
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNear blue light emitting benzimidazol-2-thione†
Bikash
Lahkar‡
 a,
Gopendra
Muduli‡
a,
Suman
Mandal‡
a,
Arushi
Rawat
b,
Abhilash
Sahu
b,
Kohsuke
Matsumoto
b,
Osamu
Tsutsumi
b and
Ganesan
Prabusankar
*a
a,
Gopendra
Muduli‡
a,
Suman
Mandal‡
a,
Arushi
Rawat
b,
Abhilash
Sahu
b,
Kohsuke
Matsumoto
b,
Osamu
Tsutsumi
b and
Ganesan
Prabusankar
*a
aOrganometallics and Materials Chemistry Lab, Department of Chemistry, Indian Institute of Technology Hyderabad, Kandi, Sangareddy, Telangana 502285, India. E-mail: prabu@chy.iith.ac.in
bDepartment of Applied Chemistry, Ritsumeikan University, Kusatsu, 525-8577, Japan. E-mail: tsutsumi@sk.ritsumei.ac.jp
First published on 17th June 2025
Abstract
The discrete organosulfur molecule possessing a high quantum yield with blue luminescence properties has significant potential as an optically active material for future applications. In this context, the sulfoxide or sulfone-based fused heterocyclic materials are known as the new generation of luminogens. The first sulfur-fused heterocyclic thioketone luminogen consisting of a donor–acceptor structure has been reported in this work, with benzimidazole and sulfur acting as the donor, whereas anthracene and pyridine moieties act as the acceptor. Herewith, we report the synthesis of 1-(9-methyl anthracene)-2-(2-methyl pyridine)-benzimidazol-2-thione (APBT). The donor and acceptor are connected through a methyl linker. Molecule APBT emits at λem = 494 nm in the crystalline state and λem = 410 nm in the solution state. Besides, molecule APBT showed an aggregation-induced emission due to its molecular packing in the crystalline state. The molecule emits in the near blue region in the crystalline state with a quantum yield of 17.82% and a luminescent lifetime of 47.19 ns. The light-emitting behaviour of crystalline APBT is comparable with APBT-coated LED. DFT calculations were performed to determine the energy of frontier molecular orbitals, and the HOMO–LUMO gap (HLG) was found to be 3.45 eV.
Introduction
Sulfur is one of the vital elements present in Earth's crust for various biological applications in living organisms.1 The history of luminescence goes back to the unintentional discovery of Bolognian stone containing BaS, the first sulfide phosphor ever discovered.2 Thereafter, in 1700, CaS phosphor was developed by Friedrich Hoffmann and followed by SrS phosphor by J. F. John in 1817. Due to its natural abundance and reactivity, it has been utilised in various pharmaceutical and material applications.3,4 One of the major contributions of sulfur is towards the organic semiconductors for light-emitting device applications.5–7The sulfoxide or sulfone-based fused heterocyclic materials have been extensively investigated as the new generation of luminogens for OLED applications.8,9 Because of the presence of the two lone pairs and the empty d-orbital in organo sulfur materials, suitable materials can be designed with electron transportation or electron injection properties.10 Due to such excellent properties, various sulfur-containing chromophores have been studied widely, namely DBT (Dibenzothiophene), DPS (Diphenyl sulfones), and BZ/NZ (Thiadiazole) were isolated with sp3 hybridized sulfur center (Scheme 1). DBT with a rigid structure is used as an electron-rich donor, while DPS with distorted confirmation is often used as a classical electron acceptor for blue-emitting materials.11,12 However, BZ and NZ have been commonly used chromophores for hot excitons.13 Also, these sulfur-containing materials are well known to possess special phenomena like aggregation-induced emission (AIE), solvatochromism, mechanochromism etc.14 In 2024, Frederic and co-workers have isolated benzo[4,5]thieno-S,S-dioxide-[3,2-b]benzofuran (BTOBF) based compounds showcasing an excellent quantum yield of 83% in the solution state and 63% in the crystalline state.15 However, fused heterocyclic thioketone luminogens with a donor–acceptor structure have not been isolated yet. Because of the large π-conjugation and high quantum yield and the access modification of their structures, fused ring systems are gaining enormous popularity in organic luminescent materials.16
 | ||
| Scheme 1 Schematic representation of orbital splitting in different hybridizations of S atom. | ||
Herewith, we report the first sulfur-fused heterocyclic thioketone luminogen, which consists of a donor–acceptor structure. A discrete benzimidazol-2-thione compound 1-(9-methyl anthracene)-2-(2-methyl pyridine)-benzimidazol-2-thione (APBT) has been synthesized and characterized. The photophysical properties of the molecule were examined, and an LED demo was also shown. Besides, the theoretical calculations were carried out to understand the structural and electronic properties.
Results and discussion
APBT was synthesized using the standard Schlenk technique under an argon atmosphere to ensure an inert environment during the chemical reactions. APBT was prepared by reacting 1-(9-methyl anthracene)-2-(2-methyl pyridine)-benzimidazolium chloride salt, K2CO3 as a base, and sulfur in the presence of methanol as a solvent under reflux conditions (Scheme 2). It was characterized by HRMS, 1H NMR, 13C NMR, FT-IR, PXRD, and SCXRD. From the 1H NMR, the disappearance of the imidazo N–CH–N proton is observed. In the 13C NMR, the characteristic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S peak is observed at 170.2 ppm. This confirms the formation of the product by the NMR technique.17 The specific stretching frequency of CS, which appeared at 1220 cm−1 was observed in the FT-IR spectrum (see ESI,† Fig. S1–S4).
S peak is observed at 170.2 ppm. This confirms the formation of the product by the NMR technique.17 The specific stretching frequency of CS, which appeared at 1220 cm−1 was observed in the FT-IR spectrum (see ESI,† Fig. S1–S4).
 | ||
| Scheme 2 Synthesis of APBT. | ||
A single crystal of APBT was isolated from a saturated DCM solution. Single crystal X-ray diffraction technique (SCXRD) was used to determine the crystalline-state structure of APBT (Fig. 1). It was crystallized in the triclinic crystal system with the space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) . The C1–S1 bond length in the molecule is 1.671(14) Å, which is comparable to the bond lengths of thione molecules reported in the literature.17 The structural parameters of APBT have been summarised in the ESI† (see ESI,† Table S1). The bond angle between N1–C1–S1 is 126.26(10)°. The bulk phase sample purity was determined by comparing the simulated powder pattern with the experimental powder XRD pattern (see ESI,† Fig. S5).
. The C1–S1 bond length in the molecule is 1.671(14) Å, which is comparable to the bond lengths of thione molecules reported in the literature.17 The structural parameters of APBT have been summarised in the ESI† (see ESI,† Table S1). The bond angle between N1–C1–S1 is 126.26(10)°. The bulk phase sample purity was determined by comparing the simulated powder pattern with the experimental powder XRD pattern (see ESI,† Fig. S5).
 | ||
| Fig. 1 (i) Solid state structure of APBT. (ii) Space-filling model of APBT. | ||
The packing diagram of APBT showed intermolecular CH⋯π interactions ranging from 2.586 Å to 4.401 Å between the methylene proton linked with pyridine and anthracene π ring (Fig. 2).18 Also, an intermolecular interaction (2.931 Å) exists between sulfur and hydrogen.19,20 This type of interaction is called sulfur-centered hydrogen bonding interaction (SCHB) and is comparable to the similar interactions reported.21,22 The percentage buried volume (%Vbur) and topographic steric map of APBT were calculated (see ESI,† Fig. S6).23
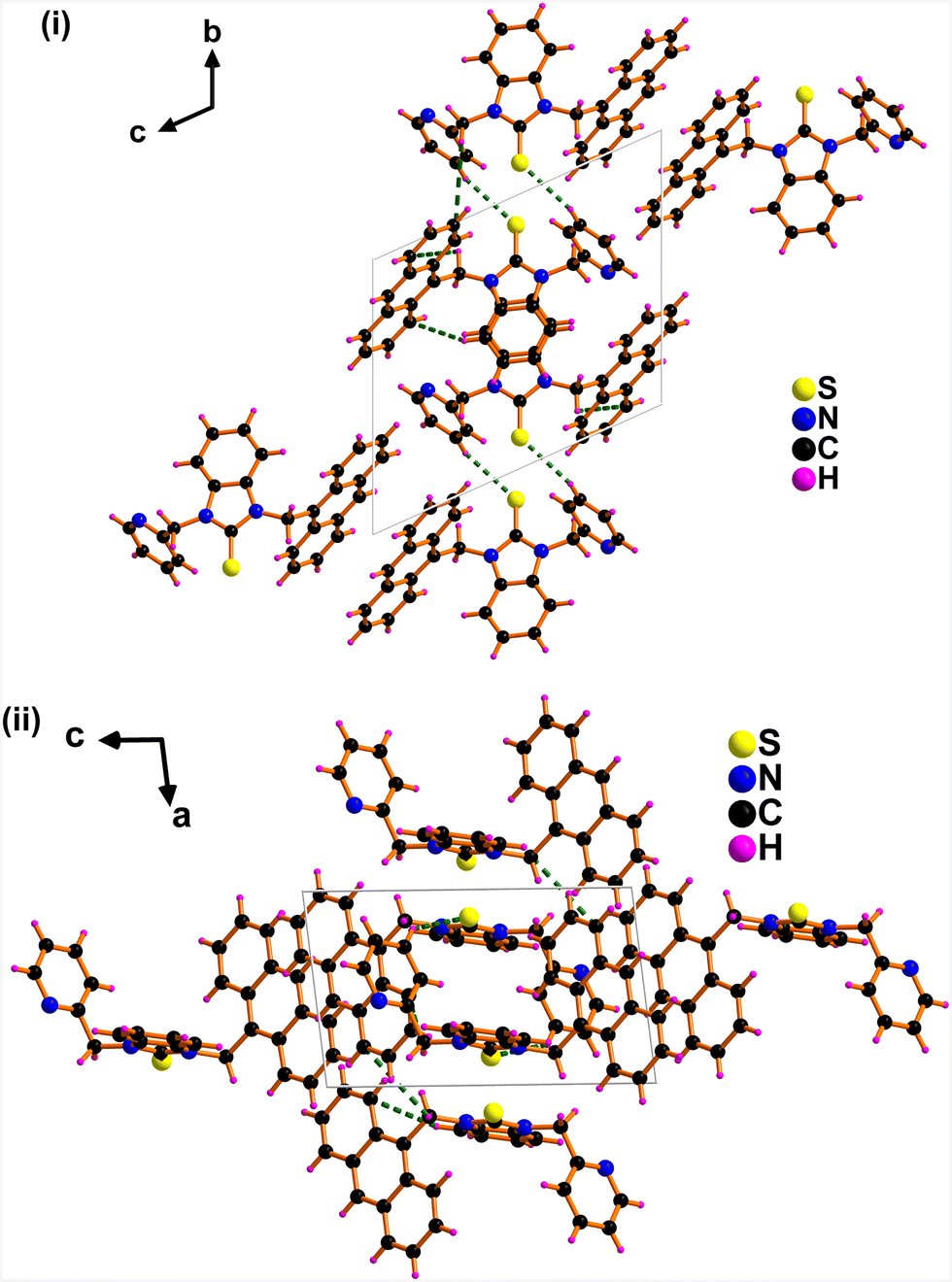 | ||
| Fig. 2 Solid-state packing of APBT consists of (C)S⋯H and CH⋯π interactions (i) view along a axis. (ii) View along b axis. | ||
The solid-state structure of the molecule suggests the presence of several interactions such as S⋯H, CH⋯CH, CH⋯π. It was designed by incorporating anthracene, pyridine, and sulfur groups into benzimidazole. An investigation was carried out to achieve greater insight into geometries, electronic properties, and electronic transitions for APBT. A comprehensive DFT computational study was carried out with the help of the B3LYP/6-31G(d,p) basis set. The ground state optimized geometry of APBT is comparable to that of the experimental X-ray crystallography structure as shown in Fig. 1. The interactions between sulfur and hydrogens get contracted to stabilize the molecule. The excited state energy of the molecule was calculated using the TD-SCF method by taking a well-optimized geometry of APBT. The ground state electronic clouds of the molecule can be seen from the ESP map with the most negative potential (red color), positive potential (blue color), and neutral potential (green and yellow) (see ESI,† Fig. S7). To discuss more intramolecular interactions in the crystal of APBT, topology analysis, and RDG-NCI analysis were carried out and the results are shown. (See ESI,† Fig. S8 and S9). The intramolecular S⋯H interactions show several bond critical points. They are mostly noncovalent characters. The Lagrangian kinetic energy at the bond critical points G(r) between sulfur and hydrogen is greater than the potential energy density V(r). The ratio of |G(r)/V(r)| is greater than or equal to 1, confirming the noncovalent nature and interactions mainly showing the van der Waals in nature.
The crystal structure of APBT and the packing diagram suggested checking the Hirshfeld surface analysis. The surface and its fingerprint plots were used to measure the intramolecular as well as intermolecular interactions. The Hirshfeld surface computation was carried out using Crystal Explorer 21.5 software.24 The term de and di refers to the closest distance from the Hirshfeld surface to molecules on the outside and inside (see ESI,† Fig. S18). The major intermolecular interaction (∼50.7%) occurs through hydrogen bonds H⋯H at a distance of de ∼ 2.033 Å. The second main interaction (∼28.4%) can be seen for C⋯H/H⋯C with a distance range of 2.695 Å. The S⋯H/H⋯S interaction accounts for ∼8.2% of the Hirshfeld area in the range of 2.816 Å. Also, various other weak interactions have been identified by mapping shape index, curvedness, fragment patch, etc.
The thermal stability of APBT was investigated by the thermogravimetric analysis (TGA) technique at a heating rate of 10 °C per minute at a temperature ranging from 30 to 700 °C (Fig. 3). The molecule shows decomposition in a single-step process. The molecule starts to decompose after 210 °C, leaving only the sulfur in the system until 400 °C (93% weight loss). This sulfur was also removed from the system at a temperature above 400 °C, which is around 7% (cal: 7.3%).25 The melting point of APBT, which is 220 °C, was obtained using the differential scanning calorimetry (DSC) technique (see ESI,† Fig. S10).
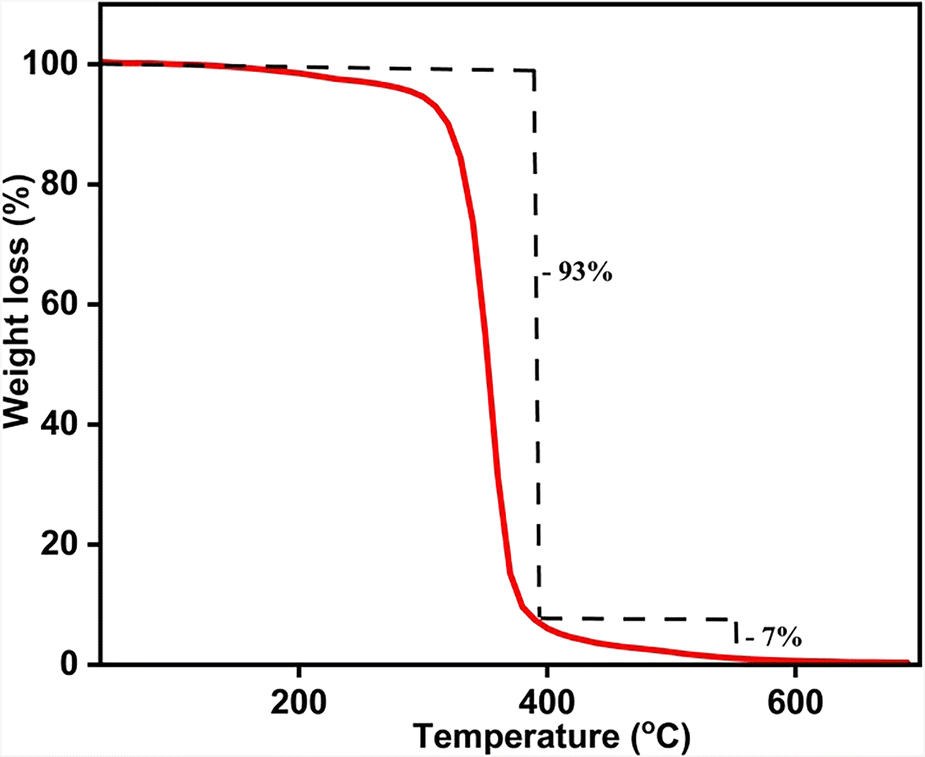 | ||
| Fig. 3 TGA plot of APBT at a heating rate of 10 °C minute−1. | ||
The photophysical characteristics of the molecule were studied. The UV-vis measurement was conducted in 1 × 10−5 M DCM solution (Fig. 4). The λabs of APBT were at 257 nm in DCM, which is due to the n → π* transition from the nitrogen center of the imidazole ring to the anthracene moieties (Table 1). The λabs peak appearing at 312 nm can be accredited for π → π* transition arising from the π conjugation of the aromatic rings. The shoulder peaks at 368 nm to 390 nm arise due to intra-ligand charge transfer.26
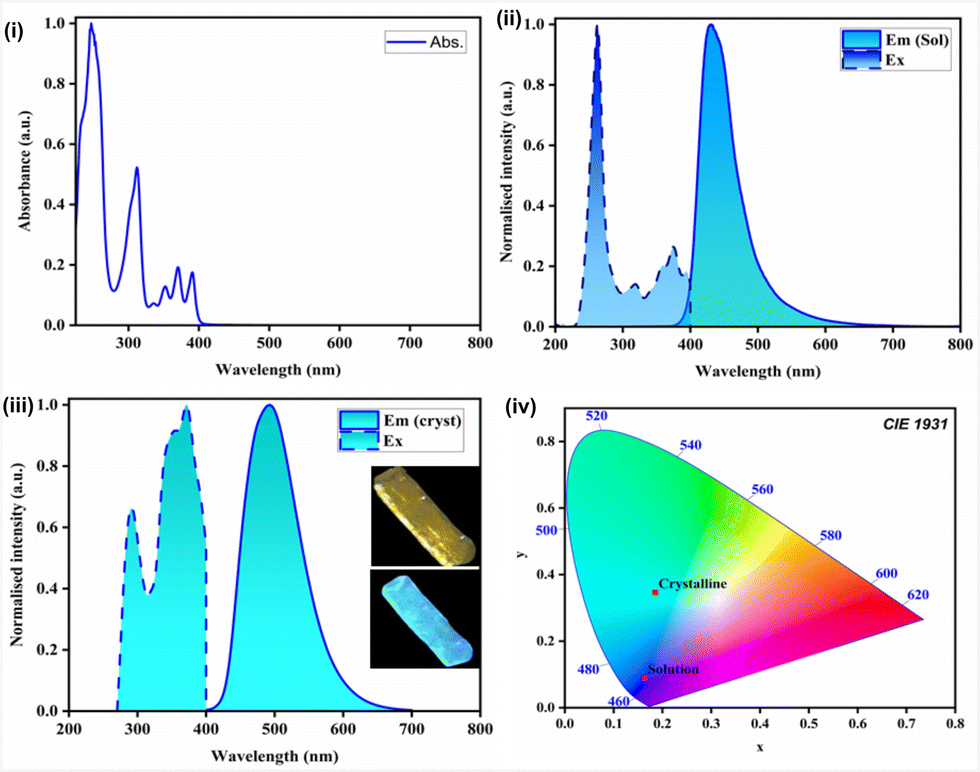 | ||
| Fig. 4 (i) UV-Vis spectrum of APBT in DCM at RT (C = 1 × 10−5 M); (ii) emission spectrum of APBT (λex = 262 nm) in DCM at RT (C = 1 × 10−5 M); (iii) emission spectrum of APBT (λex = 285 nm) as single crystals at RT; (iv) CIE diagram of APBT in crystalline and solution state. | ||
The emission spectrum of the molecule solution state was obtained in (1 × 10−5 M) DCM solution and compared with the crystalline-state emission spectra that were measured using single crystals of the molecule. In the solution state, the λem = 410 nm (excitation at 262 nm), which gets red-shifted to λem = 494 nm in the crystalline state (excitation at 285 nm) (Fig. 4 and Table 1). This is due to the difference in packing in the crystalline state. The emission arises due to the extended conjugation of the polyaromatic anthracene ring and also because of the intra-ligand charge transfer facilitated by the sulfur atom.
The photoluminescence quantum yield (PLQY) of APBT in DCM solution was calculated relative to the standard fluorophore anthracene in ethanol. The fluorescence quantum yield is 5.41%. The lifetime decay profile was found in the nanosecond range of τavg of 2.14 ns in DCM solution. Similarly, PLQY in the crystalline state was acquired using single crystals of APBT at RT. We observed a good quantum yield of 17.82%. The lifetime data was collected using the time-correlated single photon counting technique (TCSPC). The nanosecond decay profile was observed for the molecule (τavg = 47.19 ns). The obtained quantum yield of the APBT molecule in the crystalline state is found to be better than the previously reported thiophene-bridged macrocyclic pseudo-meta[2.2]paracyclophanes.27 The decay profile of APBT is fitted by a biexponential decay curve, and the nanosecond decay curve corresponds to fluorescence. The radiative rate constant (kr) was found to be 3.7 × 106 s−1 for fluorescence. This corresponds to the emission corresponding to the transition from the S1 → S0 state. The quantitative evaluation of the photoluminescence color was estimated with the Commission Internationale de L'Eclairage (CIE) plot. The CIE coordinates of observed emission in the solution state are x = 0.1594, y = 0.0468 while the emission color in the crystalline state is near blue with coordinates x = 0.1855, y = 0.3464.
To study the solvatochromism and mechanochromism behavior of APBT, we have taken solution state emission in various organic solvents (C = 1 × 10−5 M), and it is found that there is no considerable change in the emission behavior of the molecule in varying the solvent (see ESI,† Fig. S11). There is also no such change in the emissive property of the molecule under mechanical stress when we compared the emission of the crystalline sample and the ground powder sample (see ESI,† Fig. S12).
An investigation of the AIE (aggregation-induced emission) behavior of APBT in different water ratios in the THF solution was performed (Fig. 5). This investigation reveals a 10 nm red shift in the emission of APBT in 80% water in the THF solution as compared to a lower water percentage (λex = 262 nm).28,29 Notably, the emission intensity of APBT increases when we increase the content of water, decreasing the volume of APBT in THF. The emission spectra of APBT in 80% water content broadened towards the red region.
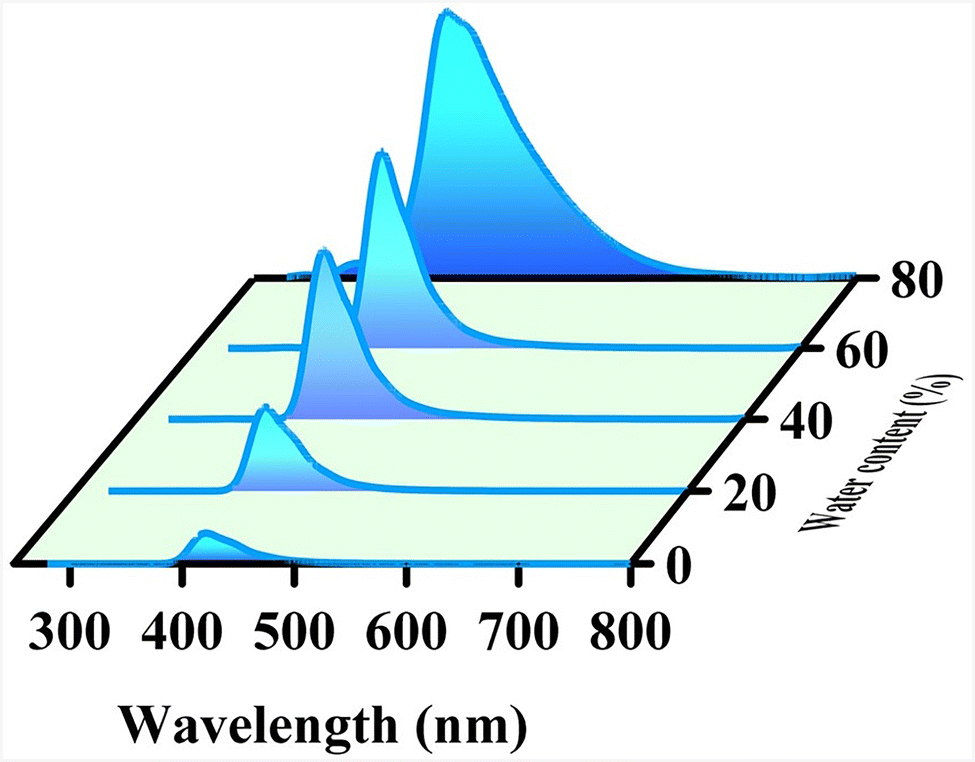 | ||
| Fig. 5 Emission of APBT in THF in different water percentages. Increases water content from front to back. | ||
The above-mentioned AIE phenomena can be enlightened by the RIM mechanism (restriction of intramolecular motion).30 In the solution state, the molecules of APBT experienced minimal intermolecular interaction with each other when encircled by the solvent molecules. Therefore, upon excitation in the solution state, they de-excited back to the ground state via non-radiative (NR) processes, causing lesser emission. But in the presence of excess water, the hydrophobic molecules of APBT start to aggregate. This induces a steric effect which restricts the free motion of the molecules, prevents the non-radiative processes, and produces comparatively higher emissions.
The frontier molecular orbital energy was explored to gain an idea about the HOMO and LUMO (Fig. 6 and Table 2). The corresponding key parameters are shown in Table S3 (ESI†) from DFT calculations. The singlet–triplet energy levels were also calculated from TD-DFT. Energy for S1 and T1 are found to be 2.947 eV and 1.745 eV, respectively, and their difference (ΔEST) is 1.202 eV. The main distribution at the HOMO is appearing from the electron-donating benzimidazole–thione moiety. The LUMO is found in the electron-accepting anthracene unit. The energy level of HOMO and LUMO are −5.32 eV and −1.87 eV, respectively. The HOMO–LUMO gap (HLG) is found to be 3.45 eV. The vertical transitions were determined by using time-dependent DFT (TD-DFT). The simulated UV-vis spectra were calculated to compare with the experimental value (see ESI,† Fig. S13).
 | ||
| Fig. 6 (i) Optimized geometry of APBT. (ii) The proposed decay scheme of LLCT. (iii) HOMO–LUMO energy gap of APBT. | ||
| Total energy | −1642.15196293 a.u. |
| Dipole moment | 3.6444 Debye |
| E HOMO (eV) | −5.328 |
| E HOMO−1 (eV) | −5.359 |
| E LUMO (eV) | −1.872 |
| E LUMO+1 (eV) | −0.844 |
| HLG (eV) | 3.456 |
| Total energy | −1642.15196293 a.u. |
The electrochemical characteristics of APBT were examined using cyclic voltammetry in a 5 mM DMF solution. An Ag/AgCl electrode served as the reference, with platinum as the working electrode and carbon as the counter electrode. The compound showed a reversible one-electron oxidation, with a half-wave potential (E1/2) of 0.20 V. The oxidation and reduction peak potentials were observed at 0.51 V and −0.09 V, respectively, resulting in an electrochemical energy gap of 0.61 V (see ESI,† Fig. S21). The HOMO and LUMO energy levels were found to be −4.91 eV and −4.30 eV. This significant deviation from the DFT calculated HOMO–LUMO gap can be due to the absence of non-covalent or secondary interactions in the solution state.
To further demonstrate the application of such light-emitting molecules in the synthesis of LEDs, we have coated the compound on the surface of a commercially available 3 V LED bulb (λem = 410 nm) by using a THF solution of polymethyl methacrylate (PMMA) and APBT in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 ratio by weight (Fig. 7). The advantages of using PMMA as a binding agent arise from its cost-effectiveness and its low optical absorbance capacity. The emission spectrum of the coated LED is obtained, and it shows emission from λem = 428 to 600 nm in the bluish-green region with emission maxima at λem = 470 nm. The coated LED emission spectrum shows a very weak peak at λem = 410 nm, showing the presence of minimal emission from the LED where the molecule APBT was coated. The emmision of coated LED is nearly comparable to that of crystalline APBT. This near-blue emitting molecule can be used for the synthesis of white LEDs by mixing with red and green emitting molecules.
:10 ratio by weight (Fig. 7). The advantages of using PMMA as a binding agent arise from its cost-effectiveness and its low optical absorbance capacity. The emission spectrum of the coated LED is obtained, and it shows emission from λem = 428 to 600 nm in the bluish-green region with emission maxima at λem = 470 nm. The coated LED emission spectrum shows a very weak peak at λem = 410 nm, showing the presence of minimal emission from the LED where the molecule APBT was coated. The emmision of coated LED is nearly comparable to that of crystalline APBT. This near-blue emitting molecule can be used for the synthesis of white LEDs by mixing with red and green emitting molecules.
 | ||
| Fig. 7 (i) The prototype LED demo with emission spectrum (λex = 410 nm); (ii) CIE diagram of its emission. | ||
Experimental
Methods and materials
All reactions have been performed using a standard Schlenk technique under an inert atmosphere to ensure an inert environment during the chemical reactions. The starting materials 9-anthracenemethanol, benzimidazole, and 2-picolyl chloride were obtained from commercial sources. Before use, the solvent underwent a drying and distillation process per industry standards. The NMR spectra were recorded on a Bruker Ultra Shield 400 MHz spectrometer at 25 oC. FT-IR data were obtained utilizing the ATR technique with a Bruker Alpha-P Fourier Transform Spectrometer. Suitable single crystals of APBT were acquired from a solution of DCM. Bruker D8 Venture Single Crystal X-ray diffractometer was used to obtain the crystal structure of APBT using the source Mo-Kα (0.71073 Å) at 298 K. Olex2.solve was used to solve the structures, and the Charge Flipping method was used to solve them.31 The structures have been refined using the Gauss–Newton minimization method with the Olex2 refinement package.32 Powder X-ray diffraction was attained utilizing a Rigaku Ultima IV instrument. The thermal stability study was carried out using the TA-SDT-Q600 instrument. UV-Vis Spectrometer LAB INDIA 2000 U was used to record electronic spectra, while the Hitachi F-4700 fluorescence spectrophotometer was used to acquire emission spectra. Quantum yield of APBT in DCM was determined relative to standard anthracene in ethanol with a QY of 27%. The refractive index of solvents was taken care of. All samples were diluted to ensure absorbance less than 0.1. The photoluminescence quantum yield (Φ) in the crystalline state was measured using a Hitachi F-7000 fluorescence spectrophotometer using a calibrated integration sphere system (Hitachi, Tokyo, Japan) using alumina oxide as the standard reference. Photoluminescence decay profiles were measured by using Quantaurus-Tau (Hamamatsu) and Hitachi Fluoromax4. The AUTOLAB 302 Modular Potentiostat electrochemical analyser was used to perform cyclic voltammetry (CV) of APBT at 298 ± 1 K. Three standard electrode configurations—a glassy carbon working electrode, a platinum plate auxiliary electrode, and an Ag/AgCl reference electrode—were used to maintain the scan rate at 20 mV s−1 during the tests, which were conducted in dimethylformamide (DMF) with 0.1 M tetrabutylammonium perchlorate (Bu4NClO4) as a supporting electrolyte.Computational methodology
The CIF file of APBT from X-ray crystallography has been utilized for computational studies. The density functional theory (DFT) for the ground state and time-dependent density functional theory (TD-DFT) for the excited states were carried out at the B3LYP function with 6-31G(d,p) basis set for C, H, N, and S using Gaussian16 package. The topological investigation of electron density, reduced density gradient (RDG), and non-covalent interactions (NCI) were analyzed by the Multiwfn program (version 3.8) and the VMD program was utilized for visualization.33–38S), for aromatic carbons 155.8, 149.5, 137.1, 131.5, 131.3, 129.9, 127.2, 125.4, 125.2, 123.0, 122.9, 122.0, 110.7, 109.8, 50.7 (CH2), 44.1 (CH2). FT-IR (cm−1, neat): 3044 (w), 2924 (m), 2154 (w), 2015 (m), 1585 (m), 1480 (m), 1398 (s), 1320 (s), 1220 (m), 1169 (m), 1020 (m), 980 (m), 832 (m), 733 (vs).
Conclusions
We have successfully synthesized and characterized near-blue light-emitting thione molecule APBT. The photophysical studies of the molecule were studied. In the solution state, it emits at 410 nm, and in the crystalline state, it emits at 494 nm. The quantum yield of APBT was found to be 17.82% with a nanosecond decay profile (τavg = 47.19 ns), indicating fluorescence type of emission. Subsequently, the LED demo was carried out where the coated LED was emitting at 470 nm. Besides, the DFT calculations were performed, and the HOMO–LUMO gap (HLG) was found to be 3.45 eV, considering the transition from sulfur and benzimidazole groups to anthracene moiety.Author contributions
BL: conceptualization of the original idea, designing methodology experimental, analysis of data, and writing the manuscript; GM: conceptualization of the original idea, designing methodology experimental, analysis of data, and writing the manuscript; SM: conceptualization of the original idea, designing methodology experimental, analysis of data, and writing the manuscript; AR: conceptualization of the analysis of data, and writing the manuscript; AS: conceptualization of the analysis of data, and writing the manuscript; MK: conceptualization of the analysis of data, and writing the manuscript; OT: conceptualization of the analysis of data, and writing the manuscript; GP: conceptualization of the original idea, supervision, fund acquisition, discussion, review and writing the manuscript.Conflicts of interest
The authors filed a patent for this work. Application No: 202441074170.Data availability
Data for this article include FT-IR, NMR, PXRD, crystal images, PL studies, HRMS, and DSC along with crystal data, structure refinement of the reported molecule, and DFT-the data for the ESI.† CCDC 2435269 (APBT) contains the supplementary crystallographic data for this paper.Acknowledgements
GP and BL gratefully acknowledges SERB-CRG CRG/2022/000714, New Delhi, India, for the financial support. SM sincerely thanks MoE, and GM thanks PMRF funding New Delhi, India, for the monetary support.Notes and references
- J. Yuan, Z. Xu and M. O. Wolf, Chem. Sci., 2022, 13, 5447–5464 RSC
.
-
E. N. Harvey, American Philosophical Society, Philadelphia, 1957
- E. A. Ilardi, E. Vitaku and J. T. Njardarson, J. Med. Chem., 2014, 57, 2832–2842 CrossRef CAS PubMed
- H. Mutlu, E. B. Ceper, X. Li, J. Yang, W. Dong, M. M. Ozmen and P. Theato, Macromol. Rapid Commun., 2019, 40, 1800650 CrossRef PubMed
- S. C. Rasmussen, S. J. Evenson and C. B. McCausland, Chem. Commun., 2015, 51, 4528–4543 RSC
- Y. Li, J.-Y. Liu, Y.-D. Zhao and Y.-C. Cao, Mater. Today, 2017, 20, 258–266 CrossRef
- K. Takimiya, S. Shinamura, I. Osaka and E. Miyazaki, Adv. Mater., 2011, 23, 4347–4370 CrossRef CAS PubMed
- P. F. Smet, I. Moreels, Z. Hens and D. Poelman, Materials, 2010, 3, 2834–2883 CrossRef CAS
- Y. Li, W. Li, J. Hu, X. Yao, L. Hua, W. Cai, S. Shi, C. Zhang, Z. Liu, S. Li, X. Chen, Z. Sun, Z. Ren, M. C. Tang, G. Wei and Z. Fei, Adv. Opt. Mater., 2023, 11, 2300298 CrossRef CAS
- Z. Feng, Z. Cheng, H. Jin and P. Lu, J. Mater. Chem. C, 2022, 10, 4497–4520 RSC
- R. Huang, N. A. Kukhta, J. S. Ward, A. Danos, A. S. Batsanov, M. R. Bryce and F. B. Dias, J. Mater. Chem. C, 2019, 7, 13224–13234 RSC
- Q. Zhang, J. Li, K. Shizu, S. Huang, S. Hirata, H. Miyazaki and C. Adachi, J. Am. Chem. Soc., 2012, 134, 14706–14709 CrossRef CAS PubMed
- J. Liu, Z. Li, T. Hu, X. Wei, R. Wang, X. Hu, Y. Liu, Y. Yi, Y. Y. Takamura, Y. Wang and P. Wang, Adv. Opt. Mater., 2019, 7, 1801190 CrossRef
- J. Guo, X.-L. Li, H. Nie, W. Luo, R. Hu, A. Qin, Z. Zhao, S. J. Su and B. Z. Tang, Chem. Mater., 2017, 29, 3623–3631 CrossRef CAS
- K. Youssef, A. Gasonoo, M. Allain, H. Melville, L. Sanguinet, G. C. Welch and F. Gohier, ACS Appl. Opt. Mater., 2024, 2, 1610–1618 CrossRef CAS
- W. Ma, G. Liu, L. Zhou, B. Li and Y. Wang, J. Mater. Chem. C, 2020, 8, 8796–8803 RSC
- M. Mannarsamy and G. Prabusankar, New J. Chem., 2021, 45, 5933–5938 RSC
- A. Fujii, S. Morita, M. Miyazaki, T. Ebata and N. Mikami, J. Phys. Chem. A, 2004, 108, 2652–2658 CrossRef CAS
- A. D. Sherry and K. F. Purcell, J. Am. Chem. Soc., 1972, 94, 1848–1853 CrossRef CAS
- F. Wennmohs, V. Staemmler and M. Schindler, J. Chem. Phys., 2003, 119, 3208–3218 CrossRef CAS
- P. Bhadoria and V. Ramanathan, J. Phys. Chem. A, 2023, 127, 8095–8109 CrossRef CAS PubMed
- S. Sarkar, M. Monu and B. Bandyopadhyay, J. Phys. Chem., 2019, 21, 25439–25448 CAS
- L. Falivene, Z. Cao, A. Petta, L. Serra, A. Poater, R. Oliva, V. Scarano and L. Cavallo, Nat. Chem., 2019, 11, 872–879 CrossRef CAS PubMed
- P. R. Spackman, M. J. Turner, J. J. McKinnon, S. K. Wolff, D. J. Grimwood, D. Jayatilaka and M. A. Spackman, J. Appl. Crystallogr., 2021, 54, 1006–1011 CrossRef CAS PubMed
- W. F. Faragher, J. C. Morrell and S. Comay, Ind. Eng. Chem. Res., 1928, 20, 527–532 CrossRef CAS
- T. M. Swager, C. J. Gil and M. S. Wrighton, J. Phys. Chem., 1995, 99, 4886–4893 CrossRef CAS
- C. Kress, E. Sidler, P. Downey, P. Zwick, O. Fuhr, D. Fenske, S. Bernhard and M. Mayor, Chem. – Eur. J., 2024, 30, e202303798 CrossRef CAS PubMed
- Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361–5388 RSC
- Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Commun., 2009, 4332–4353 RSC
- J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718–11940 CrossRef CAS PubMed
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS
- L. J. Bourhis, O. V. Dolomanov, R. J. Gildea, J. A. K. Howard and H. Puschmann, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 59–75 CrossRef CAS PubMed
- J. Contreras-García, E. R. Johnson, S. Keinan, R. Chaudret, J.-P. Piquemal, D. N. Beratan and W. Yang, J. Chem. Theory Comput., 2011, 7, 625–632 CrossRef PubMed
- E. R. Johnson, S. Keinan, P. Mori-Sánchez, J. Contreras-García, A. J. Cohen and W. Yang, J. Am. Chem. Soc., 2010, 132, 6498–6506 CrossRef CAS PubMed
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro and M. J. Bearpark, Gaussian 16, Revision A.03, Gaussian, Inc., Wallingford CT, 2016 Search PubMed
-
R. Dennington, T. A. Keith and J. M. Millam, GaussView, Version 6, Semichem Inc., Shawnee Mission, KS, 2016 Search PubMed
Footnotes |
| † Electronic supplementary information (ESI) available: FT-IR, NMR, SCXRD, PXRD, PL, DSC, HRMS, DFT, TD-DFT. CCDC 2435269. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5ma00344j |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |