
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA ladder-type organic molecule with pseudocapacitive properties enabling superior electrochemical desalination†
Yujie
Cui
a,
Yueheng
Tao
a,
Jun
Yang
 *a,
Houxiang
Wang
a,
Peipei
Zhang
a,
Guangxing
Li
a,
Minjie
Shi
*a and
Edison Huixiang
Ang
*b
*a,
Houxiang
Wang
a,
Peipei
Zhang
a,
Guangxing
Li
a,
Minjie
Shi
*a and
Edison Huixiang
Ang
*b
aSchool of Materials Science and Engineering, Jiangsu University of Science and Technology, Jiangsu 212003, P. R. China. E-mail: iamjyang@just.edu.cn; shiminjie@just.edu.cn
bNatural Sciences and Science Education, National Institute of Education, Nanyang Technological University, Singapore 637616, Singapore. E-mail: edison.ang@nie.edu.sg
First published on 10th January 2025
Abstract
The availability of clean water is fundamental for maintaining sustainable environments and human ecosystems. Capacitive deionization offers a cost-effective, environmentally friendly, and energy-efficient solution to meet the rising demand for clean water. Electrode materials based on pseudocapacitive adsorption have attracted significant attention in capacitive deionization due to their relatively high desalination capacity. In this study, a novel organic compound, PTQN, is introduced, featuring a ladder-type structure enriched with imine-based active sites, specifically designed for capacitive deionization. This advanced molecular design imparts the PTQN compound with exceptional pseudocapacitive properties, enhanced electron delocalization, and superior structural stability, which are supported by both experimental results and theoretical analyses. As an electrode, PTQN exhibits a high pseudocapacitive capacitance of 238.26 F g−1 and demonstrates excellent long-term stability, retaining approximately 100 percent of its capacitance after 5000 cycles in NaCl solution. The involvement of PTQN active sites in the Na+ electrosorption process was further elucidated using theoretical calculations and ex situ characterization. Moreover, a hybrid capacitive deionization (HCDI) device employing the PTQN electrode exhibited an impressive salt removal capacity of 61.55 mg g−1, a rapid average removal rate of 2.05 mg g−1 min−1, and consistent regeneration performance (∼97.04 percent after 50 cycles), demonstrating its potential for capacitive deionization systems. Furthermore, the PTQN electrode displayed superior removal efficiency for tetracycline. This work contributes to the rational design of organic materials for the development of advanced electrochemical desalination systems.
New conceptsThis work introduces a novel ladder-type organic molecule (PTQN) with imine-based active sites, specifically designed to enhance pseudocapacitive performance for electrochemical desalination applications. Unlike traditional electrode materials, which often suffer from insufficient electroadsorption capacity, PTQN exhibits a highly stable and efficient pseudocapacitive behavior, driven by its unique structural features and significant electron delocalization. This concept differentiates itself from existing research by integrating a tailored molecular architecture that not only improves capacitive performance but also enhances the material's stability and regeneration capabilities. Moreover, the study provides new insights into the role of imine-based active sites in Na+ electrosorption processes, combining both experimental and theoretical approaches. The development of PTQN as a highly effective electrode material demonstrates the potential of organic molecules in overcoming key limitations in capacitive deionization technologies. This new concept paves the way for the rational design of organic-based electrodes with improved performance, offering a promising strategy for advancing cleaner, more sustainable desalination technologies in materials science. |
1. Introduction
The demand for clean water has been steadily rising, driven by factors such as global population growth and rapid industrialization. Consequently, ensuring access to clean water has become a pressing global issue. Although Earth's total water volume is approximately 1.38 billion km3, only around 0.3% is suitable for human consumption. This has led to increasing attention on desalinating brackish water to produce potable water. Electrochemical desalination methods have emerged as viable alternatives, with strategies including electrodialysis, capacitive deionization, shock electrodialysis, and ion concentration polarization.1–3 Among these, capacitive deionization (CDI) has garnered interest as a relatively recent technique that relies on ion separation via capacitive electrodes. The desalination mechanism in CDI operates based on electroadsorption, where ions are attracted to oppositely charged electrodes upon the application of a voltage and subsequently released when the voltage is reversed.4,5 This approach makes CDI a cost-effective, environmentally friendly, and energy-efficient technology, offering a competitive solution for brackish water desalination.6–8Electrode materials are key to achieving efficient and rapid desalination in CDI systems, driving significant research into the design and synthesis of advanced electrode materials. Carbon-based materials, such as activated carbon, graphene, and carbon nanotubes, are commonly employed due to their high specific surface area and excellent electrical conductivity.9 These materials enable ion removal through the formation of an electric double layer at the electrode–electrolyte interface. However, the salt removal capacity of carbon materials, typically below 50 mg g−1, is often limited by their restricted specific surface area available for ion adsorption.10,11 In contrast, pseudocapacitive materials offer a promising option, relying primarily on rapid redox reactions or insertion mechanisms to store charge. In the electrochemical process, specific functional groups in the material (such as conjugated structures, redox active groups, etc.) undergo reversible electron transfer, so as to achieve charge storage and release. It has a higher specific capacitance than the double electric layer capacitor, showing enhanced salt removal capability and selectivity. These features enable pseudocapacitive materials to overcome the limitations of traditional carbon electrodes, making their development crucial for advancing capacitive deionization technologies.12
Extensive research has been conducted to explore pseudocapacitive materials for CDI systems, with a particular focus on inorganic compounds such as metal oxides, carbides, and sulfides. However, the widespread application of these inorganic materials poses challenges due to complex fabrication and recycling processes. In contrast, organic compounds, composed of readily available elements such as carbon, hydrogen, oxygen, and nitrogen, offer significant advantages in terms of molecular diversity, design flexibility, and environmental sustainability.13–16 The inherent variability of organic materials allows for molecular tailoring to meet specific performance requirements.17–19 Furthermore, the redox behavior of organic compounds enables ion removal through the coordination of functional groups without necessitating phase transitions. Recently studied desalination mechanisms of organic compounds (such as MXenes,20,21 Prussian blue22 and conductive polymers23) are based on Faraday reactions, and their applications in the field of CDI research are highly suggestive. Despite these advantages, the exploration of organic compounds for CDI systems remains in its early stages due to limitations such as poor electron mobility and low redox activity, which hinder efficient ion electroadsorption.
In this study, a novel centrosymmetric, ladder-type organic molecule, PTQN, was synthesized through the reaction of 4,5,9,10-pyrenetetrone (PTO) and 2,3-diaminoquinoline (DAQN) monomers. The C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N group acts as the active site and ion for reversible redox reaction. This high reactivity enables the organic electrode containing CN groups to achieve high specific capacitance. The remarkable structure of the PTQN molecule incorporates several advantages, particularly the extension of π-electron delocalization, which was confirmed by computational simulations, while exhibiting enhanced thermal stability, reduced energy gap, and excellent electrophilicity. When applied as an electrode in NaCl aqueous solution, PTQN demonstrated efficient and rapid Na+ adsorption with pseudocapacitive behavior. At a current density of 2 A g−1, it achieved a capacitance of 238.26 F g−1, with no degradation after 5000 cycles. Theoretical calculations confirmed its exceptional electrochemical performance, attributed to symmetrical active sites facilitating the adsorption of eight Na+ ions. When tested in HCDI devices, PTQN exhibited a high desalination rate of 7.24 mg g−1 min−1 and a desalination capacity of 61.55 mg g−1 at an operating voltage of 1.2 V. PTQN also demonstrated efficient removal of tetracycline in various concentrations, highlighting its versatility and potential in water purification applications.
N group acts as the active site and ion for reversible redox reaction. This high reactivity enables the organic electrode containing CN groups to achieve high specific capacitance. The remarkable structure of the PTQN molecule incorporates several advantages, particularly the extension of π-electron delocalization, which was confirmed by computational simulations, while exhibiting enhanced thermal stability, reduced energy gap, and excellent electrophilicity. When applied as an electrode in NaCl aqueous solution, PTQN demonstrated efficient and rapid Na+ adsorption with pseudocapacitive behavior. At a current density of 2 A g−1, it achieved a capacitance of 238.26 F g−1, with no degradation after 5000 cycles. Theoretical calculations confirmed its exceptional electrochemical performance, attributed to symmetrical active sites facilitating the adsorption of eight Na+ ions. When tested in HCDI devices, PTQN exhibited a high desalination rate of 7.24 mg g−1 min−1 and a desalination capacity of 61.55 mg g−1 at an operating voltage of 1.2 V. PTQN also demonstrated efficient removal of tetracycline in various concentrations, highlighting its versatility and potential in water purification applications.
2. Results and discussions
A one-step dehydration condensation method was employed, utilizing 4,5,9,10-pyrenetetrone (PTO, Fig. S1, ESI†) and 2,3-diaminoquinoline (DAQN) as starting materials to synthesize a fully ladder-type, centrosymmetric PTQN organic molecule featuring multiple imine-based active sites (Fig. 1(a)). In this reaction, the CO group of the PTO precursor and the –NH2 group of the DAQN precursor participated in dehydration condensation, leading to the formation of an eight-membered CN linkage within the PTQN molecule. Scanning electron microscopy (SEM) confirmed the microstructure of PTQN, revealing the presence of irregularly shaped small particles, while energy dispersive spectroscopy (EDS) analysis demonstrated a uniform distribution of carbon and nitrogen elements throughout the molecule (Fig. S2, ESI†). The 13C solid-state nuclear magnetic resonance (NMR) spectrum (Fig. 1(b)) displayed a distinct peak at 144 ppm, corresponding to the carbon atom linked to the CN bond, alongside peaks between 125 and 130 ppm associated with carbon atoms in CC bonds. The broadening of these peaks is likely attributed to the rigid π-conjugated molecular structure. Thermogravimetric analysis (TGA) (Fig. 1(c)) revealed significant weight loss of the PTQN compound below 100 °C, attributed to water evaporation. Notably, the PTQN compound exhibited substantially lower weight loss up to 500 °C compared to the PTO and DAQN precursors, indicating enhanced thermal stability due to the formation of a robust π-conjugated framework. X-ray photoelectron spectroscopy (XPS) analysis of the PTQN organic compound revealed that the high-resolution C 1s spectrum could be deconvoluted into three distinct peaks corresponding to C–C/CC, C–N, and CN bonds at binding energies of 283.9 eV, 284.9 eV, and 285.8 eV, respectively (Fig. 1(d)). An additional peak at 288.2 eV was observed, which is attributed to π–π interactions within the molecular framework. The X-ray diffraction (XRD) pattern of the PTQN organic compound displayed a prominent diffraction peak at 26.16°, indicating a d-spacing of 0.34 nm (Fig. S3, ESI†). This finding confirms the presence of significant π–π stacking interactions among the imine heterocycles within the PTQN structure. Furthermore, the reduced density gradient (RDG) simulation supported this observation. The green peak in the range of −0.02 to 0.00 a.u., indicating the existence of specific weak interaction in organic molecule, resulting in high structural integrity of PTQN organic molecule, and affecting the site selectivity and reaction rate of organic molecules (Fig. S4, ESI†). These interactions are critical in enhancing redox kinetics and facilitating rapid charge transfer within the PTQN framework. The ladder-type PTQN molecule, rich in imine groups, influences the energy levels of the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) due to an electron-withdrawing effect. This effect results in an elevation of the HOMO or a reduction in the LUMO, thereby minimizing the energy gap. The PTQN molecule exhibited the smallest energy gap (3.08 eV) compared to its precursors, PTO (3.45 eV) and DAQN (3.08 eV) (Fig. S5, ESI†). The localized orbital locator-π (LOL-π) color-filled map further demonstrated the extensive conjugation and electron delocalization within the PTQN molecule (Fig. 1(e)). This distinctive configuration, characterized by centrosymmetric chains and enhanced electron delocalization at the redox centers, plays a crucial role in improving both electron transport efficiency and overall stability.
 | ||
| Fig. 1 (a) Synthetic process of the PTQN molecule. (b) Solid-state 13C spectrum illustrating the carbon environments. (c) TGA curves demonstrating the thermal stability and decomposition profile of the samples. (d) High-resolution XPS spectrum of the C 1s region, accompanied by an inset showing the percentage distribution of the various chemical states of carbon within the PTQN structure. (e) LOL-π color-filled map depicting the electronic distribution of the PTQN molecule. | ||
The electrochemical properties of the PTQN organic electrode were assessed in a 1 M NaCl aqueous solution using a standard three-electrode testing configuration. The cyclic voltammetry (CV) curves reveal the presence of two pairs of reversible redox peaks at various scan rates. To exclude proton contribution, electrochemical tests of PTQN electrode were first conducted in 10 M NaOH solution (Fig. S6, ESI†). The peak shapes remain consistent with increasing sweep rates, indicating the availability of redox-active sites within the PTQN organic electrode that facilitate efficient Na+ capture (Fig. 2(a)). As shown in Fig. 2(b), there are minor variations in the oxidation and reduction peaks; specifically, the anodic peak shifts from −0.42 V to −0.38 V and from 0.26 V to 0.346 V, while the cathodic peak shifts from 0.176 V to 0.126 V and from −0.51 V to −0.578 V, respectively. The subtlety of these shifts suggests that significant local polarization is unlikely at the electrode interface, thereby confirming the exceptional charge transfer kinetics of the PTQN electrode. To elucidate the electrochemical mechanisms underlying the performance of the PTQN electrode, a detailed examination of the CV curves was conducted. The relationship between the observed peak current (i) and the sweep rate (v) in CV curves is typically described by the power law equation log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) i = loga + blogv, where “a” and “b” are adjustable parameters. The calculated b values for the two pairs of redox peaks are 0.71, 0.71, 0.68, and 0.80, respectively (Fig. S7, ESI†). These values indicate that the redox storage mechanism in the PTQN electrode is influenced by both diffusion-controlled and capacitive-controlled processes. To quantitatively separate the contributions from diffusion control and surface control, Trasatti analysis was employed. Furthermore, according to the proposed formula: i(v) = k1v + k2v1/2, the respective contributions of diffusion-controlled capacitance and surface-controlled capacitance can be quantified by analyzing their behavior at a specific scan rate. The quantization of the pseudo-capacitance contribution is shown in Fig. S8 (ESI†). When the incremental scan rate is 5, 10, 20 and 50 mV s−1, the pseudo-capacitance contribution is 39.05%, 45.16%, 52.84% and 66.34%, respectively. This trend suggests that the contribution of pseudocapacitance increases with the scan rate, highlighting the exceptional electrochemical responsiveness of the PTQN electrode. Additionally, the electrochemical kinetics at specific potentials during the charge and discharge processes were analyzed using ex situ electrochemical impedance spectroscopy (EIS). The nearly identical shapes of the Nyquist curves across various potentials and the relatively constant charge transfer resistance (Rct) (Fig. S9, ESI†). This result is in strong concordance with the analysis conducted using the DRT function. In DRT analysis (Fig. 2(e)), the integrated area under the peak represents the resistive contribution to the process, and each characteristic peak on the DRT curve corresponds to an electrochemical reaction behavior, highlight the rapid and efficient charge transfer dynamics between the PTQN electrode and the electrolyte. Further calculations reveal that the Na+ diffusion coefficient during the charge and discharge processes ranges from 1.35 × 10−13 to 1.45 × 10−13 cm2 s−1 (Fig. 2(c)), indicating that the PTQN electrode possesses favorable electrochemical kinetics for Na+ capture. These results reflect the potential of the PTQN electrode to advance high-performance capacitive deionization systems. The dashed line with a phase angle of 45° in the Bode diagram (Fig. 2(d)) indicates a range of eigenfrequency (f0) from 4.345 to 6.934 Hz, highlighting an exceptionally rapid frequency response (τ0) characterized by a remarkably short time interval of 0.14 to 0.23 s. The PTQN organic compound exhibits remarkable redox kinetics and rapid electrochemical responsiveness, attributable to its unique ladder-type structure enriched with imine-based active sites. Fig. S10 (ESI†) illustrates the HOMO and the LUMO of the PTQN organic compound. Theoretical frameworks suggest that holomolecular conjugation results in a reduction of the energy levels of both the HOMO and LUMO. Furthermore, the structural characteristics of conjugated systems significantly influence the reversibility of charge transport and redox reactions. Consequently, the PTQN molecule exhibits the smallest HOMO–LUMO band gap of 3.08 eV compared to previously reported organic compounds (Table S1, ESI†),24–28 underscoring its exceptional electron transfer conductivity and favorable reaction kinetics. Furthermore, the partial density of states (PDOS, Fig. 2(f)) analysis for the PTQN organic compound indicates a higher electron density near the Fermi energy (as represented by the dashed line). This phenomenon arises from the stepped conjugated configuration of PTQN, which promotes enhanced electron mobility within the molecule. Consequently, this configuration facilitates greater electron delocalization and substantially improves the conductivity of the material.
i = loga + blogv, where “a” and “b” are adjustable parameters. The calculated b values for the two pairs of redox peaks are 0.71, 0.71, 0.68, and 0.80, respectively (Fig. S7, ESI†). These values indicate that the redox storage mechanism in the PTQN electrode is influenced by both diffusion-controlled and capacitive-controlled processes. To quantitatively separate the contributions from diffusion control and surface control, Trasatti analysis was employed. Furthermore, according to the proposed formula: i(v) = k1v + k2v1/2, the respective contributions of diffusion-controlled capacitance and surface-controlled capacitance can be quantified by analyzing their behavior at a specific scan rate. The quantization of the pseudo-capacitance contribution is shown in Fig. S8 (ESI†). When the incremental scan rate is 5, 10, 20 and 50 mV s−1, the pseudo-capacitance contribution is 39.05%, 45.16%, 52.84% and 66.34%, respectively. This trend suggests that the contribution of pseudocapacitance increases with the scan rate, highlighting the exceptional electrochemical responsiveness of the PTQN electrode. Additionally, the electrochemical kinetics at specific potentials during the charge and discharge processes were analyzed using ex situ electrochemical impedance spectroscopy (EIS). The nearly identical shapes of the Nyquist curves across various potentials and the relatively constant charge transfer resistance (Rct) (Fig. S9, ESI†). This result is in strong concordance with the analysis conducted using the DRT function. In DRT analysis (Fig. 2(e)), the integrated area under the peak represents the resistive contribution to the process, and each characteristic peak on the DRT curve corresponds to an electrochemical reaction behavior, highlight the rapid and efficient charge transfer dynamics between the PTQN electrode and the electrolyte. Further calculations reveal that the Na+ diffusion coefficient during the charge and discharge processes ranges from 1.35 × 10−13 to 1.45 × 10−13 cm2 s−1 (Fig. 2(c)), indicating that the PTQN electrode possesses favorable electrochemical kinetics for Na+ capture. These results reflect the potential of the PTQN electrode to advance high-performance capacitive deionization systems. The dashed line with a phase angle of 45° in the Bode diagram (Fig. 2(d)) indicates a range of eigenfrequency (f0) from 4.345 to 6.934 Hz, highlighting an exceptionally rapid frequency response (τ0) characterized by a remarkably short time interval of 0.14 to 0.23 s. The PTQN organic compound exhibits remarkable redox kinetics and rapid electrochemical responsiveness, attributable to its unique ladder-type structure enriched with imine-based active sites. Fig. S10 (ESI†) illustrates the HOMO and the LUMO of the PTQN organic compound. Theoretical frameworks suggest that holomolecular conjugation results in a reduction of the energy levels of both the HOMO and LUMO. Furthermore, the structural characteristics of conjugated systems significantly influence the reversibility of charge transport and redox reactions. Consequently, the PTQN molecule exhibits the smallest HOMO–LUMO band gap of 3.08 eV compared to previously reported organic compounds (Table S1, ESI†),24–28 underscoring its exceptional electron transfer conductivity and favorable reaction kinetics. Furthermore, the partial density of states (PDOS, Fig. 2(f)) analysis for the PTQN organic compound indicates a higher electron density near the Fermi energy (as represented by the dashed line). This phenomenon arises from the stepped conjugated configuration of PTQN, which promotes enhanced electron mobility within the molecule. Consequently, this configuration facilitates greater electron delocalization and substantially improves the conductivity of the material.
 | ||
| Fig. 2 (a) CV curves at different scan rates, illustrating the electrochemical behavior of the PTQN compound. (b) Corresponding oxidation–reduction potentials derived from the CV curves at various scan rates. (c) Diffusion coefficients (D values) during the charging and discharging processes. (d) Bode plots displaying the frequency response characteristics of the PTQN compound. (e) DRT analysis during charging and discharging, correlating the frequency response with electrochemical performance. (f) PDOS of the PTQN organic compound, providing insights into its electronic structure. | ||
The galvanostatic charge–discharge (GCD) curves of the PTQN electrode display two charging platforms and two discharge platforms, which are correspond to the two pairs of redox peaks observed in the CV measurements (Fig. 3(a)). The PTQN electrode achieves a high specific discharge capacitance of 289 F g−1 at a current density of 1 A g−1. In addition, it maintains a capacitance of 149 F g−1 even at a high current density of 10 A g−1, demonstrating excellent rate performance. Notably, when the current density is returned to 1 A g−1 after 60 cycles, the initial capacitance is preserved (Fig. 3(b)). At lower current densities, the internal resistance decreases, allowing for a greater utilization of active sites, which enhances Na+ capture. These observations can be attributed to the fully ladder-type and centrosymmetric structure of the PTQN molecule, which exhibits low spatial hindrance, favorable electronic conductivity, and significant electron delocalization. As illustrated in Fig. 3(c), the PTQN electrode demonstrates remarkable cycling stability. At a current density of 2 A g−1, it achieves a notable capacitance of 238.31 F g−1, which is maintained at 238.26 F g−1 after 5000 cycles. The coulombic efficiency remains stable at approximately 100%, with a cycle retention rate of around 99.98%. The GCD curves before and after cycling exhibit nearly identical shapes, and the corresponding capacitance shows minimal decrease, indicating stable Na+ electrosorption performance (Fig. 3(d)). The exceptional electrochemical cycling stability of the PTQN electrode is attributed to its fully ladder-type double-chain configuration, which provides a rigid backbone that ensures inherent stability and minimal solubility in the electrolyte. Consequently, the PTQN electrode remains entirely insoluble in aqueous electrolytes, suggesting its potential for advantageous cycling performance. Real-time observations of the dissolution behavior of the PTQN electrode during charge–discharge cycles were conducted using in situ UV spectroscopy. Throughout the cycling process, no significant changes in the electrochemical signals were detected, and the NaCl electrolyte remained transparent (Fig. S11, ESI†), further confirming the absence of dissolution and the stable Na+ electrosorption performance of the PTQN electrode. In comparison to other reported organic electrodes, the cycling performance of the PTQN electrode exhibits significant advantages in both capacitance retention and lifespan. It maintains an impressive capacitance retention of approximately 99.98% over 5000 cycles, while most reported organic electrodes demonstrate capacitance retention rates below 85% and typically do not achieve 5000 cycles (Fig. 3(e) and Table S2, ESI†).29–36
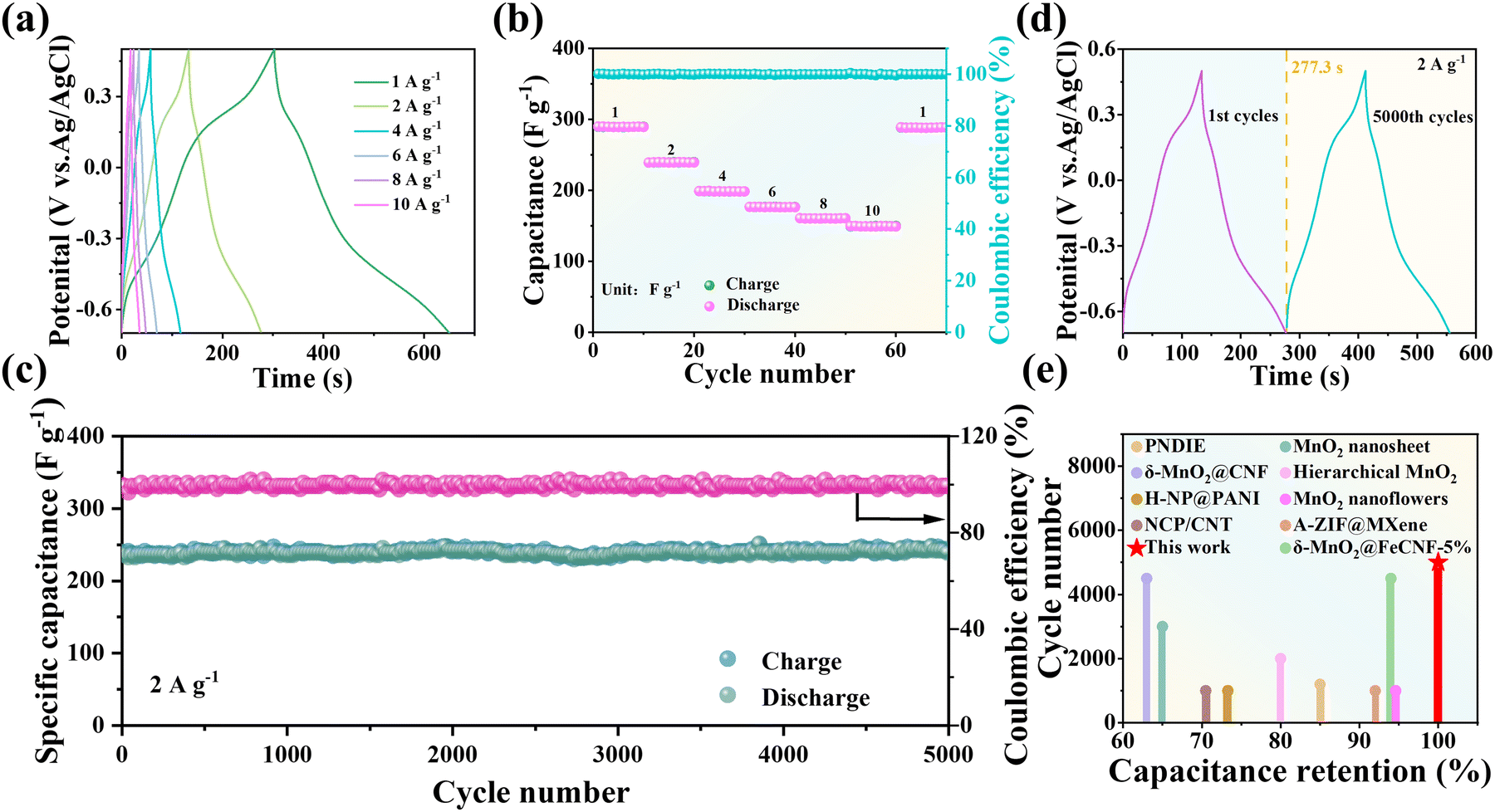 | ||
| Fig. 3 (a) GCD curves of the PTQN electrode measured across current densities ranging from 1 to 10 A g−1. (b) Specific capacitances of the PTQN electrode at varying current densities, illustrating the electrochemical performance. (c) Long-term cycling stability of the PTQN electrode assessed at a constant current density of 2 A g−1. (d) GCD curves of the PTQN electrode before and after cycling, highlighting any changes in electrochemical behavior. (e) Comparative cycling performance of the PTQN electrode alongside other reported organic electrodes, showcasing its relative stability and capacitance retention. | ||
To elucidate the Na+ electrosorption mechanism in the PTQN organic electrode, several critical potentials during the charge–discharge cycle were identified, and a series of ex situ X-ray photoelectron spectroscopy (XPS) analyses were conducted. This approach aimed to enhance the understanding of the structural changes occurring in the PTQN organic electrode while submerged in NaCl aqueous solution throughout the charging and discharging phases (Fig. 4(a)). The N 1s spectrum indicates that during discharge, the characteristic peak corresponding to the CN bond at 398.14 eV gradually diminishes, while the peak associated with the C–N bond at 399.70 eV steadily increases. Simultaneously, a peak in the Na 1s spectrum (Fig. 4(b)) emerges and becomes more pronounced, signifying the incorporation of Na+ ions into the PTQN structure, which transforms CN bonds into C–N–Na bonds. During the charging phase, the characteristic peak for the C–N bond decreases, while the peak for the CN bond rises and ultimately returns to its original state, coinciding with the disappearance of the peak in the Na 1s spectrum. These observations suggest that the CN bonds function as redox-active centers with a strong affinity for Na+ ions within the PTQN electrode. The mechanism of the PTQN electrode in the seawater desalination process was further elucidated through density functional theory (DFT) simulations (Fig. 4(c)). The molecular electrostatic potential (MESP) diagram (shown in the inset of Fig. 4(c)) reveals that the electronegativities surrounding the eight imine groups in the PTQN molecule are significantly higher than those in other regions, indicating that these sites act as redox-active centers with a strong propensity to coordinate with Na+. Detailed calculations classified the active sites in the PTQN molecule into two specific regions, designated as Site A and Site B, each containing four centrosymmetric active sites (Fig. 4(d)). This process is divided into three steps: PTQN, PTQN + 4Na, and PTQN + 8Na, which are utilized to evaluate the pathway energy and illustrate the structural evolution. The initial total energy of the newly prepared PTQN electrode was determined to be −4.5323 × 104 eV. During the discharge process, Na+ ions initially interacted with PTQN to form PTQN + 4Na (−6.2996 × 104 eV), which contains four CN bonds at Site A. Subsequently, Na+ ions further interacted to form PTQN + 8Na (−8.0660 × 104 eV), which contains four CN bonds at Site B.
 | ||
| Fig. 4 Ex situ XPS spectra of (a) N 1s and (b) Na 1s collected at selected potentials, revealing the chemical states of nitrogen and sodium within the PTQN structure (c) Calculated total energies of sodium-intercalated PTQN structures, indicating stability variations with sodium incorporation. (d) Schematic diagram illustrating the mechanism of the PTQN electrode during the desalination process, depicting key electrochemical reactions and ion transport dynamics. | ||
PTQN material was cultured by HK-2 cells (human renal tubular epithelial cells) for toxicity analysis (Fig. S12, ESI†), and the results showed no significant toxicity. Therefore, a HCDI system (Fig. 5(a)) developed in this study uses the prepared safe and non-toxic PTQN electrode for desalination experiments. The HCDI system was configured with an activated carbon (AC) electrode (Fig. S13–S15, ESI†) functioning as the cathode for capturing Cl− ions, while the PTQN electrode (Fig. S16 and S17, ESI†) served as the anode for capturing Na+ ions (PTQN||AC), thereby facilitating an efficient desalination process.37 To assess the desalination efficiency of the PTQN electrode, experiments were conducted within the HCDI system at varying voltages using a solution with an initial concentration of 500 mg L−1 NaCl. Fig. 5(b) illustrates the concentration curves over time at different voltage levels. Upon application of voltage to the HCDI device, Na+ ions in the solution are drawn toward the electrode surface due to electrostatic forces, resulting in a significant reduction in solution concentration until it stabilizes at equilibrium. When a reverse voltage is subsequently applied to the electrode, Na+ ions are released, and the concentration rapidly returns to its original level, demonstrating the excellent regeneration capability of the PTQN electrode. The transient current curves (Fig. S18 and S19, ESI†) reveal real-time variations in current associated with the continuous release and adsorption of Na+ and Cl− ions influenced by the electric field. At various applied voltages, an initial high current is observed, which quickly diminishes and stabilizes. This trend indicates that as the adsorption process progresses, the charged ions adsorbed in the HCDI system reach equilibrium in a short time frame, underscoring the rapid desalination capability of the electrodes. Future research efforts could focus on optimizing the voltage range and exploring the potential applications of PTQN electrodes in practical desalination scenarios. In addition, we tested the desalination capacity of PTQN-based HCDI system at different concentrations. As the salt concentration increases from 300 mg L−1 to 900 mg L−1, the desalination capacity of the PTQN-based HCDI unit rises from 31.58 mg g−1 to 99.87 mg g−1 (Fig. S20, ESI†), demonstrating its potential application over a wide range of salt concentrations.
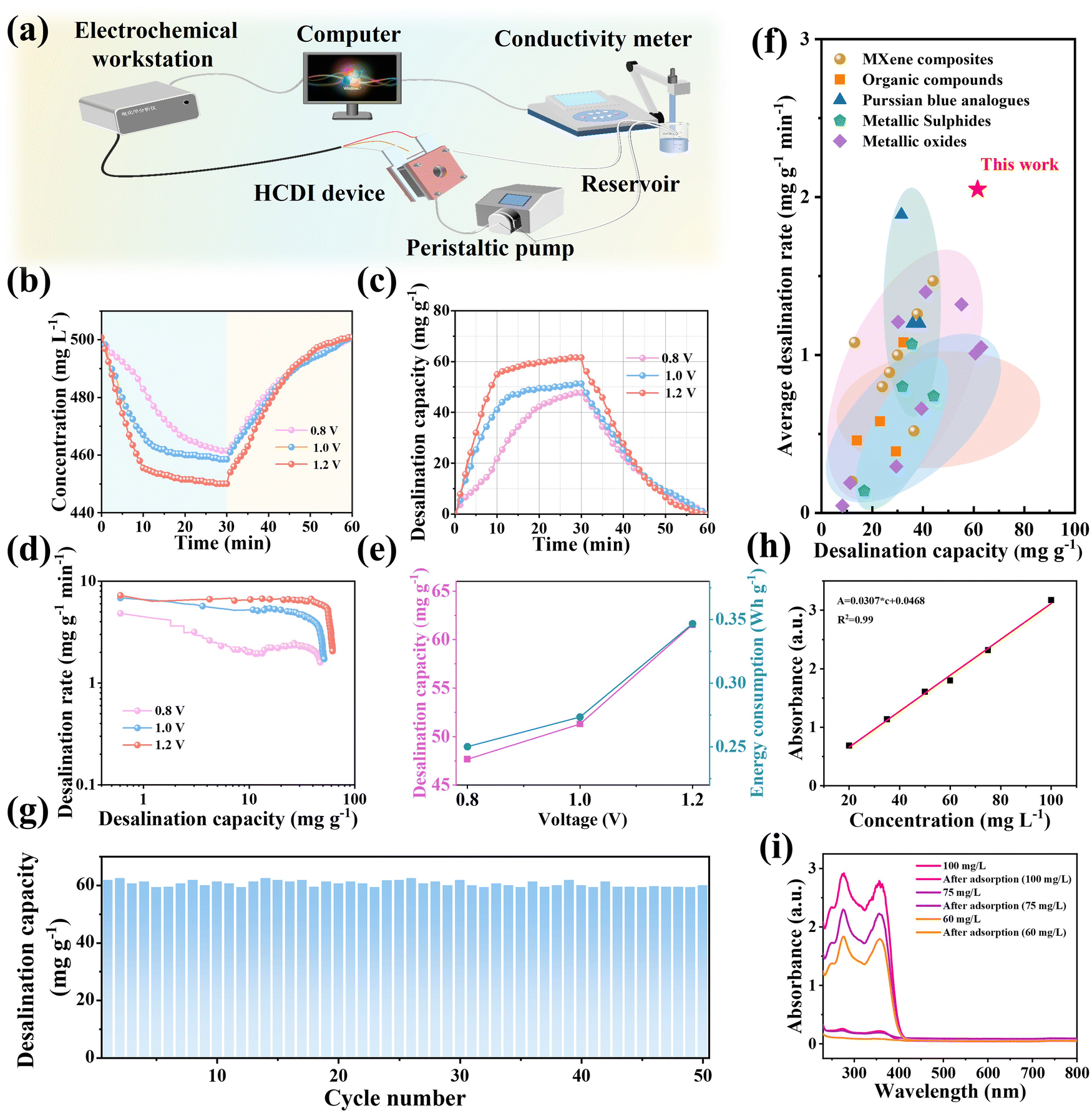 | ||
| Fig. 5 (a) Schematic representation of the desalination system, illustrating the configuration and operational principles. (b) Temporal changes in NaCl concentration during the desalination process. (c) Desalination capacity as a function of applied voltage. (d) Desalination rate at varying imposed voltages, demonstrating efficiency. (e) Desalination capacity and energy consumption at different imposed voltages. (f) Comparison of the desalination performance of the PTQN system with previously reported HCDI systems. (g) Long-term cycling performance of the desalination system in a 500 mg L−1 NaCl solution, indicating stability over time (h) Standard curve of tetracycline measured at a wavelength of 357 nm, used for quantitative analysis. (i) UV-Vis absorption spectra of tetracycline solutions at different concentrations, highlighting changes before and after adsorption. | ||
Fig. 5(c) shows the desalination capacity of the HCDI system operating under constant voltage conditions. The desalination performance of the PTQN||AC configuration significantly improves with increasing working voltage, achieving an impressive desalination capacity of 61.55 mg g−1 at a voltage of 1.2 V. The relationship between desalination rate and desalination capacity is further depicted in the Ragone plot (Fig. 5(d)). As shown, higher working voltages shift the data toward the upper right region of the plot, indicating that PTQN||AC achieves the highest desalination rate of 7.24 mg g−1 min−1 and the aforementioned capacity at an operating voltage of 1.2 V. The increase in applied voltage correlates with enhancements in both desalination capacity and rate, attributed to improved ion migration and adsorption facilitated by greater electrical driving forces that enhance pseudo-capacitance effects. Within the voltage range of 0.8 to 1.0 V, the desalination performance improves by 10.26 mg g−1, while energy consumption increases only marginally by 0.347 W h g−1 (Fig. 5(e)). Thus, taking into account both desalination capacity and energy consumption, the optimal applied voltage for the modular HCDI system is determined to be 1.2 V. We measured the pH value of the PTQN//AC system during the 1.2 V desalination process in a 500 mg L−1 NaCl solution. The pH exhibited fluctuations corresponding to the adsorption and desorption processes (Fig. S21, ESI†). Simultaneously, the concentrations of Na+ and Cl− in the solution varied in tandem with the changes in electrical conductivity observed during adsorption and desorption cycles (Fig. S22). Furthermore, kinetic models were developed based on experimental data concerning Na+ adsorption changes over desalination time. The rate-limiting step was characterized using time-dependent pseudo-first-order and pseudo-second-order kinetic analyses (Fig. S23, ESI†). To further evaluate the desalination performance of the HCDI system, Fig. 5(f) illustrates the correlation between desalination capacity and average desalination rate. Under comparable desalination conditions, the system's capability surpasses that of most previously reported HCDI systems utilizing various pseudocapacitive electrodes, including metal oxides, sulfides, Prussian blue analogs, and MXene composites. The average desalination rate of the developed HCDI system is measured at 2.05 mg g−1 min−1, exceeding that of many state-of-the-art HCDI systems, which typically report average rates below 2 mg g−1 min−1 (Table S3, ESI†).35,38–70
Long-term cycle performance, commonly referred to as regeneration stability, is a critical factor for evaluating the practical applicability of desalination systems. This performance is assessed through 50 cycles of continuous adsorption–desorption experiments conducted under a constant pressure of 1.2 V, specifically designed for optimal desalination of brackish water and seawater. As illustrated in Fig. 5(g) and Fig. S24 (ESI†), the conductivity of the solution exhibits a consistent pattern of decrease and increase, while the desalination capacity of the developed HCDI system fluctuates within a defined range. Remarkably, after 50 cycles, the system retains 97.04% of its original desalination capacity, there is no significant change in the UV of the solution before and after the desalination cycle (Fig. S25, ESI†) and FT-IR spectrum of the PTQN organic electrode before and after cycles did not show any change (Fig. S26, ESI†), demonstrating the exceptional recycling stability of the HCDI system.
Moreover, the system not only exhibits outstanding cyclic performance but also achieves an adsorption capacity of 61.55 mg g−1, surpassing the capacities reported for current organic-based HCDI systems. In addition, the study investigated the efficacy of the PTQN organic compound in removing tetracycline from water. Tetracycline possesses two chromophores, featuring conjugated benzene, enol, and ketone groups, which result in significant absorption peaks in the UV spectrum at approximately 275 nm and 357 nm. Due to the strong solvent interference and baseline issues at 275 nm, the absorbance at 357 nm was selected for quantitative analysis of tetracycline to establish the relationship between concentration and absorbance (Fig. 5(h)). The optical characteristics of tetracycline solutions at varying concentrations (60 mg L−1, 75 mg L−1, and 100 mg L−1) were examined using UV-visible spectroscopy following a 30-min adsorption period at 1.2 V within the HCDI system. The resulting absorption spectra for both original and treated tetracycline solutions are displayed in Fig. 5(i). In all treated solutions, the spectra exhibited a significant decrease in absorbance across the 250 to 800 nm range, nearing zero at lower concentrations. These findings indicate that the PTQN organic compound effectively treats wastewater contaminated with organic dyes, demonstrating its potential application in water purification processes.
3. Conclusions
A novel ladder-type conjugated organic molecule, PTQN, has been synthesized through a straightforward one-step dehydration condensation process. This molecule exhibits characteristics of centrosymmetry and electronic delocalization, with eight fully symmetrical CN active sites that facilitate uniform electron and Na+ transport. Electrochemical performance analysis in NaCl aqueous solution demonstrates that PTQN possesses high specific capacity and cycling stability, attributed to its robust structure and pseudocapacitive Na+ adsorption behavior. The reversible electrochemical adsorption of up to eight Na+ ions is achieved via a two-step redox reaction. Incorporating the PTQN electrode into a HCDI system results in a competitive desalination capacity of 61.55 mg g−1 over 50 cycles. Furthermore, the potential application of PTQN in the removal of tetracycline dye was investigated. The efficient synthesis and application of the PTQN electrode provide valuable insights for addressing critical environmental challenges, including seawater desalination and dye removal. Based on the application potential of PTQN electrodes in HCDI, the future directions and challenges mainly focus on the following aspects: (1) scaled-up synthesis of ladder-type PTQN electrode materials or the design of more organic electrode materials of a similar type; (2) enhancing the reuse or effective degradation of electrode materials to contribute to sustainability; (3) combining in situ characterization with theoretical calculations to analyze the multifaceted synergistic dynamic electrochemical mechanisms of electrode materials; (4) scaling up organic electrode-based HCDI systems and exploring more functional and practical applications.
Author contributions
Y. Cui, J. Yang, M. Shi, and E. H. Ang developed the conceptual framework and designed the experiments. Y. Cui and Y. Tao were responsible for material fabrication. Y. Cui also performed data measurement and analysis, while H. Wang created the figures. P. Zhang carried out CDI measurements, G. Li handled data analysis. The manuscript was prepared by Y. Cui, J. Yang, M. Shi, and E. H. Ang. Funding support was provided by M. Shi and E. H. Ang. All authors contributed to discussions and provided feedback on the manuscript.Data availability
All relevant data are within the manuscript and the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We greatly acknowledge the funding for this project through National Natural Science Foundation of China (no. 52002157 and 22279166), China Postdoctoral Science Foundation (2023M741471 and 2022M711686), the Postgraduate Research & Practice Innovation Program of Jiangsu Province (SJCX24_2512), Ministry of Education, Singapore, under its Academic Research Fund Tier 1 (RG10/22 and RG88/23), and National Institute of Education, Singapore, under its Academic Research Fund (RI 1/21 EAH and RI 3/23 EAH). We extend our gratitude to Yu Cai from Zhejiang Provincial People's Hospital for conducting the cytotoxicity tests on PTQN.Notes and references
- T. Ying, Y. Xiong, H. Peng, R. Yang, L. Mei, Z. Zhang, W. Zheng, R. Yan, Y. Zhang, H. Hu, C. Ma, Y. Chen, X. Xu, J. Yang, D. Voiry, C. Y. Tang, J. Fan and Z. Zeng, Adv. Mater., 2024, 2403385 CrossRef CAS PubMed.
- A. A. Obisanya, L. Ma, J. Liu, T. Yang, Z. Ren, X. Tan, F. Gao and J. Wang, Adv. Funct. Mater., 2024, 2404591 CrossRef CAS.
- Z. Q. Yang, P. Y. Yang, X. C. Zhang, H. Y. Yin, F. Yu and J. Ma, Chem. Mater., 2023, 35, 2069–2077 CrossRef CAS.
- S. D. Datar, R. Mane and N. Jha, Water Environ. Res., 2022, 94, e10696 CrossRef PubMed.
- B. Zhang, Q. Yi, W. Qu, K. Zhang, Q. Lu, T. Yan and D. Zhang, Adv. Funct. Mater., 2024, 2401332 CrossRef CAS.
- F. Yu, L. Wang, Y. Wang, X. Shen, Y. Cheng and J. Ma, J. Mater. Chem. A, 2019, 7, 15999–16027 RSC.
- M. Liang, X. Bai, F. Yu and J. Ma, Nano Res., 2021, 14, 684–691 CrossRef CAS.
- J. J. Lei, F. Yu, H. J. Xie and J. Ma, Chem. Sci., 2023, 14, 3610–3621 RSC.
- F. Zhou, T. Gao, M. Luo and H. Li, Chem. Eng. J., 2018, 343, 8–15 CrossRef CAS.
- W. Shi, X. Liu, T. Deng, S. Huang, M. Ding, X. Miao, C. Zhu, Y. Zhu, W. Liu, F. Wu, C. Gao, S. W. Yang, H. Y. Yang, J. Shen and X. Cao, Adv. Mater., 2020, 32, 1907404 CrossRef CAS PubMed.
- J. S. Kang, S. Kim, J. Kang, H. Joo, J. Jang, K. Jo, S. Park, H. I. Kim, S. J. Yoo, J. Yoon, Y. E. Sung and T. A. Hatton, Environ. Sci. Technol., 2022, 56, 12602–12612 CrossRef CAS PubMed.
- L. Yu, N. Liu, B. Liu, F. Yu and J. Ma, J. Alloys Compd., 2023, 965, 171501 CrossRef CAS.
- J. Huang, X. Dong, Z. Guo and Y. Wang, Angew. Chem., 2020, 132, 18478–18489 CrossRef.
- X. Chen, X. Feng, B. Ren, L. Jiang, H. Shu, X. Yang, Z. Chen, X. Sun, E. Liu and P. Gao, Nano-Micro Lett., 2021, 13, 1–16 CrossRef CAS PubMed.
- M. Li, R. P. Hicks, Z. Chen, C. Luo, J. Guo, C. Wang and Y. Xu, Chem. Rev., 2023, 123, 1712–1773 CrossRef CAS PubMed.
- R. Wang, J. He, C. Yan, R. Jing, Y. Zhao, J. Yang, M. Shi and X. Yan, Adv. Mater., 2024, 36, 2402681 CrossRef CAS PubMed.
- X. Feng, X. Wu, X. Chen, J. Yuan, S. Lv, B. Ren, X. Sun, E. Liu, S. Tan and P. Gao, Energy Storage Mater., 2021, 42, 454–463 CrossRef.
- T. Sun, C. Liu, X. Xu, Q. Nian, S. Zheng, X. Hou, J. Liang and Z. Tao, J. Mater. Chem. A, 2020, 8, 21983–21987 RSC.
- R. Wang, M. Shi, L. Li, Y. Zhao, L. Zhao and C. Yan, Chem. Eng. J., 2023, 451, 138652 CrossRef CAS.
- W. Bao, X. Tang, X. Guo, S. Choi, C. Wang, Y. Gogotsi and G. Wang, Joule, 2018, 2, 778–787 CrossRef CAS.
- L. Guo, X. Wang, Z. Y. Leong, R. Mo, L. Sun and H. Y. Yang, Flatchem, 2018, 8, 17–24 CrossRef CAS.
- L. Guo, R. W. Mo, W. H. Shi, Y. X. Huang, Z. Y. Leong, M. Ding, F. M. Chen and H. Y. Yang, Nanoscale, 2017, 9, 13305–13312 RSC.
- H. Kong, M. Yang, Y. Miao and X. Zhao, Energy Technol., 2019, 7, 1900835 CrossRef CAS.
- M. Shi, R. Wang, L. Li, N. Chen, P. Xiao, C. Yan and X. Yan, Adv. Funct. Mater., 2023, 33, 2209777 CrossRef CAS.
- M. Zhu, L. Zhao, Q. Ran, Y. Zhang, R. Peng, G. Lu, X. Jia, D. Chao and C. Wang, Adv. Sci., 2022, 9, 2103896 CrossRef CAS PubMed.
- T. Sun, W. Zhang, Q. Nian and Z. Tao, Chem. Eng. J., 2023, 452, 139324 CrossRef CAS.
- Y. Dai, X. Yan, J. Zhang, C. Wu, Q. Guo, J. Luo, M. Hu and J. Yang, Electrochim. Acta, 2023, 442, 141870 CrossRef CAS.
- H. Peng, J. Xiao, Z. Wu, L. Zhang, Y. Geng, W. Xin, J. Li, Z. Yan, K. Zhang and Z. Zhu, CCS Chem., 2023, 5, 1789–1801 CrossRef CAS.
- H. Tao, Y. Cheng, L. Du and X. Chen, Nanosci. Nanotechnol. Lett., 2013, 5, 1070–1075 CrossRef CAS.
- X. Bai, X. Tong, Y. Gao, W. Zhu, C. Fu, J. Ma, T. Tan, C. Wang, Y. Luo and H. Sun, Electrochim. Acta, 2018, 281, 525–533 CrossRef CAS.
- S. Zhao, T. Liu, D. Hou, W. Zeng, B. Miao, S. Hussain, X. Peng and M. S. Javed, Appl. Surf. Sci., 2015, 356, 259–265 CrossRef CAS.
- J. Li, B. Hu, P. Nie, X. Shang, W. Jiang, K. Xu, J. Yang and J. Liu, Appl. Surf. Sci., 2021, 542, 148715 CrossRef CAS.
- A. Fombona-Pascual, N. Patil, E. Garcia-Quismondo, N. Goujon, D. Mecerreyes, R. Marcilla, J. Palma and J. J. Lado, Chem. Eng. J., 2023, 461, 142001 CrossRef CAS.
- A. M. Aghdam, N. M. Chahartagh and E. Delfani, Adv. Mater. Technol., 2023, 8, 2300628 CrossRef.
- J. Guo, Y. Wang, H. Zhang, Y. Cai and R. Fang, Desalination, 2023, 548, 116305 CrossRef CAS.
- J. Guo, Y. Wang, Y. Cai, H. Zhang, Y. Li and D. Liu, Desalination, 2022, 528, 115622 CrossRef CAS.
- S. A. Hawks, A. Ramachandran, S. Porada, P. G. Campbell, M. E. Suss, P. M. Biesheuvel, J. G. Santiago and M. Stadermann, Water Res., 2019, 152, 126–137 CrossRef CAS PubMed.
- M. Torkamanzadeh, L. Wang, Y. Zhang, O. Budak, P. Srimuk and V. Presser, ACS Appl. Mater. Interfaces, 2020, 12, 26013–26025 CrossRef CAS PubMed.
- P. Srimuk, F. Kaasik, B. Kruener, A. Tolosa, S. Fleischmann, N. Jaeckel, M. C. Tekeli, M. Aslan, M. E. Suss and V. Presser, J. Mater. Chem. A, 2016, 4, 18265–18271 RSC.
- Q. Li, X. Xu, J. Guo, J. P. Hill, H. Xu, L. Xiang, C. Li, Y. Yamauchi and Y. Mai, Angew. Chem., 2021, 133, 26732–26738 CrossRef.
- Z. Chen, X. Xu, Y. Liu, J. Li, K. Wang, Z. Ding, F. Meng, T. Lu and L. Pan, Desalination, 2022, 528, 115616 CrossRef CAS.
- L. Guo, X. Wang, Z. Y. Leong, R. Mo, L. Sun and H. Y. Yang, Flatchem, 2018, 8, 17–24 CrossRef CAS.
- Z. Chen, Z. Ding, Y. Chen, X. Xu, Y. Liu, T. Lu and L. Pan, Chem. Eng. J., 2023, 452, 139451 CrossRef CAS.
- Z. Yang, P. Yang, X. Zhang, H. Yin, F. Yu and J. Ma, Chem. Mater., 2023, 35, 2069–2077 CrossRef CAS.
- K. Wang, L. Chen, G. Zhu, X. Xu, L. Wan, T. Lu and L. Pan, Desalination, 2022, 522, 115420 CrossRef CAS.
- N. Liu, L. Yu, B. Liu, F. Yu, L. Li, Y. Xiao, J. Yang and J. Ma, Adv. Sci., 2023, 10, 2204041 CrossRef CAS PubMed.
- Y. Li, Z. Ding, J. Li, K. Wang, T. Lu and L. Pan, Desalination, 2020, 481, 114379 CrossRef CAS.
- J. Chen, K. Zuo, B. Li, D. Xia, L. Lin, J. Liang and X. Li, Sep. Purif. Technol., 2023, 304, 122381 CrossRef CAS.
- D. Liu, X. Ning, Y. Hong, Y. Li, Q. Bian and J. Zhang, Electrochim. Acta, 2019, 296, 327–334 CrossRef CAS.
- H. Liu, J. Zhang, X. Xu and Q. Wang, Chem. – Eur. J., 2020, 26, 4403–4409 CrossRef CAS PubMed.
- B. W. Byles, D. A. Cullen, K. L. More and E. Pomerantseva, Nano Energy, 2018, 44, 476–488 CrossRef CAS.
- Y. Cai, Y. Wang, R. Fang and J. Wang, Sep. Purif. Technol., 2022, 280, 119828 CrossRef CAS.
- Y. Cai, W. Zhang, J. Zhao and Y. Wang, Appl. Surf. Sci., 2023, 622, 156926 CrossRef CAS.
- J. Guo, Y. Wang, Y. Cai, H. Zhang, Y. Li and D. Liu, Desalination, 2022, 528, 115622 CrossRef CAS.
- W. Cai, Z. Xiong, T. Hussain, J. Yang, Y. Wang and J. Liu, J. Electrochem. Soc., 2016, 163, A2515–A2523 CrossRef CAS.
- X. Ma, Y. A. Chen, K. Zhou, P. C. Wu and C. H. Hou, Electrochim. Acta, 2019, 295, 769–777 CrossRef CAS.
- M. A. Jaoude, E. Alhseinat, K. Polychronopoulou, G. Bharath, I. F. F. Darawsheh, S. Anwer, M. A. Baker, S. J. Hinder and F. Banat, Electrochim. Acta, 2020, 330, 135202 CrossRef CAS.
- S. Kim, J. Lee, C. Kim and J. Yoon, Electrochim. Acta, 2016, 203, 265–271 CrossRef CAS.
- W. Si and H. Li, Adv. Mater. Interfaces, 2021, 8, 2100125 CrossRef CAS.
- Z. Chen, X. Xu, K. Wang, F. Meng, T. Lu and L. Pan, Desalination, 2023, 564, 116733 CrossRef CAS.
- B. Liu, L. Yu, F. Yu and J. Ma, Desalination, 2021, 500, 114897 CrossRef CAS.
- Z. Liu, W. Ma and H. Li, Nanoscale, 2020, 12, 7586–7594 RSC.
- Z. Liu and H. Li, Energy Environ. Mater., 2023, 6, e12255 CrossRef CAS.
- Y. Liu, X. Gao, L. Zhang, X. Shen, X. Du, X. Dou and X. Yuan, Desalination, 2020, 494, 114665 CrossRef CAS.
- Z. Liu, W. Ma and H. Li, Nanoscale, 2020, 12, 7586–7594 RSC.
- W. Peng, W. Wang, G. Han, Y. Huang and Y. Zhang, Desalination, 2020, 473, 114191 CrossRef CAS.
- X. Wen, M. Zhao, Z. Zhao, X. Ma and M. Ye, ACS Sustainable Chem. Eng., 2020, 8, 7335–7342 CrossRef CAS.
- Y. Cai, Y. Wang, L. Zhang, R. Fang and J. Wang, ACS Appl. Mater. Interfaces, 2022, 14, 2833–2847 CrossRef CAS PubMed.
- Z. Zhao, J. Zhao, Y. Sun, M. Ye and X. Wen, Chem. Eng. J., 2022, 429, 132582 CrossRef CAS.
- M. Gao, W. Liang, Z. Yang, T. Ao and W. Chen, J. Colloid Interface Sci., 2023, 650, 1244–1252 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4mh01342e |
| This journal is © The Royal Society of Chemistry 2025 |