
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThiochromenes and thiochromanes: a comprehensive review of their diverse biological activities and structure–activity relationship (SAR) insights
Jatin
a,
Solai
Murugappan
a,
Shivani
Kirad
a,
Chandu
Ala
a,
Pranali Vijaykumar
Kuthe
a,
Chandra Sekhar Venkata Gowri
Kondapalli
 b and
Murugesan
Sankaranarayanan
*a
b and
Murugesan
Sankaranarayanan
*a
aMedicinal Chemistry Research Laboratory, Department of Pharmacy, Birla Institute of Technology and Science Pilani, Pilani Campus, Vidya Vihar, Pilani-333031, Rajasthan, India. E-mail: murugesan@pilani.bits-pilani.ac.in
bDepartment of Chemistry, Birla Institute of Technology and Science Pilani, Hyderabad Campus, Jawahar Nagar, Kapra Mandal, Hyderabad-500078, Telangana, India
First published on 24th March 2025
Abstract
Thiochromene and thiochromane scaffolds, sulfur containing heterocycles, have gained significant attention in medicinal chemistry due to their diverse pharmacological activities. This review provides a comprehensive analysis of their antibacterial, antifungal, antiviral, anti-parasitic, and anticancer properties, emphasizing their therapeutic potential. SAR studies highlight key molecular modifications such as electron withdrawing substituents, sulfur oxidation, and tailored ring substitutions that enhance bioactivity, potency, and target specificity. Mechanistic insights reveal their ability to inhibit microbial enzymes, disrupt cellular pathways, and modulate key biological targets. By summarizing recent advancements, this review underscores the potential of thiochromene and thiochromane based therapeutics and encourages further research to address existing limitations and enhance their drug development prospects.
1 Introduction
Sulfur is an essential and one of the most abundant elements found in nature.1,2 Despite being well-known and studied for centuries, it remains at the center of a wide range of scientific research topics. Sulfur is a common reaction site within biological systems, known for its role in a wide variety of biochemical processes and as a critical component of numerous biologically active compounds. The versatility of sulfur arises from its unique chemical properties arising from its electronic configuration and position in the periodic table. It can exhibit a wide range of oxidation states (+2 to +6), enabling diverse reactivity.3 Sulfur's high polarizability facilitates strong nucleophilicity, while its ability to form stable single, double, and delocalized bonds supports the creation of complex molecular architectures.4 The formation of disulfide bonds (R–S–S–R′) is crucial for structural stability in biomolecules. Additionally, sulfur's ability to form thiol (R–SH), thioether (R–S–R′), and sulfonyl (R–SO2–R′) groups allows for enhanced reactivity and tuneable properties in various chemical environments, which enable it to form multiple types of bonds and exhibit various oxidation states.5 These properties make sulfur a key component in the structure and function of proteins, enzymes, vitamins, and co-factors, as well as playing pivotal roles in cellular metabolism, redox balance, and enzymatic catalysis. Furthermore, sulfur is found in a variety of biologically active molecules such as anti-biotics, vitamins, and hormones, highlighting its significance in drug development and therapeutic applications.6 In biological systems, sulfur is primarily found in amino acids, such as cysteine and methionine, which are essential for protein structure and function. Moreover, sulfur-containing functional groups like thiols, sulfides, and sulfoxides contribute to the reactivity and specificity of numerous enzymes, including proteases and oxidoreductases.7 Sulfur's role extends beyond primary metabolism, with its presence in secondary metabolites and pharmaceuticals, including drugs like penicillin and cephalosporins, which contain sulfur in their core structure demonstrating sulfur's importance in therapeutic applications. These antibiotics exploit sulfur's unique properties to interfere with bacterial cell wall synthesis.8Sulfur containing heterocycles represent a pivotal class of compounds in medicinal chemistry, distinguished by their diverse chemical reactivity and broad spectrum of biological activities. The incorporation of sulfur into heterocyclic frameworks introduces significant modifications to the electronic distribution and enhances the lipophilicity of the molecules. These alterations often translate into improved physicochemical properties such as membrane permeability and bioavailability, thereby rendering sulfur containing heterocycles highly attractive scaffolds in drug discovery and development.9 Their versatility has been harnessed extensively in therapeutic areas including oncology, anti-infectives, and cardiovascular diseases.10
Among the most extensively studied sulfur containing heterocycles are thiophenes, thiazoles, and thiadiazoles, which have demonstrated a broad spectrum of pharmacological activities, including anti-microbial, anti-inflammatory, anti-tumor, and anti-oxidant properties. Notably, many FDA approved drugs feature these heterocycles in their structures (Fig. 1a), highlighting their critical role in modern therapeutic development. These activities are largely attributed to the sulfur atom's intrinsic ability to modulate electronic and steric effects, which facilitates precise tuning of molecular interactions with biological targets.11,12 This foundational understanding of sulfur heterocycles provides a framework for the design of more complex sulfur based systems.13 Building on this foundation, thiochromenes and thiochromanes have emerged as particularly noteworthy sulfur-containing heterocycles due to their expansive therapeutic potential. Structurally, thiochromenes serve as sulfur analogs of chromenes where in the oxygen atom in the chromene core is replaced by sulfur, imparting distinctive electronic and steric characteristics.14–17 Thiochromanes, as saturated derivatives of thiochromenes, further expand the structural diversity and biological relevance of this class. These scaffolds have garnered considerable interest in medicinal chemistry for their promising applications across various therapeutic domains.18
 | ||
| Fig. 1 a Sulfur containing FDA approved drugs. b Diverse biological application of thiochromene/thiochromane scaffold. | ||
Thiochromenes and thiochromanes exhibit a broad range of pharmacological activities, including anti-cancer, anti-microbial and various other activities (Fig. 1b), their ability to interact with multiple biological targets makes them versatile scaffolds for drug design. The presence of sulfur in these compounds enhances their chemical reactivity, which contributes to their ability to modulate enzyme activity and inhibit disease related processes.19,20 Several thiochromane derivatives have been reported to inhibit key enzymes involved in cancer progression, such as tyrosine kinases and carbonic anhydrases, highlighting their potential as anti-cancer agents.21,22 Additionally, thiochromene derivatives have shown promise as anti-microbial agents, targeting both bacterial and fungal pathogens. Their mechanism of action typically involves disrupting membrane integrity or interfering with nucleic acid synthesis leading to cell death. This anti-microbial activity of these compounds is particularly significant in light of rising anti-biotic resistance, as there is an urgent need for novel agents that can combat resistant strains of bacteria and fungi.23–26
The chemistry of thiochromene and thiochromane is characterized by the ability to undergo a variety of synthetic transformations, making them valuable intermediates in the development of new therapeutic agents. Thiochromenes can be synthesized through several methods, including cyclization reactions of sulfur-containing precursors and transition metal-catalyzed processes.27–29 The presence of sulfur in the heterocyclic core provides unique opportunities for functionalization, allowing for the introduction of various substituents at specific positions on the ring. The electronic properties of sulfur play a crucial role in the reactivity of thiochromenes, influencing their behaviour in electrophilic and nucleophilic substitution reactions. This reactivity facilitates design and synthesis of a wide array of thiochromene derivatives with tailored biological activities. Thiochromanes being saturated analogues offer additional flexibility in terms of stereochemistry which can be exploited to enhance drug–receptor interactions and improve pharmacokinetic properties.
The pharmacological significance of thiochromenes and thiochromanes is evident in their diverse biological activities and potential applications in drug development. Their anti-cancer activity is particularly noteworthy as several derivatives demonstrate the ability to inhibit tumor cell proliferation, induce apoptosis, and prevent metastasis. These compounds exert their anti-cancer effects through various mechanisms, including inhibiting key signalling pathways involved in cell growth and survival, such as the ERK–MAPK pathway, or through reactive oxygen species-mediated pathways.30,31 Given the increasing recognition of the therapeutic potential of sulfur-containing heterocycles, particularly thiochromenes and thiochromanes, there is a need for a comprehensive review of their chemistry and pharmacological properties. This review highlights the latest advances in the biological evaluation of thiochromene/thiochromane derivatives, focusing on their potential applications in drug discovery. By providing an overview of the current state of research, this review will serve as a valuable resource for medicinal chemists and pharmacologists interested in exploring the therapeutic implications of these compounds, thereby fostering further research and innovation in this promising field. By highlighting key areas for future research, this review aims to contribute to the ongoing efforts to develop novel sulfur-containing heterocycles for the treatment of various diseases.
In this review, thiochromene and thiochromane derivatives have been broadly categorized based on their pharmacological indications into different groups as anti-bacterial, anti-fungal, anti-viral, anti-cancer activity, anti-parasitic activities, anti-leishmanial, anti-malarial, anti-trypanosomal, activities. The following sections discuss various reported thiochromene and thiochromane derivatives, highlighting their structure–activity relationships (SAR). Additionally, the different molecular targets of these scaffolds are discussed in a later section.
2 Anti-microbial activity
2.1 Anti-bacterial activity
 | ||
| Fig. 2 Structure of thiochromane analogs as anti-bacterial agents. | ||
Building on the potential of thiochromanones, Chouchène et al. synthesized a series of spiro pyrrolidines incorporating thiochroman-4-one/chroman-4-one and oxindole/acenaphthylene-1,2-dione moieties to evaluate both anti-bacterial and anti-fungal properties. The thiochromanone scaffold showed the best biological activity, outperforming standard drugs like amoxicillin and amphotericin B.33 Among these, compound 8 (Fig. 2) demonstrated the best anti-bacterial activity among the synthesized series of compounds, with the lowest MIC values of 32 μg mL−1 against Bacillus subtilis, Staphylococcus epidermidis, Staphylococcus aureus (ATCC 25923), and Enterococcus faecalis. DFT studies further supported the increased reactivity and biological activity of the thiochromanone-containing spiro pyrrolidines due to their lower HOMO–LUMO energy gaps, suggesting a strong correlation between electronic properties and anti-microbial efficacy.
In another innovative approach, Kumar et al. explored the synthesis of novel thiochromane derivatives coupled with benzimidazole scaffolds via Sonogashira coupling reactions. This study broadened the scope of biological applications by evaluating anti-bacterial, anti-asthmatic, and anti-diabetic activities.34 Specifically the series of compound 9 (Fig. 2) derivatives exhibited moderate anti-bacterial activity against both Gram-positive and Gram-negative bacteria. Compounds of this series showed medium growth compared to controls, indicating partial inhibition against Salmonella typhimurium and S. aureus. Among these, compound 9 exhibit the highest activity with consistent inhibition at the lowest concentration of 200 μg ml−1.
Expanding on the structural versatility of thiochromanones, Xiao et al. introduced carboxamide-functionalized thiochromanone derivatives to investigate their anti-bacterial and anti-fungal activities.35 Compound 10 (Fig. 2) with a chloro substitution on the thiochromanone scaffold exhibited potent anti-bacterial activity, with EC50 values of 15 μg mL−1 for Xoo, 19 μg mL−1 for Xoc, and 23 μg mL−1 for Xac. Structure–activity relationship analysis indicated that adding an oxime ether or oxime group at the 4-position enhanced anti-bacterial activity while decreasing anti-fungal efficacy. Compounds with a chlorine substituent at 6th position generally showed better results. Furthermore, carboxamide linker at 2nd position improved both anti-bacterial and anti-fungal activities. In terms of anti-bacterial effectiveness, compound 10 outperformed bismerthiazol and thiodiazole copper, highlighting its potential as a promising candidate for further development.
Complementing these findings, Yu et al. synthesized thiochroman-4-one derivatives incorporating carboxamide and 1,3,4-thiadiazole thioether functionalities to further probe their anti-bacterial and anti-fungal properties.36 The biological assays tested their anti-bacterial and anti-fungal properties. The thiochroman-4-one scaffold was used for this purpose. Among the synthesized derivatives, compound 11 (Fig. 2) exhibited the most potent anti-bacterial activity, with EC50 values of 24 μg mL−1 against Xoo and 30 μg mL−1 against Xac. SAR analysis revealed that the presence of a chlorine group at the 6th position and a methylthio group at the terminal end of the heterocyclic linker generally enhanced anti-bacterial activity. The anti-bacterial assays indicated that compound 11 had superior activity compared to bismerthiazol and thiadiazole copper.
 | ||
| Fig. 3 Structure of thiochromene analogues as anti-bacterial agents. | ||
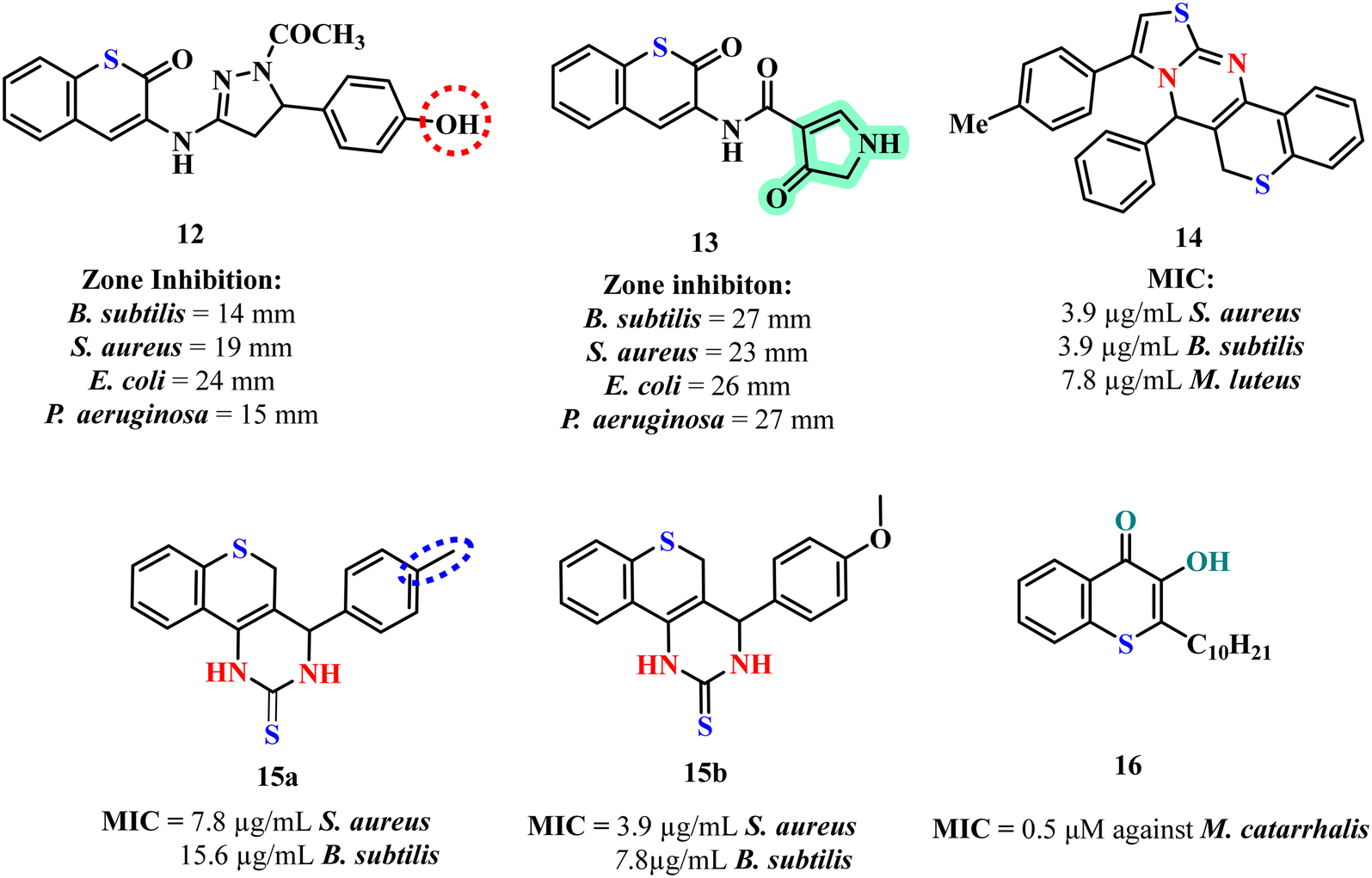
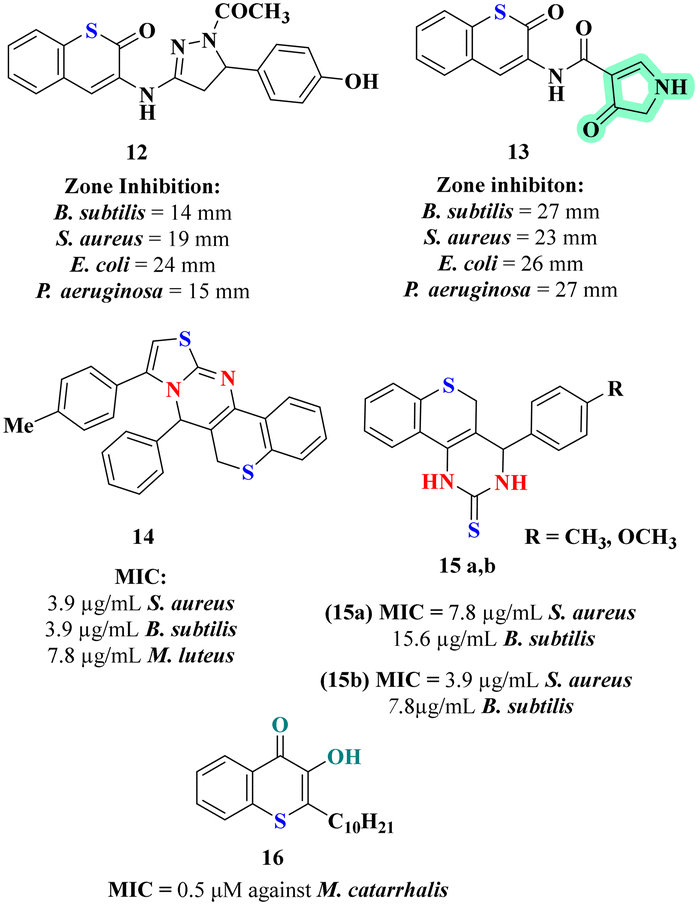
Expanding on the anti-microbial potential of thiochromene derivatives, Elkanzi et al. synthesized and evaluated new compounds featuring heterocyclic moieties and varying linker groups. These derivatives exhibited broad spectrum anti-microbial activity against both Gram-positive and Gram-negative bacteria as well as fungi, as determined by disk diffusion assays.24 The biological activity of these compounds was tested against Gram-positive bacteria (B. subtilis, S. aureus), Gram-negative bacteria (E. coli, P. aeruginosa), and fungi (A. flavus, C. albicans) using the disk diffusion method. The thiochromene derivatives were constructed with various heterocyclic moieties and linkers. Compound 13 (Fig. 3), exhibited the highest activity across all tested microbes with zones of inhibition ranging from 23 to 27 mm. Structure activity analysis demonstrated that compounds featuring smaller heterocycles and shorter linkers generally showed enhanced anti-bacterial activity. Anti-bacterial assays and molecular docking studies underscored compound 13 as a lead anti-microbial agent with exceptional binding affinity and activity at the dihydropteroate synthase (DHPS) active site. Notably, it achieved a binding energy score of −6.53 kcal mol−1, forming three pivotal hydrogen bonds with Asp184, Asn120, and Arg254. These interactions, indicative of its strong affinity for the enzyme's active site, align with its superior anti-microbial efficacy, establishing compound 13 as a promising candidate for further therapeutic exploration.
Suresh et al. developed and evaluated thiochromene-based scaffolds synthesized through a one-pot four-component cascade reaction and tested them for anti-bacterial, antibiofilm, and intracellular reactive oxygen species (ROS) accumulation activities.25 Among the synthesized compounds 14 (Fig. 3) exhibited the most promising biological activity. The SAR analysis revealed that the compound's simple thiazolo-[3,2-a]-thiochromeno-[4,3-d]-pyrimidine scaffold with a neutral hydrogen atom contributed significantly to its anti-bacterial efficacy. In the anti-bacterial assays, compound 14 displayed the lowest minimum inhibitory concentration (MIC) values 3.9 μg mL−1 against Bacillus subtilis, Staphylococcus aureus, indicating superior anti-bacterial activity against Gram positive strains. The increased accumulation of intracellular ROS in S. aureus MTCC 96 treated with compound 14 indicates that oxidative stress plays a critical role in its anti-bacterial mechanism, leading to bacterial cell death through oxidative damage. This highlights potential of compound 14 as an effective anti-microbial agent targeting both planktonic and biofilm associated bacterial infections.
The same group also explored an efficient synthesis route for thiochromeno-[3,4-d]-pyrimidine derivatives using a one-pot three component reaction, and to evaluate their biological activities.26 The thiochromeno-[3,4-d]-pyrimidine scaffold was tested for anti-bacterial, minimum bactericidal concentration (MBC), and antibiofilm activities against various bacterial strains, including S. aureus MTCC 96, S. aureus MLS16 MTCC 2940, and B. subtilis MTCC 121. Among the synthesized compounds, 15a and 15b (Fig. 3) exhibited the most promising results. Compound 15b showed the best anti-bacterial activity, with MIC values of 3.9 μg mL−1 against S. aureus MTCC 96 and MLS16 MTCC 2940, and 7.8 μg mL−1 against B. subtilis MTCC 121. In the SAR analysis, methoxy substituent on compound 15b contributed to its enhanced anti-bacterial activity, while compound 15a methyl substituent also provided significant activity, albeit lower. The main inference from the anti-bacterial assays is that both 15a and 15b demonstrated effective inhibition of bacterial growth, with 15b showing superior activity. The biofilm inhibition assay revealed that compound 15b had the highest antibiofilm activity, disrupting biofilm formation and causing significant biofilm reduction. Additionally, compound 15b induced intracellular ROS accumulation in S. aureus MTCC 96, leading to increased oxidative stress and consequent apoptotic cell death, demonstrating its potential as an effective anti-bacterial agent with dual action of biofilm disruption and oxidative damage.
Szamosvári et al. aimed to evaluate the antibiotic potential of thiochromene derivatives, particularly focusing on their activity against the Gram-negative pathogen Moraxella catarrhalis. Biological assays assessed the inhibitory effects of these compounds, with a specific emphasis on their anti-bacterial properties.37 Among the derivatives tested, 3-hydroxythiochromen-4-one exhibited the most potent activity, achieving a MIC of 0.5 μM against M. catarrhalis. The SAR analysis revealed that the presence of the 3-hydroxyl group was essential for activity, while the introduction of longer alkyl chains further enhanced the anti-bacterial efficacy. The lead compound 16 (Fig. 3) demonstrated remarkable species selectivity, effectively inhibiting M. catarrhalis while having minimal impact on other pathogens and commensals. It was found to target primary energy metabolism, causing rapid ATP depletion, and exhibit low cytotoxicity to eukaryotic cells, underscoring its potential as a highly selective and effective anti-biotic with reduced off-target effects. It was also noted that compound 16 exhibited an unexpected level of selectivity, making them potential candidates for targeted anti-biotic therapy against specific Gram-negative pathogens. The fact that compound 16 did not induce resistance in M. catarrhalis over prolonged exposure suggests its robust effectiveness and a lower likelihood of rapid resistance development. This high level of specificity and efficacy in depleting ATP highlights the potential for these compounds to be used not only as targeted treatments but also as tools for understanding and manipulating microbial energy metabolism.
2.2 Anti-fungal activity
The exploration of anti-fungal activities of thiochromene derivatives continues to yield promising results, showcasing diverse scaffolds and mechanisms of action. Wu et al. introduced spiro-heterocyclic compounds based on the spiro-indoline thiochromane scaffold.38 The anti-fungal activity of these compounds was evaluated, with compound 17 (Fig. 4) showed the most potent inhibition, particularly against invasive fungi like Candida neoformans (8 μg mL−1) and Mucor racemosa (6 μg mL−1), outperforming fluconazole. The SAR analysis indicated that methoxy groups and polar substituents in the fused pyrrolidine ring enhance anti-fungal activity. The study concluded that these compounds exhibit strong selectivity for invasive fungi with low toxicity, making them promising candidates for anti-fungal therapy. | ||
| Fig. 4 Structure of thiochromene/thiochromane analogues as anti-fungal agents. | ||
Building on this framework, Han et al. developed a series of 6-alkyl-indolo-[3,2-c]-2H-thiochroman derivatives, focusing on their anti-fungal potential.39 The biological activity tested was anti-fungal efficacy against invasive fungi, particularly C. albicans and C. neoformans. Compound 18 exhibited the best anti-fungal activity, surpassing even fluconazole and amphotericin B, with an MIC of 4 μg mL−1. SAR analysis revealed that compounds with a pyrrolidine chain linker on the indole ring significantly increased potency molecular docking indicated that Compound 18 (Fig. 4) binds effectively to the N-myristoyltransferase (NMT) active site in C. albicans, forming key hydrophobic interactions but lacking certain hydrogen bonds, which might be optimized in future studies. The anti-fungal assays confirmed high selectivity and potency, with favourable metabolic stability and low toxicity, making these compounds promising candidates for anti-fungal drug development.
Pinedo-Rivilla et al. explored the biotransformation of compound 19a (Fig. 4) and compound 19b using Trichoderma viride to produce new derivatives with anti-fungal properties against the phytopathogen Botrytis cinerea.40 The biological activity tested was anti-fungal efficacy against two strains of B. cinerea. SAR analysis revealed that the bio-transformed products retained strong anti-fungal activity, particularly those with modified functional groups like 1-oxide and 1,1-dioxide derivatives. The anti-fungal assays demonstrated significant potency and selectivity (MIC: 100–250 μg mL−1), with the biotransformation process enhancing activity. The detoxification mechanism of B. cinerea and the high enantiomeric purity of the bio transformed products suggest potential for developing selective anti-fungal agents with low toxicity.
Adding to the advancements in thiochromene based anti-fungal scaffolds, Song et al. synthesized 2-(indole-3-yl)-thiochroman-4-ones using an ionic liquid-mediated approach, specifically against various fungal strains including C. albicans, C. neoformans, M. racemosa, Microsporum gypseum, and Epidermophyton floccosum.41 The structural scaffold employed was the thiochroman-4-one core, with derivatives showing significant anti-fungal activity, particularly compound 20 (Fig. 4) which achieved MIC value at 4 μg mL−1 against C. albicans. SAR analysis indicated that electron withdrawing groups on the 6th position of thiochroman-4-one ring enhance anti-fungal activity, while substitution of halogen such as chlorine on the indole ring further improved efficacy. The anti-fungal assays demonstrated superior activity to fluconazole, highlighting the potential of these compounds as effective anti-fungal agents with favourable selectivity and low toxicity.
Parallel to these efforts, Wang et al. explored the anti-fungal potential of 4-chloro-2H-thiochromenes featuring nitrogen containing side chains, focusing on their inhibitory activities against fungal pathogens such as C. albicans, C. neoformans, and C. tropicalis.42 The structural scaffold used was the 4-chloro-2H-thiochromene core, with compounds 21a and 21b (Fig. 4) demonstrating the most potent anti-fungal activity exhibiting lowest MIC value at 0.25 μg mL−1 against C. neoformans (compound 21a) and 2 μg mL−1C. tropicalis (compound 21b). The SAR analysis revealed that the inclusion of nitrogen containing moieties, particularly aliphatic amine and azole fragments, significantly enhanced anti-fungal efficacy. The anti-fungal assays indicated that these compounds exhibited excellent selectivity with minimal toxicity on mammalian cells as evidenced by the MTT assay positioning them as promising lead structures for the development of novel anti-fungal agents.
Zhong et al. investigated the anti-fungal potential of thiochroman-4-one derivatives as inhibitors of NMT, a validated target for treating fungal infections.43 The biological activity tested included anti-fungal efficacy against strains such as C. albicans, C. neoformans, E. floccosum, Mucor racemosa, M. gypseum, and A. niger. The structural scaffold employed was the thiochroman-4-one core showing the most potent anti-fungal activity, particularly compound 22, (Fig. 4) which exhibited MIC values as low as 0.5 μg mL−1 against C. albicans. The SAR analysis revealed that unsubstituted phenyl ring at 3rd position was more potent when compared with electron withdrawing or donating groups, replacing the N-terminal with morpholinyl or furfuryl in place of benzyl or phenylethyl chain significantly decreased activity. The anti-fungal assays demonstrated that these compounds were more effective against deep fungi, with compound 22 showing comparable or superior activity to amphotericin B. Molecular docking studies confirmed the strong binding affinity of compound 22 to the active site of C. albicans NMT (CaNMT), supporting its potential as a high performance NMT inhibitor with favourable selectivity and binding efficiency.
2.3 Anti-tubercular activity
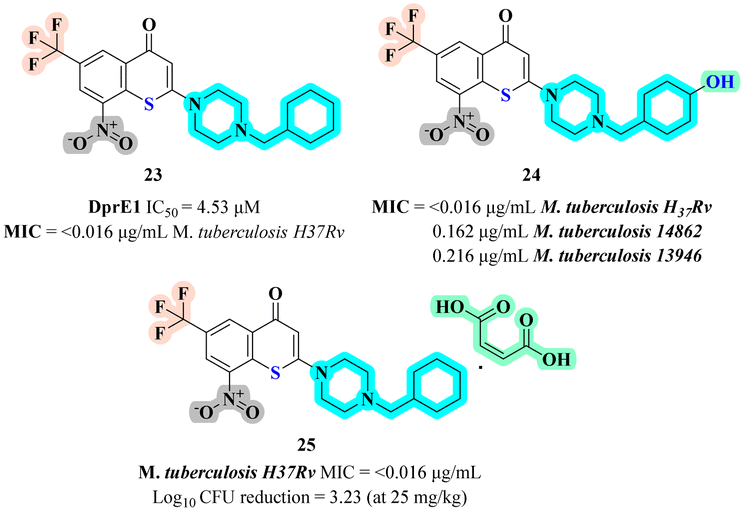
Li et al. designed and evaluated three novel series of benzoxazinone, benzothiopyranone, and benzopyranone derivatives as potential inhibitors of DprE1, targeting Mycobacterium tuberculosis.44 The biological activity tested including in vitro anti-mycobacterial efficacy against Mycobacterium tuberculosis and cytotoxicity against Vero cells. Among the scaffolds, the benzothiopyranone series exhibited the best activity with compound 23 (Fig. 5) demonstrating potent activity against both drug susceptible and drug-resistant tuberculosis strains with IC50 value of 4.53 μM and MIC of <0.016 μg mL−1 against M. tuberculosis H37Rv, low cytotoxicity, and excellent pharmacokinetic (PK) properties. The SAR analysis revealed that lipophilicity and the presence of a sulfur atom at the 1st position, substitution with cyclohexyl methyl piperazine at 2nd position was found to be most potent among the series. In anti-mycobacterial assays, compound 23 showed strong efficacy against XDR-TB strains and excellent selectivity, with favourable metabolic stability and pharmacokinetics. Docking studies confirmed that the anti-mycobacterial effect is primarily due to the inhibition of the DprE1 enzyme, making compound 23 a promising clinical candidate for tuberculosis treatment. | ||
| Fig. 5 Structure of thiochromene/thiochromane analogues as anti-tubercular agents. | ||
The same group evaluated the anti-mycobacterial activity of novel benzothiopyranone derivatives, particularly focusing on their efficacy against various M. tuberculosis strains, including drug-resistant clinical isolates.45 The structural scaffold employed in this research was derived from the active metabolite 24 (Fig. 5) of the anti-tubercular agent 23. Benzothiopyranones containing ester, sulfonate, and phosphoryl motifs were explored, containing a benzoate fragment, exhibiting the most potent in vitro activity and metabolic stability. The SAR analysis revealed that presence of hydroxy group at 4th position of 1-(cyclohexylmethyl)piperazine, significantly enhanced anti-mycobacterial potency. In various assays, compound 24 demonstrated potent activity against both drug susceptible and drug-resistant strains, with low cytotoxicity and good hepatocyte stability. Docking studies and metabolic stability assays further supported the potential of this compound as promising leads for the development of new anti-TB agents.
Continuing with their investigation, Li et al. optimized the solubility and pharmacokinetic properties of nitro benzothiopyranone derivatives, targeting the DprE1 enzyme, to identify a promising preclinical candidate for treating tuberculosis.46 The biological activity tested was the anti-mycobacterial efficacy against M. tuberculosis H37Rv, with a focus on evaluating various salts of compound 23. The maleate salt 25 (Fig. 5) demonstrated potent in vivo anti-mycobacterial activity and superior pharmacokinetic properties, including higher oral bioavailability and plasma exposure in rats displayed comparable bactericidal activity in the lungs of mice to the front-line drug isoniazid at 25 mg kg−1 (3.23 vs. 3.89![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) log10 CFU reduction) when compared with fumarate, citrate, hydrochloride salt. The SAR analysis indicated that the salt formation significantly enhanced aqueous solubility, which correlated with improved bioavailability and in vivo efficacy. Anti-mycobacterial assays confirmed that salt was highly effective, with activity comparable to the front-line drug isoniazid at certain doses. The study concluded that the maleate salt is a promising preclinical candidate with favourable stability, selectivity, and metabolic properties.
log10 CFU reduction) when compared with fumarate, citrate, hydrochloride salt. The SAR analysis indicated that the salt formation significantly enhanced aqueous solubility, which correlated with improved bioavailability and in vivo efficacy. Anti-mycobacterial assays confirmed that salt was highly effective, with activity comparable to the front-line drug isoniazid at certain doses. The study concluded that the maleate salt is a promising preclinical candidate with favourable stability, selectivity, and metabolic properties.
2.4 Anti-viral activity
Ghosh et al., evaluated a series of novel HIV-1 protease inhibitors featuring carboxamide derivatives as the P2 (ATP, ADP, UTP and UDP) ligands, with a focus on improving enzyme inhibition and anti-viral activity. Biological activity was tested through HIV-1 protease inhibition assays and anti-viral efficacy in MT-4 human T-lymphoid cells.47 The structural scaffolds employed included aminothiochromane and aminotetrahydro naphthalene derivatives, with compound 26 (Fig. 6) demonstrating the highest potency (IC50 = 47 nM). Structure–activity analysis revealed that sulfone derivatives, significantly enhance both enzyme inhibition and anti-viral activity compared to their sulfide counterparts, due to improved hydrogen bonding interactions with the protease active site. The X-ray crystal structure of compound 26 showed that effective interactions with key residues in the HIV-1 protease active site, such as Asp29 and Asp30, are crucial for high potency, while variations in the P2 ligand's stereochemistry and functional groups affect the binding and activity. These insights highlight the importance of optimizing ligand interactions to enhance anti-viral efficacy and enzyme selectivity. | ||
| Fig. 6 Structure of thiochromene/thiochromane analogues as anti-viral agents. | ||
Further expanding the anti-viral potential Sepay et al. explored the potential of benzylidene chromanones, specifically designed to inhibit the mutant SARS-CoV-2 main protease (Mpro) enzyme, using molecular docking, bioinformatics, and molecular electrostatic potential analyses.48 Among these compounds 27 (Fig. 6) demonstrated the highest binding affinity to SARS-CoV-2 Mpro, suggesting its efficacy in blocking viral replication. The addition of two hydroxyl (–OH) and one amino (–NH2) group enhances water solubility. The compound shows high gastrointestinal absorption but no blood–brain barrier (BBB) permeability. Molecular electrostatic potential (MEP) analysis of compound 27 shows increased electron density due to the electron-donating effects of –OH and –NH2. This favors strong π-stacking interactions. Docking studies with CoV Mpro proteins confirm that compound 27 binds more strongly than related compounds, highlighting its enhanced interaction with the active site.
2.5 Anti-parasitic activity
 | ||
| Fig. 7 Structure of thiochromene/thiochromane analogues as anti-leishmanial agents. | ||
Building on the anti-parasitic potential of thiochromene derivatives, the same group further explored the scaffold, substituted primarily at the 2nd or 3rd positions with functional groups like carbonyl or carboxyl groups. These modifications were aimed at enhancing anti-leishmanial activity, particularly against intracellular amastigotes of Leishmania (V) panamensis. Out of the synthesized compounds, around 12 compounds showed significant activity (EC50 < 40 μM), and four of them demonstrated high efficacy (EC50 < 10 μM) with favourable selectivity indices (SI > 2.6), despite structural similarities, compounds which contained a methyl ester at 3rd position, were much less effective, emphasizing the importance of precise substitutions for activity. Compounds 29a and 29b, (Fig. 7) featuring alkyl chains at 2nd position also exhibited strong leishmanicidal potential, while those with phenyl groups experienced reduced activity and increased cytotoxicity. The SAR across these derivatives was complex, with no clear pattern observed in terms of substitution and anti-leishmanial activity. However, the presence of electron-withdrawing groups at 2nd position, such as nitriles or trifluoromethyl groups, appeared beneficial in some cases. Moreover, the importance of carbonyl substituents at 3rd position was underscored by the inactivity of thiochroman-4-one compounds without such groups. These results suggest that steric and electronic factors play a significant role in the interaction of these compounds with potential leishmanial enzyme targets.
Ortiz et al. focused on the design and synthesis of various substituted 2H-thiochroman derivatives as potential anti-leishmanial agents. 4H-Thiochromen-4-one-1,1-dioxide derivatives for their activity against Leishmania donovani, the causative agent of visceral leishmaniasis.50 The tested cell line was J774A.1 macrophage cells, commonly used in anti-leishmanial studies to assess cytotoxicity and parasite killing efficacy. Among the synthesized compounds, compound 30 (Fig. 7) exhibited potent anti-leishmanial activity with an EC50 value of 3.96 μM against Leishmania donovani. The SAR studies revealed aromatic substituents at 3rd position with strong electron withdrawing groups (CF3) cause increase in polar area and close proximity to the heterocycle and sulfone group, is the most effective combination to selective increase potency, replacement with alkyl chain at 3rd position reduced both selectivity and potency against the leishmanial parasite.
Extending the focus to cutaneous leishmaniasis (CL), Upegui et al. synthesized thiochroman-4-one hydrazone derivatives.51 Following the synthesis and characterization of these compounds, their cytotoxicity was assessed via MTT assay, and anti-leishmanial activity was evaluated against Leishmania (Viannia) panamensis and L. (V) braziliensis amastigotes in macrophages using flow cytometry. Among the tested derivatives, compound 31 (Fig. 7) with a phenyl hydrazone linker at 4th position displayed significant anti-leishmanial activity with an EC50 value of 62.2 μg mL−1, indicating its effectiveness against Leishmania (Viannia) braziliensis intracellular amastigotes in macrophages. In addition, compound 31 exhibited potential wound healing properties, enhancing fibroblast migration and reducing inflammation. Therefore, compound 31 emerged as the superior candidate highlighting its dual role in treating CL and promoting wound healing.
Vargas et al. designed a series of thiochroman-4-one derivatives as a promising scaffold for developing effective anti-leishmanial agents against Leishmania panamensis.52 The synthesized compounds were evaluated against intracellular amastigotes using a cell viability assay protocol with human monocytes (U-937 ATCC CRL-1593.2). Among the compounds tested, those bearing a vinyl sulfone moiety demonstrated significant activity, with compound 32 exhibit EC50 of 3.24 μM and a high selectivity index (SI) of 173.24, surpassing the reference drug amphotericin B. Molecular docking studies indicated favourable interactions between compound 32 (Fig. 7) and key biological targets in Leishmania, contributing to its strong anti-leishmanial activity. The SAR analysis revealed that introducing a vinyl sulfone group significantly enhanced the anti-leishmanial activity of thiochromone derivatives. Notably, fluorine substitution at the 6th position increased leishmanicidal activity, highlighting the importance of specific modifications in optimizing therapeutic efficacy.
Further advancing the structural optimization of thiochromene derivatives, Vargas et al. synthesized acyl hydrazone derivatives to combat L. panamensis.53 They synthesized around 18 acyl hydrazone derivatives and screened them for in vitro anti-leishmanial activity against intracellular amastigotes using human monocytes (U-937 ATCC CRL-1593.2). The results showed varying activities, with compound 33, (Fig. 7) a semicarbazone derivative, demonstrating the best efficacy at an EC50 of 5.4 μM and low cytotoxicity (LC50 of 100.2 μM), resulting in a high selectivity index (SI) of 18.6. Molecular docking studies indicated favourable interactions between compound 33 and key parasite targets. The SAR analysis revealed that incorporating hydrazone moieties enhanced the activity of thiochroman-4-ones. Compound 33 had the highest activity, while specific phenyl substitutions positively influenced efficacy. These findings highlight the significance of structural optimization in developing effective and safe agents against Leishmaniasis.
 | ||
| Fig. 8 Structure of thiochromene/thiochromane analogues as anti-malarial agents. | ||
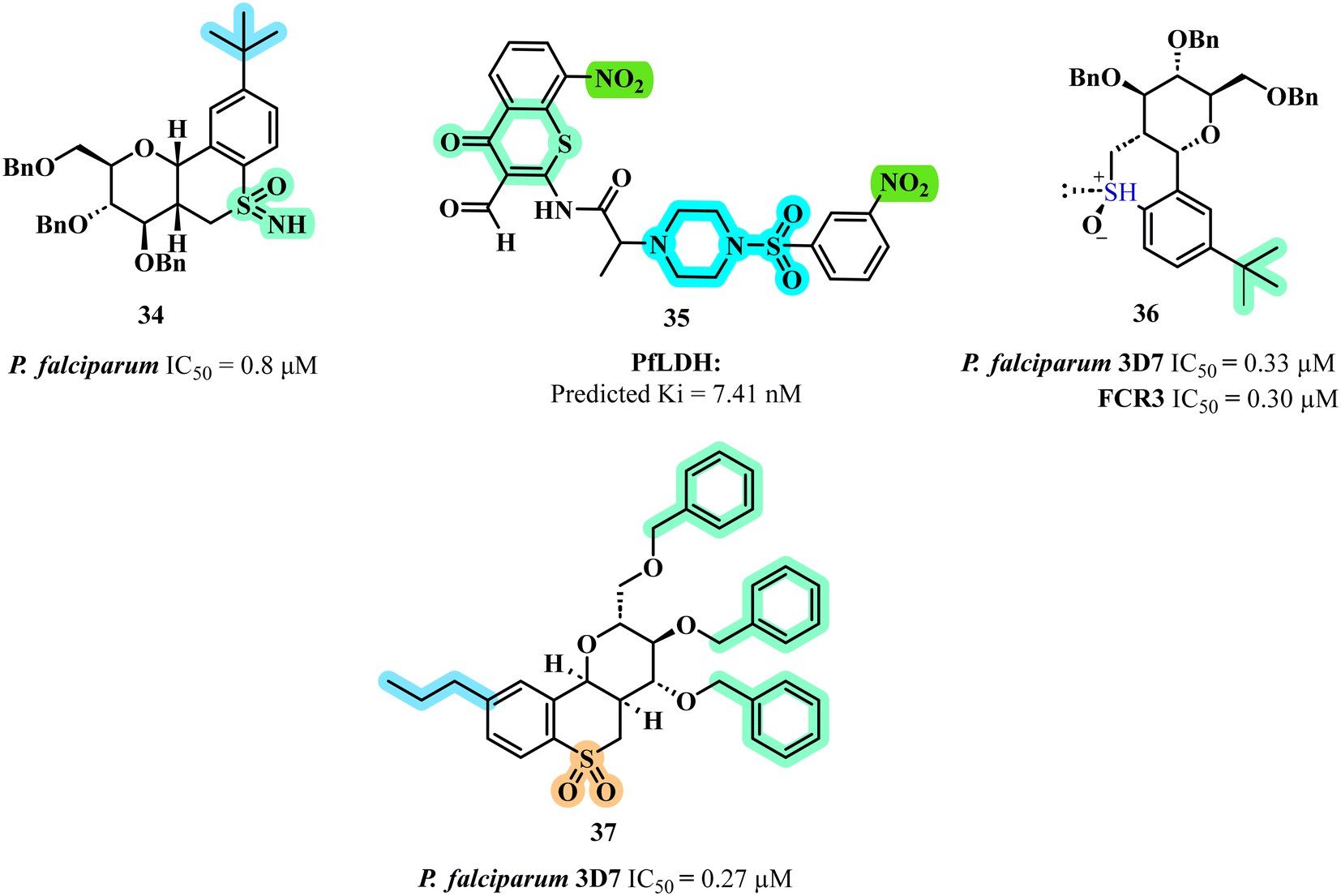
Building on the pursuit of novel anti-malarial agents, Dey et al. explored chromone and thiochromone derivatives as inhibitors of P. falciparum lactate dehydrogenase (PfLDH).55 The most promising thiochromone compound identified against malaria was compound 35 (Fig. 8) which demonstrated the highest docking score and exhibited a significant number of interactions with the PfLDH target. In silico studies revealed robust binding affinity, with a predicted Ki value of 7.41 nM, highlighting its potential as an effective therapeutic agent. The SAR analysis indicated that the presence of a nitro group at the 8th position of the thiochromone moiety, combined with a sulphonyl piperazine substitution at the 3rd position of the phenyl ring, enhanced the compound's inhibitory activity. Key interactions were observed, including hydrogen bonding with crucial amino acids such as THR 97 and HIS 195, as well as pi–alkyl interactions with PRO 250 and ILE 31, underscoring the compound's favourable binding profile. Molecular dynamics (MD) simulations further demonstrated stable conformational behaviour at the active site, with consistent interactions maintained throughout the simulation period.
Further diversifying the thiochroman scaffold, Kinfe et al. synthesized carbohydrate-fused thiochroman derivatives to combat chloroquine-sensitive and chloroquine-resistant strains of P. falciparum.56 The synthesis involved an efficient nucleophilic displacement reaction, resulting in the formation of thiochromane derivatives with various aromatic substituents. These compounds were subsequently oxidized to their sulfoxide and sulfone forms to enhance molecular diversity. The anti-malarial activity was assessed against both chloroquine-sensitive (3D7) and chloroquine-resistant (FCR3) strains of P. falciparum using a pLDH assay. Among the tested compounds, compound 36 (Fig. 8) which featured a bulky, lipophilic tert-butyl group at the para-position of the aromatic ring, exhibited the highest potency with IC50 values of 0.33 and 0.30 μM against both strains respectively. The SAR studies revealed that the oxidation state of sulfur and the presence of lipophilic substituents were critical for enhancing anti-malarial activity, with sulfoxides and sulfones demonstrating superior efficacy compared to sulfides. Furthermore, the configuration of the sugar moiety's anomeric position significantly influenced potency, underscoring the importance of optimizing both lipophilicity and polarity in the design of effective anti-malarial compounds.
In a complementary investigation, Madumo et al. synthesized carbohydrate-derived thiochromane derivatives to explore the interplay between aromatic alkyl substituents and sugar stereochemistry on anti-malarial activity.57 The compounds were synthesized and evaluated for their in vitro anti-malarial activity against the chloroquine-sensitive 3D7 strain of P. falciparum. Among the derivatives tested, compound 37 (Fig. 8) featuring a n-propyl substituent, was identified as the most potent, exhibiting an IC50 value of 0.27 μM. This compound's high activity is attributed to its short chain lipophilic substituent, the presence of a benzyl ether protecting group, and the equatorial orientation of the 4th position substituent on the sugar moiety, which is crucial for enhancing anti-malarial efficacy. The SAR analysis indicated that shorter branched alkyl chains are favourable for activity, while longer linear chains diminished effectiveness. Additionally, the findings underscored the importance of the benzyl protecting group and the glucosyl configuration in maximizing the anti-malarial potential.
 | ||
| Fig. 9 Structure of thiochromane analogues as anti-trypanosomal agents. | ||
For further exploration of anti-parasitic potential, Ortiz and co-workers evaluated 4H-thiochromen-4-one-1,1-dioxide derivatives for their anti-trypanosomal activity.50 The metacyclic trypomastigotes of Trypanosoma cruzi were used to assess the potency of these compounds in selectively targeting the parasite. Among the series, compound 39 (Fig. 9) exhibited the highest inhibition in the anti-trypanosomal assay (EC50 = 24.33 μM), however its potency against leishmaniasis was comparatively higher, with an EC50 of 9.30 μM. Structural relationship within the series shows a significant loss of activity when the aromatic groups were deactivated by the trifluoromethyl substituent as it moved away from the sulfur atom.
3 Structure activity relationship for the development of potent anti-microbial agents
3.1 SAR of thiochromane
The SAR of thiochromanes reveals how specific structural modifications influence anti-microbial activity across different pathogens (Fig. 10). Replacing the oxo group with oxime or oxime ether enhances anti-bacterial efficacy but reduces anti-fungal potency.26 The inclusion of N-methyl-2-phenyl benzimidazole at 6th position boosts activity against both Gram-positive and Gram-negative bacteria, with the preferred R2 substitution order for anti-bacterial activity being Cl > F > Me.25 Anti-fungal activity, however, is enhanced by substitutions at 8th position (F, Me, Cl) rather than 6th, and a 2-carbon side chain at 6th position is critical for anti-fungal potency, which declines when replaced by benzofuran.32,33 Substituting methoxyphenyl at 3rd position promotes stronger anti-fungal activity compared to fluorophenyl or chlorophenyl.34 The sulfur atom in the thiochromane ring is pivotal for activity against various pathogens; replacing it with sulfoxide makes the compound inactive against Leishmania species but enhances anti-malarial properties.44,48 Presence of amide side chain also boosts anti-leishmanial potency, with longer carbon side chains decreasing anti-malarial efficacy.41,49 The SAR analysis of thiochromane therefore highlights the importance of targeted substitutions for enhancing the anti-microbial activity of thiochromanes. For anti-bacterial properties, N-methyl-2-phenyl benzimidazole at 6th position and electron-withdrawing groups at R2 are crucial, while anti-fungal efficacy depends on 6th and 8th position substitutions. The sulfur atom's modification, particularly to sulfoxide, reveals an interesting trade-off between anti-malarial and leishmanial activities. | ||
| Fig. 10 Structure–activity relationship of thiochromanes: key modifications for enhanced anti-microbial activity. | ||
3.2 SAR of thiochromen-2-one analogs
The SAR of thiochromen-2-one shows that electron-withdrawing groups at 6th position enhance anti-fungal and anti-leishmanial activity, while acyclic aliphatic side chains at 3rd are more effective than cyclic amines for anti-fungal effects.14,15 Carboxamide linkers at 3rd position and pyrolo substituents improve anti-bacterial activity, while thioxapyrazole motifs decrease it. Sulfoxide substitution boosts Leishmanial activity, and aminopyrazole/pyrimidine maintains strong anti-bacterial efficacy.48 Electron-withdrawing groups at 6th position and specific side chain modifications at 3rd are crucial for optimizing anti-fungal, anti-bacterial, and anti-leishmanial properties. Sulfoxide and carboxamide linkers improve activity, while thioxapyrazole decreases effectiveness, highlighting the need for precise structural tuning to enhance potency (Fig. 11).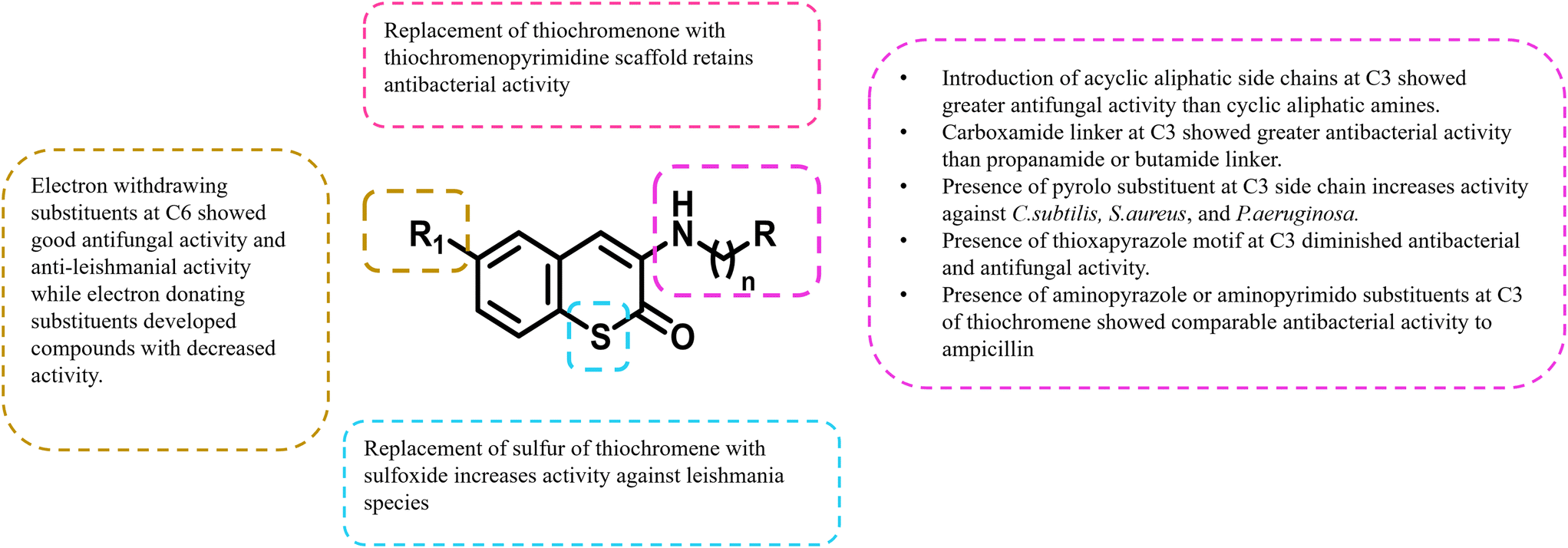 | ||
| Fig. 11 Structure–activity relationship of thiochromen-2-ones: key modifications for enhanced anti-microbial activity. | ||
3.3 SAR of thiochromen-4-one analogs
The SAR of thiochromen-4-ones highlights that an –OH group at 3rd position is essential for activity against M. catarrhalis, while sulfur oxidation decreases anti-bacterial potency.28 Cyclohexane carboxylate and tertiary amino groups at 2nd position enhance anti-mycobacterial activity, and phenylethyl or decyl substitutions at 2nd position improve anti-bacterial effects. Pyridine-4-carboxylate boosts anti-mycobacterial activity and reduces toxicity, but replacing thiochromenone with benzoxazinone or benzopyranone lowers efficacy. These findings suggest that the structural integrity of the thiochromenone ring, particularly with the –OH group at 3rd position and selective side-chain substitutions, is essential for maintaining and enhancing anti-bacterial and anti-mycobacterial activities. Careful optimization of side chains and functional groups can lead to more potent and selective anti-microbial agents (Fig. 12). | ||
| Fig. 12 Structure–activity relationship of thiochroman-4-ones: key modifications for enhanced anti-microbial activity. | ||
4 Anti-cancer activity
4.1 Thiochromane derivatives
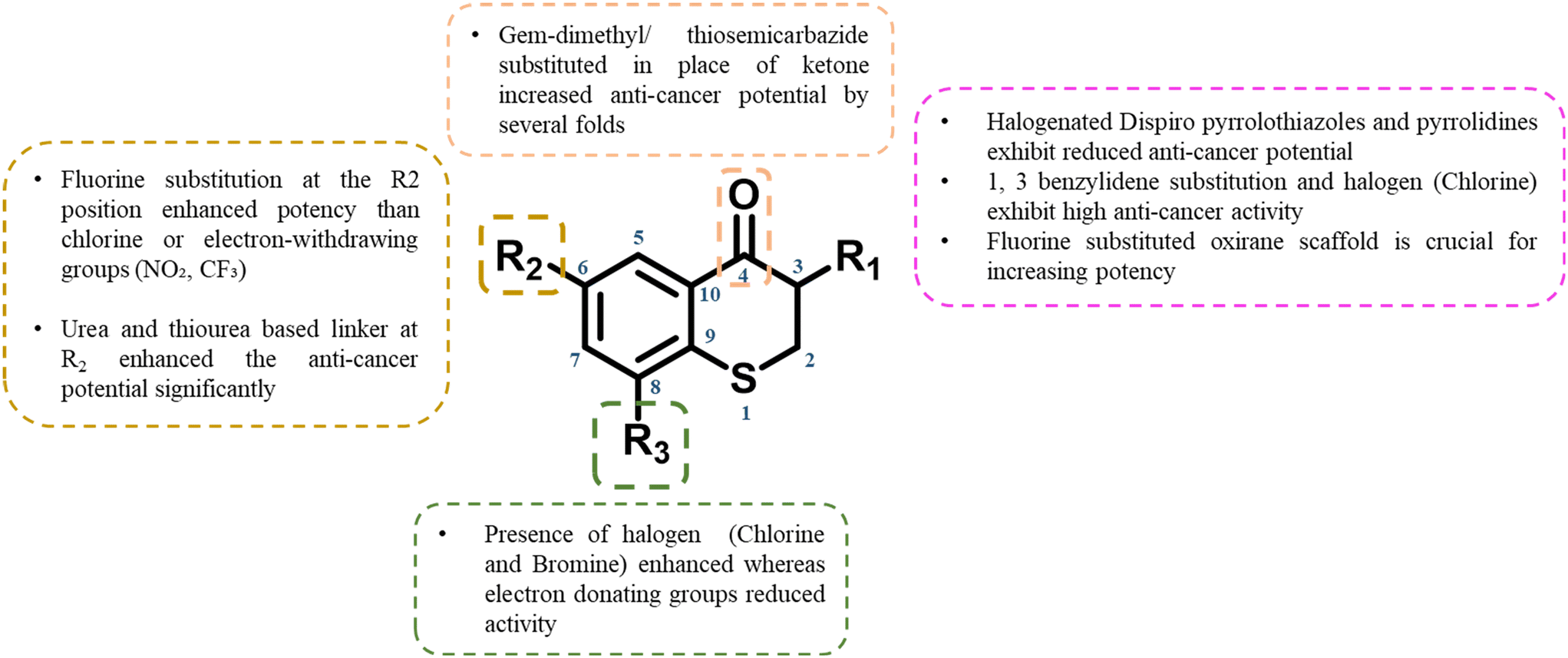
Bharkavi and co-workers, synthesized dispiro-indeno pyrrolidine/pyrrolothiazole–thiochroman hybrids and investigated their anti-cancer efficacy against the CCRF-CEM, HT29, and MCF7 cell lines, employing doxorubicin as the reference standard and DMSO as the solvent control.59 Each compound demonstrated over 50% inhibition across CCRF-CEM/HT29/MCF7 cell lines at a concentration of 50 μM. Compound 40 (Fig. 13) with an IC50 of 45.79 μM, emerged as the most potent, displaying superior activity compared to the reference drug doxorubicin (IC50 of 55.91 μM) against the CCRF-CEM cell line. From the aggregated data, it is discernible that halogen-substituted dispiropyrrolothiazoles demonstrate enhanced efficacy, with activity modulation following the trend: meta > para > ortho > ortho, para (di-substituted). The presence of alkyl and 2-thienyl groups on the aryl moiety of the di-spiroheterocycles correlates with a diminution in anti-cancer activity. | ||
| Fig. 13 Structure of thiochromane analogues as anti-cancer agents. | ||
Extending the exploration of thiochroman derivatives, Demirayak and co-workers synthesized chroman-4-one/thiochroman-4-one compounds and assessed their activity across five concentrations. The compounds exhibited tumor cell growth percentages ranging from −11.63% to 103.44%. Notably, compounds with the presence of a 1,3-benzylidene moiety, demonstrated the highest anti-proliferative activity, whereas derivatives with a 1,4-benzylidene moiety did not exhibit higher activities.60 Among the cancer types tested, leukemia, melanoma, and colon cancer cells were the most sensitive to the compounds. The lowest growth value (100.00%) was observed for compound 41 (Fig. 13) against cell lines S (leukemia), LOXIMVI (melanoma), and SW-620 (colon cancer). Compounds with growth percentages under 80% underwent further testing, and their log10 GI50 and MG-MID values were determined. The lowest concentrations (MG-MID) were reached for compound 41 was (−5.83), which correlated with its growth percentage values thereby study indicating that thiochromanone compounds containing a 1,3-disubstituted benzylidene residue exhibit exceptionally high anti-cancer activity.
Adding to the structural diversity, Farghaly and co-workers synthesized thiazole and thiadiazole-based analogs fused with thiochromane. The in vitro data suggests that thiochromane fused with substituted 1,3,4-thiadiazole nucleus demonstrated potent anti-cancer activity for tested three cancer lines.61 Compound 42 (Fig. 13) exhibited significant potential across tested cell lines (MCF-7 IC50 = 3.25 μM, HT29 IC50 = 3.12 μM, HepG2 IC50 = 2.67 μM) and further concluded that the most reactive thiadiazole derivative compound 42 which carry ester group at position 2 in thiadiazole moiety is responsible for its increased activity.
Advancing the SAR investigations, Nammalwar and co-workers synthesized SHetA2 (NSC-721689) analogs targeting ovarian cancer (A2780). The primary aim of this investigation was to elucidate a comprehensive SAR, and hypothesize the implications of these SAR findings on the binding interactions with a presumptive target protein. SHetA2 served as the benchmark for comparative analysis. Electron-withdrawing substituents at 4th position, such as trifluoromethyl and trifluoromethoxy, exhibited potency and efficacy on par with or superior to SHetA2. Conversely, electron-donating groups, the screening of various functional groups on the new derivatives facilitated the identification of several compounds exhibiting greater potency than SHetA2. Particularly, compound 43 (Fig. 13) (IC50 = 1.86 μM, 95.6% efficacy), emerged as the most potent analog of SHetA2 (IC50 = 3.17 μM, 84.3% efficacy) against the A2780 ovarian cancer cell lines. For ureas and thioureas, analogs bearing 4′ electron-withdrawing groups generally showed robust activity.62
In another notable contribution, Pandya et al. developed 3-arylspiro-[oxirane-2,3′-thiochroman]-4′-one compounds, demonstrating superior anti-cancer activity. Among the synthesized compounds, compound 44 with 3-methoxyphenyl substituent exhibited GI50 values below 10 μM, surpassing the standard anti-cancer drug adriamycin (ADR) in efficacy. These compounds demonstrated a significant inhibitory profile against the human lung cancer cell line A-549. The superior activity of compound 44 (Fig. 13) is characterized by a fluorine substituent at the 6th position of the thiochromane ring and a meta-substituted methoxy group in phenyl ring. Consequently, the structural framework of these α,β-epoxyketones represents a promising new class of anti-cancer agents.63,64
In another innovative approach, Song Ya-li et al. synthesized thiochromane bisindollylalkanes. The inhibitory effects of these compounds were evaluated across all tested carcinoma cell lines (MCF7 and A549).65 The presence of an electron-withdrawing group at the 8th position appeared to be associated with a general increase in activity. Among them, compound 45 (Fig. 13) exhibited the lowest IC50 value at 7.79 μg mL−1 for the MCF7 cell line. Inhibition of topoisomerase relaxation was evaluated using topoisomerase II with VP-16 as positive control. Compound 45 showed comparable inhibitory activity (91.1%) to VP-16 (90.3%) 100 μM concentration.
Further expanding the repertoire of thiochromane derivatives, Yali Song et al. synthesized and evaluated a novel series of compounds that combine the active pyrazoline moiety with a thiochroman-containing indole skeleton. Most of the compounds demonstrated pronounced anti-proliferative activity against MGC-803, exhibiting lower cytotoxicity on normal cells compared to the positive control. Notably, compound 46 (Fig. 13) displayed significant anti-proliferative activity (MCF-7 IC50 = 26.9 μM, Hela IC50 = 29.25 μM, MGC-803 IC50 = 26.53 μM, Bel-7404 IC50 = 23.73 μM, L929 IC50 = 133.9 μM) across all the assayed cell lines. Flow cytometric analysis revealed that this class of compounds can significantly induce apoptosis in MGC-803 cells and arrest the cell cycle at the G2/M phase. Investigation into the SAR indicated that chlorine-substituted groups play a crucial role in enhancing both enzyme inhibition and cytotoxicity of the compounds. Compound 46 underwent further analysis to elucidate its mechanism of action through cleavage reaction assays, DNA unwinding assays, and UV-titration assays. The results indicated that compound 46 acts as a non-intercalative Topo II catalytic inhibitor.66 Finally, molecular docking analysis suggested that compound 46 binds firmly within the catalytic cavity of Topo II via hydrogen bonding, halogen bonding, and hydrophobic interactions, without intercalating into DNA base pairs, consistent with DNA unwinding assay results. Molecular dynamics simulations provided mechanistic insights into the interaction phenomena between these compounds and topoisomerase.
Adding to the array of potent scaffolds, Song Jiangli and co-workers carried the synthesis and anti-cancer activity of thiosemicarbazones derivatives of thiochromanones and related scaffolds. In comparative analysis with the standard cisplatin, notably, compound 47 (Fig. 13) emerged with exceptional efficacy, displaying IC50 values of 0.42 μM against MCF-7, 0.58 μM against SK-mel-2, and 0.43 μM against DU145. The enhancement in cytotoxic activity conferred by the thiochromane scaffold with thiosemicarbazone at 4th position and presence of halogen especially fluorine was corroborated through SAR analysis. Mechanistic investigations revealed that compound 48 effectively induces cell cycle arrest at the G2/M phase in MCF-7 cells and triggers apoptosis through a dose-dependent increase in intracellular reactive oxygen species (ROS) levels.67 Consequently, thiochromanone-derived thiosemicarbazones exhibit significant potential as anti-cancer agents, leveraging ROS-mediated cytotoxic mechanisms against human breast cancer MCF-7 cells.
Thanh C. Le et al. synthesized novel heteroarotinoids with anti-cancer activity against ovarian cancer cells, the sulfur containing heteroarotinoids demonstrated significant inhibition of ovarian cancer cell proliferation within the micromolar concentration range.68 The potencies of these compounds were quantified through the determination of EC50 values, compound 49 (Fig. 13) bearing a nitro substitution and thiourea displayed highest potency against A2780 (EC50 = 1.72 μM), normal Endometrium (EC50 = 2.96 μM). Preliminary evidence from in vivo suggests that compound 49 can be achieved and maintained in micromolar concentrations in animal models while the mechanism of action to induce apoptosis occurs through direct effects on mitochondria and mitochondrial proteins. Moreover, at 3 μM concentration, the effects of compound 49 on differentiation has been observed by a normalization of the cancerous cell phenotype and induction of orderly tissue architecture.
 | ||
| Fig. 14 Structure–activity relationship (SAR) of thiochromane: key modifications for anti-proliferative activity. | ||
4.2 Thiochromene derivatives
Luque-Agudo and co-workers synthesized thiochromene derived from carbohydrates and tested their and anti-cancer potential. Notably, diastereomeric sulfa-Michael adducts both demonstrating moderate anti-proliferative effects across the six cell lines tested (A-549, HBL-100, HeLa, and SW1573 as well as T-47D (breast) and WiDr (colon)), with GI50 values ranging from 15 to 47 μM. Interestingly, a substantial divergence in activity was observed between the diastereomeric thiochromenes 49a and 49b (Fig. 15). Specifically, while compound 49a exhibited no significant activity even at the highest concentration tested (100 μM), compound 49b showed moderate anti-proliferative activity, with GI50 values ranging from 25 to 35 μM. Remarkably, compound 49b demonstrated comparable or enhanced potency relative to 5-fluorouracil. Conversely, the presence of a carbohydrate chain at the 2nd position in parent scaffold was found to attenuate anti-proliferative activity against A-549 cells compared to their unsubstituted counterparts.69 | ||
| Fig. 15 Structure of thiohromene analogues as anti-cancer agents. | ||
Building on the structural diversity of thiochromene scaffolds, Barakat et al. synthesized and evaluated the anti-cancer potential of novel spirooxindole pyrrolidine-grafted thiochromene derivatives.70 The anti-proliferative activity of the spiro-oxindole/pyrrolidine/thiochromene series against the PC3 prostate cancer cell line revealed that compound 50 (Fig. 15), which incorporates a C-5 chlorine atom on the isatin moiety and a para-trifluoromethylphenyl group, with an IC50 value of 8.4 ± 0.5 μM. However, replacing the C-5 chlorine on the isatin ring with a C-5 nitro group resulted in a complete abrogation of anti-proliferative activity. In contrast, replacing the C-5 chlorine-substituted isatin moiety with a C-5 fluoro-substituted isatin resulted in a marked decrease in anti-cancer activity, this decrease in potency was further exacerbated which features a para-bromo group instead of a para-nitrophenyl moiety.
In a complementary study, Salerno S. and co-workers investigated benzothiopyranoindole and pyridothiopyranoindole based anti-proliferative agents targeting topoisomerases. The anti-proliferative efficacy of the newly synthesized derivatives was assessed in vitro against three human tumor cell lines: HeLa (cervix adenocarcinoma), A-431 (squamous carcinoma), and MSTO-211H (biphasic mesothelioma).71 The compounds exhibited significant anti-proliferative activity across the tested cell lines, with GI50 values ranging from 0.31 to 6.93 μM, in some instances rivaling the potency of ellipticine, which served as the reference standard. Notably, the benzothiopyranoindole derivative 51 (Fig. 15) emerged as the most potent, with IC50 values spanning from 0.31 to 0.82 μM across all cell lines evaluated.71 Structurally, this enhanced activity can be ascribed to the presence of a dimethylaminoethyl side chain, along with dimethoxy groups in the indole ring, which appear to be crucial for the observed bioactivity.
Extending the exploration of thiochromene derivatives, Shen J. and colleagues synthesized thiochromen-4-one derivatives and evaluated their anti-cancer potency. The in vitro data revealed that compound 52 (Fig. 15) exhibited pronounced inhibitory activity across all tested cell lines, with IC50 values of 6.93 μM for (MGC-803), 5.01 μM (T-24), 25.20 μM (NCI-H460), 5.26 μM (HepG2), 9.58 μM (Hep-3B), and 7.34 μM (SMMC-7721). The SAR analysis of the thiochromene-2-one derivatives reveals a pronounced influence of electronic and steric factors at the para position of the aryl substituent on biological potency, with 4-nitro emerging as the most potent compound.72 Halogenated derivatives, such as 4-chloro and 4-bromo, exhibit moderate activity, suggesting a tolerance for steric bulk but reduced efficiency compared to the nitro group, while electron-donating groups like 4-methyl and 4-methoxy with comparatively diminished activity, underscoring the critical role of electron-withdrawing effects in optimizing receptor–ligand interactions. Collectively, the data suggest that para-substituted aryl groups with electron-withdrawing substituents, particularly nitro groups, are pivotal for maximizing potency, potentially through enhanced electronic complementarity and binding affinity at the molecular target.
Further enriching this discourse, L. Dalla Via et al. synthesized and evaluated 1,4-dihydrobenzothiopyrano-[4,3-c]-pyrazole derivatives for their pro-apoptotic mitochondrial targeted activity. The anti-proliferative efficacy of the newly synthesized benzothiopyranopyrazole derivatives was rigorously assessed against two human tumor cell lines, HeLa (cervix adenocarcinoma) and HL-60 (myeloid leukemia).73 The data revealed a pronounced anti-proliferative effect for derivatives, each distinguished by a methoxy substituent at the 7th position and variously substituted aryl groups – phenyl, p-chlorophenyl, or p-methoxyphenyl—anchored at the 1st position of the pyrazole ring. Among these, compound 53 (Fig. 15) emerged as the most potent, exhibiting IC50 values of 6.7 μM against HeLa cells and 3.8 μM against HL-60 cells, underscoring its superior activity across both cell lines.
D. Guo and co-workers synthesized 4,5-dihydro-1H-thiochromeno-[4,3-d]-pyrimidine derivatives as potential anti-tumor agents, the anti-tumor efficacy of the synthesized target compounds was systematically evaluated against two human cancer cell lines, A549 and H460. The majority of these compounds exhibited a spectrum of anti-tumor activities ranging from moderate to outstanding against one or both cell lines. Notably, compound 54 (Fig. 15) demonstrated remarkable potency, with IC50 values of 0.44 μM for A549 and 3.07 μM for H460, underscoring its broad-spectrum efficacy.74 Structure–activity relationship (SAR) analysis revealed critical determinants of anti-tumor activity, the methoxy substituent's position showed minimal impact on efficacy, aminocarboximidamide incorporation significantly enhanced both potency and selectivity, substitution patterns on the molecular scaffold, particularly multiple fluoro groups, were pivotal for achieving high potency and selectivity and replacing the phenyl ring with a methoxy group diminished activity, underscoring the phenyl ring's essential role in maintaining bioactivity.
 | ||
| Fig. 16 Structure–activity relationships of thiochromene: key modifications for anti-proliferative activity. | ||
5 Miscellaneous activities
5.1 Thiochromane analogs
Bang E. et al. evaluated the inhibitory activity of (Z)-3-(hydroxyl-substituted benzylidene)-thiochroman-4-one analogues on mushroom tyrosinase using kojic acid as a positive control.21 Their findings revealed that compound 55 (MHY1498) (Fig. 17) exhibited significantly stronger inhibitory activity than kojic acid, with 90.7% inhibition. The IC50 values for MHY1498 and kojic acid were 4.1 μM and 22.0 μM, respectively, indicating that MHY1498 was approximately five times more potent. Furthermore, its depigmentation efficacy was confirmed using B16F10 murine melanoma cells, demonstrating its potential in pigmentation disorders. | ||
| Fig. 17 Structure of thiochromane analogues with miscellaneous activity. | ||
Building on the theme of enzyme inhibition, Barresi E. et al. synthesized a novel series of dihydrobenzothiopyrano-[4,3-c]-pyrazole derivatives targeting physiologically relevant carbonic anhydrase (CA) isoforms: CA I, II, IX, and XII. Among these, sulfonamide compound 56 (Fig. 17) showed remarkable inhibitory potency, with Ki values of 22.5 nM, 7.1 nM, 6.3 nM, and 66.7 nM for CA I, II, IX, and XII, respectively.75 This study highlighted the potential of sulfonamide and carboxylic acid moieties fused to heterocyclic systems as a promising approach for designing isoform-selective CA inhibitors with diverse therapeutic applications.
Shifting focus to receptor potentiation, Etsè and co-workers investigated the AMPA receptor (AMPAR) potentiation activity of 2H-thiochromene-1,1-dioxide and thiochroman-1,1-dioxide analogues.76 By replacing nitrogen atoms in benzothiadiazine-1,1-dioxides with carbon, they developed AMPAR potentiator compound 57 (Fig. 17) a thiochroman-based positive allosteric modulator (PAM) with an EC50 of 1.2 μM, comparable to the benzothiadiazine modulator BPAM344. Structural analysis using X-ray crystallography revealed that compound 56 binds to the GluA2 receptor with a preference for the R-enantiomer, paving the way for further optimization in receptor-targeted therapies.
Kanbe Y. et al. extended the exploration of thiochromane derivatives by focusing on their anti-estrogenic properties, by substituting the sulfoxide group in pure anti-estrogens with carboxylic acid, sulfonamide, or sulfamide functionalities, they identified compound 58 (Fig. 17) as a standout candidate.77 This compound exhibited superior anti-estrogenic efficacy compared to ICI 182780, achieving 95% inhibition at a dosage of 10 mg kg−1 in oral administration. The enhanced absorption and metabolic stability of compound 57 contributed to its remarkable pharmacological profile, underscoring its potential as an orally active anti-estrogen.
Song J. et al. investigated thiochromanone thiosemicarbazone derivatives for their cathepsin L inhibitory activity. Among the synthesized compounds, 6,7-difluorothiochromanone thiosemicarbazone compound 59 (Fig. 17) emerged as the most potent inhibitor, with an IC50 of 46 nM. Interestingly, sulfide-based analogues consistently outperformed their sulfone counterparts.78 Structural modifications, such as introducing substituents at the 8th position or bulky groups at 6th, generally diminished activity. Nevertheless, electron-donating and electron-withdrawing groups at 6th position retained strong inhibitory efficacy, offering valuable insights into the structure–activity relationship of these inhibitors.
5.2 Thiochromene analogs
Han Q. et al. synthesized and evaluated 4H-thieno-[3,2-c]-chromene-based inhibitors of Notum pectinacetylesterase, identifying compound 60 (Fig. 18) as the most potent, with mEC50 and hEC50 values of 20 nM and 8 nM, respectively. SAR studies centered on thiochromenes revealed consistent trends. Electron-withdrawing moieties on the phenyl ring, particularly halogens, enhanced potency with fluoro analogs outperforming chloro analogs. Furthermore, di-fluoro substitutions demonstrated superior efficacy over mono-substituted counterparts, highlighting the critical role of halogenation in augmenting activity.79 | ||
| Fig. 18 Structure of thiochromene analogs with miscellaneous activity. | ||
Continuing the exploration of enzyme inhibitors, Islam S. M. and co-workers synthesized spirooxindole–pyrrolidine-clubbed thiochromene–pyrazole pharmacophores as alpha-amylase inhibitors.80 Among these, compound 61 (Fig. 18) displayed the highest potency, with an IC50 value of 111.3 nM. SAR analysis suggested that the chlorine group enhanced enzymatic activity, while the methoxy group further promoted potency through its electron-donating properties, emphasizing the combined contributions of functional groups in optimizing inhibitory activity.
Mi P. et al. contributed to the field of selective enzyme inhibitors by developing isoxazole-substituted thiochromone compounds and their corresponding S,S-dioxide analogs as hMAO-B inhibitors.81 Among the series, compound 62 (Fig. 18) exhibited exceptional selectivity and potency, with an IC50 value of 2 nM. Interestingly, oxidation of the thiochromone scaffold to its S,S-dioxide form significantly improved both solubility and inhibitory activity. The rigid isoxazole ring at the 3rd position preserved potency against hMAO-B, demonstrating the effectiveness of sulfur oxidation as a strategic modification to enhance bioactivity and drug-like properties.
Sakauchi N. et al. explored the α-1D adrenoceptor antagonist potential of 3,4-dihydro-2H-thiochromene-1,1-dioxide derivatives. Among these, compound 63 (Fig. 18) featuring a methylamino substituent, demonstrated a 2-fold higher binding affinity to α-1D-AR and exhibited over 400-fold subtype selectivity for α-1D-AR over other α-AR subtypes.82 This analog dose-dependently inhibited bladder contractions with an IC50 value of 80 nM. SAR investigations highlighted the importance of linker length between the amine and phenyl ring, with the conformational constraint of the sulfonyl group emerging as a crucial factor for enhancing selectivity and potency. These findings culminated in the identification of a highly selective α-1D-AR antagonist equivalent to the clinical compound TAKe259.
6 Molecular targets of thiochromanes/thiochromenes
Thiochromene and thiochromane derivatives have gained attention for their significant anti-bacterial activity by targeting specific molecular pathways in bacteria. A prominent target is the dihydropteroate synthase (DHPS) enzyme, crucial for folate biosynthesis. For instance, pyrazolothiochromene derivatives have demonstrated high binding affinity to DHPS, engaging in key interactions such as hydrogen bonding and arene–cation interactions (PDB ID: 4DAI). This binding effectively inhibits bacterial growth by disrupting essential metabolic processes, leading to strong anti-microbial action.Thiochromane derivatives also exhibit remarkable anti-bacterial potency by selectively targeting microbial cell walls and energy metabolism pathways. An example is the compound 3-hydroxythiochromen-4-one (16) which specifically inhibited M. catarrhalis by depleting ATP levels, a critical component of bacterial energy metabolism. Structural elements, such as hydroxyl and chlorine groups, play a crucial role in enhancing the anti-bacterial activity of these derivatives.37 Their targeted approach not only minimizes the impact on commensal bacteria but also reduces the likelihood of inducing resistance, positioning thiochromane derivatives as promising agents with a dual mechanism of biofilm disruption and oxidative damage.
In the anti-fungal arena, thiochromene and thiochromane derivatives have shown efficacy by targeting N-myristoyltransferase (NMT), an enzyme vital for fungal survival. Studies have identified that derivatives like 6-alkyl-indolo-[3,2-c]-2H-thiochromanes (18) and thiochroman-4-one inhibitors, engage in hydrophobic interactions within the active site of NMT in pathogens like C. albicans and C. neoformans (PDB ID: 5UPZ). These interactions significantly disrupt fungal cell growth, with structure–activity relationship (SAR) analyses indicating that electron-donating and methoxy groups enhance binding efficiency, providing selective anti-fungal effects while minimizing toxicity.39
Beyond their anti-bacterial and anti-fungal activities, thiochromene and thiochromane derivatives have emerged as potential anti-viral agents targeting viral proteases. Benzylidenechromanones, a subclass of these compounds, have been identified as inhibitors of the SARS-CoV-2 main protease (Mpro), an essential enzyme for viral replication. The (Z)-3-(4′-chlorobenzylidene)-thiochroman-4-one (27) derivative exhibits high binding affinity to Mpro through molecular docking studies (PDB ID: 2Q6F), suggesting its potential to block viral replication.48 The incorporation of chloro and bromo groups into the thiochromane scaffold can further enhance viral enzyme inhibition, opening avenues for the development of effective anti-viral therapies.
In addition to their anti-malarial properties, thiochromane derivatives have exhibited substantial anti-trypanosomal activity. Ben Hadda et al. focused on spiroheterocyclic compounds targeting T. cruzi, the causative agent of Chagas disease.58 Among the synthesized compound (38) displayed potent activity against both trypomastigotes and epimastigotes, with IC50 values of 1.5 μM and 1.95 μM, respectively. Structural modifications, particularly the introduction of sulfur and specific substitutions on the aromatic rings, were critical in enhancing the selectivity and efficacy of these compounds against benznidazole resistant strains of T. cruzi. This highlights the potential of thiochromane derivatives as effective anti-trypanosomal agents, emphasizing the need for continued exploration of their molecular targets in the pursuit of innovative therapeutics for malaria and Chagas disease.
The anti-cancer activity of thiochromane-based compounds and their derivatives can be attributed to various molecular mechanisms that influence cell growth, apoptosis, and signaling pathways. Bharkavi et al. work on dispiro-indeno pyrrolidine/pyrrolothiazole–thiochroman hybrids (40) demonstrated that halogen substitutions, particularly in meta and para positions, enhanced anti-cancer efficacy, suggesting that electron withdrawing groups contribute to better binding affinity and interaction with cellular targets.59 These structural modifications may strengthen interactions with enzymes or receptors involved in apoptosis and cell cycle regulation, positioning these derivatives as potent inhibitors of tumor cell proliferation. The observed IC50 values highlight the compound's potential to outperform standard treatments, such as doxorubicin indicating their promise as effective chemotherapeutic agents.
Compounds synthesized by Demirayak and colleagues underscored the significance of thiochromanone structures combined with 1,3-benzylidene moieties (41) in enhancing anti-proliferative effects-particularly against leukemia, melanoma, and colon cancer cells.60 These findings suggest that modifications at specific positions within the thiochromanone core influence key cancer related pathways, such as cell adhesion, migration, and invasion. Notably, the enhanced activity of derivatives featuring methoxy and chloro substituents aligns with their known role in modulating membrane permeability and binding interactions, potentially inhibiting critical proteases like cathepsin L that facilitate cancer progression and metastasis.
Pandya and co-workers provided further insights into the molecular interactions by investigating 3-arylspiro-[oxirane-2,3′-thiochroman]-4′-one and 5H-thiochromeno-[4,3-d]-pyrimidine derivatives (44). The structural frameworks revealed that the presence of fluorine or chlorine as substituents influenced the anti-cancer activity through modifications in electronic properties, which affect how these compounds interact with key cellular proteins and DNA. Such substitutions are believed to enhance the binding affinity for target proteins and impede the activity of enzymes that promote cancer cell survival and proliferation.63,64 The reduced activity observed with bromine substitutions underscores the delicate balance in electronic and steric effects required for optimal anti-cancer action. Collectively, these studies emphasize the importance of SAR and suggest pathways involving oxidative stress modulation, apoptosis induction, and inhibition of metastatic pathways as central mechanisms driving the anti-cancer properties of thiochromane derivatives (Fig. 19).
 | ||
| Fig. 19 Structure of thiochromane/thiochromene derivatives for various molecular targets. | ||
7 Future perspectives and challenges
The future of thiochromene and thiochromane derivatives in therapeutic applications is promising, driven by their multifaceted mechanisms of action against various pathogens. Advances in medicinal chemistry and structural biology could enhance the design and optimization of these compounds, enabling the development of more potent and selective agents. Table 1 summarizes the biological activities, structural features, and SAR insights of all the discussed compounds. Tailoring their chemical structure to improve pharmacokinetics and bioavailability may significantly increase their efficacy in clinical settings. Moreover, the potential for these derivatives to serve as scaffolds for hybrid molecules could further expand their therapeutic repertoire, allowing for the combination of different mechanisms of action against resistant strains of bacteria, fungi, and viruses.| S. no. | Compounds | Biological activity | SAR insights | Ref. |
|---|---|---|---|---|
| 1. |

|
Anti-bacterial activity (thiochromane derivatives) | 1. The presence of acyl hydrazone or oxime ether linker at 4th position significantly increased anti-bacterial activity (compound 7 & 10) | 32–36 |
| 2. Chlorine substitution at 6th position showed comparatively higher potency than unsubstituted or hetero aromatic substitutions (compound 7, 10 & 11) | ||||
| 3. Aromatic/heteroaromatic carboxamide linkage at 2nd position slightly increased anti-bacterial activity when compared with unsubstituted compounds (compound 10 & 11) | ||||
| 4. Fused spirocyclic compounds at 3nd position led to a slight reduction in potency (compound 8) | ||||
|
Anti-bacterial activity (thiochromene derivatives) | 1. Fused ring derivatives at 3rd and 4th position exhibit higher anti-bacterial potential in comparison with (–OH) group, carboxamide and secondary amine linkers (compound 14 & 15) | 24–26, 37 | |
| 2. Unsubstituted phenyl ring of thiochromene is generally preferable (compound 12, 13, 14, 15 & 16) | ||||
| 3. Presence of long alkyl chains at 2nd position offers selective anti-bacterial activity against Gram-negative bacteria (compound 16) | ||||
| 4. Nitrogen containing heterocycles at 3rd enhanced anti-bacterial potency | ||||
| 2. |

|
Anti-fungal activity | 1. Substituting bromine in place of chlorine at 6th position enhanced anti-fungal activity whereas EDG (–CH3) led to decreased activity (compound 21b) | 38–43 |
| 2. Presence of aromatic & aliphatic linker substitutions at 3rd position increased potency significantly (compound 21a, b & 22) | ||||
| 3. Replacement of chlorine in place of ketone at 4th position improved anti-fungal activity against specific anti-fungal species (compound 21b) | ||||
| 4. Fused spirocyclic compounds at 3nd and 4th position led to a slight reduction in anti-fungal activity (compound 17) | ||||
| 5. Di halogen substituted derivatives showcased better potential when compared to mono substituted compounds. (compound 20 & 21a, b) | ||||
| 3. |
|
Anti-tubercular activity | 1. Strong EWG (–NO2, CF3) at 6th and 8th is critical of DpRE1 inhibition | 44–46 |
| 2. Lipophilic substitutions (cyclohexyl, cyclopentyl group) on piperazine ring exhibit significant anti-tubercular potential (compound 24) | ||||
| 3. Salt formation of the compounds improved the ADME profile significantly. (compound 25) | ||||
| 4. |

|
Anti-viral activity | 1. Oxidation of sulfide to sulfone significantly improved the HIV-1 protease inhibition | 47, 48 |
| 2. Removal of Boc-group lead to substantial loss of activity | ||||
| 3. Hydroxy/amino substituted derivatives exhibit better water solubility as well as overall ADME profile (compound 26) | ||||
| 5. |
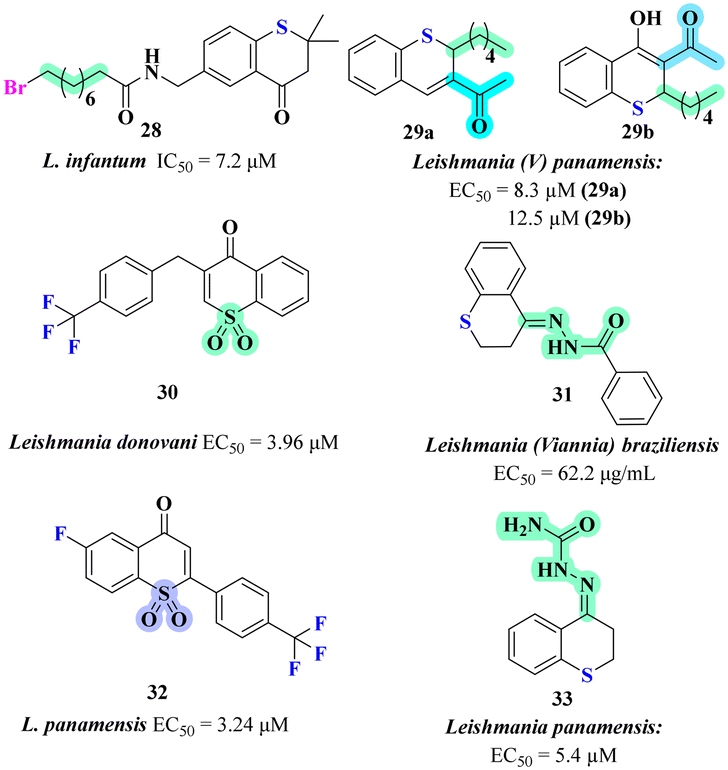
|
Anti-leishmanial activity | 1. Presence of alkyl linker chains at 2nd was crucial for membrane permeability. (compound 29a, b) | 49–53, 83 |
| 2. Sulfone containing derivatives exhibit comparatively higher anti-leishmanial activity as compared to the sulfide ones (compound 30, 32) | ||||
| 3. Semicarbazone/phenyl hydrazone linkers at 4th position in place of ketone improved potency (compound 31, 33) | ||||
| 4. Presence of EWG at 6th and 7th position was crucial for improving potency (compound 30 & 32) | ||||
| 6. |
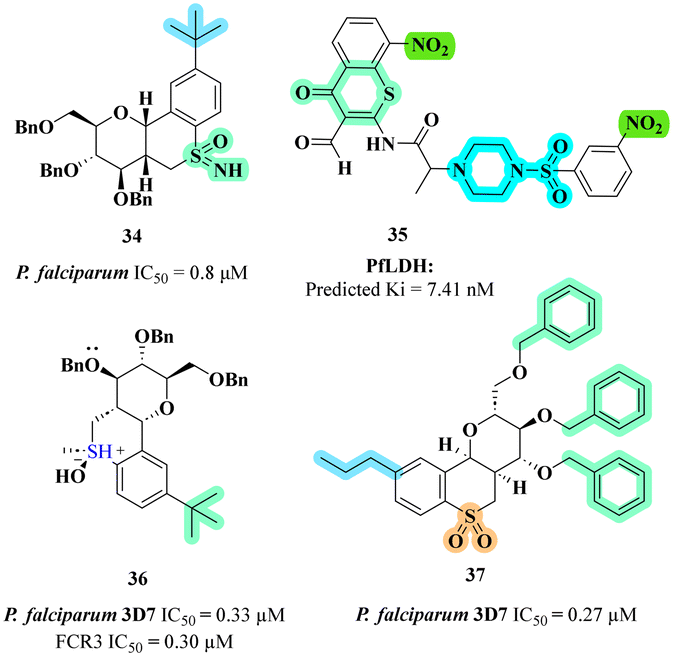
|
Anti-malarial activity | 1. Sugar tethered thiochromene derivatives exhibit greater selectivity against plasmodial parasite (compound 34, 36, 37) | 54–57 |
| 2. Oxidation of sulfide to sulfone increased the potency by several folds even when compared with sulfoximine derivatives (compound 37) | ||||
| 3. Substitution of short alkyl chain or bulky methyl groups in ring B may contribute to increased potency (compound 34 & 37) | ||||
| 7. |

|
Anti-trypanosomal activity | 1. Unsubstituted ring B is generally preferred for selectivity | 50–58 |
| 2. Sulfide containing molecule displayed significantly higher potency when compared to sulfones (compound 38) | ||||
| 8. |

|
Anti-cancer activity (thiochromane derivatives) | 1. Presence of urea/thiourea linkers enhanced anti-cancer activity by providing flexibility and enhancing binding with the receptor (compound 43 & 48) | 59–68 |
| 2. Fused ring/spiro cyclic compounds at 3rd position exhibit lower activity as compared with heteroaromatic or oxirane moiety (compound 40, 45 & 46) | ||||
| 3. Replacement of ketone at 4th position with thiosemicarbazide group increased anti-cancer activity against multiple cancer cell lines (compound 47) | ||||
| 4. gem-Dimethyl substituents at 2nd and 4th position of ring A are essential in modulating logP and logD properties and also improve the LiPE of the compound (compound 43) |
||||
| 5. Incorporation of fluorine at 6th and 8th position of ring B increased anti-cancer potential of the compounds by several folds when compared with chlorine/di-chloro/EWG (NO2, CF3) substituted analogs (compound 44 & 47) | ||||
| 9. |

|
Anti-cancer activity (thiochromene derivatives) | 1. Nitrogen containing tricyclic and tetracyclic fused ring systems on ring A exhibit significant anti-cancer potential (compound 51, 53 & 54) | 69–74 |
| 2. Presence of carbohydrate chain at 2nd position attenuates the anti-proliferative activity when compared with unsubstituted derivatives (compound 49a, b) | ||||
| 3. Mono/di halogenated derivatives specially fluorine and chlorine pronounced antiproliferative effect (compound 50, 53 & 54) | ||||
| 4. Sterically bulky substrates at 3rd position may lead to decrease in potency (compound 50) |
Despite their potential, several challenges must be addressed to realize the full therapeutic capabilities of thiochromene and thiochromane derivatives. One of the main hurdles is the need for comprehensive preclinical and clinical studies to establish safety profiles and potential side effects. The variability in biological responses among different pathogens necessitates extensive testing to determine the optimal conditions for their use. Furthermore, understanding the resistance mechanisms that may arise against these compounds is critical, as pathogens can evolve quickly to counteract new therapies. Continuous surveillance and research are essential to pre-emptively address these challenges and adapt treatment strategies accordingly.
Another significant challenge lies in the scaling up of production and formulation of thiochromene and thiochromane derivatives for widespread clinical use. The synthesis of these compounds often involves complex chemical processes that may not be easily translatable to large-scale manufacturing. Streamlining synthesis methods while maintaining product integrity and bioactivity is crucial for their commercialization. Additionally, developing effective delivery systems that enhance the targeted delivery of these derivatives to specific tissues or pathogens could improve therapeutic outcomes and minimize potential toxicity.
Finally, collaboration between academia, industry, and regulatory bodies will be vital in overcoming the challenges associated with bringing thiochromene and thiochromane derivatives to the market. Such partnerships can facilitate knowledge exchange, foster innovation, and expedite the translation of research findings into clinical applications. Establishing clear regulatory pathways for the approval of novel anti-microbial and anti-viral agents will also be essential in addressing the growing global threat of anti-microbial resistance. By leveraging collective expertise and resources, stakeholders can work towards developing effective therapies that harness the unique properties of thiochromene and thiochromane derivatives.
8 Conclusion
Thiochromene and thiochromane derivatives hold immense promise as therapeutic agents due to their broad-spectrum biological activities and potential for structural optimization. Their diverse mechanisms of action, including anti-microbial, anti-cancer, and anti-fungal effects, make them valuable scaffolds in drug discovery. SAR studies have demonstrated that tailored modifications, such as electron-withdrawing groups and sulfur oxidation, can significantly enhance their efficacy and selectivity. However, successfully translating these compounds from laboratory research to clinical application requires overcoming key challenges, including toxicity concerns, resistance mechanisms, and pharmacokinetic limitations.Future research should focus on improving the synthesis, bioavailability, and targeted delivery of these derivatives to maximize their therapeutic benefits. Advances in medicinal chemistry, structural biology, and drug formulation techniques will be crucial in refining their properties for clinical use. Additionally, interdisciplinary collaborations between researchers, pharmaceutical industries, and regulatory bodies will facilitate the development of novel strategies to optimize their safety and efficacy. Addressing these challenges will pave the way for thiochromene and thiochromane derivatives to emerge as effective therapeutic agents in combating drug-resistant infections and various diseases.
Data availability
Data sharing is not applicable to this article as no datasets were generated or analysed during the current study.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We sincerely express our gratitude to BITS-Pilani, Pilani Campus, and BITS-Pilani, Hyderabad Campus, for providing the necessary facilities to do the work. The authors SM and KVGC acknowledge the funding received from the SERB-CRG projects (Ref. No. SERB-CRG/2022/005290 and SERB-CRG/2022/001889) under the Department of Science and Technology (DST), New Delhi.References
- L. A. Komarnisky, R. J. Christopherson and T. K. Basu, Nutrition, 2003, 19, 54–61 CAS.
- L. O. Fuentes-Lara, J. Medrano-Macías, F. Pérez-Labrada, E. N. Rivas-Martínez, E. L. García-Enciso, S. González-Morales and A. Benavides-Mendoza, Molecules, 2019, 24, 2282 CAS.
- A. Francioso, A. Baseggio Conrado, L. Mosca and M. Fontana, Oxid. Med. Cell. Longevity, 2020, 2020, 8294158 Search PubMed.
- A. Vairavamurthy, B. Manowitz, G. W. Luther III and Y. Jeon, Geochim. Cosmochim. Acta, 1993, 57, 1619–1623 CAS.
- M. C. H. Gruhlke and A. J. Slusarenko, Plant Physiol. Biochem., 2012, 59, 98–107 CAS.
- M. Feng, B. Tang, S. H. Liang and X. Jiang, Curr. Top. Med. Chem., 2016, 16, 1200–1216 Search PubMed.
- J. T. Brosnan and M. E. Brosnan, J. Nutr., 2006, 136, 1636S–1640S Search PubMed.
- R. Hill, A. Shafaei, L. Balmer, J. R. Lewis, J. M. Hodgson, A. H. Millar and L. C. Blekkenhorst, Crit. Rev. Food Sci. Nutr., 2023, 63, 8616–8638 CrossRef PubMed.
- S. Gharge and S. G. Alegaon, Chem. Biodiversity, 2023, 21, e202301738 Search PubMed.
- P. Czerniawski and P. Bednarek, Front. Plant Sci., 2018, 9, 1639 CrossRef PubMed.
- A. Rusu, I.-M. Moga, L. Uncu and G. Hancu, Pharmaceutics, 2023, 15, 2554 Search PubMed.
- R. Shah and P. K. Verma, Chem. Cent. J., 2018, 12, 137 CrossRef CAS PubMed.
- K. Laxmikeshav, P. Kumari and N. Shankaraiah, Med. Res. Rev., 2022, 42, 513–575 CrossRef CAS PubMed.
- R. Shah and P. K. Verma, Chem. Cent. J., 2018, 12, 137 Search PubMed.
- S. Pathania, R. K. Narang and R. K. Rawal, Eur. J. Med. Chem., 2019, 180, 486–508 Search PubMed.
- M. Krátký and J. Vinsova, Curr. Top. Med. Chem., 2016, 16, 2921–2952 Search PubMed.
- E. B. Elkaeed, E. U. Mughal, S. Kausar, H. A. Al-ghulikah, N. Naeem, A. A. Altaf and A. Sadiq, J. Mol. Struct., 2022, 1270, 133972 Search PubMed.
- S. Murugappan, P. V. Kuthe, K. V. G. Chandra Sekhar and M. Sankaranarayanan, Org. Biomol. Chem., 2024, 22, 6045–6079 RSC.
- D. J. Wang, Z. Hou, H. Xu, R. An, X. Su and C. Guo, Bioorg. Med. Chem. Lett., 2018, 28, 3574–3578 Search PubMed.
- R. S. Lodi, X. Dong, X. Wang, Y. Han, X. Liang, C. Peng and L. Peng, Fungal Biol. Rev., 2025, 51, 100413 Search PubMed.
- E. Bang, S.-G. Noh, S. Ha, H. J. Jung, D. H. Kim, A. K. Lee, M. K. Hyun, D. Kang, S. Lee, C. Park, H. R. Moon and H. Y. Chung, Molecules, 2018, 23, 3307 Search PubMed.
- E. Barresi, S. Salerno, A. M. Marini, S. Taliani, C. La Motta, F. Simorini, F. Da Settimo, D. Vullo and C. T. Supuran, Bioorg. Med. Chem., 2016, 24, 921–927 Search PubMed.
- R. B. Bakr, I. H. El Azab and N. A. A. Elkanzi, J. Iran. Chem. Soc., 2022, 19, 1413–1423 Search PubMed.
- N. Elkanzi, I. El Azab and R. Bakr, Polycyclic Aromat. Compd., 2022, 42, 1–20 CrossRef.
- L. Suresh, P. Sagar Vijay Kumar, Y. Poornachandra, C. Ganesh Kumar, N. J. Babu and G. V. P. Chandramouli, Bioorg. Med. Chem., 2016, 24, 3808–3817 CAS.
- L. Suresh, P. Sagar Vijay Kumar, Y. Poornachandra, C. Ganesh Kumar and G. V. P. Chandramouli, Bioorg. Chem., 2016, 68, 159–165 CAS.
- X.-S. Wang, J.-R. Wu, J. Zhou and M.-M. Zhang, J. Heterocyclic Chem., 2011, 48, 1056–1060 CAS.
- B. I. Usachev, M. A. Shafeev and V. Y. Sosnovskikh, Russ. Chem. Bull., 2006, 55, 523–528 CAS.
- A. Liepa, O. Nguyen and S. Saubern, Aust. J. Chem., 2005, 58, 864–869 Search PubMed.
- T. Kataoka, S. Watanabe, E. Mori, R. Kadomoto, S. Tanimura and M. Kohno, Bioorg. Med. Chem., 2004, 12, 2397–2407 CAS.
- J. Song, R. Pan, G. Li, W. Su, X. Song, J. Li and S. Liu, Med. Chem. Res., 2020, 29, 630–642 CAS.
- P. Jiyan, Chi Lu Yu and M. Chen, Phosphorus, Sulfur Silicon Relat. Elem., 2022, 197, 13–17 Search PubMed.
- N. Chouchène, A. Toumi, S. Boudriga, H. Edziri, M. Sobeh, M. A. O. Abdelfattah, M. Askri, M. Knorr, C. Strohmann, L. Brieger and A. Soldera, Molecules, 2022, 27, 582 Search PubMed.
- R. Vinodkumar, S. D. Vaidya, B. V. Siva Kumar, U. N. Bhise, S. B. Bhirud and U. C. Mashelkar, Eur. J. Med. Chem., 2008, 43, 986–995 Search PubMed.
- L. Xiao, L. Yu, P. Li, J. Chi, Z. Tang, J. Li, S. Tan and X. Wang, Molecules, 2021, 26, 4391 Search PubMed.
- L. Yu, L. Xiao, P. Li, J. Chi, J. Li and S. Tan, J. Chem., 2022, 2022, 5354088 Search PubMed.
- D. Szamosvári, T. Schuhmacher, C. R. Hauck and T. Böttcher, Chem. Sci., 2019, 10, 6624–6628 RSC.
- F. Wu, G.-C. Liang, G. Zhou, Q.-J. Liu, C.-C. Zhang, J.-J. Yu, X.-H. Dong and Y.-L. Song, Lett. Org. Chem., 2016, 13, 206–216 CrossRef CAS.
- X.-Y. Han, Y.-F. Zhong, S.-B. Li, G.-C. Liang, G. Zhou, X.-K. Wang, B.-H. Chen and Y.-L. Song, Chem. Pharm. Bull., 2016, 64, 1411–1416 CrossRef CAS PubMed.
- C. Pinedo-Rivilla, I. G. Collado and J. Aleu, J. Nat. Prod., 2018, 81, 1036–1040 CrossRef CAS PubMed.
- Y.-L. Song, F. Wu, C.-C. Zhang, G.-C. Liang, G. Zhou and J.-J. Yu, Bioorg. Med. Chem. Lett., 2015, 25, 259–261 Search PubMed.
- D.-J. Wang, Z. Hou, H. Xu, R. An, X. Su and C. Guo, Bioorg. Med. Chem. Lett., 2018, 28, 3574–3578 CAS.
- Y. Zhong, X. Han, S. Li, H. Qi, Y. Song and X. Qiao, Chem. Pharm. Bull., 2017, 65, 904–910 Search PubMed.
- P. Li, B. Wang, X. Zhang, S. M. Batt, G. S. Besra, T. Zhang, C. Ma, D. Zhang, Z. Lin, G. Li, H. Huang and Y. Lu, Eur. J. Med. Chem., 2018, 160, 157–170 Search PubMed.
- P. Li, B. Wang, L. Fu, K. Guo, C. Ma, B. Wang, Z. Lin, G. Li, H. Huang and Y. Lu, Eur. J. Med. Chem., 2021, 222, 113603 Search PubMed.
- P. Li, K. Guo, L. Fu, B. Wang, B. Zhang, N. Gong, Y. Lu, C. Ma, H. Huang, Y. Lu and G. Li, Eur. J. Med. Chem., 2023, 246, 114993 CAS.
- A. K. Ghosh, R. D. Jadhav, H. Simpson, S. Kovela, H. Osswald, J. Agniswamy, Y.-F. Wang, S. Hattori, I. T. Weber and H. Mitsuya, Eur. J. Med. Chem., 2018, 160, 171–182 Search PubMed.
- N. Sepay, N. Sepay, A. Al Hoque, R. Mondal, U. C. Halder and M. Muddassir, Struct. Chem., 2020, 31, 1831–1840 CAS.
- S. Coll, M. Alhazmi, P. de Aguiar Amaral, S. Bourgeade-Delmas, A.-C. Le Lamer and J. W. Barlow, Molecules, 2021, 26, 2209 CAS.
- C. Ortiz, M. Breuning, S. Robledo, F. Echeverri, E. Vargas and W. Quiñones, Heliyon, 2023, 9, 17801 Search PubMed.
- Y. Upegui, K. Rios, W. Quiñones, F. Echeverri, R. Archbold, J. D. Murillo, F. Torres, G. Escobar, I. D. Vélez and S. M. Robledo, Med. Chem. Res., 2019, 28, 2184–2199 Search PubMed.
- E. Vargas, F. Echeverri, I. D. Vélez, S. M. Robledo and W. Quiñones, Molecules, 2017, 22, 2041 Search PubMed.
- E. Vargas, F. Echeverri, Y. A. Upegui, S. M. Robledo and W. Quiñones, Molecules, 2018, 23, 70 Search PubMed.
- M. Chizema, T. F. Mabasa, H. C. Hoppe and H. H. Kinfe, Chem. Biol. Drug Des., 2019, 93, 254–261 Search PubMed.
- S. Dey, B. K. Kumar, S. Johri, F. Faheem and S. Murugesan, Struct. Chem., 2022, 33, 2063–2082 CrossRef CAS.
- H. H. Kinfe, P. T. Moshapo, F. L. Makolo, D. W. Gammon, M. Ehlers and C. Schmuck, Eur. J. Med. Chem., 2014, 87, 197–202 CrossRef CAS PubMed.
- G. K. Madumo, P. T. Moshapo and H. H. Kinfe, Med. Chem. Res., 2018, 27, 817–833 Search PubMed.
- T. Ben Hadda, A. Kerbal, B. Bennani, G. Al Houari, M. Daoudi, A. C. L. Leite, V. H. Masand, R. D. Jawarkar and Z. Charrouf, Med. Chem. Res., 2013, 22, 57–69 CrossRef CAS.
- C. Bharkavi, S. Vivek Kumar, M. Ashraf Ali, H. Osman, S. Muthusubramanian and S. Perumal, Bioorg. Med. Chem., 2016, 24, 5873–5883 CrossRef CAS PubMed.
- S. Demirayak, L. Yurttas, N. Gundogdu-Karaburun, A. C. Karaburun and I. Kayagil, Saudi Pharm. J., 2017, 25, 1063–1072 CrossRef PubMed.
- T. A. Farghaly and E. M. H. Abbas, J. Chem. Res., 2012, 36, 660–664 CrossRef CAS.
- B. Nammalwar, R. A. Bunce, K. D. Berlin, D. M. Benbrook and C. Toal, Eur. J. Med. Chem., 2019, 170, 16–27 Search PubMed.
- D. Pandya and Y. Naliapara, Lett. Org. Chem., 2019, 16, 517–524 CrossRef CAS.
- D. Pandya and Y. Naliapara, Lett. Org. Chem., 2019, 17, 294–302 CrossRef.
- Y. L. Song, Y.-F. Dong, T. Yang, C.-C. Zhang, L.-M. Su, X. Huang, D.-N. Zhang, G.-L. Yang and Y.-X. Liu, Bioorg. Med. Chem., 2013, 21, 7624–7627 Search PubMed.
- Y. Song, S. Feng, J. Feng, J. Dong, K. Yang, Z. Liu and X. Qiao, Eur. J. Med. Chem., 2020, 200, 112459 Search PubMed.
- J. Song, R. Pan, G. Li, W. Su, X. Song, J. Li and S. Liu, Med. Chem. Res., 2020, 29, 630–642 CAS.
- T. C. Le, K. D. Berlin, S. D. Benson, M. A. Eastman, G. Bell-Eunice, A. C. Nelson and D. M. Benbrook, Open Med. Chem. J., 2007, 1, 11–23 Search PubMed.
- V. Luque-Agudo, J. Albarrán-Velo, J. G. Fernández-Bolaños, O. López, M. E. Light, J. M. Padrón, I. Lagunes, E. Román, J. A. Serrano and M. V. Gil, New J. Chem., 2017, 41, 3154–3162 CAS.
- A. Barakat, M. S. Islam, M. Ali, A. M. Al-Majid, S. Alshahrani, A. S. Alamary, S. Yousuf and M. I. Choudhary, Symmetry, 2021, 13, 1426 Search PubMed.
- S. Salerno, V. La Pietra, M. Hyeraci, S. Taliani, M. Robello, E. Barresi, C. Milite, F. Simorini, A. N. García-Argáez, L. Marinelli, E. Novellino, F. Da Settimo, A. M. Marini and L. Dalla Via, Eur. J. Med. Chem., 2019, 165, 46–58 CrossRef CAS PubMed.
- J. Shen, Y. Yang, X. Hou, W. Zeng, A. Yu, X. Zhao and X. Meng, Org. Biomol. Chem., 2018, 16, 3487–3494 Search PubMed.
- L. Dalla Via, A. M. Marini, S. Salerno, C. La Motta, M. Condello, G. Arancia, E. Agostinelli and A. Toninello, Bioorg. Med. Chem., 2009, 17, 326–336 CrossRef CAS.
- D. Guo, Y. Liu, T. Li, N. Wang, X. Zhai, C. Hu and P. Gong, Sci. China: Chem., 2012, 55, 347–351 CrossRef CAS.
- E. Barresi, S. Salerno, A. M. Marini, S. Taliani, C. La Motta, F. Simorini, F. Da Settimo, D. Vullo and C. T. Supuran, Bioorg. Med. Chem., 2016, 24, 921–927 CrossRef CAS PubMed.
- K. S. Etsè, J. Dorosz, K. McLain Christensen, J.-Y. Thomas, I. Botez Pop, E. Goffin, T. Colson, P. Lestage, L. Danober, B. Pirotte, J. S. Kastrup and P. Francotte, ACS Chem. Neurosci., 2021, 12, 2679–2692 Search PubMed.
- Y. Kanbe, M.-H. Kim, M. Nishimoto, Y. Ohtake, T. Tsunenari, K. Taniguchi, I. Ohizumi, S. Kaiho, Y. Nabuchi, S. Kawata, K. Morikawa, J.-C. Jo, H.-A. Kwon, H.-S. Lim and H.-Y. Kim, Bioorg. Med. Chem. Lett., 2006, 16, 4090–4094 CrossRef CAS PubMed.
- J. Song, L. M. Jones, G. D. K. Kumar, E. S. Conner, L. Bayeh, G. E. Chavarria, A. K. Charlton-Sevcik, S.-E. Chen, D. J. Chaplin, M. L. Trawick and K. G. Pinney, ACS Med. Chem. Lett., 2012, 3, 450–453 CrossRef CAS PubMed.
- Q. Han, P. K. Pabba, J. Barbosa, R. Mabon, J. P. Healy, M. W. Gardyan, K. M. Terranova, R. Brommage, A. Y. Thompson, J. M. Schmidt, A. G. E. Wilson, X. Xu, J. E. Tarver and K. G. Carson, Bioorg. Med. Chem. Lett., 2016, 26, 1184–1187 CrossRef CAS PubMed.
- M. S. Islam, A. M. Al-Majid, M. Haukka, Z. Parveen, N. Ravaiz, A. Wadood, A. U. Rehman, M. Ríos-Gutiérrez, L. R. Domingo and A. Barakat, Chem. Biol. Drug Des., 2023, 102, 972–995 CrossRef CAS PubMed.
- P. Mi, Y. Tan, S. Ye, J.-J. Lang, Y. Lv, J. Jiang, L. Chen, J. Luo, Y. Lin, Z. Yuan, X. Zheng and Y.-W. Lin, Eur. J. Med. Chem., 2024, 263, 115956 Search PubMed.
- N. Sakauchi, H. Furukawa, J. Shirai, A. Sato, H. Kuno, R. Saikawa and M. Yoshida, Eur. J. Med. Chem., 2017, 139, 114–127 CrossRef CAS PubMed.
- S. Coll, M. Alhazmi, P. de Aguiar Amaral, S. Bourgeade-Delmas, A.-C. Le Lamer and J. W. Barlow, Molecules, 2020, 25, 800 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2025 |