising for predicting the potential novel biomolecules, they fall short of covering the entire range of novel compounds, owing to their inherent homology-driven nature.3,13However, change in growth conditions and interactions with other biotic systems are some of the conditions that activate the genes involved in the biosynthesis of natural compounds.9,14,15 A comprehensive approach such as the integration of powerful functional genomics tools and a modified growth configuration would identify novel compounds and help in elucidating their cellular mechanisms. Combining untargeted metabolomics and other omics techniques such as proteomics or transcriptomics has great potential to uncover the novel compounds and their underlying biochemical processes particularly in less explored bacterial groups.
Rubrivivax benzoatilyticus strain JA2 is one such metabolically versatile phototrophic bacterium. Studies on L-tryptophan metabolism in strain JA2 resulted in the discovery of the novel biomolecule, rubrivivaxin16 and a novel enzyme tryptophan ammonia lyase.17 Furthermore, a non-canonical pathway of L-tryptophan based melanin synthesis was discovered in strain JA2.18 The strain JA2 is capable of transforming L-phenylalanine and L-tyrosine into phenolic compounds, such as phenyllactic acid, pigmented metabolites, and pyomelanin.19–22 Altered growth conditions and untargeted metabolic profiling revealed a previously unknown phenylalanine catabolic process in strain JA2 and anthocyanin-like pigments.22,23 In line with this strategy, we recently reported the L-tryptophan-dependent catabolic phenotype in strain JA2 and the biosynthesis of L-tryptophan-dependent melanin under the chemotrophic state.18 Though the strain JA2 metabolizes organic compounds under phototrophic and chemotrophic growth modes, previous studies indicate that the chemotrophic state activates unique metabolic processes and our understanding of biochemical adaptations of strain JA2 under the chemotrophic state is limited. The present investigation employs global proteomic and metabolic profiling tools to uncover the cellular and biochemical adaptations under chemotrophic conditions and L-tryptophan catabolism leading to the production of secondary metabolites in strain JA2.
2. Materials and methods
2.1 Organism and growth conditions
Rubrivivax benzoatilyticus (strain JA2T, =MTCC 7087T = JCM 13220T = ATCC BAA-35T) was grown photoheterotrophically in a 100/250 ml culture bottle (at 30 ± 1 °C, and light 2400 Lux) containing mineral media (pH 6.8 to 6.9) with malate (22 mM) and ammonium chloride (7 mM) as the carbon and nitrogen source, respectively.18,24,25 In the current study, photoheterotrophically grown log phase culture (0.3 OD. @ 660 nm) was used as an inoculum, and ammonium chloride was replaced with L-tryptophan (1 mM) as the sole source of nitrogen. Chemotrophic culture was grown in a 500 ml conical flask containing 100 ml of medium and incubated in a shaker incubator (Eppendorf Innova) operating at 180 rpm and 30 ± 1 °C. Experiments were performed in triplicate and chemotrophic cultures fed with L-tryptophan (1 mM) and control cultures without any L-tryptophan feeding were used for all the studies. Growth was measured turbidometrically at 660 nm and pigment formation was measured spectrophotometrically at 400 nm.2.2 Estimation of total indoles
Total indole formation in L-tryptophan fed and control culture supernatants was measured using Salper's reagent;26,27 1.0 ml of culture supernatant was mixed with 1.0 ml of ethyl acetate and to this 2 ml of freshly prepared Salper's reagent [1 ml of 0.5 M FeCl3 in 50 ml of 35% (v/v) perchloric acid] was added and absorbance was read at 535 nm against reagent blank.2.3 Metabolite extraction and TLC and HPLC profiling
Strain JA2 was grown with and without L-tryptophan feeding (control) in a 500 ml conical flask containing 100 ml of media to the stationary phase (for 48 hours) under chemotrophic conditions. Cultures were harvested by centrifugation (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm at 4 °C, 10 min) and the collected supernatants were acidified (pH 2.0) with 5 N HCl. Acidified supernatants were stored for two to four days at 4 °C to allow pigment precipitation; the precipitated brown pigment was separated, and the remaining supernatant was used for further metabolite extraction (Fig. S1a, ESI†). Acidified supernatants (100 ml) of L-tryptophan fed and control cultures were used for metabolite extraction, and metabolites were extracted three times with an equal volume of ethyl acetate. Ethyl acetate extracts were then pooled and evaporated under vacuum using a rotary flash evaporator (Heidolph, Germany) at 35 °C. The dried samples were then dissolved in 0.5 ml of MS grade methanol and filtered through a membrane filter (0.22 μm, Icon pall-Scientifics) and the thus obtained extracts of control and L-tryptophan fed cultures were stored at −80 °C till analysis.
000 rpm at 4 °C, 10 min) and the collected supernatants were acidified (pH 2.0) with 5 N HCl. Acidified supernatants were stored for two to four days at 4 °C to allow pigment precipitation; the precipitated brown pigment was separated, and the remaining supernatant was used for further metabolite extraction (Fig. S1a, ESI†). Acidified supernatants (100 ml) of L-tryptophan fed and control cultures were used for metabolite extraction, and metabolites were extracted three times with an equal volume of ethyl acetate. Ethyl acetate extracts were then pooled and evaporated under vacuum using a rotary flash evaporator (Heidolph, Germany) at 35 °C. The dried samples were then dissolved in 0.5 ml of MS grade methanol and filtered through a membrane filter (0.22 μm, Icon pall-Scientifics) and the thus obtained extracts of control and L-tryptophan fed cultures were stored at −80 °C till analysis.
The exometabolites obtained from control and L-tryptophan fed cultures (ethyl acetate extracts dissolved in MS grade methanol) were subjected to TLC profiling (Silica gel 60 dimension, F254, 20 × 10 cm, 0.2 mm from Merck) using a solvent system consisting of chloroform:methanol:glacial acetic acid (9:0.95:0.05). HPLC analysis was performed according to previous reports.28,29 In brief, HPLC analysis was carried out using the Shimadzu prominence HPLC system with a Phenomenex C-18 column (Luna, 5 μm, 250 × 4.6 mm). The absorption spectra of metabolites were recorded using a PDA detector and the concentration of metabolites was quantified based on the peak area of the known concentration of the authentic standard, procured from Sigma-Aldrich.
2.4 LC–MS-ESI analysis, data interpretation and identification of metabolites
The exometabolite samples (ethyl acetate extracts in triplicates) obtained from control and L-tryptophan fed chemotrophic conditions were subjected to LC–MS analysis. Mass analysis was performed on an Agilent Technologies Series 6520 LC–MS/MS TOF system equipped with a PDA detector. Metabolites were separated by reverse-phase column chromatography (Luna, C-18, Luna, 3.6 μm, 150 × 4.6 mm), and the parameters used for the chromatographic separation of metabolites, data acquisition, data analysis and metabolite identification were the same as those published in previous reports.18,21,23 Metabolites were identified based on their absorption spectra and masses compared with the available databases HMDB (https://www.hmdb.ca), Massbank (https://www.massbank.jp), METLIN (https://metlin.scripps.edu), and KEGG (https://www.genome.jp/kegg/pathway) and authentic standards available.2.5 Enzyme assay
Aminotransferase enzyme assay was performed using the cell free extract obtained from chemotrophic culture of strain JA2 fed with L-tryptophan. A clear cell free extract was prepared according to earlier studies. In brief, clear cell free extracts were used as a source of enzymes to study aminotransferase/transaminase activity, and the reaction was carried out in 4 ml of α-ketoglutarate (1 mM) and pyridoxal phosphate (PLP; 50 μM) as cofactors, with hydroxytryptophan (1 mM) as a substrate. The reaction was started with the addition of an appropriate volume of the enzyme source into the reaction mixture and the pre-denatured enzyme was used as control. The reaction mixture was incubated for 1 h at 37 °C and the reaction was terminated by adding 5 N HCl after 1.0 hour incubation. The reaction mixture was then centrifuged at 16000 rpm (at 4 °C) for 20 min, and the supernatant obtained was used to extract reaction products; metabolites were extracted thrice with equal volumes of ethyl acetate. Ethyl acetate fractions were pooled and dried by using a rotary flash evaporator. Finally, the extracted fraction was dissolved in 300 μl of methanol (MS grade). The aminotransferase assay products OH-IPyA (hydroxyindole-pyruvate) or hydroxyindole-3-acetic acid were measured by HPLC and coelution with authentic standards.
2.6 Protein extraction and iTRAQ labeling
Chemotrophic cultures of strain JA2 (biological triplicates) grown on L-tryptophan and control (without L-tryptophan) were used for proteomics studies. Control and L-tryptophan fed cultures were harvested after 48 h (centrifuged at 4 °C, 10000 rpm for 10 min) and the cell pellet was collected. Control and L-tryptophan fed culture proteomes were extracted as per previous reports28,29 and the thus obtained protein samples were lyophilized (dry freezer for moisture removal) and stored at −80 °C for further analysis. To check the quality of proteins, proteomic samples were analyzed on 10% SDS-PAGE. Further, iTRAQ based quantitative proteomic analysis (triplex control and L-tryptophan fed conditions) was outsourced to the University of California, San Diego, USA. Lyophilized protein samples (100 μg) were resuspended in 600 μl of buffer (100 mM Tris buffer pH 8.0, 8 M urea), vortexed for 5 to 10 min and TCEP [tris(2-carboxyethyl)] phosphine was added at a final concentration of 10 mM. Chloroacetamide solution (40 mM) was added and vortexed. Optimal conditions for protein estimation were chosen: an equal volume of 50 mM buffer (Tris pH 8) was added to dilute the urea concentration to 4 M, and the resulting solution was then subjected to enzymatic digestion. For LysC digestion, the LysC enzyme was added at 1:500 to the protein sample. The protein sample was incubated at 37 °C in a shaker incubator for a period ranging from 4 to 6 hours. Similarly, for trypsin digestion, another equal volume of 50 mM buffer (Tris pH 8) was added to reduce the urea concentration to 2 M. Subsequently, the trypsin enzyme was added at a ratio of 1:50 to the protein sample for the digestion. Trypsinized samples (6 samples) were labelled with isobaric tags (iTRAQ8, ABSCIEX30), where each sample was labelled with a specific tag (113, 114, 115 for control and 116, 117, 118 for L-tryptophan fed conditions) according to the manufacturer's instructions. The iTRAQ8 (Sciex 4390812 used first 6 tags) labelled peptides from each set were pooled in the ratio of 1:1:1:1:1:1 and fractionated using high pH reverse phase chromatography as described previously.28
3. Results
3.1 Growth, pigment and indole production by strain JA2
Strain JA2 utilized L-tryptophan as the sole source of nitrogen for growth under chemotrophic conditions and concomitant pigment formation was observed only in the L-tryptophan-fed chemotrophic state but not under control conditions (Fig. 1A and B). Indole formation was found to be significantly higher under L-tryptophan fed conditions compared to the control (Fig. 1B). Further, chemotrophic conditions influence L-tryptophan biotransformation as evident from the indole derivatives and brown color pigment formation in the culture supernatant. The culture supernatants from L-tryptophan fed culture turned pigmented over a period of time (Fig. 1A) compared to the control indicating the possible pigmented exometabolite accumulation in the medium (Fig. S1A, ESI†). Furthermore, the TLC profiling of the ethyl acetate fraction obtained from the culture supernatants of the L-tryptophan fed chemotrophic cultures of strain JA2 showed an array of color bands (yellow, red, and pink band) and brown color metabolites as compared to control (Fig. 2A). Exometabolite HPLC profiling detected the array of indole derivatives under the L-tryptophan fed conditions but not under control conditions (Fig. S1C). The purified major pigments from preparative TLC were subjected to HPLC analysis and they exhibited significant absorbance in the UV-vis range (270 to 290 and 400 to 500 nm; Fig. 2B). | ||
| Fig. 1 Growth, pigment and indole production in strain JA2 under L-tryptophan fed and control conditions. (A) Control and tryptophan (Trp) fed cultures and their respective supernatants; (B) growth, pigment and indole formation in L-tryptophan fed culture and indole production in control (yellow triangles) in strain JA2. | ||
 | ||
| Fig. 2 Pigmented metabolites’ TLC and HPLC analysis. (A) TLC profiling of ethyl acetate fractions of control and L-tryptophan (Trp) fed conditions; (B) HPLC chromatogram of pink and yellow pigments and the insert shows the absorbance spectrum of respective pigments. | ||
3.2 HPLC based exometabolite profiling
HPLC-based exometabolite profiles of extracts obtained from L-tryptophan fed cultures were distinct from control, wherein more metabolites with specific absorbance at 270, 280, and 290 nm were found under L-tryptophan fed conditions (Table 1). Metabolites showing strong absorbance at 280–290 nm are indicative of the production of indole derivatives in L-tryptophan fed cultures, while they were not found in control (Fig. S1C, ESI†). The LC–MS analysis of ethyl acetate extracts of L-tryptophan fed conditions detected more numbers of metabolic features in negative ionization mode as compared to positive (Table 1). LC–MS/MS analysis revealed a total of thirty-six metabolic features from L-tryptophan fed cultures (Table 1), wherein thirteen metabolites were confirmed, three metabolites were putatively identified, and the remaining were unidentified (Table 1). Metabolites Rt, absorption and molecular ion masses are presented in Table 1 and some metabolites were identified based on mass fragmentation, absorption spectrum, and authentic standards. The metabolite peak at Rt 7.9 min had absorption maxima at 275 nm with an ion mass of 219 [M−] and was identified as a hydroxytryptophan. Rt at 10.9 min had absorption maxima of 260 and 290 nm and an ion mass of 207 [M−] and was identified as 5,6-dihydroxy indole-acetic acid (IAA). Peak Rt at 10.9 min had absorption maxima at 270, 280, and 290 nm with a molecular mass of 207 [M−], and was identified as dihydroxy methyl indole carboxylic acid. Rt at 15.6 min having absorption maxima at 270, 272, and 288 nm with a molecular mass of 175 [M−] was identified as 5-hydroxy indole acetaldehyde. Peak [Rt] at 14.3 min having absorption maxima at 275 and 288 nm with a molecular mass of 191 [M−] was identified as 5-hydroxy indole acetic acid, while Rt at 15.5 min having an absorption of 260 and 292 nm with a molecular mass of 219 [M−] was identified as hydroxy indole pyruvate. Rt at 16.3 min having absorption maxima of 290 nm with a molecular mass of 147 [M−] was identified as hydroxymethyl indole. Rt at 18 min had absorption maxima at 254 nm with a molecular mass of 137 [M−] and was identified as 4-hydroxybenzoic acid, Rt at 22 min with absorption maxima at 280 nm with a molecular mass of 175 [M−] was identified as hydroxy indole acetic acid, and Rt at 22 min with absorption maxima of 275 and 300 nm with a molecular mass of 175 [M−] was identified as 5-hydroxyindole acetaldehyde (Table 1). Some of the metabolites remained unidentified due to their complex molecular formulas, multiple hits, and due to lack of authentic standards (Table 1).| S. no. | R t | Generated formula | Exact mass | Neg. mode | Pos. mode | ppm | Absorbance [nm] | Metabolites |
|---|---|---|---|---|---|---|---|---|
| a These metabolites are confirmed by the retention time, mass fragmentation through the authentic standards and by comparison with the available database. b These metabolites are putatively identified on the basis of molecular formula, absorption spectra, and mass in comparison with the database.UN, unidentified; no suitable match of molecular formula in the database; Rt, retention time; Neg., negative ionization mode; Pos., positive ionization mode. 5-OH-IAA, 5-hydroxyindole-3-acetic acids; OH-KYNA, hydroxyl-kyneuric acid; 5-OH IAA, 5-hydroxy indole-3-acetic acid; 5-OHIPyA, 5-hydroxy indole-3-pyruvic acids; IAA, indole-3-acetic acid; OH me-indole, hydroxy-methylindole. | ||||||||
| 1 | 5.36 | C9H11NO3 | 181 | 180 | −0.75 | 272, 280, 288 | UN | |
| 2 | 7.22 | C8H7NO3 | 165 | 164 | −0.41 | 262, 290 | UN | |
| 3a | 7.9 | C11H12N2O3 | 220 | 219.77 | −0.75 | 275 | 5-Hydroxytryptophan | |
| 4a | 10.9 | C10H9NO4 | 207 | 206 | −0.45 | 260, 290 | 1H-Indole-3-acetic acid, 5,7-dihydroxy | |
| 5a | 10.9 | C11H9NO4 | 207 | 206 | −0.86 | 270, 280, 288 | 5,6-Dihydroxy IAA/dihydroxy methyl indole carboxylic acid | |
| 6 | 14.3 | C10H9NO3 | 191 | 190 | −0.34 | 275, 288 | UN | |
| 7 | 14.5 | C10H7NO3 | 189 | 188 | −4.25 | 240, 262, 305 | Kynurenic acid | |
| 8 | 15 | C10H9NO2 | 175 | 174 | −5 | 256, 305 | UN | |
| 9 | 15.7 | C8H7N | 118 | 117 | −0.02 | 256, 300 | UN | |
| 10a | 15.5 | C11H9NO4 | 119 | 218 | −1.42 | 260, 292 | Hydroxyindole pyruvate | |
| 11a | 15.6 | C10H9NO2 | 175 | 174 | −3.23 | 272, 288, 290 | 5-Hydroxyindole acetaldehyde | |
| 12a | 16.3 | C9H9NO | 147 | 146 | −1.36 | 290 | OH methyl-indole | |
| 13 | 17.25 | C10H7NO | 157 | 156 | −2.48 | 260, 290 | UN | |
| 14a | 18 | C7H6NO3 | 138 | 137 | −1.9 | 254 | Hydroxybenzoic acid | |
| 15 | 19.2 | C10H7NO2 | 173 | 172 | −1.08 | 300 | UN | |
| 16 | 19.36 | C21H17N3O4 | 376 | 377 | −1.7 | 260, 270 | UN | |
| 17 | 20.6 | C27H19N3O3 | 433 | 434 | −1.8 | 272, 278, 290 | UN | |
| 18b | 20.6 | C25H17N3 | 359 | 360 | −2.39 | 270, 280, 290 | 3,3′-(3H-Indol-3-ylidenemethylene)bis(1H-indole) | |
| 19 | 21.8 | C10H9N | 175 | 174 | −1.42 | 260, 290, 300 | UN | |
| 20a | 22 | C10H9NO3 | 191 | 190 | −4.26 | 275, 295 | 5-Hydroxy IAA | |
| 21a | 22 | C10H9NO2 | 175 | 174 | 0.36 | 260, 300 | 5-Hydroxyindole acetaldehyde | |
| 22a | 22.1 | C11H11NO3 | 205 | 204 | 3.11 | 225, 260, 290 | Indole lactic acid | |
| 23b | 23 | C18H12N2O2 | 289 | 287 | 2.11 | 272, 280, 288 | Di-indol-3-yl-ethanedione | |
| 24 | 23.8 | C20H19N3O3 | 349 | 350 | 350 | −2.5 | 280, 290, 300 | UN |
| 25 | 24.1 | C10H9NO3 | 161 | 160 | 1.88 | 258, 286 | UN | |
| 26a | 24.39 | C9H7NO2 | 161 | 160 | 1.34 | 260, 290 | Indole carboxylic acid | |
| 27 | 25.52 | C9H7NO | 145 | 144 | 0.39 | 260, 300 | UN | |
| 28a | 26.22 | C10H9NO2 | 175 | 174 | 3 | 270, 280, 290 | IAA | |
| 29 | 29.5 | C16H12N2O2 | 264 | 263 | 1.24 | 260, 290 | UN | |
| 30 | 34.3 | C21H17N3O3 | 359 | 358 | 0.84 | 258, 290 | UN | |
| 31 | 35 | C19H12N2O3 | 316 | 315 | 0.57 | 260, 290 | UN | |
| 32 | 35.01 | C22H19N2O2 | 358 | 358 | 359 | −2.23 | 270, 280, 290 | UN |
| 33b | 48.36 | C26H19N3O | 389 | 388 | 0.72 | 290 | Indole trimer | |
| 34 | 37.2 | C18H12N2O2 | 288 | 289 | 289 | −2.54 | 270, 280, 290 | UN |
| 35 | 37.3 | C20H17N3O3 | 347 | 346 | −1.17 | 260, 290 | UN | |
| 36 | 40.14 | C20H10N2O2 | 310 | 311 | 312 | 2.61 | 260, 270 | UN |
3.3 Hydroxytryptophan catabolism by aminotransferase enzyme activity
Hydroxytryptophan catabolism was further validated by the key enzyme aminotransferase activity using cell-free extracts and the activity was confirmed by the detection of hydroxyindole-3-acetic and hydroxyindole-3-pyruvic acids through HPLC and coelution with the authentic standard (Fig. S2A and B, ESI†). Hydroxytryptophan aminotransferase assay can also lead to hydroxyindole-3-acetic acid formation through the IPyA pathway (Fig. S2A and B, ESI†). Collectively, our results revealed that strain JA2 transformed L-tryptophan to hydroxytryptophan which was further catabolized through aminotransferase to hydroxyindole derivatives under chemotrophic conditions.3.4 Proteomic inventory of strain JA2
To decipher the molecular response of strain JA2 grown on L-tryptophan as the sole source of nitrogen under chemotrophic conditions, an iTRAQ-based quantitative proteomic study was carried out. SDS-PAGE preliminary analysis of global proteomic samples obtained from control and L-tryptophan fed cells under chemotrophic conditions showed a distinct protein band pattern (Fig. S3A and B, ESI†). Further, the proteomic samples were subjected to iTRAQ analysis and the workflow of the study is depicted in Fig. S3C (ESI†). Based on the set parameters mentioned in the materials and methods section on iTRAQ analysis, global proteome iTRAQ analysis detected 2411 proteins against the reference database of strain JA2 (coding 3898 proteins in the genome). iTRAQ analysis could capture 61.86% of the total theoretical proteome of strain JA2 (Fig. S3D, ESI†).Total proteins detected by iTRAQ analysis were subjected to principal component analysis (PCA), an unsupervised reductive statistical method to discriminate control and L-tryptophan fed conditions. PCA analysis could explain 92% of the variation between control and L-tryptophan-fed proteomes, wherein component one (PC1) resolved 81% of the variation and component two (PC2) 11%, respectively (Fig. 3A). The PCA score plot separated control and L-tryptophan fed samples into distinct clusters indicative of variations in proteomes (Fig. 3A).
 | ||
| Fig. 3 PCA analysis and functional classification of iTRAQ quantified proteins. (A) PCA scatter plot of iTRAQ quantified proteins from control and tryptophan-fed chemotrophic states; prediction ellipses indicate 0.95 probability; (B) functional classification of deferentially regulated proteins; proteins with significance threshold score ≥20 above and fold change >1.25 and <0.8 were considered as differentially regulated and these proteins were functionally classified according to the KEGG database. | ||
A total of 210 differentially regulated proteins were classified into different categories, based on their molecular and cellular function, according to the KEGG pathway (Fig. 3B). These proteins were further grouped into eighteen functional categories including membrane transport, signal response, central carbon metabolism, aromatic amino acid metabolism, carbohydrate metabolism, vitamins and cofactors, folding and sorting, hypothetical proteins, metabolism of amino acids, energy metabolism, nucleotide metabolism, transcription, and translation (Fig. 3B). Furthermore, the highly upregulated proteins were grouped into eighteen categories including signaling sensors, membrane signal histidine kinases, and membrane transport proteins (Fig. 3B). These upregulated proteins were observed in conjunction with aromatic compound metabolism, carbohydrate metabolism, amino acid metabolism, vitamins and cofactors, nucleotide metabolism, energy-related processes, transcription, translation, as well as hypothetical proteins (Fig. 3B). Hypothetical proteins showed the highest differential regulation among all categories (Fig. 3B) under L-tryptophan-fed conditions. The percentage ratio of differentially upregulated proteins was as follows: membrane proteins related to signalling 10.4%, aromatic amino acid metabolism 3.44%, lipid metabolism 5.7%, carbohydrate metabolism 6.8%, metabolism 3.4%, stress 5.7%, vitamins and cofactors 4.6%, transcription 6.8%, translation 3.4%, membrane transport 11.5% and hypothetical proteins 25.2% (Fig. 4A). The percentage ratio of downregulated proteins was as follows: signal related 8.1%, amino acid metabolism 5.7%, fatty acid metabolism 6.5%, lipid metabolism 1.62%, carbohydrate metabolism 5.71%, metabolism 7.31%, DNA repair 2.3%, stress 1.62%, vitamins and cofactors 7.31%, photosystem 8.13%, transcription 1%, translation 5.7%, membrane transport 8.13%, electron transport chain (ETC) 8.1%, antioxidant 4%, shikimate pathway 0.83%, and hypothetical proteins 18.7% (Fig. 4B).
 | ||
| Fig. 4 Pie chart representing functional categories of upregulated proteins (A) and downregulated proteins (B). Proteins with significance threshold score ≥20 and fold change >1.25 and <0.8 were considered as differentially regulated and these proteins were functionally classified according to the KEGG database. | ||
4. Discussion
Strain JA2 is unable grow at the expense of aromatic compounds as the sole source of carbon; it uses aromatic amino acids as a nitrogen source under phototrophic (anaerobic) as well as chemotrophic (aerobic) growth modes. However, much of the previous reports on aromatic compound metabolism explored the remarkable biotransformation potential of strain JA2 under phototrophic conditions.19,29,32–34 Recent studies on the chemotrophic metabolism of aromatic amino acids like L-phenylalanine and L-tryptophan by strain JA2 revealed hitherto unknown new metabolic processes and novel metabolite production. These studies suggest that strain JA2 uses different metabolic and molecular processes to metabolize aromatic amino acids under varying growth conditions. The present study elucidates the molecular responses of strain JA2 when grown on L-tryptophan under chemotrophic conditions using an integrated global proteomic and metabolic profiling approach. Proteomics and metabolomics are two powerful functional genomic tools that can capture different layers of cellular function and their integration thus reveals a true snapshot of the molecular phenotype of cells.4.1 Molecular response of strain JA2 to L-tryptophan-fed chemotrophic conditions
We employed metabolite profiling to identify the metabolites that represent the final products of gene expression, thus providing a snapshot of the underlying molecular processes. It is widely acknowledged that alteration in growth conditions can alter the metabolic processes, and in agreement with this pigment formation was observed in strain JA2 cultures that were grown on L-tryptophan under chemotrophic conditions (Fig. 1A and B). Similarly, we reported previously the production of novel pigments from L-phenylalanine-fed chemotrophic cultures of strain JA2.21,22 There is increasing evidence that the dynamic changes in environmental exposure to aromatic compounds, including aromatic amino acids like L-tryptophan, can modulate microbial physiology and potentially rewire the metabolism.19,21,22,29,32 Sensing the extracellular cues is pivotal for survival and bacteria sense and thereby modulate their metabolic process; in this regard we observed significant upregulation of proteins related to signal transduction such as histidine kinase-like protein, PAS/PAC sensor hybrid histidine kinase, and diguanylate cyclase/phosphodiesterase (Table 2 and Fig. 5), which are involved in the modulation of gene expression in response to changes in the external environment.29,35 Several transcriptional factors, including the TetR family, YidE/YbjL duplication, Sel1 domain-containing protein, transcriptional activator domain-containing protein, silent information regulator protein Sir2, transcription regulator proteins, and FliA/WhiG family RNA polymerase sigma 28 subunits, were upregulated indicating the orchestration of gene expression required to adapt to the metabolic demands of cells under chemotrophic conditions (Table 2).| Accession no. | Description of proteins | Avg. FC (tryptophan fed/control) | log2_Fc (tryptophan fed/controls) | SD (standard dev.) |
|---|---|---|---|---|
| Protein ratios (L-tryptophan fed/control) identified under chemotrophic conditions were log2 transformed, and values are mean ± standard deviation of three independent experiments. Proteins discovered in at least two out of three replicates with two peptide and one unique peptide and P-value ≤0.05 (significance threshold of peptide score >20) are all presented in the table. Protein fold change (FC) ratios (tryptophan fed/control) of identified proteins were log2 transformed and the values presented are average of log2FC ratios obtained from biological triplicate experiments (as mentioned in the Materials and methods section). Protein fold change ≥1.25 (log2FC ≥ 0.22) was considered upregulation and fold change ≥0.8 (log2FC ≥ −0.22) was considered downregulation. Bold denotes up-regulated proteins and italic denotes down-regulated proteins. |
||||
| Signal related proteins | ||||
| giI332110811 | Signal recognition particle protein | 1.285062 | 0.250807 | 0.113059 |
| giI332107759 | Kinase-like protein | 1.258988 | 0.230294 | 0.179213 |
| giI332107765 | Putative signal peptide protein | 17.399429 | 2.856437 | 0.190705 |
| giI332110892 | Twitching motility signal transduction protein | 1.530321 | 0.425476 | 0.147247 |
| giI332110828 | YidE/YbjL duplication | 3.200660 | 1.163357 | 0.439744 |
| giI332111620 | Response regulator | 2.259650 | 0.815210 | 0.101321 |
| giI332112187 | PAS/PAC sensor hybrid histidine kinase | 1.553856 | 0.440741 | 0.060059 |
| giI332107914 | TetR family transcriptional regulator | 4.816176 | 1.571980 | 0.552208 |
| giI332109371 | Diguanylate cyclase/phosphodiesterase | 1.479512 | 0.391712 | 0.262764 |
| giI332108263 | Crp/FNR family transcriptional regulator | 0.724264 | −0.322613 | 0.013171 |
| giI332108264 | Cyclic glucan phosphoglycerol modification protein | 1.219412 | 0.198369 | 0.030267 |
| giI332111653 | Multi-sensor hybrid histidine kinase | 0.650858 | −0.429461 | 0.163594 |
| giI332110816 | N-Acetyl-anhydromuranmyl-L-alanine amidase | 0.729942 | −0.314792 | 0.040106 |
| giI332109370 | Putative high potential iron–sulfur (hipip) signal peptide protein | 0.743768 | −0.296032 | 0.044503 |
| giI332110893 | Twitching motility protein | 0.777192 | −0.252071 | 0.271268 |
| giI332111874 | AraC family transcriptional regulator | 0.769219 | −0.262383 | 0.183645 |
| giI332111446 | Methyl-accepting chemotaxis sensory transducer | 0.524714 | −0.644931 | 0.240615 |
| giI332110825 | CheW protein | 0.424778 | −0.856192 | 0.065022 |
| giI332111479 | Response regulator receiver protein | 0.444271 | −0.811321 | 0.067809 |
| giI332111651 | Osmosensitive K+ channel signal transduction histidine kinase | 0.296164 | −1.216841 | 0.018921 |
| giI332111476 | Methyl-accepting chemotaxis sensory transducer | 0.688727 | −0.372912 | 0.654661 |
| giI332111446 | Methyl-accepting chemotaxis sensory transducer | 0.524712 | −0.644921 | 0.240615 |
| Amino acid/aromatic amino acid | ||||
| giI332111318 | Putative amidase | 1.623396 | 0.484520 | 0.3009423 |
| giI332107776 | Phenylalanine 4-monooxygenase | 1.619804 | 0.482305 | 0.0951972 |
| giI332112186 | Tryptophanase/L-cysteine desulfohydrase PLP-dependent | 21.740121 | 3.079161 | 0.0774442 |
| giI332108048 | Carbamoyl-phosphate synthase L chain ATP-binding protein | 0.428892 | −0.846552 | 0.048883 |
| giI332109992 | Carboxyl transferase | 0.604910 | −0.502671 | 0.075871 |
| giI332108280 | Urease accessory protein UreD | 0.756294 | −0.279323 | 0.034407 |
| giI332111194 | Glycine cleavage system H protein | 0.662492 | −0.411752 | 0.252108 |
| giI332109142 | FAD linked oxidase domain-containing protein | 0.786068 | −0.240715 | 0.084512 |
| giI332112222 | S-Adenosyl-L-homocysteine hydrolase | 0.530532 | −0.633871 | 0.078485 |
| Lipid metabolism | ||||
| giI332109908 | Geranyltranstransferase | 1.524236 | 0.421493 | 0.218178 |
| giI332110050 | Putative phospholipase A1 | 1.421203 | 0.351504 | 0.042938 |
| giI332112215 | Lipid A biosynthesis acyltransferase | 1.562746 | 0.446445 | 0.250501 |
| giI332108133 | Lipoprotein YaeC family | 1.551964 | 0.439521 | 0.039473 |
| giI332109922 | Hydroxyneurosporene synthase | 1.290276 | 0.254856 | 0.169613 |
| giI332108566 | Esterase/lipase/thioesterase family protein | 1.206379 | 0.187623 | 0.198624 |
| giI332110872 | Acyltransferase WS/DGAT/MGAT | 1.207434 | 0.188498 | 0.056543 |
| giI332109204 | Pullanase-associated protein | 1.235948 | 0.211838 | 0.006892 |
| giI332108038 | Acetyl-CoA acetyltransferase | 0.755801 | −0.279983 | 0.006328 |
| giI332108563 | Modular polyketide synthase | 0.795594 | −0.228672 | 0.051645 |
| giI332109100 | Acyltransferase | 0.755801 | −0.279981 | 0.075156 |
| giI332108035 | Isovaleryl-CoA dehydrogenase | 0.427782 | −0.849143 | 0.023989 |
| giI332111406 | Acyl carrier protein ACP | 0.738686 | −0.302881 | 0.096934 |
| giI332108046 | Enoyl-CoA hydratase/isomerase | 0.529848 | −0.635163 | 0.067127 |
| giI332108044 | Propionyl-CoA carboxylase | 0.420041 | −0.867432 | 0.102905 |
| giI 332108049 | Hydroxymethylglutaryl-CoA lyase | 0.791216 | −0.234181 | 0.027697 |
| giI332108049 | Hydroxymethylglutaryl-CoA lyase | 0.791216 | −0.234183 | 0.027697 |
| giI332108116 | Propionyl-CoA carboxylase subunit alpha | 0.592150 | −0.524723 | 0.041921 |
| giI332109504 | Butyryl-CoA dehydrogenase | 0.555028 | −0.588741 | 0.338557 |
| giI332109978 | Metallophosphoesterase | 0.798696 | −0.224782 | 0.037705 |
| Carbohydrate metabolism | ||||
| giI332107698 | Radical SAM family protein | 1.489623 | 0.398523 | 0.158851 |
| giI332112596 | Blue (type 1) copper domain protein | 2.501581 | 0.916923 | 0.211158 |
| giI332108733 | Methyltransferase FkbM family protein | 1.750670 | 0.559998 | 0.145732 |
| giI332112236 | Dihydroneopterin aldolase | 1.405922 | 0.340694 | 0.149005 |
| giI332108628 | Precorrin-2 C20-methyltransferase | 1.944139 | 0.664819 | 0.066500 |
| giI 332108584 | Glycosyl transferase group 1 | 1.415214 | 0.347281 | 0.273457 |
| giI332108769 | Group 1 glycosyl transferase | 1.452036 | 0.372966 | 0.163735 |
| giI332108575 | Galactosamine-containing minor teichoic acid biosynthesis | 1.518870 | 0.417966 | 0.127898 |
| giI332111130 | Polyhydroxyalkanoate depolymerase intracellular | 1.275586 | 0.243406 | 0.219222 |
| giI332110443 | Poly-beta-hydroxybutyrate polymerase-like protein | 1.499534 | 0.405154 | 0.722681 |
| giI332108738 | Inositol monophosphatase | 1.492906 | 0.400724 | 0.024237 |
| giI332109093 | 6,7-Dimethyl-8-ribityllumazine synthase | 1.208715 | 0.189558 | 0.063707 |
| Metabolism related proteins | ||||
| giI332107698 | Radical SAM family protein | 1.489623 | 0.398523 | 0.158851 |
| giI332112596 | Blue (type 1) copper domain protein | 2.501581 | 0.916923 | 0.211158 |
| giI332108733 | Methyltransferase FkbM family protein | 1.750670 | 0.559998 | 0.145732 |
| giI332111446 | Methyl-accepting chemotaxis sensory transducer | 0.524712 | −0.644912 | 0.240615 |
| giI332110825 | CheW protein | 0.424778 | −0.856193 | 0.065022 |
| giI332111479 | Response regulator receiver protein | 0.444271 | −0.811321 | 0.067809 |
| giI332111651 | Osmosensitive K+ channel signal transduction histidine kinase | 0.296164 | −1.216843 | 0.018912 |
| giI332111971 | Bifunctional 3-demethylubiquinone-9 3-methyltransferase/2-octaprenyl-6-hydroxy phenol methylase | 0.778571 | −0.250292 | 0.011096 |
| Stress related protein | ||||
| giI332111778 | Heat-inducible transcription repressor | 1.700926 | 0.531172 | 0.255731 |
| giI332108229 | Cysteine proteinase | 1.426278 | 0.355068 | 0.241143 |
| giI332108269 | Transthyretin | 1.384174 | 0.325103 | 0.092155 |
| giI332108410 | NmrA-like protein | 1.265308 | 0.235316 | 0.180224 |
| giI332107928 | CcmE/CycJ protein | 0.726850 | −0.319032 | 0.131878 |
| giI332109993 | Membrane ATPase/protein kinase | 0.666412 | −0.405853 | 0.034699 |
| giI332108331 | co-Chaperonin GroES | 0.745458 | −0.293762 | 0.192393 |
| giI332108280 | Urease accessory protein UreD | 0.756294 | −0.279321 | 0.034406 |
| giI332110848 | Alkyl hydroperoxide reductase/thiol specific antioxidant/Mal allergen | 0.641842 | −0.443413 | 0.010722 |
| giI332110906 | Alkyl hydroperoxide reductase/thiol specific antioxidant/Mal allergen | 0.700231 | −0.356342 | 0.144799 |
| giI332111629 | Multiple antibiotic resistance (MarC)-like protein | 0.528754 | −0.637231 | 0.254592 |
| giI332108118 | Glyoxalase/bleomycin resistance protein/dioxygenase | 0.659913 | −0.415654 | 0.142838 |
| giI332109995 | GntR family transcriptional regulator | 0.699616 | −0.357221 | 0.02108 |
| Vitamins and cofactors metabolism | ||||
| giI332108336 | TMAO/DMSO reductase | 0.711854 | −0.339882 | 0.032776 |
| giI332110824 | Biotin carboxyl carrier protein | 0.671958 | −0.397561 | 0.182103 |
| giI332108117 | Biotin synthase | 0.783324 | −0.244213 | 0.063372 |
| giI332112748 | Adenosylcobinamide-phosphate synthase | 0.780775 | −0.247471 | 0.313741 |
| giI332112223 | 5,10-Methylenetetrahydrofolate reductase | 0.713350 | −0.337781 | 0.009864 |
| giI332109937 | Coenzyme B12-binding aerobic repressor | 0.786061 | −0.240723 | 0.066481 |
| Photosystem and electron transport chain related proteins | ||||
| giI332112043 | F0F1 ATP synthase subunit delta | 1.260569 | 0.231556 | 0.113997 |
| giI332109917 | Photosynthetic reaction center subunit | 1.259735 | 0.230901 | 0.078051 |
| giI332108428 | Pyruvate flavodoxin/ferredoxin oxidoreductase domain protein | 1.302798 | 0.264514 | 0.067028 |
| giI332112736 | PUCC protein | 1.329158 | 0.284546 | 0.203301 |
| giI332109448 | Oxidoreductase | 1.790259 | 0.582360 | 0.416203 |
| giI332111162 | NADH dehydrogenase (quinone) | 1.255974 | 0.227912 | 0.248718 |
| giI332112043 | F0F1 ATP synthase subunit delta | 1.260561 | 0.231556 | 0.113997 |
| giI332109917 | Photosynthetic reaction center subunit L | 1.259735 | 0.230901 | 0.078051 |
| giI332111893 | Cytochrome d1 heme region | 0.795332 | −0.228991 | 0.072877 |
| giI332109128 | Cytochrome b/b6 domain-containing protein | 0.758833 | −0.275972 | 0.170348 |
| giI332111159 | NADH-ubiquinone oxidoreductase subunit J | 0.674932 | −0.393143 | 0.113572 |
| giI332107504 | Oxidoreductase FAD/NAD(P)-binding protein | 0.563364 | −0.573831 | 0.203011 |
| giI332109911 | Chlorophyllide reductase iron protein subunit X | 0.789292 | −0.236621 | 0.096708 |
| giI332109144 | Ubiquinone/menaquinone biosynthesis methyltransferase | 0.787392 | −0.239031 | 0.032531 |
| giI332109504 | Butyryl-CoA dehydrogenase | 0.555028 | −0.588741 | 0.338557 |
| giI332109142 | FAD linked oxidase domain-containing protein | 0.786068 | −0.240712 | 0.084500 |
| giI332107928 | CcmE/CycJ protein | 0.726850 | −0.319032 | 0.131878 |
| giI332111971 | Bifunctional 3-demethylubiquinone-9 3-methyltransferase/2-octaprenyl-6-hydroxy phenol methylase | 0.795332 | −0.228991 | 0.011096 |
| giI332111893 | Cytochrome d1 heme region | 0.758834 | −0.275973 | 0.072877 |
| giI332109128 | Cytochrome b/b6 domain-containing protein | 0.674932 | −0.393141 | 0.170348 |
| giI332111159 | NADH-ubiquinone oxidoreductase subunit J | 0.563364 | −0.573832 | 0.113572 |
| giI332107504 | Oxidoreductase FAD/NAD(P)-binding protein | 0.778571 | −0.250291 | 0.011096 |
| Transcription | ||||
| giI332107591 | FliA/WhiG family RNA polymerase sigma 28 subunit | 1.670856 | 0.513336 | 0.236045 |
| giI332108120 | Transcription regulator protein | 1.640899 | 0.495244 | 0.336495 |
| giI332112137 | Nucleoside diphosphate kinase regulator | 1.609610 | 0.475992 | 0.182085 |
| giI332112180 | Sel1 domain-containing protein | 1.542559 | 0.433442 | 0.216528 |
| giI332109398 | Transcriptional activator domain-containing protein | 1.421898 | 0.351993 | 0.253212 |
| giI332107624 | Silent information regulator protein Sir2 | 1.901756 | 0.642777 | 0.808419 |
| giI332110749 | Chromosome segregation and condensation protein ScpA | 0.725120 | −0.321426 | 0.100932 |
| giI332109844 | Transcriptional regulatory protein | 0.464472 | −0.766855 | 0.167103 |
| giI332111449 | Putative two-component response-regulatory protein YehT | 0.753769 | −0.282673 | 0.080353 |
| giI332108330 | Transcriptional regulator NifA | 0.723226 | −0.324031 | 0.088084 |
| giI332109946 | Zinc-binding alcohol dehydrogenase | 0.787379 | −0.239043 | 0.133698 |
| giI332107887 | Preprotein translocase subunit SecY | 0.760956 | −0.273181 | 0.124596 |
| giI332109483 | Integration host factor alpha-subunit | 0.766809 | −0.265523 | 0.034022 |
| giI332111850 | Heavy metal translocating P-type ATPase | 0.765950 | −0.266645 | 0.081188 |
| giI332107543 | Nucleoid protein Hbs | 0.785261 | −0.241743 | 0.282477 |
| giI332109092 | Putative N utilization substance B | 0.785261 | −0.241741 | 0.161277 |
| giI332109844 | Transcriptional regulatory protein | 0.464472 | −0.766851 | 0.167103 |
| giI332111449 | Putative two-component response-regulatory protein YehT | 0.753769 | −0.282673 | 0.080353 |
| Protein synthesis | ||||
| giI32109861 | 50S ribosomal protein L33 | 1.271606 | 0.240281 | 0.551598 |
| giI332108000 | Sigma 54 modulation protein/ribosomal protein S30EA | 1.556705 | 0.442571 | 0.047306 |
| giI332111487 | GTP-binding protein | 1.556705 | 15.567050 | 0.214125 |
| giI332109232 | 50S ribosomal protein L34 | 4.447049 | 1.492240 | 0.616053 |
| giI332107870 | 30S ribosomal protein S19 | 0.707446 | −0.34609 | 0.184761 |
| giI332107880 | 30S ribosomal protein S14 | 0.780216 | −0.24818 | 0.096944 |
| giI332112450 | Ribosome recycling factor | 0.777953 | −0.25109 | 0.099912 |
| giI332108097 | Ribosomal protein l11 | 0.787738 | −0.23859 | 0.048772 |
| giI332108090 | 30S ribosomal protein S12 | 0.798265 | −0.22531 | 0.172108 |
| giI332112098 | ABC transporter-like protein | 0.797229 | −0.22661 | 0.070808 |
| giI 332111866 | TPR domain-containing protein | 0.793412 | −0.23141 | 0.165118 |
| giI 332110456 | HPr kinase | 0.791272 | −0.23411 | 0.103097 |
| Transporter and membrane proteins | ||||
| giI332109251 | RND family efflux transporter MFP subunit | 1.317720 | 0.275903 | 0.059969 |
| giI332107915 | Efflux transporter RND family MFP subunit | 1.385910 | 0.326357 | 0.048904 |
| giI332112091 | Putative ABC transporter ATP-binding protein | 1.417498 | 0.348893 | 0.222226 |
| giI332109313 | Ferrous iron transport protein B | 1.279440 | 0.246422 | 0.109868 |
| giI332111700 | Putative transport system ATP-binding protein | 1.264302 | 0.234520 | 0.041694 |
| giI332111371 | Putative transmembrane sensor histidine kinase transcription regulator protein | 1.259109 | 0.222431 | 0.178917 |
| giI332111172 | TRAP dicarboxylate transporter subunit DctP | 1.311781 | 0.271386 | 0.081982 |
| giI332112665 | Putative lipoprotein | 1.365804 | 0.311743 | 0.128253 |
| giI332109251 | RND family efflux transporter MFP subunit | 1.317720 | 0.275903 | 0.059968 |
| giI332107915 | Efflux transporter RND family MFP subunit | 1.385910 | 0.326357 | 0.048904 |
| giI332112091 | Putative ABC transporter ATP-binding protein | 1.417498 | 0.348893 | 0.222226 |
| giI332109313 | Ferrous iron transport protein B | 1.279440 | 0.246422 | 0.109868 |
| giI332107916 | RND efflux system outer membrane lipoprotein | 1.596582 | 0.467864 | 0.149292 |
| giI332107926 | Periplasmic protein thiol | 0.707790 | −0.345612 | 0.044628 |
| giI332111814 | Putative polar amino acid transport system ATP-binding protein | 0.773599 | −0.256731 | 0.176880 |
| giI332107951 | TonB-dependent siderophore receptor family protein 13 | 0.689158 | −0.372283 | 0.069959 |
| giI332108054 | Acetate permease | 0.779919 | −0.248561 | 0.079658 |
| giI332110494 | Putative transmembrane protein | 0.729962 | −0.314762 | 0.072124 |
| giI332110561 | Potassium transporter | 0.770433 | −0.260832 | 0.042607 |
| giI332107546 | Anaerobic c4-dicarboxylate membrane transporter family protein | 0.744580 | −0.294931 | 0.103902 |
| giI332108262 | Molybdenum cofactor sulfurylase | 0.702288 | −0.353412 | 0.173396 |
| giI332112098 | ABC transporter-like protein | 0.797229 | −0.226613 | 0.070807 |
| giI332109993 | Membrane ATPase/protein kinase | 0.666412 | −0.405852 | 0.034699 |
| Electron transport chain proteins | ||||
| giI332111669 | Cytochrome d ubiquinol oxidase subunit II | 0.522634 | −0.648871 | 0.159393 |
| giI332109185 | Formate dehydrogenase subunit alpha | 0.680776 | −0.384532 | 0.104479 |
| giI332109907 | Cytochrome c-552 precursor | 0.670386 | −0.399921 | 0.052807 |
| Antioxidant | ||||
| giI332110848 | Alkyl hydroperoxide reductase/thiol specific antioxidant/Mal allergen | 0.641842 | −0.443412 | 0.010721 |
| giI332110906 | Alkyl hydroperoxide reductase/thiol specific antioxidant/Mal allergen | 0.700231 | −0.356341 | 0.144798 |
| giI332111629 | Multiple antibiotic resistance (MarC)-like protein | 0.528753 | −0.637232 | 0.254592 |
| giI332110560 | Benzoate transporter | 0.655899 | −0.421753 | 0.076028 |
| giI332108118 | Glyoxalase/bleomycin resistance protein/dioxygenase | 0.659913 | −0.415652 | 0.142837 |
| DNA replication, repair and synthesis | ||||
| giI332112171 | DNA-directed DNA polymerase | 1.360696 | 0.307996 | 0.150143 |
| giI332111924 | Holliday junction DNA helicase RuvA | 1.286508 | 0.251932 | 0.273658 |
| giI332110436 | Anaerobic ribonucleoside triphosphate reductase | 0.791772 | −0.233481 | 0.217804 |
| giI332108691 | Phosphoribosylaminoimidazole carboxylase ATPase subunit | 0.735638 | −0.307022 | 0.114037 |
| Shikimate pathway | ||||
| giI332109290 | Isochorismate synthase | 0.732438 | −0.311382 | 0.053731 |
| giI332111971 | Bifunctional 3-demethylubiquinone-9 3-methyltransferase/2-octaprenyl-6-hydroxy phenol methylase | 0.778571 | −0.250291 | 0.011096 |
| Cell wall proteins | ||||
| giI332109978 | Metallophosphoesterase | 0.798696 | −0.224781 | 0.037705 |
| giI332107758 | Peptidyl-dipeptidase Dcp | 0.790719 | −0.234812 | 0.089857 |
| giI332109465 | Hydrolase | 0.476220 | −0.741871 | 0.165655 |
| giI332111521 | Phospho-N-acetylmuramoyl-pentapeptide-transferase | 0.788817 | −0.237223 | 0.108067 |
| giI332108581 | Polysaccharide deacetylase | 0.550108 | −0.597641 | 0.114778 |
| giI332108759 | Mannose-1-phosphate guanylyltransferase/mannose-6-phosphate isomerase | 0.798645 | −0.224843 | 0.071808 |
| giI332110816 | N-Acetyl-anhydromuranmyl-L-alanine amidase | 0.729942 | −0.314792 | 0.040107 |
| giI332112579 | HAD-superfamily hydrolase subfamily IA variant 3 | 0.762906 | −0.270621 | 0.040375 |
| giI332108541 | Alpha/beta hydrolase fold protein | 0.765301 | −0.267492 | 0.079825 |
| Hypothetical proteins | ||||
| giI332109514 | Hypothetical protein RBXJA2T_08925 | 1.279060 | 0.246126 | 0.104975 |
| giI332112531 | Hypothetical protein RBXJA2T_18423 | 1.307658 | 0.268238 | 0.085321 |
| giI332111655 | Hypothetical protein RBXJA2T_14968 | 1.380884 | 0.322724 | 0.083241 |
| giI332110463 | Hypothetical protein RBXJA2T_10691 | 1.331688 | 0.286447 | 0.054846 |
| giI332111444 | Hypothetical protein RBXJA2T_13904 | 1.670387 | 0.513056 | 0.227013 |
| giI332108437 | Hypothetical protein RBXJA2T_04998 | 1.357358 | 0.305541 | 0.215566 |
| giI332111125 | Hypothetical protein RBXJA2T_12552 | 1.353201 | 0.302472 | 0.175941 |
| giI332109166 | Hypothetical protein RBXJA2T_07160 | 1.350034 | 0.300131 | 0.169409 |
| giI332107851 | Hypothetical protein RBXJA2T_02040 | 1.328880 | 0.284336 | 0.176266 |
| giI332111706 | Hypothetical protein RBXJA2T_15223 | 1.346939 | 0.297828 | 0.239591 |
| giI332109490 | Hypothetical protein RBXJA2T_08805 | 1.317135 | 0.275412 | 0.224887 |
| giI332108504 | Hypothetical protein RBXJA2T_05333 | 1.720186 | 0.542432 | 0.246681 |
| giI332112575 | Hypothetical protein RBXJA2T_18643 | 1.774418 | 0.573472 | 0.210987 |
| giI332110761 | Hypothetical protein RBXJA2T_11453 | 2.892699 | 1.062191 | 0.312293 |
| giI332107824 | Hypothetical protein RBXJA2T_01905 | 2.426420 | 0.886418 | 0.329965 |
| giI332111907 | Hypothetical protein RBXJA2T_16222 | 1.609389 | 0.475848 | 0.123126 |
| giI332108499 | Hypothetical protein RBXJA2T_05308 | 1.536876 | 0.429752 | 0.226361 |
| giI332112648 | Hypothetical protein RBXJA2T_19014 | 1.312625 | 0.272029 | 0.086227 |
| giI332112608 | Hypothetical protein RBXJA2T_18814 | 1.285558 | 0.251192 | 0.161866 |
| giI332109471 | Hypothetical protein RBXJA2T_08710 | 1.302381 | 0.264194 | 0.086941 |
| giI332111436 | Hypothetical protein RBXJA2T_13864 | 1.256437 | 0.220289 | 0.120844 |
| giI332107518 | Hypothetical protein RBXJA2T_00345 | 1.199880 | 0.182221 | 0.047647 |
| giI332107846 | Hypothetical protein RBXJA2T_02015 | 0.800367 | −0.222684 | 0.025080 |
| giI332112597 | Hypothetical protein RBXJA2T_18753 | 0.789716 | −0.236082 | 0.082459 |
| giI332108659 | Hypothetical protein RBXJA2T_06120 | 0.786881 | −0.239678 | 0.092913 |
| giI332111328 | Hypothetical protein RBXJA2T_13329 | 0.777793 | −0.251293 | 0.097477 |
| giI332107821 | Hypothetical protein RBXJA2T_01890 | 0.776611 | −0.252814 | 0.044973 |
| giI332109359 | Hypothetical protein RBXJA2T_08148 | 0.776455 | −0.253016 | 0.069663 |
| giI332111066 | Hypothetical protein RBXJA2T_12257 | 0.773731 | −0.256530 | 0.047320 |
| giI332109904 | Hypothetical protein RBXJA2T_09407 | 0.773582 | −0.256723 | 0.082530 |
| giI332108050 | Hypothetical protein RBXJA2T_03051 | 0.773230 | −0.257178 | 0.178172 |
| giI332112705 | Hypothetical protein RBXJA2T_19301 | 0.772483 | −0.258145 | 0.112544 |
| giI332108447 | Hypothetical protein RBXJA2T_05048 | 0.762806 | −0.270752 | 0.184806 |
| giI332110026 | Hypothetical protein RBXJA2T_10029 | 0.761009 | −0.273109 | 0.036106 |
| giI332107818 | Hypothetical protein RBXJA2T_01875 | 0.743329 | −0.296616 | 0.098524 |
| giI332112191 | Hypothetical protein RBXJA2T_17636 | 0.732628 | −0.311118 | 0.135147 |
| giI332111402 | Hypothetical protein RBXJA2T_13694 | 0.725228 | −0.321268 | 0.042410 |
| giI332111415 | Hypothetical protein RBXJA2T_13759 | 0.720756 | −0.327454 | 0.017046 |
| giI332111212 | Hypothetical protein RBXJA2T_12989 | 0.717391 | −0.332133 | 0.081103 |
| giI332110876 | Hypothetical protein RBXJA2T_12032 | 0.708705 | −0.344316 | 0.116150 |
| giI332111846 | Hypothetical protein RBXJA2T_15917 | 0.708024 | −0.345278 | 0.260682 |
| giI332107981 | Hypothetical protein RBXJA2T_02702 | 0.690276 | −0.370663 | 0.078981 |
| giI332110540 | Hypothetical protein RBXJA2T_11076 | 0.676633 | −0.390624 | 0.324398 |
| giI332109437 | Hypothetical protein RBXJA2T_08540 | 0.588054 | −0.530936 | 0.085965 |
| giI332110899 | Hypothetical protein RBXJA2T_12147 | 0.578375 | −0.547531 | 0.290891 |
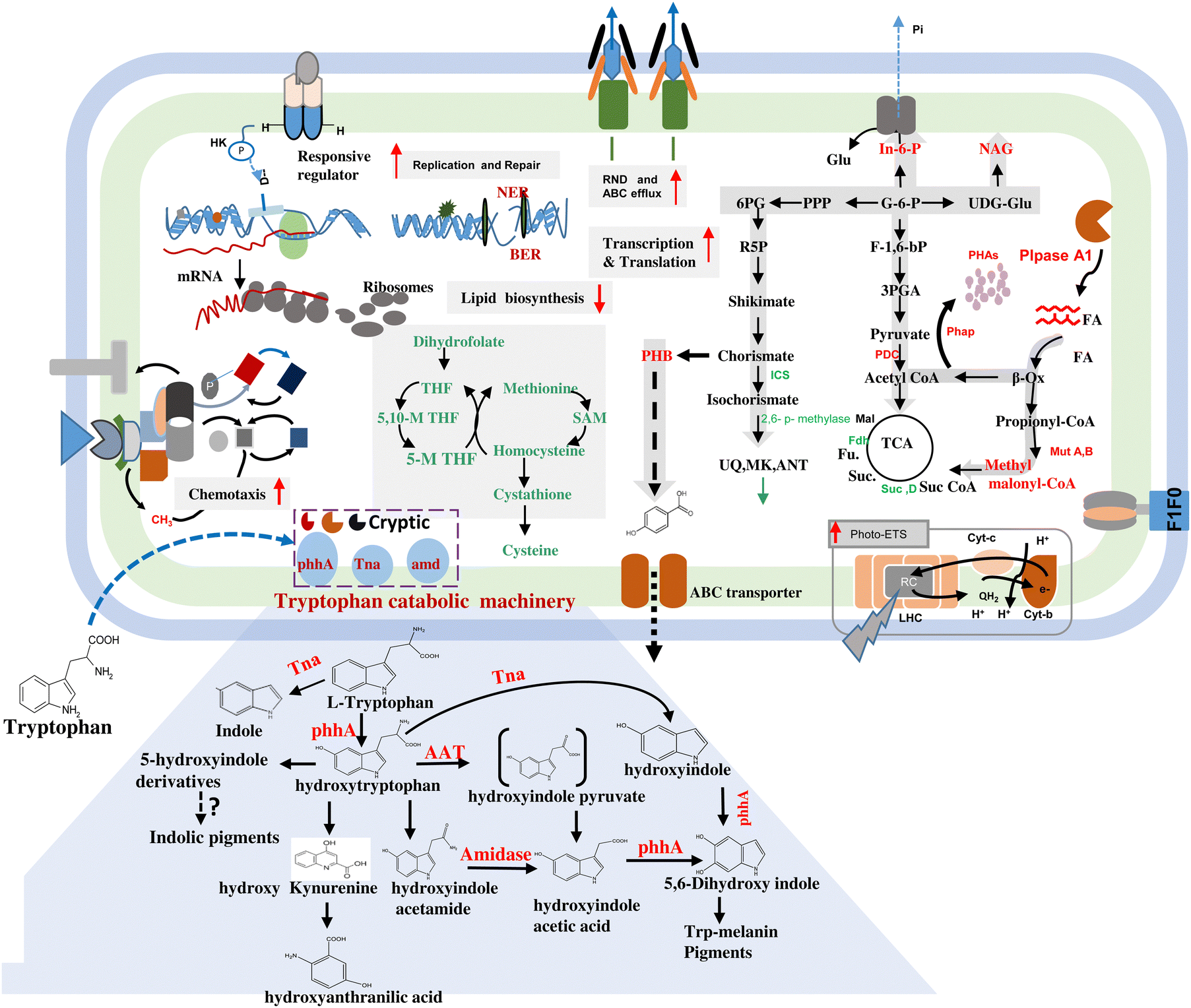 | ||
| Fig. 5 Model illustrating the molecular phenotype of strain JA2 grown on L-tryptophan under the chemotrophic state. Solid up arrows in red indicate upregulation and down arrows in green indicate downregulation of the metabolic process. Proteins in red indicate upregulation, green indicates downregulation and black indicates proteins whose levels were unchanged. Dotted arrows denote multiple steps in the pathway; metabolites which are in high levels are indicated in red and those in low levels in green. SucC, SucD, succinate synthase subunits; SdhC, D, succinate dehydrogenase subunits; Fdh, fumarate dehydrogenase; ICS, isochorismate synthase; 5,6 bp methylase, bifunctional 3-demethylubiquinone-9 3-methyltransferase/2-octaprenyl-6-hydroxy phenol methylase; THF, tetrahydrobiopterine; 5,6 THF, 5,10-methylenetetrahydrofolate reductase; 5-M THF, 5-methyltetrahydrobiopterine; SAM, S-adenosyl methionine; NER, nucleotide excision repair; BER, base excision repair; In-6-p, inositol-6-phosphate; NAG, N-A-acetyl galactosamine; PHB, parahydroxybenzoic acid; UQ, ubiquinone; Mk6, menaquinone; ANT, anthraquinone; Phap, polyhydroxyalkanoate polymerization; phad, polyhydroxyalkanoate depolymerisation; Plpase, phospholipase; Mut A & B, methylmelonyl A and methylmelonyl B; pphA, phenylalanine 4-monooxygenase; Tna, tryptophanase; AAT, aromatic amino transferase; amd, amidase. | ||
Proteins involved in translation initiation and fidelity, such as 30S ribosomal proteins S21 (RBXJA2T_12532), S1 (RBXJA2T_16502), S8 (RBXJA2T_02190), S4 (RBXJA2T_02245), S5 (RBXJA2T_02205), and S3 (RBXJA2T_02145), remained unaffected. However, 50S ribosomal protein L34, sigma 54 modulation protein/ribosomal protein S30EA, and GTP-binding protein, which play an important role in the functional translation machinery, particularly in response to various environmental signals including stress, were upregulated.3,36,37
Membrane transport-related proteins were highly differentially regulated indicating the dynamic metabolite/ion transport facilitating the cell survival (Table 2) particularly upon exposure to aromatic compounds. Bacteria upregulate proteins related to efflux pumps in response to aromatic compound exposure to prevent cell damage. In line with this, membrane efflux pump proteins such as ABC transporters, and related proteins, as well as proteins related to RND pumps (Table 2) were upregulated, strongly suggesting the involvement of an active efflux system in tryptophan exposure. This correlates with the detection of an array of indole/hydroxyindole derivatives in the culture supernatant (Table 1) and indole derivatives are known to cause cellular damage at high concentrations;38,39 thus strain JA2 activates the efflux system to avoid toxicity. Detection of 35% of transmembrane proteins (hydrophobic) in GRAVY analysis (Fig. S5A, ESI†) may further supports the active membrane transport system.
DNA gyrase A and DNA topoisomerase IV subunit A, which play a role in the unwinding and decatenation of DNA, showed no significant change; on the other hand, DNA repair proteins RadA and Holliday junction DNA helicase RuvB were upregulated (Table 2). These results imply a possible DNA damage in cells grown on L-tryptophan and this is because the high concentrations of indolic metabolites derived from L-tryptophan may cause DNA damage as reported earlier.38–41 Further downregulation of nucleotide biosynthesis proteins phosphoribosylaminoimidazole carboxylase ATPase subunit, chromosome segregation and condensation protein ScpA, and anaerobic ribonucleoside triphosphate reductases in L-tryptophan fed cells (Table 2) possibly aimed at minimizing the cellular energy burden as these mechanisms temporally may not offer fitness benefit to the cell.28,32,34,42
Strain JA2 shows a remarkable ability to survive under different growth modes; surprisingly in the present study photosynthetic bacterial PUCC protein, necessary for the formation of the light-harvesting (LH) complex, and photosynthetic proteins reaction center subunit L (Fig. 4A) were up-regulated (Table 2), indicating active assembly of photosystems. This observation is consistent with the findings that anoxygenic photosynthetic bacteria display light-harvesting complex assembly under aerobic cultures possibly as an energy-adaptive mechanism. Further major proteins related to the electron transport chain (ETC) were largely unaffected and NADH dehydrogenase, the alternate flavoprotein (α/β-subunit), and FoF1 ATP synthase were highly upregulated (Fig. 4A) indicating sustained energy generation to meet the cellular demands.
Furthermore, upregulation of the heat-inducible transcription repressor which negatively controls the expression of heat shock proteins28,43 is expected as there is no heat stress, and repressing heat shock proteins may be an energy conservation mechanism (Fig. 4B). Proteins related to stress response, such as cysteine proteinase, transthyretin, and NmrA-like protein, were significantly upregulated under L-tryptophan fed conditions, indicating that external L-tryptophan or its derived metabolites may act as stressors (Table 2).
4.2 Proteomics reveals extensive rewiring of metabolism
Proteins related to vitamin and cofactor biosynthesis, such as radical SAM family protein, blue (type 1) copper domain protein, methyltransferase FkbM family protein, and dihydroneopterin aldolase, showed significant upregulation (Table 2). These proteins are characterized by their roles in iron–sulfur cluster protein and play diverse roles in processes such as SAM synthesis, enzyme activation, peptide modification, transcription, and post-translational modification. Additionally, the blue (type 1) copper domain protein44 is involved in regulating various processes including lipid metabolism, metabolite biosynthesis, antibiotic production, and coenzyme/cofactor biosynthesis.44 Considering their role in various cellular processes, upregulation of these proteins suggests that they may play a pivotal role in cell survival under L-tryptophan-fed conditions.The majority of proteins related to central carbon metabolism in strain JA2 such as glycolysis (EMP), Entner–Doudoroff (ED) pathway45 and pentose phosphate pathway (PPP)46 remained unaltered in the present study (Table 2). However, protein-related oxidoreductases such as NADH dehydrogenase protein was highly upregulated along with the protein pyruvate flavodoxin/ferredoxin oxidoreductase domain protein in L-tryptophan fed cells (Table 2), indicating that pyruvate to acetyl CoA conversion may provide the substrate for the TCA cycle or PHA granular formation. Additionally, galactosamine-containing proteins, implicated in cell wall formation, were upregulated (Fig. 4), which is consistent with the observation that proteins related to cell wall biogenesis, including the glycosyl transferase group, group 1 glycosyl transferase, and CDP-glycerol glycerophosphotransferase, were upregulated (Table 2), highlighting their involvement in cell wall biogenesis. These findings strongly suggest that aromatic compound (L-tryptophan) stress may have triggered the cell well reinforcement machinery of strain JA2; a similar kind of cell wall reinforcement under aromatic compound stress was reported in bacteria.28,34,47–49
In our study, upregulation of phenylalanine 4-monooxygenase correlates with the detection of hydroxytryptophan and other hydroxyindole derivatives and this suggests the role of phenylalanine 4-monooxygenase in the synthesis of hydroxytryptophan (Fig. 5), and subsequent catabolism of hydroxytryptophan may have resulted in the formation of hydroxy indole derivatives (Fig. 5). This is further supported by identification of similar types of hydroxyindole derivatives from both L-tryptophan and hydroxytryptophan fed chemotrophic cultures of strain JA2 (unpublished data). Moreover, the demonstration of aromatic aminotransferase enzyme activity wherein the substrate, hydroxytryptophan, is catabolized through the IPA pathway, leading to the formation of hydroxyindole acetic acid (Fig. S2A and B, ESI†), strongly supports the view of tryptophan conversion to hydroxytryptophan and subsequently to hydroxyindoles. Phenylalanine 4-monooxygenase is known to express in aerobic cultures and upregulation of phenylalanine 4-monooxygenase and detection of hydroxyindoles suggest oxidative catabolism of tryptophan in chemotrophic state.
In addition, the identification of kynurenic acid (Table 1) in metabolic profiling indicates that L-tryptophan is catabolized through the kynurenine pathway. Under aerobic conditions, bacteria catabolize tryptophan via the kynurenine pathway24 and the detection of kynurenic acids hints at L-tryptophan catabolism through this pathway in strain JA2. These findings strongly suggest differential catabolism of L-tryptophan by strain JA2 under chemo and phototrophic states, suggesting that metabolic adaptation of strain JA2 to utilizes L-tryptophan under varying growth modes. Interestingly, the present study showed an accumulation of L-tryptophan-derived pigments (Fig. 2A and B) and these pigments remained unidentified. L-Tryptophan-derived pigments were also reported in fungi Cryptococcus neoformans and C. glabrata in the presence of oxygen (chemotrophic state).57 So far identified L-tryptophan-derived pigments from microorganisms are dimers or trimers of indole derivatives, and we speculate that some of these indolic derivatives formed under L-tryptophan-fed conditions may have reacted and converted into pigments. These pigmented metabolites were not formed under anaerobic (photoheterotrophic) conditions, indicating the role of the oxidative enzyme machinery in pigment formation. Similarly, chemotrophic metabolism of phenylalanine in strain JA2 led to anthocyanin-like pigment23 formation and these results suggest that strain JA2 may harbor hitherto unknown aromatic oxidative metabolism. In the present study, metabolic profiling revealed an array of indolic metabolites many of which remain unidentified, indicating possible diverse L-tryptophan catabolic processes; based on proteomic and metabolite profiling we propose L-tryptophan chemotrophic catabolism (Fig. 5) wherein some amount of L-tryptophan is converted to hydroxytryptophan which acts as a central metabolite from which catabolic routes branch off into (1) the Ehrlich pathway leading to hydroxyindole acids and (2) pigment formation. Some of the tryptophan directly converts to indole through tryptophanase activity, undergoes oxidative catabolism via the kynurenine pathway, or follows the unidentified catabolic route(s). Although metabolites of the Ehrlich and kynurenine pathway were found, corresponding proteins were not detected possibly due to their temporal expression and a time series study may provide more details. However, we could not correlate any up-regulated proteins to pigment synthesis possibly due to a combination of reasons such as lack of structural confirmation of pigments, some housekeeping/non-specific enzyme involvement or our study did not capture the entire expressed proteome due to technical limitations. Moreover, because of the diverse array of indolic metabolites detected and limited genomic information available on aromatic catabolism in strain JA2, the role of possible cryptic metabolic processes in tryptophan catabolism is suggested which needs further investigation. Changing growth conditions in microorganisms are known to activate such cryptic or silent pathways which are otherwise not detected under normal growth conditions.10,12,21,58–60 Interestingly, a large number of hypothetical proteins are upregulated in the current study and we speculate that some of these may have a role in tryptophan metabolism which needs further investigation. Here in the present study the growth conditions were altered (chemotrophic) against the preferred growth mode (phototrophic) and thus may have induced alternative tryptophan processes leading to the formation of a diverse array of tryptophan-derived metabolites. The present study highlights the importance of integrating functional omics such as metabolite and proteomic profiling to decipher the molecular responses of living systems. The current study through proteomic profiling captured the underlying molecular adaptations to the chemotrophic state while metabolite profiling revealed tryptophan catabolic diversity in strain JA2. Integration of proteomic and metabolic profiling captured the molecular phenotype of strain JA2 under tryptophan fed conditions which is otherwise difficult to capture from a single omics study. Since adaptation to changing environmental conditions involves a complex interplay between different functional layers such as genome, transcriptome, proteome and metabolome, understanding these processes also requires a more comprehensive approach such as integrated omics. From a generalistic point of view our study highlights that organisms may possess a rich biochemical repertoire to thrive under ever changing environmental conditions and understanding these processes enhances our knowledge about the functioning of biological systems.
5. Conclusions
Anoxygenic photosynthetic bacteria grow under various growth modes using diverse organic substrates, including aromatic compounds. Despite considerable interest in aromatic metabolism in phototrophic growth mode, chemotrophic metabolism and the underlying molecular adaptations remain largely unexplored. The present investigation revealed strategies of strain JA2 to thrive under L-tryptophan fed chemotrophic conditions. Strain JA2 displayed active signal transductions that control gene expression and membrane transport, which is important for survival in a changing environment. These indicate adaptive strategies of cell survival against possible DNA and envelope damage under tryptophan-fed conditions. Strain JA2 also showed extensive rewiring of metabolic pathways, including certain amino acids, fatty acid/lipid oxidation, the TCA cycle, reduced menaquinone synthesis, and possible metabolic adaptations. The metabolic flux towards PHA synthesis and active energy conservation by downregulation of certain obsolete molecular processes suggest a general stress repose strategy and that L-tryptophan-fed chemotrophic conditions may be inducing stress. Furthermore, the study revealed versatile L-tryptophan catabolism evident from a few catabolic routes predicted such as Ehrlich, kynurenine, indole, and hydroxyindole formation, which is distinct from the phototrophic catabolic footprint. The hydroxylation of L-tryptophan by the up-regulated phenylalanine 4-monooxygenase is a key biotransformation step that results in the formation of hydroxy indoles, and their subsequent reactions may result in the formation of pigments. Due to the limitations in identifying unknown metabolites and capturing possible cryptic metabolic processes, the current study provided a partial picture of molecular and metabolic adaptations of L-tryptophan catabolism. Nevertheless, our study suggests that an alerted growth environment combined with integrated metabolomics and proteomics can capture novel biochemical processes. Renewed future efforts, such as time-series transcriptional studies, may provide new insights into the catabolic processes in strain JA2.Author contributions
S. A., M. M., L. P. M., and V. R. C. conceived and designed the research. S. A. performed the experiments. S. A., M. M., L. P. M., and V. R. C. discussed and analyzed the data. S. A., M. M., and M. L. P. drafted the manuscript. S. A., M. M., M. L. P., V. R. C., and S. C. edited the manuscript. All authors have read and approved the manuscript.Data availability
The mass spectrometry proteomics data have been deposited in the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD055514. Data supporting this article have been included as an ESI† – separate PDF file.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
Shabbir Ahmad acknowledges the University Grants Commission, Govt. of India, for the JRF/SRF fellowship (File No. F1-17.1/201415/MANF-2014-15-MUS-JHA-35979) and SERB NPDF 2022 (PDF/2022/000700).References
- M. D. Lebar, J. L. Heimbegner and B. J. Baker, Cold-water marine natural products, Nat. Prod. Rep., 2007, 24, 774–797 RSC.
- A. P. Lipton, V. S. Pramitha and J. J. Jose, Marine secondary metabolites (MSM) from macro algae enhance bacterial clearance in hemolymph of Penaeus monodon, Isr. J. Aquac. – Bamidgeh., 2009, 61(1), 42–47 Search PubMed.
- S. A. Jackson, L. Crossman, E. L. Almeida, L. M. Margassery, J. Kennedy and A. D. W. Dobson, Diverse and abundant secondary metabolism biosynthetic gene clusters in the genomes of marine sponge derived Streptomyces spp. Isolates, Mar. Drugs, 2018, 16(2), 67–83 CrossRef PubMed.
- C. Kulandaisamy Venil, P. Lakshmanaperumalsamy, C. Kulandaisamy Venil and P. Lakshmanaperumalsamy, An Insightful Overview on Microbial Pigment, Prodigiosin Solid state fermentation of microorganism for pigment production View project An Insightful Overview on Microbial Pigment, Prodigiosin, Electron. J. Biol., 2009, 5(3), 49–61 Search PubMed.
- N. Sutthiwong, M. Fouillaud, A. Valla, Y. Caro and L. Dufossé, Bacteria belonging to the extremely versatile genus Arthrobacter as novel source of natural pigments with extended hue range, Food Res. Int., 2014, 65(PB), 156–162 CrossRef CAS.
- S. R. Pombeiro-Sponchiado, G. S. Sousa, J. C. R. Andrade, H. F. Lisboa and R. C. R. Gonçalves, Production of Melanin Pigment by Fungi and Its Biotechnological Applications, in Melanin, InTech, 2017 DOI:10.5772/67375.
- M. Dieser, M. Greenwood and C. M. Foreman, Carotenoid pigmentation in Antarctic heterotrophic bacteria as a strategy to withstand environmental stresses, Arctic, Antarct Alp Res., 2010, 42(4), 396–405 CrossRef.
- R. S. Celedón and L. B. Díaz, Natural pigments of bacterial origin and their possible biomedical applications, Microorganisms, 2021, 9, 739–748 CrossRef.
- L.-E. Petersen, M. Y. Kellermann and P. J. Schupp, Secondary Metabolites of Marine Microbes: From Natural Products Chemistry to Chemical Ecology, in YOUMARES 9 - The Oceans: Our Research, Our Future, ed. S. Jungblutt, V. Liebich and M. Bode-Dalby, Springer, 2020, pp. 159–180 DOI:10.1007/978-3-030-20389-4_8.
- A. K. R. Abadio, E. S. Kioshima, N. F. Martins, B. Maigret, M. S. S. Felipe and S. M. Freitas, et al., A pharmacophore-based virtual screening approach for the discovery of Trypanosoma cruzi GAPDH inhibitors, J. Chem. Inf. Model., 2015, 5, 2019–2039 Search PubMed.
- R. Srinivasan, A. Kannappan, C. Shi and X. Lin, Marine bacterial secondary metabolites: a treasure house for structurally unique and effective antimicrobial compounds, Mar. Drugs, 2021, 19, 530–539 CrossRef CAS.
- A. P. Tyurin, V. A. Alferova and V. A. Korshun, Chemical elicitors of antibiotic biosynthesis in actinomycetes, Microorganisms, 2018, 6, 52–59, DOI:10.3390/microorganisms6020052.
- E. Zazopoulos, K. Huang, A. Staffa, W. Liu, B. O. Bachmann and K. Nonaka, et al., A genomics-guided approach for discovering and expressing cryptic metabolic pathways, Nat. Biotechnol., 2003, 21(2), 187–190 CrossRef CAS.
- E. Pichersky and D. R. Gang, Genetics and biochemistry of secondary metabolites in plants: an evolutionary perspective, Trends Plant Sci., 2000, 5, 439–445 CrossRef CAS PubMed.
- N. Gurnani, D. Mehta, M. Gupta and B. K. Mehta, Natural Products: Source of Potential Drugs Natural Products Lab, School of Studies in Chemistry & Bio-Chemistry, Afr. J. Basic Appl. Sci., 2014, 6(6), 171–176 Search PubMed.
- N. K. Ranjith, C. V. Ramana and C. Sasikala, Rhodethrin: a novel indole Activities, ether produced by Rhodobacter sphaeroides has cytotoxic and phytohormonal activities, Biotechnol. Lett., 2007, 29, 1399–1402 CrossRef CAS.
- R. Kumavath, C. V. Ramana, C. Sasikala, D. Barh, A. Kumar and V. Azevedo, Isolation and Characterization of L-Tryptophan Ammonia Lyase from Rubrivivax benzoatilyticus Strain JA2, Curr. Protein Pept. Sci., 2015, 16(8), 775–781 CrossRef CAS.
- S. Ahmad, M. Mohammed, L. P. Mekala, S. Chintalapati and V. R. Chintalapati, Tryptophan, a non-canonical melanin precursor: new L-tryptophan based melanin production by Rubrivivax benzoatilyticus JA2, Sci. Rep., 2020, 10(1), 8925 CrossRef CAS.
- R. N. Kumavath, C. V. Ramana and C. Sasikala, L-Tryptophan catabolism by Rubrivivax benzoatilyticus JA2 occurs through indole 3-pyruvic acid pathway, Biodegradation, 2010, 21(5), 825–832 CrossRef CAS PubMed.
- M. Lakshmi Prasuna, M. Mujahid, C. Sasikala and C. V. Ramana, L-Phenylalanine catabolism and L-phenyllactic acid production by a phototrophic bacterium, Rubrivivax benzoatilyticus JA2, Microbiol. Res., 2012, 167(9), 526–531 CrossRef CAS PubMed.
- L. P. Mekala, M. Mohammed, S. Chintalapati and V. R. Chintalapati, Stable Isotope-Assisted Metabolic Profiling Reveals Growth Mode Dependent Differential Metabolism and Multiple Catabolic Pathways of L-Phenylalanine in Rubrivivax benzoatilyticus JA2, J. Proteome Res., 2018, 17(1), 189–202 CrossRef CAS PubMed.
- L. P. Mekala, M. Mohammed, S. Chinthalapati and V. R. Chinthalapati, Pyomelanin production: insights into the incomplete aerobic L-phenylalanine catabolism of a photosynthetic bacterium, Rubrivivax benzoatilyticus JA2, Int. J. Biol. Macromol., 2019, 126, 755–764 CrossRef CAS.
- L. P. Mekala, M. Mohammed, S. Chintalapati and V. R. Chintalapati, Precursor-feeding and altered-growth conditions reveal novel blue pigment production by Rubrivivax benzoatilyticus JA2, Biotechnol. Lett., 2019, 41(6–7), 813–822 CrossRef CAS PubMed.
- S. Ahmad, M. Mohammed, L. P. Mekala, R. Anusha, C. Sasikala and C. V. Ramana, Stable isotope-assisted metabolite profiling reveals new insights into L-tryptophan chemotrophic metabolism of Rubrivivax benzoatilyticus, World J. Microbiol. Biotechnol., 2023, 39(4), 98–103 CrossRef CAS PubMed.
- M. Mohammed, A. Isukapatla, L. P. Mekala, R. P. E. V. Venkata, S. Chintalapati and V. R. Chintalapati, Genome sequence of the phototrophic betaproteobacterium Rubrivivax benzoatilyticus strain JA2 T, J. Bacteriol., 2011, 193, 2898–2899 CrossRef CAS PubMed.
- S. Ahmad, M. Mohammed, L. P. Mekala, S. Chintalapati and V. R. Chintalapati, Tryptophan, a non-canonical melanin precursor: new L-tryptophan based melanin production by Rubrivivax benzoatilyticus JA2, Sci. Rep., 2020, 10(1), 8925 CrossRef CAS.
- S. A. Gordon and R. P. Weber, Colorimetric estimation of indoleacetic acid, Plant Physiol., 1951, 26(1), 192–195 CrossRef CAS PubMed.
- D. Gupta, M. Mohammed, L. P. Mekala, S. Chintalapati and V. R. Chintalapati, iTRAQ-based quantitative proteomics reveals insights into metabolic and molecular responses of glucose-grown cells of Rubrivivax benzoatilyticus JA2, J. Proteomics, 2019, 194, 59 CrossRef.
- M. Mujahid, M. L. Prasuna, C. Sasikala and C. V. Ramana, Integrated metabolomic and proteomic analysis reveals systemic responses of rubrivivax benzoatilyticus JA2 to aniline stress, J. Proteome Res., 2015, 14(2), 711–727 CrossRef CAS PubMed.
- P. L. Ross, Y. N. Huang, J. N. Marchese, B. Williamson, K. Parker and S. Hattan, et al., Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents, Mol. Cell. Proteomics, 2004, 3(12), 1154–1169 CrossRef CAS.
- D. Povero, A. Eguchi, H. Li, C. D. Johnson, B. G. Papouchado and A. Wree, et al., Circulating extracellular vesicles with specific proteome and liver microRNAs are potential biomarkers for liver injury in experimental fatty liver disease, PLoS One, 2014, 9(12), e113651 CrossRef.
- M. Mohammed, L. P. Mekala, S. Chintalapati and V. R. Chintalapati, New insights into aniline toxicity: aniline exposure triggers envelope stress and extracellular polymeric substance formation in Rubrivivax benzoatilyticus JA2, J. Hazard. Mater., 2020, 385, e87503 CrossRef.
- M. Mujahid, C. Sasikala and C. V. Ramana, Production of indole-3-acetic acid and related indole derivatives from L-tryptophan by Rubrivivax benzoatilyticus JA2, Appl. Microbiol. Biotechnol., 2011, 89(4), 1001–1008 CrossRef CAS.
- M. Mujahid, C. Sasikala and V. C. Ramana, Aniline is an inducer, and not a precursor, for indole derivatives in Rubrivivax benzoatilyticus JA2, PLoS One, 2014, 9(2), 121571 Search PubMed.
- T. Mascher, J. D. Helmann and G. Unden, Stimulus Perception in Bacterial Signal-Transducing Histidine Kinases, Microbiol. Mol. Biol. Rev., 2006, 70(4), 910–938 CrossRef CAS PubMed.
- M. Theunis, H. Kobayashi, W. J. Broughton and E. Prinsen, Flavonoids, NodD1, NodD2, and Nod-Box NB15 modulate expression of the y4wEFG locus that is required for indole-3-acetic acid synthesis in Rhizobium sp. strain NGR234, Mol. Plant-Microbe Interact., 2004, 17(10), 1153–1161 CrossRef CAS.
- D. Dodd, M. H. Spitzer, W. Van Treuren, B. D. Merrill, A. J. Hryckowian and S. K. Higginbottom, et al., A gut bacterial pathway metabolizes aromatic amino acids into nine circulating metabolites, Nature, 2017, 551, 648–652 CrossRef CAS PubMed.
- R. Sonowal, A. Swimm, A. Sahoo, L. Luo, Y. Matsunaga and Z. Wu, et al., Indoles from commensal bacteria extend healthspan, Proc. Natl. Acad. Sci. U. S. A., 2017, 114(36), e7506 CrossRef CAS PubMed.
- R. Sonowal, A. I. Swimm, F. Cingolani, N. Parulekar, T. L. Cleverley and A. Sahoo, et al., A microbiota and dietary metabolite integrates DNA repair and cell death to regulate embryo viability and aneuploidy during aging, Sci. Adv., 2023, 9(8), eade8653 CrossRef CAS PubMed.
- B. Bommarius, A. Anyanful, Y. Izrayelit, S. Bhatt, E. Cartwright and W. Wang, et al., A Family of Indoles Regulate Virulence and Shiga Toxin Production in Pathogenic E. coli, PLoS One, 2013, 8(1), e54456 CrossRef CAS.
- A. Swimm, C. R. Giver, Z. DeFilipp, S. Rangaraju, A. Sharma and A. U. Antonova, et al., Indoles derived from intestinal microbiota act via type I interferon signaling to limit graft-versus-host disease, Blood, 2018, 132(23), 2506–2519 CrossRef CAS PubMed.
- D. Gupta, C. Sasikala and C. V. Ramana, Dynamics of proteo-metabolome from Rubrivivax benzoatilyticus JA2 reveals a programmed switch-off of phototrophic growth, leading to a non-cultivable state as a hyperglycemic effect, J. Proteomics, 2022, 260, 104569 CrossRef CAS PubMed.
- A. Segura, L. Molina, S. Fillet, T. Krell, P. Bernal and J. Muñoz-Rojas, et al., Solvent tolerance in Gram-negative bacteria, Curr. Opin. Biotechnol, 2012, 23, 415–421 CrossRef CAS PubMed.
- L. Xie, L. Zhang, C. Wang, X. Wang, Y. M. Xu and H. Yu, et al., Methylglucosylation of aromatic amino and phenolic moieties of drug-like biosynthons by combinatorial biosynthesis, Proc. Natl. Acad. Sci. U. S. A., 2018, 115(22), 4980–4989 CrossRef PubMed.
- X. Chen, K. Schreiber, J. Appel, A. Makowka, B. Fähnrich and M. Roettger, et al., The Entner-Doudoroff pathway is an overlooked glycolytic route in cyanobacteria and plants, Proc. Natl. Acad. Sci. U. S. A., 2016, 113(19), 5441–5446 CrossRef CAS.
- M. Mohammed, A. Isukapatla and L. P. Mekala, Venkata RPEV, Chintalapati S, Chintalapati VR. Genome sequence of the phototrophic betaproteobacterium Rubrivivax benzoatilyticus strain JA2 T, J. Bacteriol., 2011, 193, 2898–2899 CrossRef CAS.
- K. Ghattavi, A. Homaei, E. Kamrani and S. K. Kim, Melanin pigment derived from marine organisms and its industrial applications, Dyes Pigm., 2022, 201, 110214 CrossRef CAS.
- C. S. Kim, J. H. Li, B. Barco, H. B. Park, A. Gatsios and A. Damania, et al., Cellular Stress Upregulates Indole Signaling Metabolites in Escherichia coli, Cell Chem. Biol., 2020, 27(6), 698–707 CrossRef CAS PubMed.
- A. Luna-Bulbarela, M. T. Romero-Gutiérrez, R. Tinoco-Valencia, E. Ortiz, M. E. Martínez-Romero and E. Galindo, et al., Response of Bacillus velezensis 83 to interaction with Colletotrichum gloeosporioides resembles a Greek phalanx-style formation: a stress resistant phenotype with antibiosis capacity, Microbiol. Res., 2024, 280, 127592 CrossRef CAS PubMed.
- R. Breitling, D. Vitkup and M. P. Barrett, New surveyor tools for charting microbial metabolic maps, Nat. Rev. Microbiol., 2008, 6(2), 156–161 CrossRef CAS.
- K. Sasikala, C. V. Ramana, P. R. Rao and K. L. Kovacs, Anoxygenic Phototrophic Bacteria: Physiology and Advances in Hydrogen Production Technology, Adv. Appl. Microbiol., 1993, 38(C), 211–295 CAS.
- M. Bergkessel, D. W. Basta and D. K. Newman, The physiology of growth arrest: uniting molecular and environmental microbiology, Nat. Rev. Microbiol., 2016, 14, 549–562 CrossRef CAS.
- S. J. Krebs and R. K. Taylor, Nutrient-dependent, rapid transition of Vibrio cholerae to coccoid morphology and expression of the toxin co-regulated pilus in this form, Microbiology, 2011, 157(10), 2942–2953 CrossRef CAS PubMed.
- B. Muszyńska, K. Sułkowska-Ziaja and H. Ekiert, Analysis of indole compounds in methanolic extracts from the fruiting bodies of Cantharellus cibarius (the Chanterelle) and from the mycelium of this species cultured in vitro, J. Food Sci. Technol., 2013, 50(6), 1233–1237 CrossRef PubMed.
- J. A. Mora-Villalobos and A. P. Zeng, Synthetic pathways and processes for effective production of 5-hydroxytryptophan and serotonin from glucose in Escherichia coli, J. Biol. Eng., 2018, 12(1), 2018 CrossRef.
- Y. Chen, J. Tang, L. Wang, Z. Tian, A. Cardenas and X. Fang, et al., Creation of Bacterial Cells with 5-Hydroxytryptophan as a 21st Amino Acid Building Block, Chem, 2020, 6(10), 2717–2727 CAS.
- S. Brunke, K. Seider, R. S. Almeida, A. Heyken, C. B. Fleck and M. Brock, et al., Candida glabrata tryptophan-based pigment production via the Ehrlich pathway, Mol. Microbiol., 2010, 76(1), 25–47 CrossRef CAS PubMed.
- M. R. Seyedsayamdost, High-Throughput platform for the discovery of elicitors of silent bacterial gene clusters, Proc. Natl. Acad. Sci. U. S. A., 2014, 111(20), 7266–7271 CrossRef CAS PubMed.
- J. S. Zarins-Tutt, T. T. Barberi, H. Gao, A. Mearns-Spragg, L. Zhang and D. J. Newman, et al., Prospecting for new bacterial metabolites: a glossary of approaches for inducing, activating and upregulating the biosynthesis of bacterial cryptic or silent natural products, Nat. Prod. Rep., 2016, 33, 54–72 RSC.
- D. B. B. Trivella and R. de Felicio, The Tripod for Bacterial Natural Product Discovery: Genome Mining, Silent Pathway Induction, and Mass Spectrometry-Based Molecular Networking, mSystems, 2018, 3(2), e00160-17 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4mo00170b |
| ‡ Current address: DBT – The Institute for Stem Cell Science and Regenerative Medicine (DBT-InStem), Bangalore 560065, Karnataka, India. |
| § Current address: Department of Botany, Bharathidasan Government College for Women, Puducherry U.T. – 605003, India. |
| ¶ Current address: Department of Botany, Avvaiyar Government College for Women, Karaikal, Puducherry- U.T 609 602, India. |
| This journal is © The Royal Society of Chemistry 2025 |