
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAloe vera-assisted synthesis of MnCo2O4 as a battery-type material for hybrid supercapacitor applications
Mohit
Bhatt
 *a,
Kajal
Gautam
b and
Anil Kumar
Sinha
*a
*a,
Kajal
Gautam
b and
Anil Kumar
Sinha
*a
aDepartment of Physics, School of Advanced Engineering, UPES, Dehradun, 248007, India. E-mail: mhtt.mb@gmail.com; anil.sinha@ddn.upes.ac.in
bDepartment of Chemistry, School of Advanced Engineering, UPES, Dehradun, 248007, India
First published on 17th October 2025
Abstract
Material advancement in sustainable energy storage is increasingly driven by green chemistry approaches. In this context, we have investigated an Aloe vera-mediated synthesis route for MnCo2O4 nanostructures as a battery-type electrode material for hybrid supercapacitor applications. The material was synthesized via an eco-conscious, biogenic route using Aloe vera gel as a natural reducing and stabilizing agent. High-resolution synchrotron XRD and TEM analyses confirmed the formation of single-phase spinel nanocrystals with an average size of ∼12.7 nm, while FESEM revealed aggregated mesoporous clusters indicative of secondary particle formation. BET analysis demonstrated a moderate specific surface area of 43.27 m2 g−1 and a dominant pore size around 10 nm, supporting rapid ion transport and efficient electrolyte access. XPS spectra revealed the coexistence of Mn3+/Mn4+ and Co2+/Co3+ oxidation states, along with abundant oxygen vacancies and/or oxygen interstitials, collectively contributing to enhanced redox kinetics and charge storage capability. The material exhibited excellent electrochemical performance, delivering a high specific capacitance of 680 F g−1 at 1 A g−1 and retaining 282.7 F g−1 even at 80 A g−1 and 365.7 F g−1 at a high scan rate of 200 mV s−1. Furthermore, it showed remarkable long-term stability, with 84.5% capacitance retention after 5000 cycles. These findings establish the Aloe vera-derived MnCo2O4 as a green-engineered, high-rate capable, and durable electrode material for hybrid supercapacitor applications.
1. Introduction
Environmental concerns have driven the need for sustainable, green synthesis methods over conventional approaches involving toxic chemicals and harsh conditions.1,2 In response, green synthesis strategies, which utilize natural, biodegradable, and renewable resources under mild conditions have gained prominence.2,3 These approaches align with green chemistry principles by reducing energy consumption, waste, and ecological impact.2 Among them, plant extract-based synthesis has emerged as a practical and eco-conscious method, leveraging phytochemicals like polyphenols, flavonoids, enzymes, and sugars that serve as both reducing and stabilizing agents.4 Aloe vera, a succulent known for its medicinal properties, is a promising candidate for green synthesis due to its rich composition of bioactive compounds capable of reducing metal precursors and controlling nanoparticle formation.1,5,6 Beyond its chemical advantages, Aloe vera is extremely resilient and widely adaptable, thriving in diverse climates and surviving 20–30 days without water, making it a sustainable green resource even in resource-limited settings.7,8 Among various plant-derived reducing agents, Aloe vera stands out for its unique biochemical composition and favorable physicochemical properties.1,5 Its gel contains polysaccharides (e.g., acemannan), phenolic compounds, amino acids, vitamins, and enzymes that act synergistically as reducing and stabilizing agents, enabling controlled nanoparticle formation under mild conditions.1,9 The near-neutral to slightly alkaline pH (6.5–7.5) of Aloe vera creates a stable reaction environment, unlike acidic extracts (e.g., lemon juice), which may introduce organic acids that hinder electronic conductivity and ion diffusion,8,10,11 which creates an ideal reaction environment for nanoparticle formation.12,13 Moreover, the gel matrix of Aloe vera provides a clean, enclosed biochemical environment that protects the extract from external contaminants such as airborne particulates or heavy metals.14,15 As the gel is internally located within the succulent leaf, it is inherently shielded from direct contact with air and environmental pollutants.15 This intrinsic protection minimizes the risk of contamination by heavy metal ions, which is a common concern in other green synthesis routes, where leaf or root extracts may uptake environmental toxins from soil or water.14,15 Studies suggest that heavy metals tend to accumulate in the outer epidermal layers of the Aloe vera leaf, not in the inner gel, thereby ensuring the chemical purity of extract.6 Thus, Aloe vera offers several integrated advantages, biocompatibility, chemical purity, mild pH, and multifunctionality,5,6 which make it a superior and sustainable choice for green synthesis of metal oxide nanostructures.Bimetallic transition metal oxides (TMOs), particularly those with spinel structures such as AB2O4 (where A is a divalent metal cation (2+), B is a trivalent metal cation (3+), and O is an oxygen anion (2−)), have garnered considerable attention as electrode materials for energy storage applications due to their high theoretical capacitance, multiple oxidation states enabling rich redox activity, robust structural stability, and synergistic interactions between metal ions.16,17 These properties contribute to enhanced electrochemical performance, including higher specific capacitance, better rate capability, and excellent cycling stability.17,18 Various spinel-type bimetallic oxides have demonstrated outstanding electrochemical performance. For example, NiCo2O4 exhibits a high specific capacitance of 1254 F g−1 at 2 A g−1,19 while FeCo2O4 reaches 1690.14 F g−1 at 0.5 A g−1.20 Similarly, ZnCo2O4 delivers 331.2 C g−1 at 2 A g−1,21 CuCo2O4 achieves 393.66 C g−1 at 1 A g−1,22 and CoMn2O4 exhibits 807.02 F g−1 at 1 A g−1.23 Another promising example is NiMn2O4, which demonstrates a specific capacitance of 930 F g−1 at a scan rate of 5 mV s−1,24 the combination of nickel and manganese offers complementary redox couples (Ni2+/Ni3+ and Mn3+/Mn4+), which facilitate multi-electron transfer processes and contribute to enhanced faradaic reactions, electrical conductivity, and improved electrochemical reversibility. Cobalt-manganese-based spinel oxide (MnCo2O4) offers the synergistic advantage of high theoretical capacitance of Mn and excellent redox activity of Co ions. This combination results into significantly improved energy storage capacity.25,26 Both Mn and Co offer multiple oxidation states leading to multiple faradaic redox reactions and contributing to improved capacitance25,26 MnCo2O4 has a spinel structure, which is a robust three-dimensional electrical framework which improves electronic and ion conductivity, electrochemical stability, and charge storage capacity, compared to other TM oxides.27 This dual-metal configuration enables a broader range of reversible redox reactions compared to monometallic oxides, leading to superior electrochemical performance.28,29 Furthermore, the synergistic effect between manganese and cobalt ions enhances not only the electrical conductivity but also the structural integrity of the material during repeated charge–discharge cycles.28 This interaction also facilitates faster charge transfer and improved ion diffusion, which are critical for achieving high power density and long-term cycling stability.28 Despite these advantages, the practical utilization of MnCo2O4 and other spinel oxides still encounters limitations, including particle agglomeration, restricted rate performance at higher current densities, and challenging synthesis conditions that may involve toxic chemicals or high-temperature processing.18,30,31 Addressing these challenges through green synthesis strategies, nano-architecture design, and morphology optimization remains crucial for unlocking the full potential of spinel bimetallic TMOs in next-generation supercapacitor and battery technologies.
To overcome these challenges, the present work introduces, environmentally benign approach for synthesizing bimetallic MnCo2O4 using Aloe vera gel as a natural reducing and structuring agent. This green synthesis route eliminates the need for toxic chemicals and high-temperature calcination, offering a safer and more sustainable alternative for material fabrication. The as-synthesized Aloe vera gel assisted MnCo2O4 nanoparticles (Alv–MnCo2O4 NPs) nanoparticles was systematically characterized using advanced techniques. Electrochemical evaluation revealed exceptional performance, with a high specific capacitance of 680 F g−1 at 1 A g−1 and 282.67 F g−1 even at an ultra-high current density of 80 A g−1, along with outstanding cycling stability, 84.5% retention after 5000 cycles at 20 A g−1. This study not only demonstrates the potential of Aloe vera-assisted green synthesis in producing high-performance electrode materials but also contributes to the ongoing quest for sustainable and scalable solutions in the field of energy storage. The integration of bio-based synthesis with advanced electrochemical applications represents a step forward in harmonizing technological progress with ecological responsibility.
2. Experimental section
2.1. Characterization
The synthesized Alv–MnCo2O4 NPs were thoroughly characterized to examine their structural, chemical, thermal, and morphological properties.2.2. Chemicals and reagents
The chemicals used in this work were of analytical grade and were utilized without any further purification. Manganese acetate (Mn(CH3COO)2, Loba Chemie, Extra Pure Grade, 98.5%) and cobalt nitrate hexahydrate (Co(NO3)2·6H2O, Loba Chemie, Extrapure, 97%) served as the primary metal precursors for the synthesis of the Alv–MnCo2O4 NPs. Aloe vera extract, obtained from the University of Petroleum and Energy Studies (UPES) nursery, Dehradun, India, was used as a natural reducing and stabilizing agent. N-Methyl-2-pyrrolidone (NMP, Sigma-Aldrich, anhydrous grade, assay 99.5%) acted as the solvent during electrode preparation, while carbon black (CB, Sigma-Aldrich) and polyvinylidene fluoride (PVDF, Sigma-Aldrich) were employed as the conductive additive and binder, respectively. Nickel foam (Vritratech, New Delhi, India, >99% purity, 1.5 mm thickness, 95–98% porosity) was used as the current collector. For electrochemical studies, potassium hydroxide (KOH, Loba Chemie, Extrapure, ≥85% purity) was used as the electrolyte. Ethanol (Loba Chemie, 99.9%) and acetone (Loba Chemie, 99.9%) were utilized for cleaning purposes. Deionized (DI) water was employed throughout the synthesis and washing processes.2.3. Synthesis of Alv–MnCo2O4 NPs
Fresh Aloe vera leaves were sourced from the polyhouse at UPES, Dehradun. The leaves were thoroughly washed multiple times with double deionized (DDI) water to eliminate dust and surface impurities. About 30 g of Aloe vera gel was carefully extracted from the cleaned leaves and transferred into a beaker. This gel was then homogenized with 150 mL of DI water using a clean grinder mixer to obtain a uniform, viscous solution. The resulting Aloe vera gel mixture was heated in a hot air oven at 85 °C for 2 hours to ensure uniform mixing. After cooling to room temperature, 5 g of manganese acetate tetrahydrate (Mn(CH3COO)2·4H2O) and 10 g of cobalt nitrate hexahydrate (Co(NO3)2·6H2O) were dissolved into the prepared Aloe vera gel under continuous magnetic stirring. Here, the Aloe vera gel functioned as a natural reducing and stabilizing agent, aiding in the reduction of metal ions and controlled nucleation. The mixture was stirred continuously at 80 °C for 1–2 hours until a dark, viscous gel formed, indicating the progression of the reaction. The resulting gel was subsequently calcined in a muffle furnace at 400 °C for 2.5 hours, with a ramp rate of 5 °C min−1, to yield Alv–MnCo2O4 NPs.34 A schematic representation of the synthesis procedure is shown in Fig. 1 | ||
| Fig. 1 Schematic representation of the green synthesis route for Alv–MnCo2O4 NPs, involving Aloe vera extract preparation, precursor mixing, and thermal treatment to obtain the final nanomaterial (created in Biorender.com). | ||
2.4. Electrode preparation and electrochemical measurements
Electrochemical performance was evaluated using electrodes fabricated with Alv–MnCo2O4 NPs, conductive carbon black (CB), and polyvinylidene fluoride (PVDF) in an 8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 weight ratio. The homogeneous slurry was coated onto nickel foam (NF) (1 × 1 cm2), which was pre-cleaned by sequential washing with acetone and deionized (DI) water, followed by bath sonication for 10 min. It was then rinsed with ethanol, immersed in 3 M HCl solution for 30 min to remove surface oxides, sonicated again in DI water, and finally washed with ethanol. The cleaned NF was dried in a vacuum oven at 60 °C for 2 h to ensure complete removal of moisture,35 and then the coated electrodes (Alv-MnCo2O4@NF) were dried in a vacuum oven at 80 °C overnight. The active material loading on NF was 0.4 mg. The prepared electrodes served as working electrodes, while a platinum mesh (2 × 2 cm2) and a saturated calomel electrode (SCE) were used as counter and reference electrodes, respectively. Aqueous 1 M KOH was employed as the electrolyte. Electrochemical measurements were conducted using a Biologic SP-150e electrochemical workstation. Cyclic voltammetry (CV) was performed at various scan rates. Galvanostatic charge–discharge (GCD) tests were carried out at different current densities, with stability and coulombic test. Electrochemical impedance spectroscopy (EIS) was measured in the frequency range of 10 mHz to 100 kHz to assess the charge transfer resistance and ion diffusion characteristics. Fig. 2 illustrates the schematic representation of the electrode preparation process and the electrochemical measurement setup.
:1:1 weight ratio. The homogeneous slurry was coated onto nickel foam (NF) (1 × 1 cm2), which was pre-cleaned by sequential washing with acetone and deionized (DI) water, followed by bath sonication for 10 min. It was then rinsed with ethanol, immersed in 3 M HCl solution for 30 min to remove surface oxides, sonicated again in DI water, and finally washed with ethanol. The cleaned NF was dried in a vacuum oven at 60 °C for 2 h to ensure complete removal of moisture,35 and then the coated electrodes (Alv-MnCo2O4@NF) were dried in a vacuum oven at 80 °C overnight. The active material loading on NF was 0.4 mg. The prepared electrodes served as working electrodes, while a platinum mesh (2 × 2 cm2) and a saturated calomel electrode (SCE) were used as counter and reference electrodes, respectively. Aqueous 1 M KOH was employed as the electrolyte. Electrochemical measurements were conducted using a Biologic SP-150e electrochemical workstation. Cyclic voltammetry (CV) was performed at various scan rates. Galvanostatic charge–discharge (GCD) tests were carried out at different current densities, with stability and coulombic test. Electrochemical impedance spectroscopy (EIS) was measured in the frequency range of 10 mHz to 100 kHz to assess the charge transfer resistance and ion diffusion characteristics. Fig. 2 illustrates the schematic representation of the electrode preparation process and the electrochemical measurement setup.
 | ||
| Fig. 2 Fabrication of Alv–MnCo2O4 electrode and electrochemical testing process, including slurry preparation, coating on nickel foam, vacuum drying, and CV analysis in a three-electrode setup (created in Biorender.com). | ||
3. Result and discussion
3.1. Morphological, structural and electronic structural characterisation
The phase purity and structural properties of the synthesized Alv–MnCo2O4 NPs were analysed using high-resolution synchrotron X-ray diffraction (XRD) with a monochromatic wavelength of 0.81262 Å. The diffraction pattern (Fig. 3a) displays broad yet well-resolved peaks that can be indexed to the face-centred cubic spinel structure with the Fd![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m space group (no. 227).25 The lattice parameters were found to be a = b = c = 0.81262, a = b = c = 8.2196 Å and α = β = γ = 90°, confirming the cubic symmetry of the structure. The most intense reflections corresponding to the (111), (220), (311), (400), (511), and (440) planes further validate the formation of a single-phase cubic MnCo2O4.25 The absence of any secondary or impurity peaks supports the phase purity of the sample. The synthesis route, involving aloe vera gel as a natural stabilizing and reducing agent, likely contributed to the clean phase formation and uniform nucleation under mild conditions. The broadening of the diffraction peaks, as evident from the pattern, suggests the formation of nanoscale crystallites with possible lattice strain.
m space group (no. 227).25 The lattice parameters were found to be a = b = c = 0.81262, a = b = c = 8.2196 Å and α = β = γ = 90°, confirming the cubic symmetry of the structure. The most intense reflections corresponding to the (111), (220), (311), (400), (511), and (440) planes further validate the formation of a single-phase cubic MnCo2O4.25 The absence of any secondary or impurity peaks supports the phase purity of the sample. The synthesis route, involving aloe vera gel as a natural stabilizing and reducing agent, likely contributed to the clean phase formation and uniform nucleation under mild conditions. The broadening of the diffraction peaks, as evident from the pattern, suggests the formation of nanoscale crystallites with possible lattice strain.
 | ||
| Fig. 3 (a) Synchrotron XRD pattern of Alv–MnCo2O4 NPs recorded in transmission mode using a wavelength of 0.08262 nm. (b) Williamson–Hall plot used to calculate the crystallite size and microstrain in Alv–MnCo2O4 NPs. (c) Thermogravimetric analysis (TGA) showing mass loss behaviour of the material. (d) FTIR spectra of Aloe vera extract (red line)34 and Alv–MnCo2O4 NPs (black line). | ||
To quantify these effects, Williamson–Hall (W–H) analysis was performed to decouple the contributions from crystallite size and microstrain. The W–H equation is expressed as following eqn (1):36
| βtotal = βcrystallitesize + βmicrostrain (β: peak broadening) | (1) |
The broadening caused by crystallite size can be calculated using the Scherrer equation:
 | (2) |
β = 4ε![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) tanθ tanθ | (3) |
Both eqn (2) and (3) are incorporated into eqn (1).
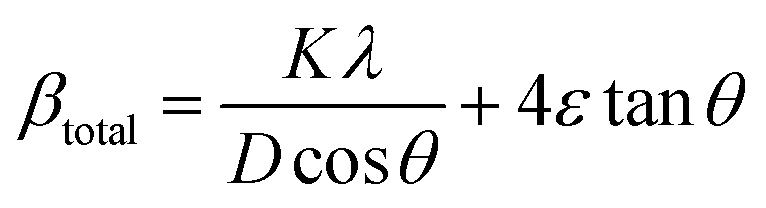

 | (4) |
Here, this takes the form of a straight-line equation: y = mx + c, where m(ε) is the slop and 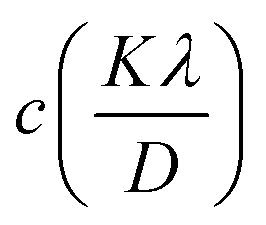 is the intercept. In eqn (4), βtotal is the full width at half maximum (FWHM) in radians, λ is the X-ray wavelength (0.81262 Å), K is the shape factor (assumed to be 0.9), D is the crystallite size, and ε is the microstrain.
is the intercept. In eqn (4), βtotal is the full width at half maximum (FWHM) in radians, λ is the X-ray wavelength (0.81262 Å), K is the shape factor (assumed to be 0.9), D is the crystallite size, and ε is the microstrain.
The corresponding W–H plot (Fig. 3b) of βcosθ versus 4sinθ showed a moderate positive slope of 0.00384 ± 0.00171, representing the microstrain, and an intercept of 0.00837 ± 0.00151, from which the average crystallite size was calculated to be approximately 9.13 nm. This nanoscale dimension is consistent with the observed peak broadening in the XRD pattern. Based on this crystallite size, the dislocation density (δ), was estimated using the following relation.37

The thermal stability of the Alv–MnCo2O4 NPs was evaluated using thermogravimetric analysis (TGA), as shown in Fig. 3c. The TGA curve displays a minimal weight loss of approximately 5.5% when the sample was heated from room temperature to 800 °C under a nitrogen atmosphere. The slight mass reduction below 200 °C is attributed to the removal of adsorbed moisture and residual volatile organic species from Aloe vera, such as surface-bound hydroxyls, carboxylates, or polysaccharide fragments.14,34 Beyond this range, the material exhibits remarkable thermal stability, with no significant weight loss observed up to 800 °C, indicating the formation of a robust and thermally stable spinel phase. This low mass loss reflects the successful calcination of organic residues during synthesis and confirms the structural integrity of the MnCo2O4 framework at elevated temperatures. Previous reports indicate that Aloe vera-derived organics decompose primarily between 200–400 °C, leaving negligible carbon residue above this temperature.40,41 This low mass loss reflects the successful calcination of organic residues during synthesis and confirms the structural integrity of the MnCo2O4 framework at elevated temperatures.40,41 Such thermal robustness is particularly advantageous for electrochemical energy storage applications, where electrode materials are expected to endure high-temperature processing and prolonged cycling without degradation.
To study the functional groups involved in the synthesis and surface chemistry of the material, we performed Fourier-transform infrared (FTIR) spectroscopy on both Aloe vera extract and the resulting Alv–MnCo2O4 NPs, as shown in Fig. 3d. The Aloe vera spectrum displays a broad and intense band in the range of 3400–3600 cm−1, attributed to the O–H stretching vibrations from hydroxyl groups present in phenolic compounds, carboxylic acids, and bound water molecules.35 These bands persist in the MnCo2O4 spectrum but are sharper with a shoulder at 3426 cm−1, suggesting retention of surface hydroxyl groups, only because of adsorbed moisture. Additionally, symmetric and asymmetric C–H stretching vibrations (in CH2 or CH3 group) appear near 2930 cm−1 and 2852 cm−1, respectively in both Aloe vera and MnCo2O4 spectra, indicative of aliphatic chain remnants or surface-bound biomolecules.9 A key band near 1634 cm−1 is observed in both spectra. In MnCo2O4, this is primarily assigned to the bending vibration (H–O–H) of surface-adsorbed water molecules, common in spinel-type metal oxides,26 whereas in Aloe vera, this band may also correspond to amide I (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching) or antisymmetric –O–CO− stretching from proteins and organic acids.9 Additional bands at 1386 cm−1 and near 1100 cm−1 are associated with symmetric carboxylate stretching and C–O/C–OH vibrations, which originate from residual Aloe vera polysaccharides and esters.26 These organic functional groups confirm active involvement of aloe vera in complexing and templating during synthesis. Significantly, the MnCo2O4 spectrum exhibits two strong absorption bands at 659 cm−1 and ∼570 cm−1, corresponding to Co–O and Mn–O stretching vibrations in tetrahedral environment, respectively characteristic of the spinel lattice.42 The presence of these sharp metal-oxygen bands affirms successful formation of crystalline MnCo2O4 nanostructures, while the attenuated but observable organic peaks indicate partial surface capping by Aloe vera-derived biomolecules. Collectively, the FTIR results support the role of Aloe vera as a green, dual-functional agent, acting as a reductant and stabilizer, facilitating the formation of MnCo2O4 nanostructures with enriched surface functionalities. These surface groups are anticipated to improve electrolyte accessibility and interface wettability, thereby benefiting the electrochemical behaviour of the material for energy storage applications.
O stretching) or antisymmetric –O–CO− stretching from proteins and organic acids.9 Additional bands at 1386 cm−1 and near 1100 cm−1 are associated with symmetric carboxylate stretching and C–O/C–OH vibrations, which originate from residual Aloe vera polysaccharides and esters.26 These organic functional groups confirm active involvement of aloe vera in complexing and templating during synthesis. Significantly, the MnCo2O4 spectrum exhibits two strong absorption bands at 659 cm−1 and ∼570 cm−1, corresponding to Co–O and Mn–O stretching vibrations in tetrahedral environment, respectively characteristic of the spinel lattice.42 The presence of these sharp metal-oxygen bands affirms successful formation of crystalline MnCo2O4 nanostructures, while the attenuated but observable organic peaks indicate partial surface capping by Aloe vera-derived biomolecules. Collectively, the FTIR results support the role of Aloe vera as a green, dual-functional agent, acting as a reductant and stabilizer, facilitating the formation of MnCo2O4 nanostructures with enriched surface functionalities. These surface groups are anticipated to improve electrolyte accessibility and interface wettability, thereby benefiting the electrochemical behaviour of the material for energy storage applications.
The morphological characteristics and elemental composition of the Alv–MnCo2O4 NPs were systematically analysed using transmission electron microscopy (TEM), field emission scanning electron microscopy (FESEM), energy-dispersive X-ray spectroscopy (EDS), and elemental mapping techniques as shown in Fig. 4. The TEM images (Fig. 4a and b) reveal quasi-spherical, nanoscale particles with good dispersion. The corresponding particle size distribution histogram (Fig. 4c) shows an average particle size of approximately 12.7 nm, which closely matches the average crystallite size estimated from XRD (∼9.2 nm), indicating that the material is largely crystalline at the nanoscale. FESEM image (Fig. 4d) displays a more aggregated morphology, where the individual particles appear clustered into larger secondary structures. The particle size histogram derived from FESEM (Fig. 4f) shows a broader distribution with an average size of 26.2 nm, which is noticeably larger than the TEM-based size. This difference can be attributed to the agglomeration of small nanocrystallites, a well-documented phenomenon in nanomaterials.43 Nanoparticles with reduced crystallite sizes possess high surface energies, which drive them to aggregate during the drying or annealing steps in order to minimize total surface energy.43,44
 | ||
| Fig. 4 (a) and (b) TEM images of Alv–MnCo2O4 NPs showing quasi-spherical and well-dispersed nanoparticles. (c) Particle size distribution histogram obtained from TEM analysis, showing an average particle size of ∼12.7 nm. (d) FESEM micrographs illustrating agglomerated secondary particles, formed due to the aggregation of primary nanocrystallites, (e) the EDS spectrum displays the elemental composition, revealing a slightly cobalt-rich stoichiometry, which is favourable for redox-based energy storage. (f) Corresponding particle size distribution from FESEM showing an average size of ∼26.2 nm. (g) Elemental mapping images confirming the homogeneous distribution of Mn, Co, and O in the nanostructure. | ||
The EDS spectrum (Fig. 4e) confirms the presence of Mn, Co, and O elements in the sample, with a weight percentage of Co (53.33%), O (25.69%), and Mn (20.98%), and corresponding atomic percentages of 31.29%, 55.51%, and 13.20%, respectively. The slightly cobalt-rich composition in material, suggests a favourable stoichiometric deviation that enhances electrochemical redox activity, likely due to the increased contribution of Co2+/Co3+ redox couples.45 Furthermore, the elemental mapping images (Fig. 4g) demonstrate a uniform and homogeneous distribution of Mn, Co, and O across the surface, validating the compositional consistency of the synthesized nanostructures. Importantly, the Aloe vera-assisted synthesis route used in this study plays a pivotal role in achieving such a fine nanostructure. Bioactive compounds present in Aloe vera, such as polysaccharides and phenolic compounds, act as natural capping and reducing agents, controlling the nucleation and limiting the growth of crystallites during synthesis.5,34 This green and sustainable approach contributes significantly to the reduction in crystallite size, which is beneficial for electrochemical applications, as it provides more active surface area and redox-active sites for charge storage.
The surface area and porosity of the Alv–MnCo2O4 NPs were assessed via nitrogen adsorption–desorption isotherms and BJH pore size distribution analysis. As presented in Fig. 5a, the adsorption/desorption curves show hysteresis and the shape suggest type IV (h3) isotherm. This reveals mesoporous nature of the nanomaterial with slit like pores.46 BET analysis yielded a specific surface area of 43.27 m2 g−1, while the corresponding BJH analysis (Fig. 5b) revealed a narrow pore size distribution centred around 10 nm, with a cumulative pore volume of 0.142 cm3 g−1. These parameters confirm the formation of a mesoporous architecture with moderate surface area and well-defined pore structure. Such mesoporosity is particularly advantageous for electrochemical energy storage applications, as it promotes efficient electrolyte penetration and facilitates rapid ion diffusion throughout the electrode matrix.47 Moreover, the accessible surface area provides abundant electroactive sites, enhancing faradaic redox interactions. The interconnected mesoporous channels are expected to mitigate ion transport limitations at high current densities, thereby improving rate performance and stability during prolonged cycling.48 Collectively, high surface area (43.27 m2 g−1), slit like pore structure, favourable morphology, surface rich in functional groups and Co rich stoichiometry of the synthesized Alv–MnCo2O4 NPs material contribute significantly to its electrochemical performance.
 | ||
| Fig. 5 (a) Nitrogen adsorption–desorption isotherms of the synthesized Alv–MnCo2O4 NPs show a typical type IV isotherm with a clear H3-type hysteresis loop, indicating mesoporous characteristics. The BET-specific surface area of the material is calculated to be 43.27 m2 g−1. (b) BJH pore size distribution curve shows both cumulative and differential pore volume as a function of pore diameter, with a dominant pore size around 10 nm and a cumulative pore volume of approximately 0.142 cm3 g−1, further confirming the mesoporous nature of the material. | ||
The XPS analysis was carried out to investigate the surface composition and oxidation states of the elements present in Alv–MnCo2O4 NPs (Fig. 6). The C 1s spectrum (Fig. 6a) was deconvoluted into three peaks located at 284.8 eV, 286.3 eV, and 288.7 eV, corresponding to C–C, C–O, and O–CO bonds, respectively.34,49,50 Since the adventitious C–C binding energy is found at 284.8 eV, we rule out any significant charging effect in the sample, and the binding energy (BE) of all the edges are used without any modification.34 The high-resolution O 1s spectrum (Fig. 6b) exhibits three peaks at 530.1 eV, 531.8 eV, and 533.2 eV, corresponding to lattice oxygen (M-O), oxygen vacancies (O-V), and surface-adsorbed oxygen species or hydroxyl groups (O-ads), respectively.27 The oxygen vacancies in the electrode materials act as functional sites for improved capacity.34 The Mn 2p high-resolution spectrum (Fig. 6c) shows spin–orbit split peaks around 642 eV (Mn 2p3/2) and 653 eV (Mn 2p1/2), respectively. A spin–orbit splitting of about 11 eV is in agreement with literature.27 The Mn 2p3/2 and 2p1/2 peaks are further fitted with two Gaussians each and the 2p3/2 peaks are centred at 642.1 and 644.6 eV. These peaks are attributed to Mn3+ and Mn4+, respectively.51 Similarly, for 2p1/2 band the peaks at 653.5 and 655.9 eV represent Mn3+ and Mn4+, respectively, indicating a mixed valence state of Mn.52 A deviation of oxidation state from nominal oxidation states of either +2 or +3 for Mn is well documented in literature,51 and a surface rich in Mn4+ also indicates oxygen vacancies at the surface, which is in agreement with FTIR analysis, where absorption modes corresponding to oxygen vacancies are observed.
 | ||
| Fig. 6 High-resolution XPS spectra of Aloe vera-assisted MnCo2O4 nanorods showing (a) C 1s spectrum deconvoluted into peaks corresponding to C–C, CC, and O–CO functional groups; (b) O 1s spectrum showing lattice oxygen (O-L), oxygen vacancies (O-V), and adsorbed oxygen species (O-ads); (c) Mn 2p spectrum indicating the presence of Mn3+, and Mn4+ oxidation states; and (d) Co 2p spectrum confirming the coexistence of Co2+ and Co3+ along with satellite features. | ||
The Co 2p spectrum (Fig. 6d) exhibits two spin–orbit split peaks around 781 eV (Co 2p3/2) and 795 eV (Co 2p1/2), along with satellite features. Deconvolution of 2p3/2 peak reveals peaks at 780.1 and 781.5 eV, characteristics of Co2+ and Co3+ species, respectively. Two shake-up satellite features at ∼786 and 791 eV, attributed to Co2+ and Co3+, are also in agreement with literature.27,51 MnCo2O4 is a direct spinel and hence Co2+ occupies entire tetrahedral site, whereas, Mn3+ and Co3+ occupy the octahedral sites. The presence of +4 oxidation state for Mn introduces oxygen vacancies and/or oxygen interstitials, which serve as electroactive sites and are beneficial for faradaic charge storage. Thus, the synergy between Co2+/Co3+ and Mn3+/Mn4+ species contributes significantly to the improved electrochemical behaviours of the Alv–MnCo2O4 electrode.52
3.2. Electrochemical characterisation
The electrochemical performance of Alv–MnCo2O4 NPs was systematically evaluated using cyclic voltammetry (CV) and galvanostatic charge–discharge (GCD) techniques, demonstrating its remarkable potential for supercapacitor applications. In the CV curves (Fig. 7a), recorded at scan rates ranging between 1 to 200 mV s−1, exhibit well-defined redox peaks characteristic of battery-type faradaic behaviour.35 At a scan rate of 5 mV s−1, the anodic peak at 0.38 V and cathodic peak at 0.29 V correspond to the oxidation and reduction of Mn and Co cations to their respective oxyhydroxides and oxides.53 The nanostructured electrode facilitates fast electron transfer between the transition metal ions.53 A slight positive shift of the anodic peak and a negative shift of the cathodic peak with increasing scan rate were observed, which is a common phenomenon in supercapacitor materials.54 This shift can be attributed to internal resistance (IR) drop and the kinetic limitations of ion diffusion at higher sweep rates, which restrict full faradaic interaction within the limited time frame.55 Despite this shift, the overall shape of the curves remains stable, indicating good electrochemical reversibility and structural stability under rapid charge–discharge conditions. The specific capacitance calculated from CV using the eqn (5),56 yielded values of 639.2, 637.0, 596.3, 527.8, 481.5, 451.4, 428.9, 411.6, 391.5, and 365.7 F g−1 at scan rates of 5, 10, 20, 40, 60, 80, 100, 120, 150, and 200 mV s−1, respectively. Even at a high scan rate of 200 mV s−1, the material retained a capacitance of 365.7 F g−1, demonstrating excellent surface-controlled capacitive behaviour and effective ion transport.34 The gradual decline in capacitance at higher scan rates is mainly due to the limited diffusion time of electrolyte ions, which restricts the utilization of deeper active sites for faradaic reactions.35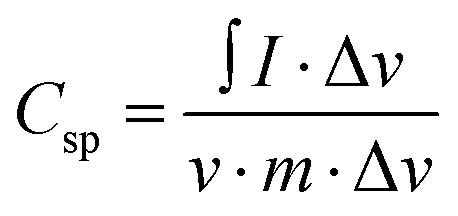 | (5) |
 | ||
| Fig. 7 (a) CV curves of Alv–MnCo2O4 NPs electrode at various scan rates from 5 to 200 mV s−1 showing stable redox peaks with minimal shift, indicating consistent redox behaviour. (b) GCD profiles recorded at current densities from 1 to 80 A g−1 demonstrating excellent rate capability and symmetric charge–discharge characteristics. (c) Anodic peak current vs. scan rate plot showing a b-value of ∼0.78, confirming mixed charge storage behaviour (supercapacitor). (d) Bar plots showing capacitive and diffusion-controlled contributions to charge storage at different scan rates. | ||
Complementing the CV analysis, GCD curves (Fig. 7b) of the Alv–MnCo2O4 electrode at current densities from 1 to 80 A g−1 exhibited nearly symmetric charge–discharge profiles, further confirming the pseudocapacitive nature of the material. The specific capacitance values calculated from the GCD data using eqn (6),56 were 680, 640, 604.4, 568.9, 506.7, 497.8, 311.1, 357.3, and 282.7 F g−1 at 1, 2, 4, 8, 12, 16, 20, 40, and 80 A g−1, respectively. Even at the extremely large current density of 80 A g−1, a decent capacitance of 282.7 F g−1 was maintained, underscoring excellent rate capability of the electrode. This performance showcases the Alv–MnCo2O4 NPs as a highly efficient electrode material, with high-rate capability for high-power applications. The consistency in redox peak behaviour and high capacitance retention at elevated scan rates and current densities strongly affirms its potential for next-generation supercapacitor applications.
 | (6) |
To further understand the charge storage mechanism, the relationship between the peak current (ip) and the scan rate (v) was analysed using the power law eqn (7):56
| ip = avb | (7) |
As shown in Fig. 7c, the calculated b value is approximately 0.78, indicating a mixed charge storage behaviour. b value close to 0.5 typically signifies a diffusion-controlled process, while a value near 1.0 suggests a surface-controlled capacitive process. The intermediate b value of 0.78 suggests that the Alv–MnCo2O4 electrode exhibits a synergistic supercapacitor behaviour, combining both battery-type faradaic reactions and capacitive surface-controlled mechanisms. At lower scan rates, the material primarily demonstrates diffusion-controlled behaviour due to the sufficient time available for ion diffusion into the bulk of the electrode. However, as the scan rate increases, the charge storage becomes increasingly dominated by surface redox reactions, where fast electron transfer and surface-accessible active sites govern the process. This transition highlights the ability of material to adapt across different charge–discharge regimes, making it highly suitable for hybrid energy storage systems demanding both high energy and power densities.
Additionally, the capacitive and diffusion contributions to the total current were separated using the eqn (8):57
| i(V) = k1v + k2v1/2 = Icapacitive + Idiffusion | (8) |
 | (9) |
The slope (k1) and intercept (k2) of the plot of  versus v1/2 (Fig. 7d) provide insights into the capacitive and diffusion contributions. Using these constants, the capacitive contribution percentage was calculated as eqn (10).58
versus v1/2 (Fig. 7d) provide insights into the capacitive and diffusion contributions. Using these constants, the capacitive contribution percentage was calculated as eqn (10).58
 | (10) |
The diffusion contribution can similarly be calculated as eqn (11):
| Diffusion contribution (%) = 100% − Capacitive contribution (%) | (11) |
Fig. 7d illustrates the trend of the capacitive and diffusion contributions as a function of scan rate. It is evident from the data that the capacitive contribution increases with increasing scan rate, highlighting the surface-controlled nature of the charge storage at higher scan rates. This trend is further supported by Fig. 8a–d, which compare the CV profiles at 5 mV s−1, 40 mV s−1, 100 mV s−1 and 200 mV s−1 respectively. At lower scan rates, diffusion-controlled processes dominate, as the ions have sufficient time to access the entire electrode surface. In contrast, at higher scan rates, the capacitive contribution increases as the process becomes more surface-controlled, resulting in faster ion exchange at the electrode–electrolyte interface.58,59
 | ||
| Fig. 8 Capacitive and diffusion contribution analysis of Alv–MnCo2O4 N at different scan rates: (a) at 5 mV s−1, capacitive contribution is 31% (b) at 40 mV s−1, capacitive contribution is 55%; (c) at 100 mV s−1, capacitive contribution is 66%; (d) at 200 mV s−1, capacitive contribution increases to 74%, highlighting surface-dominated behavior at higher scan rates. | ||
Fig. 9a illustrates the variation of specific capacitance with current density, while Fig. 9b presents the trend with scan rate. Both plots exhibit a consistent decline in specific capacitance with increasing current density and scan rate, which is typical due to limited ion diffusion and incomplete electrode utilization at high rates. Notably, the material retains a significant portion of its capacitance even at very high current densities, highlighting its excellent rate capability and robustness, an essential criterion for practical energy storage applications. Fig. 9c depicts the cycling performance of the Alv–MnCo2O4 NPs electrode over 5000 continuous charge–discharge cycles at a high current density of 20 A g−1. Remarkably, it exhibited a stable capacitance retention of 84.5% after 5000 cycles. Interestingly, an initial sudden decay of approximately 16.5% was observed within the first ∼800 cycles, likely due to surface reconstruction or structural rearrangements. However, after this initial phase, the capacitance remained nearly constant, indicating the stabilization of the electrode–electrolyte interface and mechanical integrity of the material during long-term cycling.
 | ||
| Fig. 9 (a) Specific capacitance vs. current density trend showing excellent retention at high current densities; (b) specific capacitance vs. scan rate trend indicating strong performance even at elevated scan rates; (c) long-term cycling performance showing 84.5% capacitance retention after 5000 cycles, reflecting excellent stability; (d) Ragone plot of the Alv–MnCo2O4 electrode showing the relationship between energy density and power density at various current densities, highlighting high-energy and high-power performance for supercapacitor applications. | ||
The corresponding Ragone plot (Fig. 9d) illustrates the energy–power performance of the electrode. The energy density (E) in Wh kg−1 and power density (P) in W kg−1 were calculated using the eqn (12) and (13):60
 | (12) |
 | (13) |
000 W kg−1 at 80 A g−1, demonstrating the excellent high-power performance and suitability of the electrode for supercapacitor applications.
The EIS analysis of the Alv–MnCo2O4 electrode is presented in Fig. 10, with the inset showing the equivalent circuit used for fitting. The circuit (Fig. 10 inset)60 was fitted using Z-view software with data obtained from the EC-lab module of the Biologic SP-150e electrochemical workstation. The Nyquist plot reveals a small equivalent series resistance (Rs) of ∼0.56 Ω, indicating low intrinsic resistance and good electrical conductivity. The nearly vertical line in the low-frequency region signifies ideal capacitive behavior and efficient ion diffusion. Together, these results confirm the fast charge transfer kinetics and excellent electrochemical performance of the material.
 | ||
| Fig. 10 Nyquist plot from EIS analysis showing low equivalent series resistance (∼0.56 Ω), confirming fast charge transfer kinetics and low internal resistance. | ||
The electrochemical performance of various MnCo2O4-based electrodes reported in recent literature is summarized in Table 1. The superior electrochemical performance demonstrated by the MnCo2O4 electrode material synthesized using Aloe vera pulp as a natural reducing and capping agent is a direct outcome of: (i) the formation of small (∼9 nm) and well-dispersed nanoparticles with minimal agglomeration, as confirmed by synchrotron XRD and TEM analyses, along with a mesoporous, slit-like pore structure and high surface area (43.27 m2 g−1), as revealed by BET analysis, which improves wettability and electrolyte accessibility; (ii) the presence of a thin and stable solid electrolyte interphase (SEI), as indicated by the low series resistance in EIS measurements; and (iii) the existence of surface oxygen vacancies, as confirmed by XPS and FTIR results. The optimized physicochemical characteristics revealed through preceding structural, morphological, and surface analyses. The nanostructured architecture, mesoporosity, and tailored surface chemistry collectively facilitate rapid ion transport, abundant electroactive sites, and stable redox kinetics. The presence of multi-valent Co and Mn species, as identified via XPS, enhances faradaic charge storage through multiple redox transitions, while the mesoporous texture observed from BET analysis ensures effective electrolyte penetration and mitigates diffusion resistance. Furthermore, the nanoscale features and controlled crystallinity provide an ideal balance between conductivity and structural integrity, promoting high-rate capability and prolonged cycling stability. These pre-electrochemical insights form a strong foundation that justifies the excellent GCD results and further establish the Alv–MnCo2O4 NPs as a well-engineered, high-performance electrode material for energy storage applications.
| Materials | Morphology | Synthesis | Electrolyte | Specific capacitance | Stability | Substrate | Ref. |
|---|---|---|---|---|---|---|---|
| MnCO2O4 | Nanoneedles | Hydrothermal | 2 M KOH | 420 F g−1 at 5 A g−1 | 99% after 10000 cycles |
Ni-foam | 61 |
| MnCO2O4 composite | Nanospheres | Hydrothermal | 6 M KOH | 654 F g−1 at 2 A g−1 | 92.3% after 5000 cycles | Ni-foam | 62 |
| MnCO2O4 composite | Nanosheets | Electrodeposition | 6 M KOH | 668 F g−1 at 1 A g−1 | 90% after 5000 cycles | Cu-foil | 63 |
| MnCO2O4 | Pod like porous microstructure | Solvothermal | 2 M KOH | 321 F g−1 at 1 A g−1 | 87% after 4000 cycles | Ni-foam | 64 |
| MnCO2O4 composite | Nanoplates | Hydrothermal | 2 M KOH | 271.06 C g−1 at 2 A g−1 | 81.96% at 10 A g−1 | Ni-foam | 65 |
| MnCO2O4 composite | Nanoflowers | Hydrothermal | 2 M KOH | 232.27 C g−1 at 2 A g−1 | 70.32% at 10 A g−1 | Ni-foam | 65 |
| MnCO2O4 | Falke like spherical structure | Hydrothermal | 1 M KOH | 671 F g−1 at 5 mV s−1 | 92% after 1000 cycles | Ni-foil | 66 |
| MnCO2O4 | Cuboidal micro flakes | Hydrothermal | 3 M LiOH | 600 F g−1 at 0.5 A g−1 | ∼126% after 3000 cycles | Ni-foam | 67 |
| MnCO2O4 | Nanoroads | Hydrothermal | 2 M KOH | 187.5 F g−1 at 0.25 A g−1 | 90% after 5000 cycles | Ni-foil | 42 |
| MnCO2O4 | Nanosheets | Solvothermal | 6 M KOH | 346 F g−1 at 1 A g−1 | 88% after 2500 cycles | Ni-foil | 68 |
| MnCO2O4 | Nanoparticles | Simple polymer solution | 6 M KOH | 208 F g−1 at 1 A g−1 | 70% at 1000 cycles | Ni-foam | 52 |
| MnCO2O4 | Tubular nanowires | Electro-spinning | 6 M KOH | 500.91 F g−1 at 0.5 A g−1 | 79% after 2500 cycles | Carbon fiber paper | 69 |
| MnCO2O4 composite | 3-D porous | Hydrothermal | 3 M KOH | 503 F g−1 at 1 A g−1 | 97.4% after 5000 cycles | Ni-foam | 70 |
| MnCO2O4 composite | Nanosheets | Hydrothermal | 2 M KOH | 508.3 F g−1 at 2 A g−1 | 89.7% after 2000 cycles | Ni-foam | 71 |
| CoMoO4 | Spherical | Green synthesis (bioorganic derived) | 3 M KOH | 294 F g−1 at 2 mV s−1 and 153.2 F g−1 at 0.5 A g−1 | — | Ni-foam | 72 |
| NiCo2O4 | Nanoparticles clusters | Green synthesis (orange peel assisted) | 3 M KOH | 581.25 F g−1 at 2.5 A g−1 | ∼100% after 30000 cycles |
Ni-foam | 73 |
| NiCo2O4 | Nanostructure | Green synthesis (moringa oleifera extracts) | 3 M KOH | 402 F g−1 at 1 A g−1 | ∼85% after 8000 cycles | Ni-foam | 74 |
| NiCo2O4 | Nanorods | Green synthesis (cellulose derived) | 6 M KOH | 1302 F g−1 at 1 A g−1 | 92% after 5000 cycles | Ni-foam | 75 |
| ZnMn2O4 | Nanoparticles | Green synthesis (corallocarpus epigaeus derived) | 1 M KCl | 306 F g−1 at 0.6 A g−1 | 63% after 1000 cycles | ITO substrate | 37 |
| MgFe2O4 | Nanoparticles | Green synthesis (P. macrosolen L. leaf extract) | 2 M Na2SO4 | 585 F g−1 at 3 A g−1 | 95.1% after 5000 cycles | Carbon paper | 76 |
| C@MnCo2O4 | Nanocomposite | Green synthesis | 1 M KOH | ∼726 mF cm−2 at 0.5 mV s−1 | 61.4% after 5000 cycles | Carbon | 77 |
| CuCo2O4 | Nanocomposite | Green synthesis | 3 M KOH | 421 mAh g−1 at 10 mV s−1 | ∼99% after 10000 cycles |
— | 78 |
| MnCO2O4 | Nanoparticles | Green synthesis (Aloe vera assisted) | 1 M KOH | 680 F g−1 at 1 A g−1 | 84.5% after 5000 cycles | Ni-foam | This work |
4. Conclusion
In this study, we demonstrated a sustainable, Aloe vera-assisted green synthesis of spinel-type MnCo2O4 nanostructures for high-performance hybrid supercapacitor applications. The material was synthesized via an eco-friendly route and comprehensively characterized. Synchrotron XRD confirmed the formation of a single-phase cubic spinel structure with an average crystallite size of ∼9.13 nm, and TEM/FESEM analyses revealed well-dispersed, quasi-spherical nanoparticles ranging from ∼12.7 to ∼26.2 nm. BET analysis confirmed a mesoporous structure with a moderate surface area (43.27 m2 g−1) and ∼10 nm pore size, ideal for rapid ion transport. XPS and FTIR analyses revealed the presence of oxygen vacancies and/or oxygen interstitials, and the coexistence of Mn3+/Mn4+ and Co2+/Co3+ oxidation states, providing multiple redox-active sites and improved charge storage kinetics. A b-value of ∼0.78 and increasing capacitive contributions at higher scan rates demonstrated mixed charge storage behaviour (battery-type and capacitive), supporting the supercapacitor characteristics of material. The material exhibited excellent electrochemical performance, delivering a high specific capacitance of 680 F g−1 at 1 A g−1, 506.7 F g−1 at 12 A g−1 (retaining 74.5% of initial value), and 282.7 F g−1 at 80 A g−1, based on GCD measurements; additionally, a capacitance of 365.7 F g−1 was obtained at a high scan rate of 200 mV s−1 from CV analysis, confirming excellent rate capability. The electrode maintained 84.5% of its initial capacitance after 5000 cycles at 20 A g−1. Together, these structural, chemical, and electrochemical features highlight the synergistic impact of biogenic synthesis, multi-valent redox couples, mesoporosity, and nanoscale architecture. This study provides a promising, scalable, and environmentally friendly route toward energy storage materials for hybrid supercapacitor applications.Author contributions
M. B.: conceptualization, investigation, data curation, formal analysis, methodology, visualization, writing – original draft. K. G.: conceptualization, writing – review & editing. A. K. S.: supervision, project administration, formal analysis, writing – review & editing.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper. The authors declare the following financial interests/personal relationships which may be considered as potential competing interests.Data availability
The datasets generated during and/or analysed during the current study are not publicly available due to their inclusion in the author's ongoing PhD work but are available from the authors upon reasonable request.Acknowledgements
M.B and K.G acknowledges CIC and R&D at UPES for providing the necessary research infrastructure and JRF fellowship. MB Acknowledges Dr Archna Sagdeo, BL-12, RRCAT, Indore, India for XRD characterization.References
- M. Bulla, V. Kumar, R. Devi, S. Kumar, A. K. Sisodiya, R. Dahiya and A. K. Mishra, Sci. Rep., 2024, 14, 7389 CrossRef CAS PubMed.
- J. Meena, S. S. Sivasubramaniam, E. David and Santhakumar K., RSC Sustainability, 2024, 2, 1224–1245 RSC.
- H. Ashraf and B. D. Karahan, Mater. Res. Bull., 2024, 169, 112492 CrossRef CAS.
- F. Ulaş, E. Yüksel, D. Dinçer, A. Dababat and M. İmren, Sustainability, 2025, 17, 4152 CrossRef.
- A. Surjushe, R. Vasani and D. Saple, Indian J. Dermatol., 2008, 53, 163 CrossRef PubMed.
- D. I. Sánchez-Machado, J. López-Cervantes, R. Sendón and A. Sanches-Silva, Trends Food Sci. Technol., 2017, 61, 94–102 CrossRef.
- J. P. Delatorre-Castillo, J. Delatorre-Herrera, K. S. Lay, J. Arenas-Charlín, I. Sepúlveda-Soto, L. Cardemil and E. Ostria-Gallardo, Plants, 2022, 11, 1523 CrossRef CAS PubMed.
- W. E. Luligo-Montealegre, S. Prado-Alzate, A. Ayala-Aponte, D. F. Tirado and L. Serna-Cock, Horticulturae, 2024, 10, 797 CrossRef.
- K. Gautam, M. Bhatt, S. Dutt, A. Sagdeo and A. K. Sinha, Sci. Rep., 2025, 15, 11506 CrossRef CAS PubMed.
- L. Suriati and I. M. S. Utama, J. Phys.: Conf. Ser., 2019, 1402, 066021 CrossRef CAS.
- K. Patruni, S. Chakraborty and S. R. Pavuluri, Int. J. Biol. Macromol., 2018, 120, 414–421 CrossRef CAS PubMed.
- S. Kumar, A. Tahira, A. L. Bhatti, M. A. Bhatti, R. H. Mari, N. M. Shaikh, M. Y. Solangi, A. Nafady, M. Emo, B. Vigolo, A. Infantes-Molina, A. Vomiero and Z. H. Ibupoto, RSC Adv., 2023, 13, 18614–18626 RSC.
- S. Hussain, S. A. Al-Thabaiti and Z. Khan, Bioprocess Biosyst. Eng., 2014, 37, 1727–1735 CrossRef CAS PubMed.
- A. Hadi, A. Nawab, F. Alam and K. Zehra, J. Food Process Preserv., 2022, 62(7), 2111–2118 CAS.
- S. Rahman, P. Carter and N. Bhattarai, JFB, 2017, 8, 6 CrossRef PubMed.
- M. A. Rafiq, A. Javed, M. N. Rasul, M. Nadeem, F. Iqbal and A. Hussain, Mater. Chem. Phys., 2021, 257, 123794 CrossRef CAS.
- J. Kwon, J.-H. Kim, S.-H. Kang, C.-J. Choi, J. A. Rajesh and K.-S. Ahn, Appl. Surf. Sci., 2017, 413, 83–91 CrossRef CAS.
- S. Yuvaraj, R. K. Selvan and Y. S. Lee, RSC Adv., 2016, 6, 21448–21474 RSC.
- Y. Zhu, X. Ji, Z. Wu, W. Song, H. Hou, Z. Wu, X. He, Q. Chen and C. E. Banks, J. Power Sources, 2014, 267, 888–900 CrossRef CAS.
- K. Samatha and R. Sagar, Bull. Mater. Sci., 2024, 47, 163 CrossRef CAS.
- H. Sun, Y. Miao, G. Wang, X. Han, Y. Wang, Z. Zhang, C. Luo, X. Liu, C. Xu and H. Chen, J. Energy Storage, 2024, 76, 109780 CrossRef.
- J. Sun, X. Tian, C. Xu and H. Chen, J. Materiomics, 2021, 7, 1358–1368 CrossRef.
- M. Murugesan, N. Nallamuthu, R. Ranjithkumar, M. Krishnakumar, P. Devendran and K. Ramesh, Electron. Mater. Lett., 2023, 19, 108–118 CrossRef CAS.
- S. D. Dhas, P. N. Thonge, S. D. Waghmare, G. K. Kulkarni, S. K. Shinde, D.-Y. Kim, T. M. Patil, M. A. Yewale, A. V. Moholkar and D. Kim, J. Energy Storage, 2023, 71, 108168 CrossRef.
- K. Kotalgi, A. Kanojiya, A. Tisekar and P. H. Salame, Chem. Phys. Lett., 2022, 800, 139660 CrossRef CAS.
- J. Xu, Y. Sun, M. Lu, L. Wang, J. Zhang, E. Tao, J. Qian and X. Liu, Acta Mater., 2018, 152, 162–174 CrossRef CAS.
- A. Ashok, A. Kumar, J. Ponraj and S. A. Mansour, J. Electrochem. Soc., 2020, 167, 054507 CrossRef CAS.
- S. Azizi, M. Seifi, M. T. Tourchi Moghadam, F. Jamali, P. Salarizadeh and M. B. Askari, Diamond Relat. Mater., 2023, 137, 110061 CrossRef CAS.
- X. Shi, Z. Liu, Y. Zheng and G. Zhou, Colloids Surf., A, 2017, 522, 525–535 CrossRef CAS.
- I. T. Papadas, A. Ioakeimidis, G. S. Armatas and S. A. Choulis, Adv. Sci., 2018, 5, 1701029 CrossRef PubMed.
- N. P. Zentefis, A. Fiore and Y. Yang, Energy Fuels, 2025, 39, 7167–7181 CrossRef CAS.
- A. K. Sinha, A. Sagdeo, P. Gupta, A. Upadhyay, A. Kumar, M. N. Singh, R. K. Gupta, S. R. Kane, A. Verma and S. K. Deb, J. Phys.: Conf. Ser., 2013, 425, 072017 CrossRef CAS.
- A. Hammersley, Google Scholar There is no corresponding record for this reference.
- M. Bhatt, K. Gautam, A. Verma and A. K. Sinha, Mater. Adv., 2025, 6(16), 5618–5632 RSC.
- M. Bhatt, B. Gupta and A. K. Sinha, Sci. Rep., 2025, 15, 2192 CrossRef CAS PubMed.
- M. Kawsar, M. Sahadat Hossain, N. M. Bahadur, D. Islam and S. Ahmed, Mater. Adv., 2025, 6, 3889–3902 RSC.
- J. Uma, S. Banumathi, R. Maheswaran, N. Senthilkumar and B. Balraj, J. Supercond. Nov. Magn., 2021, 34, 817–823 CrossRef CAS.
- Y. Xu, X. Wang, C. An, Y. Wang, L. Jiao and H. Yuan, J. Mater. Chem. A, 2014, 2, 16480–16488 RSC.
- T. Nguyen, M. Boudard, L. Rapenne, O. Chaix-Pluchery, M. J. Carmezim and M. F. Montemor, RSC Adv., 2015, 5, 27844–27852 RSC.
- J. Saha, Md. I. H. Mondal, F. Ahmed and M. Rahman, Arabian J. Chem., 2023, 16, 105087 CrossRef CAS.
- I. Solaberrieta, A. Jiménez, I. Cacciotti and M. C. Garrigós, Polymers, 2020, 12, 1323 CrossRef CAS PubMed.
- M. Haripriya, A. M. Ashok, S. Hussain and R. Sivasubramanian, Ionics, 2021, 27, 325–337 CrossRef CAS.
- S. Shrestha, B. Wang and P. Dutta, Adv. Colloid Interface Sci., 2020, 279, 102162 CrossRef CAS PubMed.
- A. Cartwright, K. Jackson, C. Morgan, A. Anderson and D. W. Britt, Agronomy, 2020, 10, 1018 CrossRef CAS.
- A. Tomar, C. Vankani, S. P. Singh, M. Winter and A. K. Rai, Phys. Chem. Chem. Phys., 2024, 26, 3516–3524 RSC.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. W. Sing, Pure Appl. Chem., 2015, 87, 1051–1069 CrossRef CAS.
- Y. Yan, G. Chen, P. She, G. Zhong, W. Yan, B. Y. Guan and Y. Yamauchi, Adv. Mater., 2020, 32, 2004654 CrossRef CAS PubMed.
- T. Y. Ma, Y. Zheng, S. Dai, M. Jaroniec and S. Z. Qiao, J. Mater. Chem. A, 2014, 2, 8676–8682 RSC.
- M. C. Biesinger, B. P. Payne, A. P. Grosvenor, L. W. M. Lau, A. R. Gerson and R. S. C. Smart, Appl. Surf. Sci., 2011, 257, 2717–2730 CrossRef CAS.
- W. Konicki, M. Aleksandrzak, D. Moszyński and E. Mijowska, J. Colloid Interface Sci., 2017, 496, 188–200 CrossRef CAS PubMed.
- W. Wang, L. Kuai, W. Cao, M. Huttula, S. Ollikkala, T. Ahopelto, A. Honkanen, S. Huotari, M. Yu and B. Geng, Angew. Chem., Int. Ed., 2017, 56, 14977–14981 CrossRef CAS PubMed.
- O. Kalawa, P. Kidkhunthod, N. Chanlek, J. Khajonrit and S. Maensiri, Ionics, 2020, 26, 457–469 CrossRef CAS.
- M. Haripriya, A. M. Ashok, S. Hussain and R. Sivasubramanian, Ionics, 2021, 27, 325–337 CrossRef CAS.
- L. Deehan, A. K. Kaushik, G. R. Chaudhary, P. Papakonstantinou and N. Bhalla, ACS Meas. Sci. Au, 2024, 4, 599–605 CrossRef CAS PubMed.
- Y. Liu, K. J. Aoki and J. Chen, Electrochemistry, 2023, 4, 460–472 CAS.
- S. Kumar, B. K. Satpathy and D. Pradhan, Mater. Adv., 2024, 5(6), 2271–2284 RSC.
- N. Poompiew, P. Pattananuwat and P. Potiyaraj, ACS Omega, 2021, 6, 25138–25150 CrossRef CAS PubMed.
- Y. Lan, H. Zhao, Y. Zong, X. Li, Y. Sun, J. Feng, Y. Wang, X. Zheng and Y. Du, Nanoscale, 2018, 10, 11775–11781 RSC.
- H. Su, T. Xiong, Q. Tan, F. Yang, P. B. S. Appadurai, A. A. Afuwape, M.-S. Jie, T. Balogun, Y. Huang and K. Guo, Nanomaterials, 2020, 10, 1141 CrossRef CAS PubMed.
- X. Guo, X. Diao and W. Wang, Ceram. Int., 2021, 47, 22240–22245 CrossRef CAS.
- P. M. Anjana, S. R. Sarath Kumar and R. B. Rakhi, Mater. Today Commun., 2021, 28, 102720 CrossRef CAS.
- B. Naresh, T. V. M. Sreekanth, K. Yoo and J. Kim, Mater. Lett., 2024, 377, 137520 CrossRef CAS.
- N. M. Shinde and M. Pumera, ACS Appl. Electron. Mater., 2024, 6, 7339–7345 CrossRef CAS PubMed.
- H. Chen, X. Du, J. Sun, Y. Wang, Y. Zhang and C. Xu, Int. J. Hydrogen Energy, 2020, 45, 3016–3027 CrossRef CAS.
- H. Chen, F. Tian, X. Han, H. Sun, G. Wang, Z. Zhang, J. Zhu and C. Xu, J. Energy Storage, 2025, 112, 115587 CrossRef.
- R. Tholkappiyan, A. N. Naveen, S. Sumithra and K. Vishista, J. Mater. Sci., 2015, 50, 5833–5843 CrossRef CAS.
- S. G. Krishnan, M. H. A. Rahim and R. Jose, J. Alloys Compd., 2016, 656, 707–713 CrossRef CAS.
- N. Padmanathan and S. Selladurai, Ionics, 2014, 20, 479–487 CrossRef CAS.
- A. Krittayavathananon, T. Pettong, P. Kidkhunthod and M. Sawangphruk, Electrochim. Acta, 2017, 258, 1008–1015 CrossRef CAS.
- H. Wang, C. Shen, J. Liu, W. Zhang and S. Yao, J. Alloys Compd., 2019, 792, 122–129 CrossRef CAS.
- B. Cheng, W. Zhang, M. Yang, Y. Zhang and F. Meng, Ceram. Int., 2019, 45, 20451–20457 CrossRef CAS.
- I. Shaheen, K. S. Ahmad, C. Zequine, R. K. Gupta, A. Thomas and M. A. Malik, RSC Adv., 2020, 10, 8115–8129 RSC.
- S. Kumar, A. Tahira, A. L. Bhatti, M. A. Bhatti, Z. A. Ujjan, U. Aftab, S. Kumar, A. A. Al-Kahtani, A. Nafady, E. Dawi, M. Emo, B. Vigolo, A. Infantes-Molina and Z. H. Ibupoto, J. Energy Storage, 2024, 77, 109994 CrossRef.
- S. Nayak, A. A. Kittur and S. Nayak, J. Electron. Mater., 2023, 52, 1437–1447 CrossRef CAS.
- S. Sumathy, M. Govindhan and M. Parthasarathy, J. Mater. Sci.: Mater. Electron., 2025, 36, 1336 CrossRef CAS.
- R. S. Salama, M. S. Gouda, M. F. A. Aboud, F. T. Alshorifi, A. A. El-Hallag and A. K. Badawi, Sci. Rep., 2024, 14, 8223 CrossRef CAS PubMed.
- J. Mu, C. Li, J. Zhang, X. Song, S. Chen and F. Xu, Green Energy Environ., 2023, 8, 1479–1487 CrossRef CAS.
- S. K. Shinde, S. S. Karade, N. C. Maile, H. M. Yadav, G. S. Ghodake, A. D. Jagadale and D.-Y. Kim, J. Mol. Liq., 2021, 334, 116390 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2025 |