
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceApplication of a bivalent “click” approach to target tyrosyl-DNA phosphodiesterase 1 (TDP1)†
Xue Zhi
Zhao
 *a,
Wenjie
Wang
b,
Md Rasel Al
Mahmud
b,
Keli
Agama
b,
Yves
Pommier
b and
Terrence R.
Burke
Jr.
a
*a,
Wenjie
Wang
b,
Md Rasel Al
Mahmud
b,
Keli
Agama
b,
Yves
Pommier
b and
Terrence R.
Burke
Jr.
a
aChemical Biology Laboratory, Center for Cancer Research, National Cancer Institute, Frederick, MD, USA. E-mail: xuezhi.zhao@nih.gov
bDevelopmental Therapeutics Branch & Laboratory of Molecular Pharmacology, Center for Cancer Research, National Cancer Institute, Bethesda, MD, USA
First published on 21st February 2025
Abstract
Although inhibiting the DNA repair enzyme tyrosyl-DNA phosphodiesterase 1 (TDP1) synergizes with topoisomerase type I (TOP1) inhibitors in anticancer therapy, development of TDP1 inhibitors has been highly challenging. This may be due to the open and shallow nature of the TDP1 catalytic site and the necessity of competing with a large and highly extended substrate. The toolbox available to chemical biologists for studying TDP1 could be significantly enhanced by introducing the ability to selectively eliminate TDP1 using protein degraders. Our current work starts from phenyl imidazopyridine-based TDP1 inhibitors previously developed from small molecule microarrays (SMMs). Using crystal structures of lead inhibitors bound to TDP1, we designed and synthesized a series of bivalent proteolysis-targeting chimeras (PROTACs). The focus of our current work is to explore synthetic approaches that permit installation of E3 ligase-targeting functionality, while retaining the TDP1 binding. We employed copper-catalyzed azide–alkyne cycloaddition (CuAAC) “click” reactions to assemble PROTAC constituents with 1,2,3-triazole-containing linkers. With the addition of the relatively large parts of the linkers and E3-targeting moieties, we retained the ability to inhibit TDP1. The successful development of TDP1-directed PROTACS would yield a new therapeutic class that could potentially enhance the efficacy and selectivity of TOP1 inhibitors including those used as payloads in antibody drug conjugates (ADCs).
Introduction
DNA repair enzymes are targets for anticancer therapy. DNA supercoiling can occur during replication and transcription and numerous other important cellular processes.1 Enzymes, such as topoisomerases, regulate the degree of DNA supercoiling. The human topoisomerase type I (TOP1) supresses supercoiling by relaxing the DNA helix.2 TOP1 removes supercoils by forming single-strand DNA breaks in a reversible process, which involves the nucleophilic attack of the TOP1 catalytic tyrosyl Y723 hydroxyl onto the phospho-backbone of a target DNA chain to form a 3′-phosphodiester bond with release of the 5′-terminal chain. The resulting covalent TOP1–DNA cleavage complex (TOP1cc) permits the freed 5′-DNA end to undergo rotation and relax supercoils. Once supercoiling has been removed, the cut DNA 5′-strand can attack the 3′-end phosphotyrosyl bond to regenerate the DNA backbone and release TOP1.3 This process is the target of anticancer drugs, such as the TOP1 inhibitors irinotecan and topotecan, and their derivative exatecan used in antibody drug conjugates (ADCs), as they trap TOP1cc intermediates. Subsequent double-strand breaks kill cancer cells.4 The DNA repair enzyme tyrosyl-DNA phosphodiesterase 1 (TDP1) hydrolyzes phosphodiester bonds between 3′-terminal DNA-phosphates.5,6 Human TDP1 is a 608 residues 68 kDa enzyme that serves important roles in repairing DNA lesions by hydrolyzing phosphodiester bonds between DNA 3′-phosphates and a diversity of adducts.4,7 In cells, TDP1 hydrolyzes phosphodiester bonds of nicked double-stranded DNA (dsDNA) linked to polypeptides that result from proteolytic digestion of TOP1cc complexes.8–10 Cleavage of the phosphodiester bond between the TOP1 Y723 residue and the DNA 3′-phosphate group takes place after proteolytic degradation of the TOP1ccs. By cleaving these phosphodiester bonds and enabling DNA repair, TDP1 limits the effectiveness of TOP1 inhibitors.5,11–13 Genetic inactivation of TDP1 blocks the degradation of therapeutically critical TOP1–DNA adducts and synergizes with TOP1-targeted drugs.14,15 Importantly, loss of TDP1 function is well tolerated in preclinical models.16 Thus, chemical inhibition of TDP1 could potentially retard the degradation of therapeutically critical TOP1ccs induced by TOP1 inhibitors and increase their anticancer activity.7,15,17,18 This has made development of TDP1 inhibitors a compelling line of research. Development of TDP1 inhibitors is made highly challenging by the open and shallow nature of the TDP1 catalytic site.Although TDP1 inhibitors are recognized as valid therapeutic targets for use with TOP1 inhibitors,6 there are no TDP1 inhibitors currently in clinical trials. This may be due to the challenges posed by the open and shallow nature of the TDP1 catalytic site.12,19–24 As a member of the phospholipase D (PLD) superfamily, the TDP1 catalytic apparatus contains conserved groups of histidine, lysine, and asparagine residues in close proximity (HKN motifs, H263/K265/N283 and H493/K495/N516) (Fig. 1C). These form a conserved catalytic pocket, in which a nucleophilic attack is mediated on the substrate 3′-DNA phosphodiester bond.12,25 The nature of the catalytic machinery was revealed by crystal structures of the TDP1 (Δ1–148) C-terminal domain bound to a complex of TOP1-derived peptide, DNA substrate and a vanadate transition state mimetic (PDB code: 1NOP) (Fig. 1B).12,19,25,26 These structures highlight a three-component architecture consisting of a substrate DNA strand (green sticks in Fig. 1B), a tyrosyl-containing peptide (blue sticks in Fig. 1B) and a vanadate moiety in middle that serves to mimic the geometry of the TDP1-bound phosphate transition state complex. The three major binding components of the substrate are defined by (1) the bottom of catalytic cleft formed by the two HKN motifs; (2) an extended positively charged cleft extending from one side of the active site along which lays a single-stranded DNA (green sticks in Fig. 1B) bearing the 3′-phospho adduct and (3) a more open region that accommodates the TOP1-derived peptide (blue sticks in Fig. 1B) that projects from the catalytic pocket in a direction opposite to the DNA-binding channel. Although the phospho-binding pocket is well defined, it is relatively shallow as are the open, extended topologies of the DNA and peptide-binding regions. The large macromolecular nature of the substrate and the shallow catalytic pocket pose challenges to developing potent TDP1 inhibitors.27,28 After significant effort, several groups have reported a spectrum of structurally diverse TDP1 inhibitors. These include natural product-derived inhibitors,29–39 noncompetitive inhibitors,38,40,41 potential allosteric peptide inhibitors,42 and irreversible inhibitors.40,43 The structural bases for how these inhibitors interact with TDP1 are generally unknown and many reported TDP1 inhibitors exhibit physiochemical properties that could potentially endow them with promiscuous mechanisms of inhibition, which would make them of questionable value for further development.44
 | ||
| Fig. 1 Structures of imidazopyridine TDP1 inhibitors and the TDP1 enzyme. (A) Structures of lead imidazopyridine TDP1 inhibitors (1–3). (B) Crystal structure surface rendering of TDP1(Δ1–148) with compound 2 bound in the catalytic pocket (PDB code: 6W7K). A vanadate complex of a TOP1-derived peptide (blue sticks) and DNA substrate (green sticks) (PDB code: 1NOP) are superimposed as mimics of phospho-substrate. The surfaces of N-terminal domain residues (149–350) and C-terminal domain residues (351–608) are shown in grey and brown, respectively. (C) The amino acid sequence of human TDP1 with the N-terminal and C-terminal domain residues 1–350 and 351–608 highlighted in black and tan, respectively. Positions of the catalytic ‘HKN’ motifs are highlighted in red and lysine residues (52 in total) are underlined. | ||
In a qualitative departure from previous efforts, we interrogated the ability of ligands to bind to TDP1 using a small molecule microarray (SMM) of more than 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 ligands. This led to the identification of 2-phenylimidazo[1,2-a]pyridines as TDP1-binding motifs (1a, 1b and 2, Fig. 1A).22 More recently, we applied oxime diversification strategies to a subset of these SMM-derived platforms to develop trivalent ligands (3) intended to engage residues within TDP1's catalytic pocket, while extending into the DNA and peptide-binding channels.24 X-ray crystal structure of inhibitor 2 bound within the TDP1 catalytic pocket shows that key inhibitor carboxylic acid groups form hydrogen bonds with residues of the catalytic HKN motif (Fig. 1B and 2B). The pyridine rings of the imidazopyridine unit point toward the relatively narrow and positively charged DNA–substrate-binding pocket, while the phenyl rings are directed toward the TOP1-peptide binding pocket (Fig. 1B).
000 ligands. This led to the identification of 2-phenylimidazo[1,2-a]pyridines as TDP1-binding motifs (1a, 1b and 2, Fig. 1A).22 More recently, we applied oxime diversification strategies to a subset of these SMM-derived platforms to develop trivalent ligands (3) intended to engage residues within TDP1's catalytic pocket, while extending into the DNA and peptide-binding channels.24 X-ray crystal structure of inhibitor 2 bound within the TDP1 catalytic pocket shows that key inhibitor carboxylic acid groups form hydrogen bonds with residues of the catalytic HKN motif (Fig. 1B and 2B). The pyridine rings of the imidazopyridine unit point toward the relatively narrow and positively charged DNA–substrate-binding pocket, while the phenyl rings are directed toward the TOP1-peptide binding pocket (Fig. 1B).
 | ||
| Fig. 2 PROTAC structural schematic and the binding interactions of an imidazopyridine TDP1 inhibitor. (A) PROTAC induced proximity and preparation using click chemistry. TDP1 inhibitor (grey triangle) linked with recruiter (blue oral) leads to induced proximity between TDP1 and an E3 ligase that leads to enhanced ubiquitination of TDP1. (B) Crystal structure of TDP1-bound imidazopyridine inhibitor 2 (carbon atoms are shown in yellow) binding at the catalytic site. The catalytic residues of the HKN motifs are shown in grey and orange (PDB code: 6W7K). Positions on inhibitor 2 labeled “a” and “b” point toward the solution phase serve as potential sites to introduce linkers attached to E3 recruiting functionality. | ||
Bivalent constructs have been designed to simultaneously bind distinct domains on two different targets
Proteolysis-targeting chimeras (PROTACs) employ bivalent constructs that bring into proximity a target protein and a degradation component. This can lead to the targeted degradation of the protein (Fig. 2A).45 One element of the PROTAC binds specifically to a protein that is the target of degradation, while the other component binds to an E3 ubiquitin ligase or other effector of cellular degradation.46 The induced proximity of target protein to the E3 ligase allows the E3 ligase to ubiquitinate the target protein. Once ubiquitinated, the target protein can be recognized by the proteasome and degraded into smaller peptides. This process effectively reduces the cellular levels of the target protein, representing an alternative to traditional down-regulation of function by small molecule inhibitors. PROTACs are a relatively new class of therapeutic molecules designed to selectively modulate protein function by inducing ubiquitination and degrading cellular target proteins (Fig. 2A).47–49 Protein degraders have potential advantages over other small-molecule inhibitors used for cancer treatment.50 Unlike traditional small molecules that inhibit protein function, PROTACs harness the cellular ubiquitin–proteasome system to tag the target protein for degradation. They can potentially lead to more complete and durable therapeutic effects with higher selectivity as compared to classic enzymic inhibitors. The chemical biology toolbox for studying TDP1 could be significantly enhanced by having ability to selectively reduce the levels of TDP1 using protein degraders. Such agents could potentially synergize with current TOP1 inhibitors and enhance potency and selectivity. The focus of our current work is to explore synthetic approaches to modifying known TDP1 inhibitors in ways that permit installation of E3 ligase-targeting functionality, while maintaining the ability of the ligands to retain the ability to bind to TDP1.Results and discussion
Enzyme degradation represents a new approach to address problems faced in developing TDP1 inhibitors
Members of the PROTAC class of molecular degraders typically have three key components: (A) a ligand that binds to the protein being targeted for degradation; (B) a E3 ligase-binding motif and (C) suitable linker functionality that joins these two components (Fig. 2A). E3 ubiquitin ligases are protein complexes that covalently modify other proteins with ubiquitin. Once tagged with ubiquitin, the target protein is recognized by the proteasome for degradation. Following ubiquitination, the PROTAC molecule can dissociate to target additional proteins. Highly efficient PROTACs could be very powerful tools in chemical biology and represent potential platforms for drug discovery.51The von Hippel–Lindau (VHL) and cereblon (CRBN) E3 ubiquitin ligases are extensively used for PROTAC design.46 VHL-containing E3 ubiquitin ligase complexes target proteins for degradation, particularly under low oxygen conditions. Hydroxyproline-containing small molecules can mimic the hydroxyproline residues found in the cognate VHL-binding domain of hypoxia-inducible factor (HIF-α). Appending these motifs to ligands can facilitate the targeted degradation normally induced by HIF-α. The E3 ubiquitin ligase CRBN plays a critical role in protein degradation by recruiting immunomodulatory drugs (IMiDs), such as thalidomide and its derivatives lenalidomide and pomalidomide. These IMiDs bind to CRBN and promote the degradation of specific substrates.
In the process of ubiquitination, lysine residues on the target protein serve as crucial anchor roles in the formation of the polyubiquitin chains that are essential for marking the protein for degradation. TDP1 contains a total of 52 lysine residues and many of them may potentially be positioned to serve as anchors for ubiquitination (Fig. 1C). Linkers joining the target protein and E3 ligase binding components are key factors in determining their efficacy, specificity, and overall performance of PROTACs.52 The length and flexibility of the linker can significantly influence the binding efficiency and the orientation of the two components. The linkers can provide the necessary conformational freedom for the PROTAC to adopt an optimal shape for engaging both the target protein and the E3 ligase. This is essential for effective ubiquitination and subsequent degradation. Fine-tuning the linker can improve the selectivity for the target protein, reduce off-target effects and enhance overall efficacy. Linkers can also influence the stability and solubility of PROTACs. A balanced linker design helps ensure that the compound remains stable in biological environments and maintains adequate solubility for cellular uptake, which improve the overall drug-like properties of the molecule.53
Design of bivalent TDP1 PROTACs based on insights derived from the X-ray cocrystal structures of TDP1 inhibitors bound at the catalytic site
We previously employed a crystallographic fragment cocktail screen of more than 600 small molecule fragments to discover phthalic acids and quinolones that bind within the TDP1 catalytic hot spot.20 Recently, we also combined small molecule microarray (SMM) and oxime diversification strategies to develop more extended ligands (1–3) intended to engage residues within the catalytic pocket, while extending into the DNA and peptide-binding channels (Fig. 1A).20,22,24 Certain of these constructs showed micromolar inhibitory values in in vitro assays. However, inhibitory efficacies in whole cell systems were less than what would be expected based on the results from the in vitro assays. Crystal structures of the lead compounds, such as phenyl imidazopyridine 2 (Fig. 1A), showed that the phthalic acid component replicate aspects of substrate phosphates by forming several direct hydrogen bonds with the key catalytic residues (Fig. 1B and 2B).22 In further work, we used oxime diversification strategies to develop a series of analogues having Ra or Rb substituents at the pyridine or phenyl rings (3, Fig. 1A).22,24The structural complexity and dynamics of degradation complexes render it challenging to predict which combination of anchor-linker-warhead will lead to optimal degradation. The linkers play particularly crucial roles in the design and function of PROTACs. A diverse range of linkers have been applied to design PROTACs. These include polyethylene glycol (PEG) and alkyl chains, and chains containing glycol, alkyne, triazole, piperazine, and piperidine components.52 Based on the binding mode of our phenyl imidazopyridine-based TDP1 inhibitors as revealed in ligand-bound crystal structures (Fig. 1B and 2B), we designed a series of TDP1 PROTACs 4 and 5 with linkers of varying lengths (Fig. 2A and 3). The TDP1 PROTACs 4 and 5 incorporate VHL recruiters and CRBN recruiters on either side of the phenyl imidazopyridine-based TDP1 catalytic pocket-binding core. Placement of E3-recruiting functionality was designed to situate linkers pointing away from the TDP1 protein and toward the solution phase.
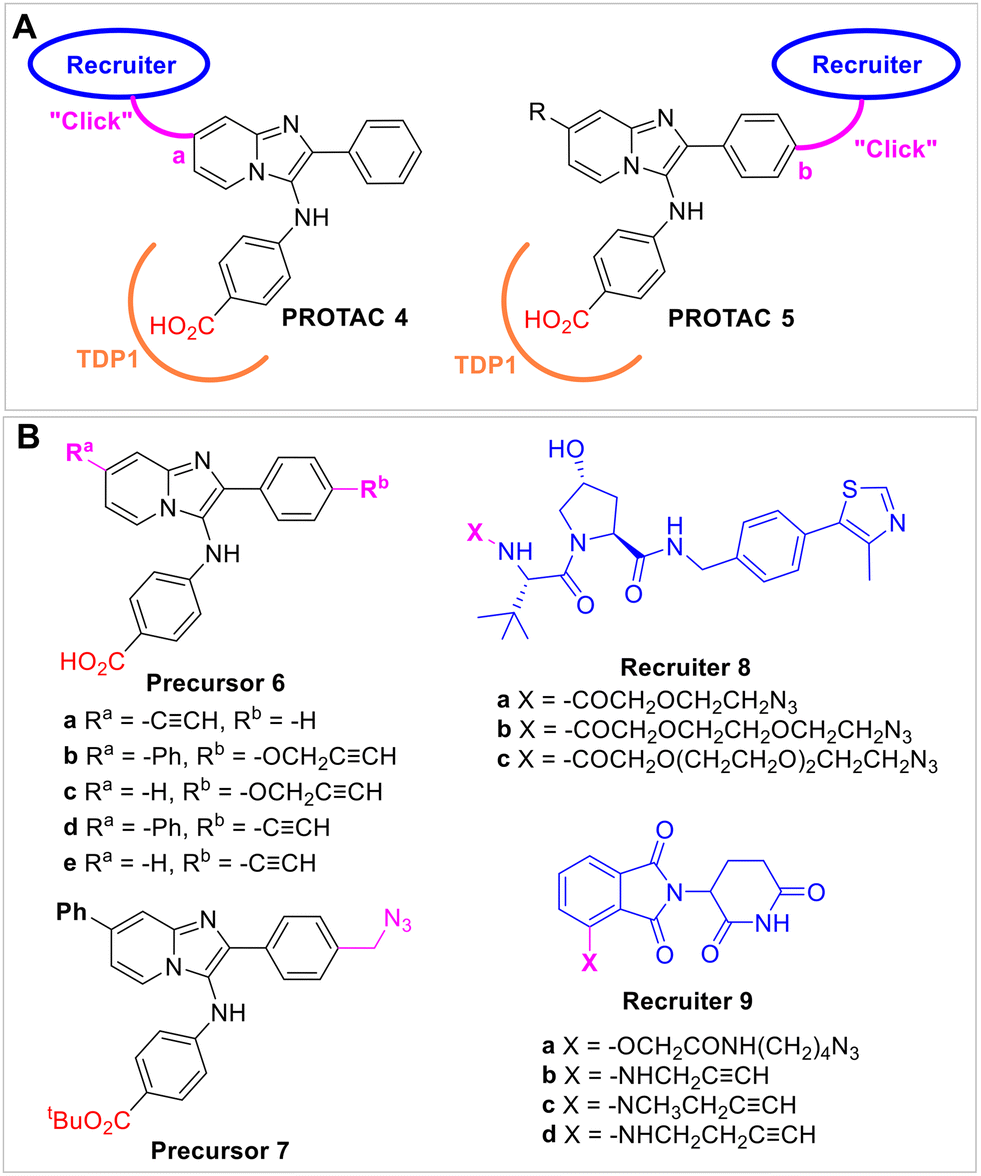 | ||
| Fig. 3 Design of TDP1 PROTACs that employ alkyne and azide precursors. (A) Structures of PROTACs (4 and 5) that append the recruiter linkers at the pyridine ring site “a” and the phenyl ring site “b” positions, respectively. (B) Structures of “click” functionality-bearing intermediates; phenyl imidazopyridine-derived TDP1-binding analogs (6a–6e and 7); and VHL-recruiter hydroxyproline-derived precursors (8a–8c) and CRBN-recruiter thalidomide-derived precursors (9a–9d). | ||
“Click chemistry” is widely used, particularly in expediting drug discovery and optimization processes. It has also been used to conjugate linkers and components in the synthesis of PROTACs.54,55 In our current work, we employed Huisgen copper-catalyzed azide–alkyne cycloaddition (CuAAC) click chemistry to couple azides and terminal alkynes to give 1,2,3-triazoles. The triazole groups in the resulting TDP1 inhibitors were formed from alkyne-labeled phenyl imidazopyridine precursors 6a–6e and azide-labeled phenyl imidazopyridine precursor 7 as well as the azide-labeled VHL-recruiter precursors 8a–8c, the azide-labeled CRBN-recruiter precursor 9a and the alkyne-labeled CRBN-recruiter precursors 9b–9d. We are not aware of prior reports detailing TDP1-targeting PROTACs.
Synthesis
We have previously prepared TDP1 inhibitors that contain triazole moieties as isosteric replacements of our lead oxime-based TDP1 inhibitors.24 The syntheses of these compounds employed key one-pot Groebke–Blackburn–Bienaymé multicomponent reactions (GBBR) together with copper-catalyzed azide–alkyne cycloaddition (CuAAC) click chemistry.24 As shown in Scheme 1, we used this chemistry to prepared alkyne-labeled precursor 6a. Starting from the 4-ethynylpyridin-2-amine 10, benzaldehyde 11 and methyl 4-isocyanobenzoate 12, we prepared ester 13 having alkyne functionality on the pyridine ring. Hydrolysis of 13 using sodium hydroxide in methanol afforded the alkyne-labeled acid precursor 6a. Click coupling of alkyne 6a with commercially available azide-labeled VHL recruiters 8a–8c having different length glycol linkers, yielded final bivalent analogs 4a–4c. Analogue 4d was prepared from alkyne 6a and commercially available alkyne-labeled CRBN-recruiter 9a using the same CuAAC click reaction catalyzed by tris((1-benzyl-4-triazolyl)methyl)amine (TBTA) (Scheme 1).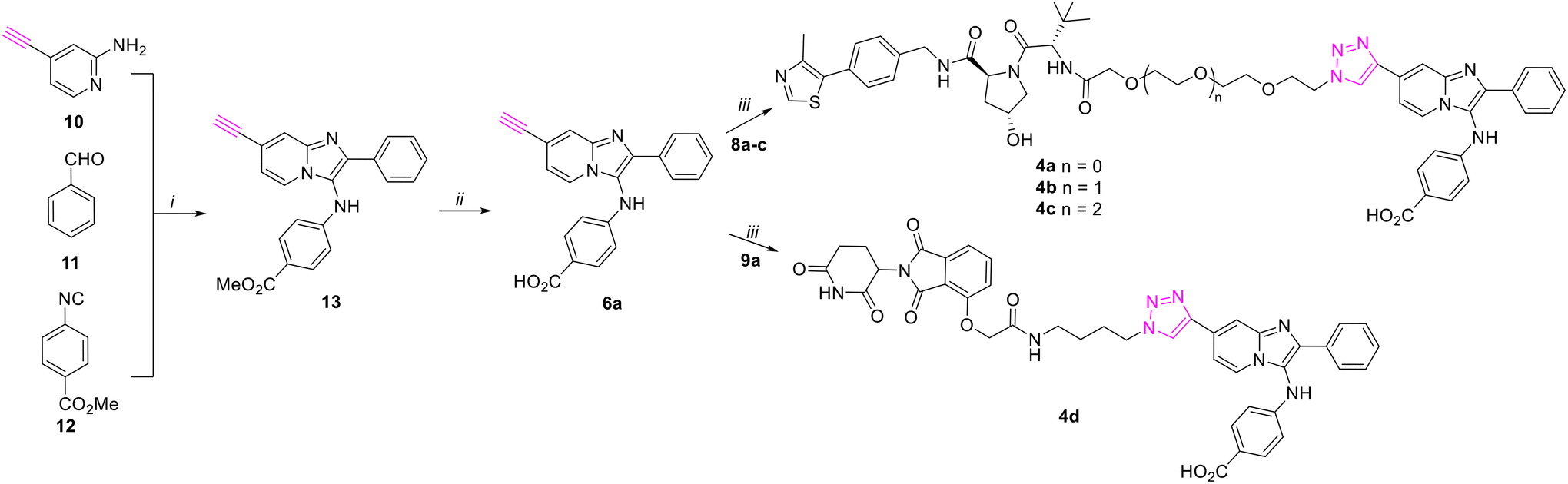 | ||
| Scheme 1 Synthesis of compounds 4a–4d using key GBBR multi-component reaction and CuAAC “click” reaction. Reagents and conditions: (i) HOAc, MeOH; (ii) NaOH, MeOH; (iii) TBTA, CuSO4–5H2O, sodium L-ascorbate, DMSO, H2O, rt. | ||
We have previously reported the preparation of alkyne-labeled phenyl imidazopyridine analogues 6b–6e using GBBR reactions.24 As shown in Scheme 2, the alkyne-labeled phenyl imidazopyridine analogues 6b–6e were easily coupled with the azide-labeled VHL recruiters 8a–8c and the alkyne-labeled CRBN-recruiter 9a to afford bivalent analogs 5a–5g.
 | ||
| Scheme 2 Synthesis of compounds 5a–5g using CuAAC click reaction. Reagents and conditions: (i) TBTA, CuSO4–5H2O, sodium L-ascorbate, DMSO, H2O, rt. | ||
Methyl ester hydrolysis to the corresponding carboxylic acids using sodium hydroxide in methanol was not compatible with the imide functionality in CRBN-recruiter thalidomide derivatives and precursors. Therefore, we changed the protection groups from methyl esters to tert-butyl esters. As shown in Scheme 3, we prepared tert-butyl ester 17 using the GBBR reaction of 4-phenylpyridin-2-amine 14, 4-(hydroxymethyl)benzaldehyde 15, and tert-butyl 4-isocyanobenzoate 16. Application of the Appel reaction using tetrabromide carbon and triphenylphosphine in acetonitrile converted the hydroxyl in 17 to afford bromide 18.56 This was followed by displacement of the bromide using sodium azide in acetone to yield azide-labeled precursor 7. The alkyne-labeled pomalidomides 9b–9d were prepared by SNAr reactions of fluoride 19 with a variety of amine-containing alkynes that included prop-2-yn-1-amine, N-methylprop-2-yn-1-amine or but-3-yn-1-amine.57 CuAAC coupling of azide-labeled precursor 7 and alkyne-labeled pomalidomides 9b–9d afforded 20a–20c. Deprotection of the acid-labile tert-butyl groups in 20a–20c using TFA provided the final bivalent analogs 5h–5j (Scheme 3).
 | ||
Scheme 3 Synthesis of compounds 5h–5j using key GBBR multi-component reaction and CuAAC “click” reaction. Reagents and conditions: (i) HOAc, MeOH; (ii) CBr4, Ph3P, CH3CN, rt; (iii) NaN3, CH3COCH3, 55 °C; (iv) amines (a, CH![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CCH2NH2; b, CHCCH2NHCH3; c, CHCCH2CH2NH2), DMSO, 130 °C; (v) TBTA, CuSO4–5H2O, sodium L-ascorbate, DMSO, H2O, rt. (vi) TFA, DCM. CCH2NH2; b, CHCCH2NHCH3; c, CHCCH2CH2NH2), DMSO, 130 °C; (v) TBTA, CuSO4–5H2O, sodium L-ascorbate, DMSO, H2O, rt. (vi) TFA, DCM. | ||
Inhibition of TDP1 in gel-based fluorescence assays
We evaluated the in vitro TDP1 inhibitory potencies of triazole-linked PROTAC conjugates 4a–4d and 5a–5j using gel-based fluorescence assay with TDP1 (Tables 1 and S1, ESI†). We previously reported that the parent compound 1a shows single-digital micromolar inhibitory potency in vitro (1a,TDP1 IC50 = 8.72 ± 1.81 μM).22,24 As shown in Table 1, addition of different length PEG-linked hydroxyproline-containing VHL recruiters at the “a” position of compound 1a, gave conjugates 4a–4c, which fail to show TDP1 inhibitory potencies (4a–4c, TDP1 IC50 > 100 μM). Addition of alkyl-linked thalidomide CRBN recruiters at the “a” position of 1a gave conjugate 4d, which also failed to show inhibitory potency within the tested range (4d, TDP1 IC50 > 100 μM). Parent compound 1b with a phenyl at “a” position of 1a shows slightly better TDP1 inhibitory potency than 1a.22,24 We moved the PEG-linked hydroxyproline-containing VHL recruiters in conjugates 4a–4c to the “b” position of compound 1b to yield compounds 5a–5c. Although the resulting conjugates 5b and 5c still failed to exhibit TDP1 inhibitory potency within the range tested, conjugate 5a having the shortest PEG linker showed moderate inhibitory potency (5a, TDP1 IC50 = 70.3 ± 2.4 μM, Table 1). Next, we replaced the PEG-linked hydroxyproline-containing VHL moieties to alkyl-linked CRBN-binding fragments to yield 5d–5g. Although conjugates 5d, 5e and 5g did not show TDP1 inhibitory potency within the range tested, the inhibitory potency of conjugate 5f (TDP1 IC50 = 33.1 ± 7.2 μM) was greater than 5a (Table 1). These results suggest that the best combination of features would be to use the CRBN warhead (as for 5f) along with shorter length linkers (as seen with 5a) attached at the “b” position of 1b. Based on this, we prepared the conjugates 5h–5j having shorter triazole-linked pomalidomide CRBN-targeting functionality. Conjugates 5h–5j showed better TDP1 inhibitory potency than both 5a and 5f (5h, TDP1 IC50 = 7.41 ± 0.19 μM; 5i, TDP1 IC50 = 18.5 ± 1.55 μM; 5j, TDP1 IC50 = 12.34 ± 1.5 μM, Table 1). Encouragingly, conjugate 5h displayed an IC50 values against TDP1 that was comparable to the parent compounds 1a and 1b (5h, TDP1 IC50 = 7.41 ± 0.19 μM vs.1a, TDP1 IC50 = 8.72 ± 1.81 μM and 1b, TDP1 IC50 = 2.98 ± 0.24 μM).| Compound | TDP1 IC50 (μM) |
|---|---|
| a The half maximal inhibitory concentration (IC50) based on gel-based TDP1 fluorescence assay. Except where indicated, assays were conducted using full-length TDP1. b IC50 values have been reported previously.22,24 c Fold-loss of IC50 values against truncated TDP1(148–608) vs. wild-type TDP1. | |
| 1a | 8.72 ± 1.81b |
| 1b | 2.98 ± 0.24b |
| 4a | >100 |
| 4b | >100 |
| 4c | >100 |
| 4d | >100 |
| 5a | 70.3 ± 2.4 |
| 5b | >100 |
| 5c | >100 |
| 5d | >100 |
| 5e | >100 |
| 5f | 33.1 ± 7.2 |
| 5g | >100 |
| 5h | 7.41 ± 0.19 |
| 5i | 18.5 ± 1.55 |
| 5j | 12.34 ± 1.5 |
| 1b [TDP1(148–608)] | 6.6×c |
| 5h [TDP1(148–608)] | 1.2×c |
Unless indicated otherwise, we conducted the gel-based fluorescence assay using full-length TDP1 (Table 1).22,24 However, in our SMM screen and crystallography studies we used truncated TDP1(148–608), which contains the catalytic domain but lacks the N-terminal flexible domain (residues 1–147).22,24 The N-terminal domain has been shown to play important roles in the cellular actions of TDP1 (ref. 58) and this domain has also been shown to be critical for allosteric inhibition by the recently discovered macrocyclic peptide.42 Although the structure of full-length TDP1 has remained elusive, its presence can significantly impact the function of TDP1. We evaluated 5h and 1b against truncated TDP1(148–608). Parent compound 1b showed 6.6-fold lower IC50 values against truncated TDP1(148–608) as compared with against wild-type full-length TDP1 (Table 1). This suggests a potential role for the N-terminal domain in the action of 1b. However, conjugate 5h showed a more modest 1.2-fold reduction in IC50 value against truncated TDP1(148–608) as compared with wild-type full-length TDP1 (Table 1). This indicates that 5h can retain binding to TDP1 catalytic domain even without the N-terminal flexible loop. This data suggests that conjugate 5h could have greater tolerance for changes in the TDP1 tertiary structure.
Comparison of TDP1 inhibitory potencies with TDP2 inhibitory potencies
TDP2 is a structurally different tyrosyl-DNA phosphodiesterase enzyme involved in the repair of TOP2ccs and TOP3ccs.10,17,59,60 TDP1 and TDP2 were discovered and named based on the fact they process 3′- and 5′-DNA ends by excising irreversible protein tyrosyl-DNA complexes involving topoisomerases I and II, respectively.6 Both enzymes have an extended spectrum of activities. Human TDP2 is smaller than TDP1 with a molecular mass of 41 kDa (362 amino acid residues). The repair function of TDP2 is devoted to the excision of topoisomerase II- and potentially topoisomerases III-DNA adducts. Like TDP1, TDP2 is a two-domain protein with the catalytic domain in the C-terminus and an N-terminal domain that is thought to play a regulatory role. Unlike TDP1, TDP2 catalysis requires divalent metals but does not form a transient covalent catalytic intermediate.As a counter screen to the in vitro TDP1 assay, we determined the inhibitory potencies of compounds 1a, 1b, 5a, 5f, 5h–5j against full-length TDP2 in gel-based in vitro fluorescence assays and compared these values with full-length TDP1 inhibitory potencies (Table 2). Parent compounds 1a and 1b showed single-digital micromolar TDP1 inhibitory potencies but failed to inhibit TDP2 within the maximum assay concentration of 100 μM (Table 2). However, the lead conjugates 5a, 5f, and 5h–5j showed only slightly reductions in inhibitory potencies against TDP2 as compared with against TDP1. Since conjugates 5f and 5h–5j showed micromolar inhibitory potencies against both TDP1 and TDP2 in vitro, this may potentially suggest that promiscuous inhibition may be at work.44
| Compound | TDP1 IC50a (μM) | TDP2 IC50b (μM) | TDP1 selectivityc |
|---|---|---|---|
| a Half maximal inhibitory concentration (IC50) based on gel-based TDP1 fluorescence assay. b Half maximal inhibitory concentration (IC50) based on gel-based TDP2 fluorescence assay. c TDP1 selectivity based on the ratio of IC50 values of TDP2 vs. TDP1. | |||
| 1a | 8.72 ± 1.81 | >100 | >11 |
| 1b | 2.98 ± 0.24 | >100 | >33 |
| 5a | 70.3 ± 2.4 | >100 | 1.4 |
| 5f | 33.1 ± 7.2 | 37.3 ± 4.5 | 1.1 |
| 5h | 7.41 ± 0.19 | 11.2 ± 0.55 | 1.5 |
| 5i | 18.5 ± 1.55 | 21.2 ± 0.15 | 1.1 |
| 5j | 12.34 ± 1.5 | 16.8 ± 0.3 | 1.4 |
The bivalent analogs do not appear to get into cells
In cellular assays, compound 5h failed to show significant cytotoxicity when used as single agents (CC50 ∼193 μM in HCT116 cell line, Table S2, ESI†). We were unable to observe degradation effects of 5h and 5i in the HCT116 cell line compared with reference glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (Fig. 4A and B). Compound 5h also failed to show synergy when used in combination with the TOP1 inhibitor camptothecin (CPT) in either HCT116 or U2OS cell lines (Fig. 4C). We interpreted this to mean that the compounds did not get into cells. Even though both of warhead components bind well to the target proteins, the absence of cellular activity is most reasonably explained by lack of cell penetration.51 PROTACs are often affected by poor cellular permeability due to their high molecular weight and large exposed polar surface area.61 This is a common challenge presented by PROTACs.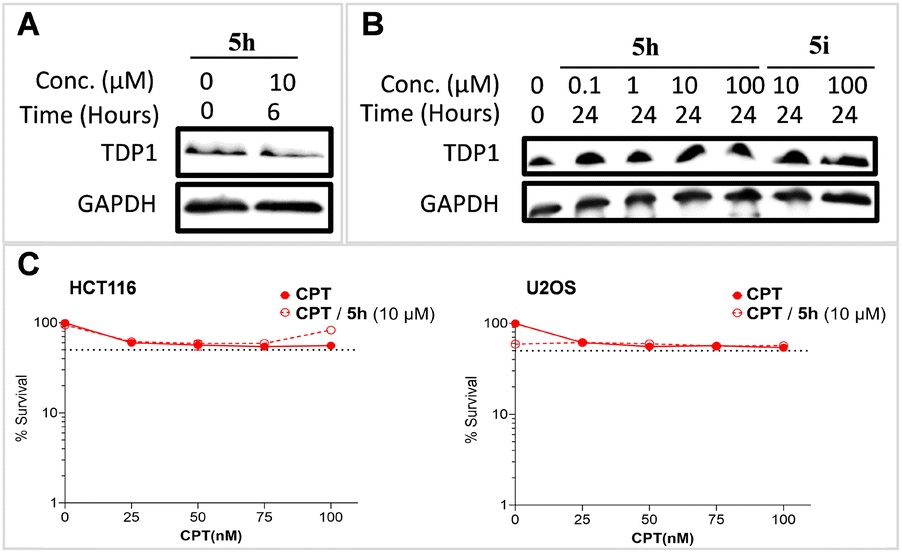 | ||
| Fig. 4 TDP1 degradation and synergy result of compound 5h. (A) TDP1 degradation of compound 5h at 10 μM compared with GAPDH within 6 hours in the HCT116 cell line. (B) TDP1 gel bands of compound 5h at 0.1, 1, 10, 100 μM and 5i at 10, 100 μM separately compared with GAPDH within 24 hours in TDP1 degradation in the HCT116 cell line. (C) Synergy curves of compound 5h at 10 μM with TOP1 inhibitor CPT in HCT116 and U2OS cell lines separately. | ||
PROTAC modeling
PROTACs induce ternary complexes that lead to ubiquitination and degradation of target proteins. A variety of methodologies have been employed to generate ternary structure of E3-PROTAC-target complexes.62 In our current work we linked lead imidazopyridine TDP1 inhibitor (1b) together with thalidomide (21) using triazole-forming “click” chemistry to provide bivalent conjugate (5h) as putative PROTAC analog (Fig. 5A). We endeavored to employ in silico modeling to explore potential geometries of PROTAC-induced proximity positioning of TDP1 with an E3 ligase. We start from the structures of the DDB1-CRBN (DNA damage-binding protein 1-cereblon) E3 ubiquitin ligase in complex with thalidomide (21, PDB code: 4CI1)63 and TDP1 bound with imidazopyridine inhibitor (2, PDB code: 6W7K).22 Using our ICM MolSoft Pro 64 (icm 3.9–4) software,64 we formed the ternary structure of E3-TDP1 complexes bound to lead conjugate 5h. We employed the following protocol: (1) read into ICM the target PDB files for TDP1 “6W7K” and CRBN “4CI1” (Fig. 5B and C); (2) delete portions of ligand structures not needed and convert both assemblies into ICM objects; (3) read in a .sdf file of molecule 5h in chemical table; (4) run PROTAC Modeling using protein–protein docking PROTAC model builder in ICM module. This provided a model of the PROTAC complex having the conjugate 5h bound to the CRBN (shown in cyan ribbon) and TDP1 (shown in grey ribbon) with the triazole linker bridging the imidazopyridine TDP1 inhibitor and thalidomide (Fig. 5D and E). This suggests that the PROTAC conjugate 5h could reasonably induce spatial proximity of both TDP1 and CRBN (Fig. 5F and G). The linker of compound 5h locates at the peptide binding groove side, points toward the opposite of the TDP1 catalytic site and extends to the CRBN binding site, which is what we excepted. | ||
| Fig. 5 Docking modes of conjugate 5h with TDP1 and CRBN. (A) Structures of lead imidazopyridine TDP1 inhibitor 2, thalidomide 21 and conjugate 5h. (B) Crystal structure of CRBN (shown in cyan ribbon) bound with thalidomide 21 (shown in blue carbons) (PDB code: 4CI1). (C) Crystal structure of TDP1 (shown in grey ribbon) bound with imidazopyridine TDP1 inhibitor 2 (shown in brown carbons) (PDB code: 6W7K). (D) Close view of thalidomide 21 and imidazopyridine TDP1 inhibitor 2 (shown in brown carbons) binding at the interface of CRBN and TDP1 enzymes. (E) Overlaid mode of conjugate 5h with thalidomide 21 and imidazopyridine TDP1 inhibitor 2 at the interface of CRBN and TDP1 enzymes. (F) Binding mode of conjugate 5h (carbons are shown in yellow spheres) at the interface of CRBN enzyme (shown in cyan ribbon) and TDP1 enzyme (shown in grey ribbon). (G) Binding mode of conjugate 5h (carbons are shown in yellow balls) at the interface of CRBN enzyme (shown in cyan ribbon) and TDP1 enzyme (shown in grey ribbon). | ||
Conclusions
The development of clinical TDP1 inhibitors has been highly challenging. The chemical biology toolbox for studying TDP1 could be significantly enhanced by including the ability to selectively degrade TDP1 using protein degraders. Guided by the crystal structures of TDP1 bound to phenyl imidazopyridine (2), we designed a series of bivalent constructs with different length linkers (4a–4d and 5a–5j) that were intended to function as TDP1 PROTACs. These TDP1 PROTACs employ E3 ligase-targeting components VHL recruiter and CRBN recruiter linked to both sides of the phenyl imidazopyridine-based TDP1-binding core. We prepared these from azide and alkyne precursors using straightforward CuAAC click chemistry. The primary objective of our current work has been to explore synthetic approaches to modifying known TDP1 inhibitors in ways that would permit installation of E3 ligase-targeting functionality, while retaining the ability of the ligands to bind to TDP1. We found among the conjugates 5h–5j that shorter linkers retain micromolar inhibition against TDP1 in gel-based fluorescence assays in vitro. Conjugate 5h shows single-digital micromolar TDP1 inhibitory potency. In cellular assays, these bivalent constructs fail to show biological activity. We interpret this to indicate extremely poor cellular uptake. None-the-less, our work validates approaches to designing and synthesizing bivalent ligands having key molecular features needed for functional TDP1 PROTACs. These results should enrich to chemical biology toolbox for studying TDP1 function.Materials and methods
TDP1 and TDP2 gel-based assay in vitro
The inhibition assays for full-length TDP1, N-terminally truncated TDP1(148–608) and TDP2 were performed following previously described gel-based methods.22,24,43 Briefly, 1 nM of the DNA substrate (N14Y, 5′Cy5-GATCTAAAAGACTT-pY-3′) was incubated with 40 pM full-length recombinant TDP1 or truncated TDP1(148–608) in the absence or presence of inhibitors for 15 min at room temperature (rt) in TDP1 reaction buffer (50 mM Tris-HCl, pH 7.5, 80 mM KCl, 2 mM EDTA, 1 mM DTT, 40 μg mL−1 BSA and 0.01% Tween 20). Similarly, 1 nM of DNA substrate (YN18, 5′-pY-TCCGTTGAAGCCTGCTTT-Cy5-3′) was incubated with 40 pM recombinant TDP2 under identical conditions, except using TDP2 reaction buffer (50 mM Tris-HCl, pH 7.5, 80 mM KCl, 5 mM MgCl2, 0.1 mM EDTA, 1 mM DTT, 40 μg mL−1 BSA, and 0.01% Tween 20). The reactions were stopped by adding an equal volume of gel loading buffer (99.5% (v/v) formamide, 5 mM EDTA). The samples were then subjected to a 20% denaturing PAGE gel followed by gel scanning using a Typhoon FLA 9500 scanner (GE Healthcare). The IC50 values of the TDP1 inhibitors were calculated by comparing the percentage of the cleavage product (N14P, 5′Cy5-GATCTAAAAGACTT-p-3′) produced to that in the DMSO control. The IC50 values of the TDP2 inhibitors were calculated by comparing the percentage of the cleavage product (PN18, 5′-p-TCCGTTGAAGCCTGCTTT-Cy5-3′) produced to that in the DMSO control.Survival curve and cytotoxicity24
MCF7 or HCT116 cells were seeded into 384-well black-clear plate until 30% confluency and then incubated with a two-fold serial dilution of TDP1 inhibitors for 72 h at 37 °C (Table S2, ESI†). The cell numbers were counted from the brightfield images taken by Biotek Cytation 5. The cell cytotoxicity was calculated based on the concentration required for 50% cell survival (CC50), using DMSO as a control.Synergy assay of TDP1 inhibitors with camptothecin (CPT)
The synergistic effects of the TDP1 inhibitors with CPT were tested in the human colon cancer HCT116 cell line and osteosarcoma U2OS cell line based on cell viability.22,24,43 Cells were first seeded into 384-well opaque plate at a density of 2 × 103 cells per well and then were cultured for 72 hours in a media containing the drugs. Cell viability was assessed by using CellTiter-Glo Luminescent Cell Viability Assay (Promega, no. G7570) according to the manufacturer's instructions and luminescence intensity was measured using SpectroMaxi3, CellTiter-Glo cell proliferation program.Western blot for TDP1 degradation
Cells treated with or without inhibitors were lysed in 100 μL sodium dodecyl sulfate (SDS) buffer containing 25 mM Tris-HCl (pH 6.5), 1% SDS, 0.24 mM β-mercaptoethanol, 0.1% bromophenol blue and 5% glycerol and cOmplete™, Mini, EDTA-free protease inhibitor cocktail (Roche, Cat# 11836170001). Whole-cell extracts were separated by SDS-PAGE gel, transferred onto polyvinylidene difluoride (PVDF) membranes, and blocked in 5% skimmed milk dissolved in 0.1% Tween-20 in phosphate buffer saline (PBS). Membranes were incubated with primary antibodies overnight at 4 °C followed by washing with 0.1% Tween-20 in PBS. Primary antibodies used in this study are as follows: rabbit polyclonal anti-TDP1 (dilution 1:1000, ABCAM, Cat# ab227144) and rabbit mAb anti-GAPDH (dilution 1:2000, Cell Signaling Technology, Cat# 2118, Clone 14C10). Membranes were incubated with anti-rabbit IgG ECL, HRP conjugated (dilution 1:4000, GE Healthcare, Cat# NA9340) at rt for 1 h and washed thrice and signals were detected by ECL chemiluminescence reaction (SuperSignal™ West Femto Maximum Sensitivity Substrate, Thermo Scientific, Waltham, MA, Cat# 34095).
General procedures
Proton (1H) and carbon (13C) NMR spectra were recorded on a Varian 400 MHz spectrometer or a Varian 500 MHz spectrometer and are reported in ppm relative to TMS and referenced to the solvent in which the spectra were collected. Solvent was removed by rotary evaporation under reduced pressure, and anhydrous solvents were obtained commercially and used without further drying. Purification by silica gel chromatography was performed using Combiflash with EtOAc–hexanes solvent systems. Preparative high pressure liquid chromatography (HPLC) was conducted using a Waters Prep quaternary gradient module 2535 system having photodiode array detector 2998, fraction collector III, autosampler 2707 and Phenomenex C18 columns (catalogue no. 00G-4436-P0-AX, 250 mm × 21.2 mm 10 μm particle size, 110 Å pore) at a flow rate of 10 mL min−1 or 20 mL min−1. Binary solvent systems consisting of A = 0.1% aqueous TFA and B = 0.1% TFA in acetonitrile were employed with gradients as indicated. Products were obtained as amorphous solids following lyophilization. Electrospray ionization-mass spectrometric (ESI-MS) were acquired with an Agilent LC/MSD system equipped with a multimode ion source. High resolution mass spectrometric (HRMS) were acquired by LC/MS-ESI using LTQ-Orbitrap-XL at 30 K resolution.1H NMR (500 MHz, DMSO-d6) 1H NMR (500 MHz, DMSO) δ 12.45 (brs, 1H), 9.08 (s, 1H), 8.97 (s, 1H), 8.60 (t, J = 6.1 Hz, 1H), 8.33 (d, J = 7.1 Hz, 1H), 8.19 (s, 1H), 8.09 (s, 1H), 7.93–7.86 (m, 4H), 7.80–7.76 (m, 2H), 7.69 (d, J = 7.2 Hz, 1H), 7.59 (dd, J = 8.3, 6.6 Hz, 2H), 7.53 (dd, J = 8.4, 6.1 Hz, 1H), 7.43 (d, J = 9.3 Hz, 1H), 7.38 (s, 4H), 7.20 (d, J = 9.0 Hz, 2H), 6.76 (d, J = 8.2 Hz, 2H), 5.18 (s, 2H), 4.56 (d, J = 9.6 Hz, 1H), 4.51 (t, J = 5.2 Hz, 2H), 4.43 (t, J = 8.2 Hz, 1H), 4.36 (dd, J = 15.7, 6.2 Hz, 2H), 4.24 (dd, J = 15.8, 5.7 Hz, 1H), 3.94 (s, 2H), 3.79 (t, J = 5.2 Hz, 2H), 3.66 (dd, J = 10.7, 3.9 Hz, 2H), 3.61–3.50 (m, 9H), 2.43 (s, 3H), 2.06 (ddd, J = 9.5, 7.1, 4.3 Hz, 1H), 1.89 (ddt, J = 13.1, 8.9, 4.4 Hz, 1H), 0.92 (s, 9H). ESI-MS m/z: 1105.2 (MH+). HRMS calcd. for C59H65N10O10S (MH+), 1105.4600; found, 1105.2327. HRMS calcd. for C59H66N10O10S (MH2)2+, 553.2337; found, 553.2327.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
XZ and TB conceived the study. XZ designed and synthesized the compounds. WW, MM, KA, and YP performed the biological studies. XZ, WW, MM, KA, YP and TB interpreted the data. XZ and TB took the lead in writing the manuscript. All authors have provided critical feedback and approved the final manuscript.Conflicts of interest
The authors declare that there is no conflict of interest.Acknowledgements
This work was supported in part by a Staff Scientist/Staff Clinician Research Award (SSSC-RA) and the Intramural Research Program of the NIH, Center for Cancer Research, National Cancer Institute, National Institutes of Health (Z01-BC 006150 and Z01-BC 006198).References
- S. Li, C. Vemuri and C. Chen, Curr. Opin. Struct. Biol., 2024, 87, 102868 CrossRef CAS PubMed.
- B. C. Soren, J. B. Dasari, A. Ottaviani, F. Iacovelli and P. Fiorani, Cancer Drug Resist., 2020, 3, 18–25 CAS.
- Y. Pommier, Y. Sun, S.-y. N. Huang and J. L. Nitiss, Nat. Rev. Mol. Cell Biol., 2016, 17, 703–721 CrossRef CAS PubMed.
- A. Tubbs and A. Nussenzweig, Cell, 2017, 168, 644–656 CrossRef CAS PubMed.
- H. U. Barthelmes, M. Habermeyer, M. O. Christensen, C. Mielke, H. Interthal, J. J. Pouliot, F. Boege and D. Marko, J. Biol. Chem., 2004, 279, 55618–55625 CrossRef CAS PubMed.
- S. N. Huang and Y. Pommier, Int. J. Mol. Sci., 2019, 20, 3015 CrossRef CAS PubMed.
- S. S. Laev, N. F. Salakhutdinov and O. I. Lavrik, Bioorg. Med. Chem., 2016, 24, 5017–5027 CrossRef CAS PubMed.
- L. Debéthune, G. Kohlhagen, A. Grandas and Y. Pommier, Nucleic Acids Res., 2002, 30, 1198–1204 CrossRef PubMed.
- Y. Sun, L. K. Saha, S. Saha, U. Jo and Y. Pommier, DNA Repair, 2020, 94, 102926 CrossRef CAS PubMed.
- Y. Sun, S. Saha, W. Wang, L. K. Saha, S. N. Huang and Y. Pommier, DNA Repair, 2020, 89, 102837 CrossRef CAS PubMed.
- J. J. Pouliot, K. C. Yao, C. A. Robertson and H. A. Nash, Science, 1999, 286, 552–555 CrossRef CAS PubMed.
- H. Interthal, J. J. Pouliot and J. J. Champoux, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 12009–12014 CrossRef CAS PubMed.
- G. L. Beretta, G. Cossa, L. Gatti, F. Zunino and P. Perego, Curr. Med. Chem., 2010, 17, 1500–1508 CrossRef CAS PubMed.
- E. Q. Comeaux and R. C. A. M. van Waardenburg, Drug Metab. Rev., 2014, 46, 494–507 CrossRef CAS PubMed.
- R. Gao, B. B. Das, R. Chatterjee, O. D. Abaan, K. Agama, R. Matuo, C. Vinson, P. S. Meltzer and Y. Pommier, DNA Repair, 2014, 13, 1–9 CrossRef CAS PubMed.
- J. Murai, S.-y. N. Huang, B. B. Das, T. S. Dexheimer, S. Takeda and Y. Pommier, J. Biol. Chem., 2012, 287, 12848–12857 CrossRef CAS PubMed.
- Y. Pommier, S.-y. N. Huang, R. Gao, B. B. Das, J. Murai and C. Marchand, DNA Repair, 2014, 19, 114–129 CrossRef CAS PubMed.
- Z. Hu, H.-w. Wang and L.-k. An, Yaoxue Xuebao, 2016, 51, 215–225 CAS.
- F. J. Flett, E. Ruksenaite, L. A. Armstrong, S. Bharati, R. Carloni, E. R. Morris, C. L. Mackay, H. Interthal and J. M. Richardson, Nat. Commun., 2018, 9, 1–13 CrossRef CAS PubMed.
- G. T. Lountos, X. Z. Zhao, T. R. Burke, E. Kiselev, Y. Pommier, J. E. Tropea, D. Needle and D. S. Waugh, Nucleic Acids Res., 2019, 47, 10134–10150 CrossRef CAS PubMed.
- Y. Zhang, Y. Li, C. Sun, X. Chen, L. Han, T. Wang, J. Liu, X. Chen and D. Zhao, Cancers, 2021, 13, 4002 CrossRef CAS PubMed.
- X. Z. Zhao, E. Kiselev, G. T. Lountos, W. Wang, J. E. Tropea, D. Needle, T. A. Hilimire, J. S. Schneekloth, D. S. Waugh, Y. Pommier and T. R. Burke, Chem. Sci., 2021, 12, 3876–3884 RSC.
- X. Z. Zhao, W. Wang, G. T. Lountos, J. E. Tropea, D. Needle, Y. Pommier and T. R. Burke, Front. Chem., 2022, 10, 910953 CrossRef CAS PubMed.
- X. Z. Zhao, W. Wang, G. T. Lountos, E. Kiselev, J. E. Tropea, D. Needle, Y. Pommier and T. R. Burke, RSC Chem. Biol., 2023, 4, 334–343 RSC.
- D. R. Davies, H. Interthal, J. J. Champoux and W. G. J. Hol, Structure, 2002, 10, 237–248 CrossRef CAS PubMed.
- D. R. Davies, H. Interthal, J. J. Champoux and W. G. J. Hol, J. Mol. Biol., 2002, 324, 917–932 CrossRef CAS PubMed.
- S. T. Dexheimer, S. Antony, C. Marchand and Y. Pommier, Anti-Cancer Agents Med. Chem., 2008, 8, 381–389 CrossRef PubMed.
- R. A. Copeland, Evaluation of Enzyme Inhibitors in Drug Discovery, John Wiley & Sons, Inc., Hoboken, New Jersey, 2nd edn, 2013 Search PubMed.
- A. Zakharenko, N. Dyrkheeva and O. Lavrik, Med. Res. Rev., 2019, 39, 1427–1441 CrossRef CAS PubMed.
- E. V. Koldysheva, A. P. Men'shchikova, E. L. Lushnikova, N. A. Popova, V. I. Kaledin, V. P. Nikolin, A. L. Zakharenko, O. A. Luzina, N. F. Salakhutdinov and O. I. Lavrik, Bull. Exp. Biol. Med., 2019, 166, 661–666 CrossRef CAS PubMed.
- A. L. Zakharenko, O. A. Luzina, D. N. Sokolov, V. I. Kaledin, V. P. Nikolin, N. A. Popova, J. Patel, O. D. Zakharova, A. A. Chepanova, A. Zafar, J. Reynisson, E. Leung, I. K. H. Leung, K. P. Volcho, N. F. Salakhutdinov and O. I. Lavrik, Eur. J. Med. Chem., 2019, 161, 581–593 CrossRef CAS PubMed.
- O. Luzina, A. Filimonov, A. Zakharenko, A. Chepanova, O. Zakharova, E. Ilina, N. Dyrkheeva, G. Likhatskaya, N. Salakhutdinov and O. Lavrik, J. Nat. Prod., 2020, 83, 2320–2329 CrossRef CAS PubMed.
- A. L. Zakharenko, M. S. Drenichev, N. S. Dyrkheeva, G. A. Ivanov, V. E. Oslovsky, E. S. Ilina, I. A. Chernyshova, O. I. Lavrik and S. N. Mikhailov, Molecules, 2020, 25, 3694 CrossRef CAS PubMed.
- T. M. Khomenko, A. L. Zakharenko, A. A. Chepanova, E. S. Ilina, O. D. Zakharova, V. I. Kaledin, V. P. Nikolin, N. A. Popova, D. V. Korchagina, J. Reynisson, R. Chand, D. M. Ayine-Tora, J. Patel, I. K. H. Leung, K. P. Volcho, N. F. Salakhutdinov and O. I. Lavrik, Int. J. Mol. Sci., 2019, 21, 126 CrossRef PubMed.
- E. D. Gladkova, I. V. Nechepurenko, R. A. Bredikhin, A. A. Chepanova, A. L. Zakharenko, O. A. Luzina, E. S. Ilina, N. S. Dyrkheeva, E. M. Mamontova, R. O. Anarbaev, J. Reynisson, K. P. Volcho, N. F. Salakhutdinov and O. I. Lavrik, Int. J. Mol. Sci., 2020, 21, 7162 CrossRef CAS PubMed.
- O. V. Salomatina, I. I. Popadyuk, A. L. Zakharenko, O. D. Zakharova, A. A. Chepanova, N. S. Dyrkheeva, N. I. Komarova, J. Reynisson, R. O. Anarbaev, N. F. Salakhutdinov, O. I. Lavrik and K. P. Volcho, Steroids, 2021, 165, 108771 CrossRef CAS PubMed.
- N. S. Dyrkheeva, A. S. Filimonov, O. A. Luzina, A. L. Zakharenko, E. S. Ilina, A. A. Malakhova, S. P. Medvedev, J. Reynisson, K. P. Volcho, S. M. Zakian, N. F. Salakhutdinov and O. I. Lavrik, Biomolecules, 2021, 11, 973 CrossRef CAS PubMed.
- A. L. Zakharenko, O. A. Luzina, A. A. Chepanova, N. S. Dyrkheeva, N. F. Salakhutdinov and O. I. Lavrik, Int. J. Mol. Sci., 2023, 24, 5781 CrossRef CAS PubMed.
- O. V. Salomatina, T. E. Kornienko, A. L. Zakharenko, N. I. Komarova, C. Achara, J. Reynisson, N. F. Salakhutdinov, O. I. Lavrik and K. P. Volcho, Molecules, 2024, 29, 581 CrossRef CAS PubMed.
- A. Bermingham, E. Price, C. Marchand, A. Chergui, A. Naumova, E. L. Whitson, L. R. H. Krumpe, E. I. Goncharova, J. R. Evans, T. C. McKee, C. J. Henrich, Y. Pommier and B. R. O'Keefe, SLAS Discovery, 2017, 22, 1093–1105 CrossRef CAS PubMed.
- X. Wei, F. T. Wang, M. X. Si-Tu, H. Fan, J. S. Hu, H. Yang, S. Y. Guan, L. K. An and C. X. Zhang, Mar. Drugs, 2022, 20, 443 CrossRef PubMed.
- L. R. H. Krumpe, B. A. P. Wilson, C. Marchand, S. N. Sunassee, A. Bermingham, W. Wang, E. Price, T. Guszczynski, J. A. Kelley, K. R. Gustafson, Y. Pommier, K. J. Rosengren, C. I. Schroeder and B. R. O'Keefe, J. Am. Chem. Soc., 2020, 142, 21178–21188 CrossRef CAS PubMed.
- X. Z. Zhao, I. A. Barakat, G. T. Lountos, W. Wang, K. Agama, M. R. A. Mahmud, K. F. Suazo, T. Andresson, Y. Pommier and T. R. Burke, Commun. Chem., 2024, 7, 208 CrossRef CAS PubMed.
- S. L. McGovern, E. Caselli, N. Grigorieff and B. K. Shoichet, J. Med. Chem., 2002, 45, 1712–1722 CrossRef CAS PubMed.
- K. M. Sakamoto, K. B. Kim, A. Kumagai, F. Mercurio, C. M. Crews and R. J. Deshaies, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 8554–8559 CrossRef CAS PubMed.
- Y. Xue, A. A. Bolinger and J. Zhou, Expert Opin. Drug Discovery, 2023, 18, 467–483 CrossRef CAS PubMed.
- A. C. Lai and C. M. Crews, Nat. Rev. Drug Discovery, 2017, 16, 101–114 CrossRef CAS PubMed.
- X. Sun, H. Gao, Y. Yang, M. He, Y. Wu, Y. Song, Y. Tong and Y. Rao, Signal Transduction Targeted Ther., 2019, 4, 64 CrossRef PubMed.
- R. Li, M. Liu, Z. Yang, J. Li, Y. Gao and R. Tan, Molecules, 2022, 27, 8828 CrossRef CAS PubMed.
- D. Chirnomas, K. R. Hornberger and C. M. Crews, Nat. Rev. Clin. Oncol., 2023, 20, 265–278 CrossRef CAS PubMed.
- M. Békés, D. R. Langley and C. M. Crews, Nat. Rev. Drug Discovery, 2022, 21, 181–200 CrossRef PubMed.
- R. I. Troup, C. Fallan and M. G. J. Baud, Explor. Target. Anti-tumor Ther., 2020, 1, 273–312 Search PubMed.
- B. Mostofian, H. J. Martin, A. Razavi, S. Patel, B. Allen, W. Sherman and J. A. Izaguirre, J. Chem. Inf. Model., 2023, 63, 5408–5432 Search PubMed.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS PubMed.
- C. Yang, R. Tripathi and B. Wang, RSC Chem. Biol., 2024, 5, 189–197 RSC.
- R. Appel, Angew. Chem., Int. Ed. Engl., 1975, 14, 801–811 CrossRef.
- D. K. Brownsey, B. C. Rowley, E. Gorobets, B. S. Gelfand and D. J. Derksen, Chem. Sci., 2021, 12, 4519–4525 RSC.
- E. J. Brettrager, S. M. Cuya, Z. E. Tibbs, J. Zhang, C. N. Falany, S. G. Aller and R. C. A. M. van Waardenburg, Sci. Rep., 2023, 13, 1377 CrossRef CAS PubMed.
- S. Saha, Y. Sun, S. N. Huang, S. A. Baechler, L. S. Pongor, K. Agama, U. Jo, H. Zhang, Y. C. Tse-Dinh and Y. Pommier, Cell Rep., 2020, 33, 108569 CrossRef CAS PubMed.
- L. K. Saha, S. Saha, X. Yang, S.-y. N. Huang, Y. Sun, U. Jo and Y. Pommier, Nat. Commun., 2023, 14, 1925 CrossRef CAS PubMed.
- C. Cecchini, S. Pannilunghi, S. Tardy and L. Scapozza, Front. Chem., 2021, 9, 672267 CrossRef CAS PubMed.
- E. Rovers and M. Schapira, Methods Enzymol., 2023, 690, 311–340 CAS.
- E. S. Fischer, K. Böhm, J. R. Lydeard, H. Yang, M. B. Stadler, S. Cavadini, J. Nagel, F. Serluca, V. Acker, G. M. Lingaraju, R. B. Tichkule, M. Schebesta, W. C. Forrester, M. Schirle, U. Hassiepen, J. Ottl, M. Hild, R. E. Beckwith, J. W. Harper, J. L. Jenkins and N. H. Thomä, Nature, 2014, 512, 49–53 CrossRef CAS PubMed.
- A. O. Ruben Abagyan, Eugene Raush, and Maxim Totrov, ICM-Pro User Guide v.3.9, https://www.molsoft.com/icmpro/.
- R. P. Wurz, K. Dellamaggiore, H. Dou, N. Javier, M.-C. Lo, J. D. McCarter, D. Mohl, C. Sastri, J. R. Lipford and V. J. Cee, J. Med. Chem., 2018, 61, 453–461 CrossRef CAS PubMed.
- C. Steinebach, I. Sosič, S. Lindner, A. Bricelj, F. Kohl, Y. L. D. Ng, M. Monschke, K. G. Wagner, J. Krönke and M. Gütschow, MedChemComm, 2019, 10, 1037–1041 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4md00824c |
| This journal is © The Royal Society of Chemistry 2025 |