
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTowards additive manufacturing of semiconducting polymers: hot-melt extrusion of PCL:P3HT blends†
Jiayi
Chen
,
Nahel
Blanc
and
Audrey
Laventure
 *
*
Department of Chemistry, Université de Montréal. 1375 Avenue Thérèse-Lavoie-Roux, Montréal, QC H2V 0B3, Canada. E-mail: audrey.laventure@umontreal.ca
First published on 28th May 2025
Abstract
Additive manufacturing of organic π-conjugated compounds via dry processing remains an ongoing challenge due to the unique thermo-mechanical properties of these materials. In this study, we propose the use of poly(ε-caprolactone) (PCL), a thermoplastic polymer well-suited for hot-melt extrusion, as a matrix to facilitate the extrusion of poly(3-hexylthiophene-2,5-diyl) (P3HT). We investigate the PCL–P3HT blends as a model to better understand its behavior during hot-melt extrusion. Thermal and rheological characterizations enable us to identify a “printability window”, defined as the temperature range (80–140 °C) where the blends exhibit optimal rheological properties for extrusion. Within this temperature range, the blends demonstrate shear thinning and thixotropic behavior, ensuring suitable printability and high print fidelity. The blends exhibit a composite-like behavior, with P3HT maintaining its aggregated state throughout the processing within this temperature range. The microstructure of the extruded architectures is characterized using UV-visible spectroscopy, revealing characteristic bands at 520, 580, and 610 nm, indicative of aggregated P3HT. Further confirmation of the P3HT aggregation state is provided by optical and fluorescence microscopy, as well as AFM-IR analyses conducted on both the surface and cross-section of the extruded architectures highlighting the sub-micron-scale phase separation between PCL and P3HT. Our results demonstrate the effectiveness of blending an organic π-conjugated compound, traditionally challenging to process via dry processing, with a thermoplastic matrix commonly used in hot-melt extrusion. This approach successfully combines the flexibility and ease of processing offered by PCL with the semiconducting properties of P3HT, thereby paving the way for the fabrication of more complex and functional 3D-printed optoelectronic devices.
Introduction
Organic π-conjugated materials, especially semiconducting ones, are increasingly valued for their advantages over their traditional inorganic counterparts for specific applications where lightness and conformability stand as targeted properties. As a result, they have become indispensable in applications such as organic photovoltaics (OPVs),1,2 organic light-emitting diodes (OLEDs),3,4 and organic field-effect transistors (OFETs).5,6 However, despite these benefits, the processing of organic semiconductors into active layers in functional devices remain challenging, encouraging the scientific community to explore innovative strategies to improve their processability.As a matter of fact, π-conjugated materials are inherently difficult to process due to their rigid molecular structures and low solubility in most common organic solvents. These characteristics result in a limited range of processability, even with conventional manufacturing techniques. A commonly used method is solvent casting,7,8 such as spin-coating, blade-coating and slot-die coating, which has shown good performance in processing π-conjugated materials into thin films for active layers of devices. These methods often rely on harsh halogenated solvents for the dissolution of the π-conjugated materials. However, the properties of the solutions prepared using π-conjugated materials for solvent casting-based methods, such as its low viscosity,9,10 makes difficult the transition towards less conventional processing techniques requiring higher viscosity values, such as 3D printing. While inkjet printing11 and direct-ink writing12–14 have been used to print the active layer of some organic optoelectronic devices as a proof-of-concept, these methods are not commonly used. Moreover, the relatively high melting points and (semi)crystalline nature of the π-conjugated compounds limit their use in the context of melt-based 3D printing, further restricting their use in freeform design processing methods. This situation creates a bottleneck for designing complex architectures for advanced applications involving organic π-conjugated compounds.
To address these challenges, various strategies have been employed. One approach involves redesigning the molecular structure of the π-conjugated materials to introduce moieties that reduce their melting point and/or promote an amorphous phase.15 Such synthetic modifications improve the solution-based and/or melt processability of these compounds. Melt-processing techniques such as melt-drawing,16 melt-pressing17 and channel-die pressing18 eliminate the need for the use of harsh solvents. However, these methods are generally limited to the formation of simple architectures, failing to support freeform and complex three-dimensional (3D) design. To overcome these limitations, the use of a thermoplastic polymer as a matrix for the π-conjugated component19,20 has emerged as a versatile solution. Numerous examples report the blending of a thermoplastic polymer and a π-conjugated compound, allowing for increased formulation viscosity contributing to the widening of the processability window, enhanced resulting mechanical properties, along with charge mobilities close to that observed for thin films of pristine π-conjugated compound.21–23 Preserving the semiconductor's charge mobility after blending it with a thermoplastic matrix opens several promising practical applications discussed above, including optoelectronics. Moreover, thermoplastic materials offer excellent processability, as they can be easily moulded or extruded under controlled thermal conditions, making them compatible with a wide range of manufacturing techniques. Among these polymers, poly(ε-caprolactone) (PCL) stands out as a particularly promising matrix. Widely utilized in biomedical applications, PCL is frequently employed for 3D printing scaffolds using hot-melt extrusion (HME) due to its biodegradability, biocompatibility, and ease of processing.24 These characteristics make it an attractive candidate for integrating π-conjugated materials into it to lead to freeform and/or complex designs using hot-melt extrusion.
Herein, we explore the use of poly(ε-caprolactone) (PCL) as a matrix to enhance the printability of π-conjugated materials. More specifically, we focus on model system comprised of a benchmark organic semiconductor, i.e. poly(3-hexylthiophene) (P3HT), to integrate it into a PCL matrix, allowing for the 3D printing of this blend using a solvent free approach, i.e. hot-melt extrusion. To assess the effectiveness of this approach, we employ several characterization techniques to better understand the physical and chemical properties of the PCL–P3HT blend, to subsequently optimize its printability for 3D printing applications. First, thermal analyses are conducted to evaluate the thermal behavior of the blends to identify an appropriate printability window. Then, rheology tests, such as shear thinning and thixotropy, are carried out to identify appropriate printing conditions and thus ensure appropriate printability during the HME process. To evaluate whether the 3D printed architectures maintain the intended dimensions, print fidelity tests are performed using stereomicroscopy and profilometry, allowing to highlight a correlation between the print fidelity and the thixotropic behavior of the formulations. Print fidelity thus serves as a witness of the material printability. Finally, UV-vis spectroscopy, combined to atomic force microscopy coupled to infrared spectroscopy (AFM-IR) analyses provided insights into the microstructure of the resulting 3D printed architectures.
Results and discussion
Formulation of the PCL–P3HT blends
The term “hot melt extrusion” (HME) generally refers to any process where heat and mechanical forces are used to melt and push material through a die. A common HME method employs a twin-screw extruder, where the mechanical turning of screws generates shear forces and pushes the material to be extruded. Compressed air can also be used in certain HME processes to assist in material transport through a die. This methodology offers advantages such as a uniform pressure application and a precise control of the material flow, while minimizing material degradation due to low shear. HME also allows to print solvent-free formulations, which means that the solidification process of the formulation after its extrusion does not involve the solvent evaporation kinetics aspect as it is the case in direct-ink writing.25–27 The model system studied herein for HME consist of a semiconducting polymer, poly(3-hexylthiophene) (P3HT), blended within a polymer matrix, poly(ε-caprolactone) (PCL), at concentrations ranging from 0.1 to 10 wt% P3HT, labeled herein as PCL–P3HT-X where X corresponds to the P3HT wt%. Fig. 1 shows the HME processing of the PCL–P3HT blends, where the formulation, contained in a heated stainless-steel syringe, is extruded though a nozzle via compressed air technology. | ||
| Fig. 1 Schematic illustration of the hot-melt extrusion of the P3HT-PCL blend. | ||
The initial blending involves the solubilization of the individual components of the blends, PCL and P3HT, in chloroform to ensure the homogeneity of the mixture. The chloroform is evaporated after the blending process of the two solutions. The solidified blended polymer film is cut into small pieces to be used for printing. Fig. S1 (ESI†) illustrates the step-by-step formulation process of the PCL–P3HT blend Fig. S2 (ESI†) presents the optical images of the P3HT solution and the PCL–P3HT dry film, demonstrating that P3HT dissolves well in chloroform and is evenly distributed within the film. Thermogravimetric analyses (TGA) are performed on all the different blends to verify the traces of residual solvent in the pieces (Fig. S3, ESI†). There is almost no observable weight loss before 350 °C, which means that the solvent is mostly evaporated during the formulation process, before the printing step. The first dramatic weight loss occurring at 350 °C is attributed to the decomposition of the PCL under a nitrogen atmosphere.
Thermal behavior of the PCL–P3HT blends
Understanding the thermal behavior of the PCL–P3HT blends is critical in the context of HME, in which, by definition, the formulation is printed in its melt state. The first step to ensure that the blend is not subject to decomposition upon printing is to proceed to the thermogravimetric analyses of the PCL–P3HT blends under air atmosphere. As shown in Fig. 2A, the PCL decomposes at a lower temperature than the P3HT, 300 °C and 450 °C respectively, which means that the upper printing temperature limit of this formulation is defined by the PCL. To further investigate the thermal stability of the formulation, isothermal TGA experiments of the pristine PCL, which is the compound having the lower degradation temperature of the blend, were conducted under different temperatures within the 100–250 °C temperature range. The experimental conditions, including temperature, atmosphere, and isothermal time, are chosen to replicate the environment experienced by the material during the HME process. Fig. 2B shows that after 60 min of isotherm at 220 °C, the PCL begins to decompose (3–4% weight loss). Therefore, 220 °C is the temperature at which decomposition begins, as per the detection limit of the TGA, and the printing process should ideally be conducted below this temperature. It is important to note that an HME experiment conducted during this study usually takes 60 minutes in the 80–140 °C temperature range, which is an amount of time that is well below the time at which decomposition was observed for these temperatures. | ||
| Fig. 2 (A) Thermogravimetric analyses of PCL and P3HT. (B) Isothermal thermogravimetric analyses under air atmosphere of PCL at different temperatures for 60 min. | ||
Fig. 3 shows the differential scanning calorimetry (DSC) of the pristine PCL and P3HT, along with the PCL–P3HT-1, -4 and -10 blends. Since PCL and P3HT are semi-crystalline polymers, they both present a melting and a crystallization event upon heating and cooling, respectively. In the HME context, identifying the melting temperature of the formulation is important as it impacts it rheology and thus, its printability. In the context of the PCL–P3HT blends, the melting temperature of PCL is the most relevant one because P3HT melts around 237 °C, which is already beyond the decomposition temperature of PCL under an air atmosphere. Thus, the lower printing temperature limit of the formulation is defined by the melting temperature of the PCL which is around 60 °C. The DSC traces of the different PCL–P3HT blends are very similar to the DSC trace of the pristine PCL because the amount of P3HT is relatively low, making the detection of the P3HT thermal events difficult using DSC. A 50 wt% P3HT blend was prepared to detect the transition temperatures of P3HT in the PCL–P3HT blend (Fig. S4, ESI†). For this blend, the transition peaks for P3HT are much more visible than for the other blends containing a lower P3HT content. These peaks remain at the same place as those of the pristine P3HT. This observation stands as a first piece of evidence that there is a phase separation between the PCL and the P3HT in the blends.
 | ||
| Fig. 3 Differential scanning calorimetry (DSC) analyses of PCL, P3HT and the PCL–P3HT-1, -4 and -10 blends. The wider temperature range corresponding to the printability window defined by DSC and TGA is shaded in orange while the printability window defined by rheology is shaded in blue. | ||
According to the melting temperature of the PCL observed by DSC, a range of printing temperature for the blends can be defined, referred to as the printability window. The lower limit of this window is theoretically 60 °C, corresponding herein to the melting temperature of PCL, but 80 °C, a temperature well above the melting point of PCL, it needed to ensure a minimal adequate flowability (vide infra for rheology results and discussion). The upper limit is 200 °C, as no decomposition was observed at this temperature after 1 h in the TGA isothermal experiment (Fig. 2B). One needs to note that TGA primarily detects degradation via mass loss, meaning that other mechanisms leading to degradation, such as chain scission, oxidation and/or cross-linking, are not fully captured. DSC and rheology measurements (vide infra) conducted on our system did not indicate significant changes in thermal transitions or rheological properties, suggesting that, although non-mass-loss degradation processes can occur, their impact is minimal in our system. One feature of these PCL–P3HT blends that need to be taken into consideration is that the printing must be done at a temperature lower than the melting point of P3HT to avoid the decomposition of the PCL. Printing at this temperature means that P3HT aggregates can clog the printing nozzle, even at percentage of P3HT as low as 10 wt% An easy way around this situation is to use a printing nozzle with a larger diameter. However, as reported by Reichmanis et al.,28 blends prepared using a poly(dimethylsiloxane) (PDMS) matrix and a semiconducting polymer such as P3HT can contain as little as 0.25 wt% P3HT and still present charge mobility values close to that of pristine P3HT in organic field-effect transistors. A similar trend was observed in the work of Sirringhaus et al.,29 where blends incorporating a semicrystalline matrix like HDPE achieved charge mobility values comparable to that of pristine P3HT in organic field-effect transistors with only ∼2 wt% of P3HT. Printing a blend in which the semiconducting component is not melted can present a few unexpected yet interesting advantages. It means that the aggregation process of the P3HT can be controlled at the solidification step of the formulation process without being perturbed significantly upon printing. Since the aggregation impacts the charge mobility of the semiconducting component, having the possibility of controlling it before printing via formulation simplifies the control of the solidification process of the 3D printed sample, which, in this case, would involve the crystallization kinetics of both the PCL and the P3HT.
Assessing the printability of the PCL–P3HT blends using rheology
The printability of the PCL–P3HT blends can be assessed using variable-temperature rheology. Rheology is an essential step to define the lower and the upper temperatures of the printability window identified using TGA and DSC with the constraints of rheology. First, shear thinning tests were conducted on PCL and the PCL–P3HT-1, PCL–P3HT-4, and PCL–P3HT-10 blends across a temperature range of 80 °C to 140 °C, in 20 °C increments, as illustrated in Fig. 4. As discussed in the previous section, 80 °C is established as the lower temperature limit to ensure sufficient flowability of the material. The upper temperature limit is 140 °C because at this temperature, the viscosity of the blend becomes too low, causing the material to flow even under minimal pressure. This low viscosity makes it challenging to maintain consistent print reproducibility. In Fig. 4, for all systems under study, it is observed, as expected, that viscosity increases at lower temperatures. Therefore, at low temperatures, a low shear rate is sufficient to induce the shear thinning effect, in line with the fact that a polymer material undergoes shear thinning once the shear rate applied is faster than the chain relaxation time. For example, PCL at 80 °C undergoes shear thinning at a shear rate of ca. 40 s−1 (Fig. 4A). In contrast, at higher temperatures, the polymer chains have greater mobility,30 leading to lower viscosity and faster relaxation times. As a result, a higher shear rate is required to observe the shear thinning effect. For instance, PCL at 140 °C undergoes shear thinning at a shear rate of ca. 200 s−1 (Fig. 4A). The same behavior is observed in Fig. 4B–D for the PCL–P3HT-1, -4 and -10 blends. Indeed, for PCL–P3HT-10 at 80 °C, shear thinning occurs at a shear rate of ca. 100 s−1 and ca. 400 s−1 at 140 °C (Fig. 4D). | ||
| Fig. 4 Viscosity flow curves of (A) PCL, (B) PCL–P3HT-1, (C) PCL–P3HT-4 and (D) PCL–P3HT-10 blends measured at 80, 100, 120 and 140 °C. The drop of the viscosity with increasing shear rate shows that all formulations exhibit a shear thinning behavior. | ||
Two additional observations can be made in Fig. 4. First, a slightly negative slope in the zero-shear plateau is observed in Fig. 4B–D. The slope also increases with the P3HT ratio, from 1 wt% to 10 wt%. Second, the viscosity of the formulation tends to increase as the P3HT content increases. These observations can be linked to the presence of P3HT aggregates in the formulation. Indeed, within the examined temperature range, P3HT remains aggregated, leading the blends to exhibit composite-like behavior. Anisotropic particles, such as carbon nanotubes (1D) and clays (2D), have a greater impact on the nonlinearity of the viscoelastic properties of polymer composites compared to spherical particles (3D).31 Nonlinear behavior is characterized by a reduction in viscosity under high deformation, leading to curvature in the viscosity curve as the shear rate increases. In the formulations studies herein, P3HT generally behaves like 1D anisotropic particles, like nanofibers and/or nanowires.32,33 The observed nonlinear behavior at low shear rates is primarily due to the reorganization of the material's microstructure, particularly the alignment of P3HT aggregates under shear. As deformation increases, the aggregates reorient, altering the composite's viscoelastic properties and resulting in nonlinear stress response behavior. The viscosity flow curves presented in Fig. 4 informs on the printability of the formulations. As mentioned above, the viscosity of a molten polymer increases as the temperature decreases. During printing, this rise in viscosity reduces the shear rate experienced by the polymer. Although polymers exhibit relatively low shear-viscosity under printing conditions due to their shear thinning nature, a decrease in printing temperature can still increase their viscosity. At a certain point, this increased viscosity requires more energy to overcome resistance and drive flow, ultimately hindering the printing process. The same effect is observed with pressure: at a constant temperature, reducing the applied pressure leads to an increase in shear-viscosity. This reduction in pressure also lowers the shear rate experienced by the polymer, making flow more difficult and requiring greater energy to sustain the printing process. This is why the prints at 80 °C, with printing pressures of 400 and 500 kPa was not possible as can be seen in Tables S1–S3 (ESI†). Returning to the “printability window” in Fig. 3, the lower temperature limit is also delimited by the rheology tests. For the formulations studied herein, this lower limit is approximately 80 °C. However, it is important to note that this printability window is determined from the parameters of the printer: if the instrument can achieve higher printing pressures and/or if a nozzle with a larger diameter is used, this limit could be reduced even further.
Thixotropy tests are then conducted on pristine PCL and the PCL–P3HT blends. This rheological test measures the viscosity of the material at a fixed shear rate during a certain amount of time to reproduce the sequence that is being experienced by the formulation upon printing. A typical thixotropy test has three steps. The first step at low shear rate reproduces what happens in the syringe when the molten material is at rest. The second step at high shear rate rather reproduces what happens at the level of the printing nozzle where the material passes through a small orifice undergoing a shearing force. The third step comes back at the low shear rate, reproducing what happens once the material is extruded on the print bed. This test was first conducted on pristine PCL across the printability window from 80 °C to 140 °C (Fig. S5, ESI†), demonstrating consistent and reliable viscosity recovery after the application of these shear cycles. For the blends, all thixotropy tests are performed at 120 °C (Fig. 5), as this temperature provides the optimal print fidelity (vide infra). The applied shear rate is represented in gray in Fig. 5, and each plateau of the applied shear rate corresponds to a step in the shear cycle. The shear rate at rest is theoretically 0 s−1, but for conducting the thixotropy tests, the low shear rate value is set to 1 s−1 (because a rheometer requires a nonzero shear rate to measure viscosity) but this value is low enough to approximate the formulation's behavior in a near-rest environment. The shear rate range for the extrusion method is between 100–103![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) s−1.34 To highlight the thixotropic effect of our system and to replicate the shearing environment experienced by the material during the HME process, the high shear rate value is set at 103 s−1.
s−1.34 To highlight the thixotropic effect of our system and to replicate the shearing environment experienced by the material during the HME process, the high shear rate value is set at 103 s−1.
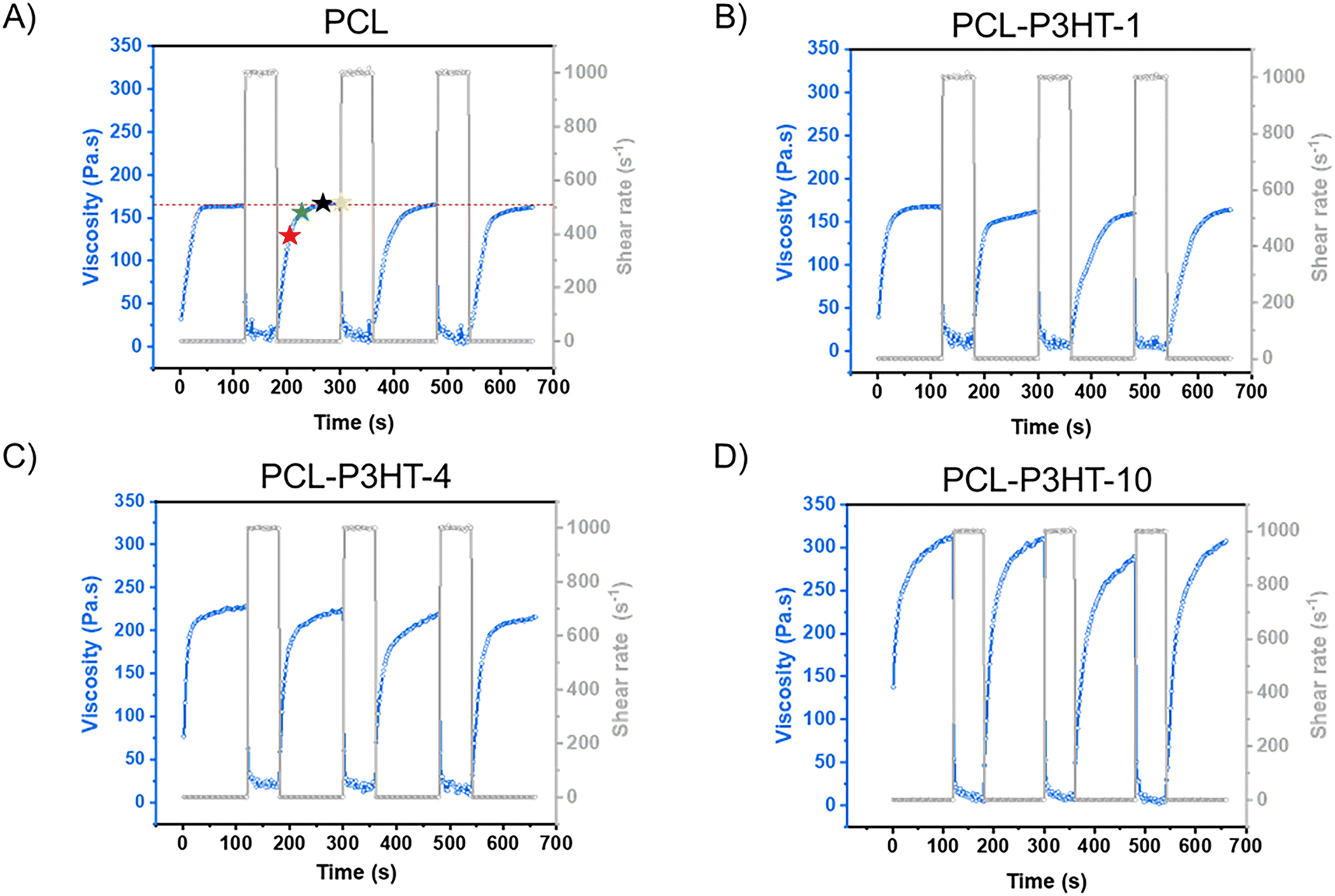 | ||
| Fig. 5 Thixotropy tests at 120 °C for (A) PCL, (B) PCL–P3HT-1, (C) PCL–P3HT-4, (D) PCL–P3HT-10 blends. In (A), the red star indicates the viscosity recovery 30 s after shear, the green star 60 s after shear, the black star 90 s after shear and the yellow star 120 s after shear. | ||
All the thixotropy tests have a total of seven steps, which includes two full cycles of the three steps described above. The first three steps are the most important, and the remaining ones are for reproducibility and properties retention assessment purposes. Indeed, some material exhibits irreversible thixotropy, meaning that the material does not fully recover its original viscosity even after the applied shear stress is removed. In such cases, the material's internal structure undergoes some permanent changes due to the shear stress and they are often referred to as “shear history dependent”35 or “shear degraded”36 materials.
As observed in Fig. 5, an increase in viscosity is observed as a function of the P3HT content in step 1. However, for all samples, this viscosity value drops to a very low value of approximately ∼10 Pa s when a shear rate of 103 s−1 is applied. The viscosity values can be directly correlated with those reported in Fig. 4 at shear rates from 1 s−1 to 1000 s−1. Due to instrument instability at these high shear rates, the viscosity measurements show some fluctuations. Low-viscosity samples such as PCL, PCL–P3HT-1, and PCL–P3HT-4 exhibit even greater instability compared to PCL–P3HT-10. This is because the low torque generated during shear makes the measured signal more sensitive to noise and external disturbances. In contrast, PCL–P3HT-10, which has a higher inherent viscosity, shows a more stable viscosity variation under high shear condition.
According to these thixotropy tests, all the formulations have reversible thixotropy after several low-shear and high-shear cycles, meaning that the extruded materials do not experience major shear history dependency. The quantification of the recovery percentage of the viscosity is calculated using the results of the third step. For an average, the value is also taken at the 6th step and the 9th step. Moreover, it is worth noting that each thixotropy measurement are made in triplicate. Table 1 shows the recovery percentage of the materials during 2 min, calculated at a 30 s interval. The stars in Fig. 5A illustrate the viscosity values that were used to calculate the viscosity recovery 30 s (red star), 60 s (green star), 90 s (black star) and 120 s (yellow star) after shear has been applied. The viscosity recovery percentage after 60 s for PCL–P3HT-10 at 120 °C is 86%, attributed to its relatively high viscosity (∼315 Pa s) and its composite-like behavior. A similar phenomenon is observed with PCL at 80 °C, where the viscosity recovery after 60 s is 91%, owing to its higher viscosity (∼550 Pa s). The recovery percentage is less for PCL–P3HT-10 at 120 °C than for pristine PCL at 80 °C because the addition of P3HT not only increases the overall viscosity of the blend, but somehow hinders the re-arrangement of the PCL polymer chains. In Table 1, a trend can be observed for the average recovery percentage at 30 s for the PCL. At 140 °C, the recovery is faster by about 18% compared to that at 80 °C. Thus, a higher temperature increases the recovery rate for a same formulation. Lower percentage of P3HT contributes to increase the recovery percentage as well. The average recovery percentage increases from 56% to 83% for PCL–P3HT-10 and PCL–P3HT-1 respectively, at 120 °C, 30 s after shear has been applied. This trend is maintained for the average recovery percentage at 60 s. However, beyond 60 s, the average recovery percentage nearly converges to 100% for all samples. Values greater than 100% are primarily due to measurement uncertainties, but the theoretical value of 100% falls within the uncertainty range for all values exceeding 100%. Since PCL–P3HT-1 and PCL–P3HT-4 are both low in terms of P3HT content, the recovery percentage at any time after the application of the high shear rate step is similar to that of PCL at 120 °C. The time associated to specific recovery percentages informs on the time one should wait for printing another layer on top of the previous layer to obtain a self-supporting structure. Indeed, the first layer must have already recovered most of its initial viscosity not to collapse following the addition of the second layer and so on.
| Samples | Recovery percentage at 30 s (%) | Recovery percentage at 60 s (%) | Recovery percentage at 90 s (%) | Recovery percentage at 120 s (%) |
|---|---|---|---|---|
| PCL_80 °C | 66 ± 7 | 91 ± 8 | 96 ± 6 | 98 ± 4 |
| PCL_100 °C | 82 ± 1 | 96 ± 3 | 96 ± 3 | 97 ± 4 |
| PCL_120 °C | 82 ± 3 | 97 ± 1 | 101 ± 4 | 102 ± 5 |
| PCL_140 °C | 84 ± 1 | 99 ± 4 | 101 ± 4 | 101 ± 3 |
| PCL–P3HT-1_120 °C | 83 ± 5 | 94 ± 3 | 96 ± 2 | 97 ± 0 |
| PCL–P3HT-4_120 °C | 84 ± 1 | 95 ± 3 | 99 ± 3 | 100 ± 2 |
| PCL–P3HT-10_120 °C | 56 ± 6 | 86 ± 1 | 97 ± 1 | 102 ± 2 |
Print fidelity assessments of the 3D printed samples
Assessing the print fidelity in 3D printing is essential for ensuring that the dimensions of the printed sample match the digital design. Print fidelity depends on many printing parameters like the extrusion pressure, the print bed temperature, the syringe temperature, the nozzle size, and the print speed, and it also depends on the properties of the printed materials. For instance, different rheological properties also lead to different extrudability. In HME, print fidelity can be quantified using uniformity parameter (U):37 | (1) |
 | (2) |
Stereomicroscopy, along with optical microscopy, stand as useful techniques to assess the overall morphology of the 3D printed sample. As it can be seen in the stereomicroscopy pictures in Fig. 6A–C, for the same printing parameters 120 °C_700 kPa_4 mm s−1 but different P3HT content, the overall appearance of the printed filament is different. The addition of P3HT in the blend exacerbates discontinuities in the 3D printed filament up to certain breaking points as it can be observed in the PCL–P3HT-4 sample in Fig. 6C. These breaking points may occur in regions containing a greater amount of P3HT. Considering that P3HT is not melted, the aggregates can potentially cause clogging in the printing nozzle. As the viscosity of the formulation increases with higher P3HT content, such as in PCL–P3HT-1, PCL–P3HT-4, and PCL–P3HT-10, the printing process becomes progressively more challenging.
 | ||
| Fig. 6 Stereomicroscopy images of (A) PCL, (B) PCL–P3HT-1 and (C) PCL–P3HT-4 digital snakes printed using the 120 °C_700 kPa_4 mm s−1 parameters. Scale bars are 4 mm. (D) Profilometry 3D mapping of the sample presented in (A). | ||
The discontinuity caused by the addition of P3HT, when aggregated, significantly affects its mechanical properties. As a proof-of-concept, Fig. S9 (ESI†) presents the stress–strain curves of PCL, PCL–P3HT-1, and PCL–P3HT-4 printed using 100 °C_500 kPa_4 mm s−1, while Table S4 (ESI†) summarizes their Young's modulus, ultimate tensile strength (UTS), and elongation at break values. The reported values are in the concordance with other mechanical properties assessment conducted on PCL samples (compression molded samples and electrospun fibers, 80 kg mol−1).39 It shows that PCL, PCL–P3HT-1, and PCL–P3HT-4 have similar Young's modulus values, indicating that they exhibit comparable stiffness under elastic deformation. However, both the UTS and elongation at break decrease progressively from PCL to PCL–P3HT-4, suggesting that the blend becomes mechanically weaker with increasing P3HT content. This could be attributed to the phase separation within the blend, leading to the creation of weak interfacial regions, further compromising mechanical strength. Additionally, PCL–P3HT-4 exhibits a significantly lower elongation at break, one order of magnitude lower, likely due to the presence of large P3HT aggregates, which introduce non-uniformity in the system and act as stress concentration points, increasing the probabilities of fracture in the printed filament. Interestingly, the mechanical behavior of these 3D printed materials is similar to that observed for films of π-conjugated compound blended in a thermoplastic matrix40 or in a diblock copolymer system, as reported, for example, for P3HT-poly(ethylene),41 in which the brittle behavior is associated to the π-conjugated moiety.
The optical microscopy images presented in Fig. S10 (ESI†) do not provide more information, but are helpful to better understand the fluorescence images, since P3HT is fluorescent. These images indicate an appropriate distribution of P3HT at the micron scale within the filament for all the blends. When it comes to the PCL–P3HT-10 blend (Fig. S10, ESI†), it gets hard to print. Extreme conditions of temperature and pressure become necessary to extrude the formulation. The parameters used for printing PCL–P3HT-10 are 140 °C_700 kPa_4 mm s−1 with a 400 μm nozzle diameter (compared to 200 μm nozzle diameter for the other blends) to avoid the clogging of the printing nozzle. It is for this reason that printing PCL–P3HT-10 with the same parameters as those used for PCL, PCL–P3HT-1 and PCL–P3HT-4 is not possible. Therefore, the profilometry results of the PLC-P3HT-10 prints are not discussed below. Fig. 6D presents an example of profilometry, showcasing a 3D mapping of PCL printed at 120 °C_700 kPa_4 mm s−1. Compared to stereomicroscopy, profilometry provides greater accuracy and reproducibility for calculating the uniformity (eqn (1)) and roundness (eqn (2)) parameters, reported in Tables S1–S3 (ESI†). In most cases, the width average of the printed PCL, PCL–P3HT-1 and PCL–P3HT-4 follows an increasing trend with increasing printing temperature and pressure (ex: from 179 μm at 100 °C_400 kPa to 277 μm at 120 °C_700 kPa). Uniformity parameter takes only the width of the sample into account, the print fidelity is better evaluated referring to the roundness. Even if this parameter is called “roundness”, the value of 1 does not necessarily mean that the printed filament is perfectly round: it can be a different shape, for instance a cuboid or another complex structure.
By comparing PCL with PCL–P3HT-1 and PCL–P3HT-4, it is observed that filaments printed from blends with higher P3HT content appear to be similar in width or even wider than PCL. For example, at 120 °C_700 kPa_4 mm s−1, the filament width is 288 μm for PCL–P3HT-1 compared to 277 μm for PCL. This result is counterintuitive, as the viscosity increases with higher P3HT content, which should result in less material being extruded. This phenomenon can be attributed to differences in thixotropic behavior. Indeed, the filament height (H) must also be considered for the roundness evaluation. Under the same conditions (120 °C_700 kPa_4 mm s−1), the height is 159 μm for PCL–P3HT-1 and 167 μm for PCL. Using eqn (2), the roundness values are calculated as 0.55 for PCL–P3HT-1 and 0.60 for PCL, the latter being closer to the ideal value of 1. The roundness can tell us to which extent the filament sags on itself. As it can be observed in Tables S1–S3 (ESI†), all the roundness values are lower than 1, which means that all the filaments sag due to the gravity, and the sag rate depends on the thixotropic behavior of the materials. It can be observed that the roundness is relatively larger for higher temperature within the 80 °C to 140 °C range and it can be correlated to the values of viscosity recovery in Table 1. The average recovery percentage of PCL at 30 s is significantly higher at 140 °C compared to lower temperatures, with approximately 18% greater recovery than at 80 °C. The same trend is observed for the PCL–P3HT blends printed at 120 °C, but because the P3HT content is only 1 and 4 wt%, the change is not obvious when comparing the recovery percentage at 30 s, so it is necessary to also have a look at the recovery percentage at 60 s in Table 1. The viscosity is higher for the PCL–P3HT-1 compared to PCL at 120 °C (∼170 Pa s for PCL–P3HT-1 and ∼160 Pa s for PCL), and the recovery percentage at 60 s is lower (94% for PCL–P3HT-1 and 97% for PCL), so the roundness is smaller of about 0.05 at 120 °C_700 kPa (Tables S1 and S2, ESI†).
From the above discussion, it can be concluded that it is challenging to identify an ideal set of printing parameters to optimize the print fidelity of the blended formulations. While the optimal uniformity parameter of 1 is achieved for PCL printed at 100 °C and 500 kPa, the addition of P3HT increases viscosity, shifting the optimal parameters to 100 °C and 500–600 kPa. However, roundness should also be considered. According to our measurements, a moderate printing speed of 4 mm s−1 is selected as it leads to the best roundness. The 1, 2 and 3 mm s−1 speeds were also tested, and as theoretically expected, a slower printing speed results in wider filaments, leading to worse uniformity parameters. A speed greater than 4 mm s−1 yields much thinner filaments which also deviate from the ideal value of 1.
Microstructure analysis of the 3D printed samples
The hot-melt extruded samples are digital snake shaped architectures, and they are very opaque even for blends containing 1 wt% of P3HT due to the high extinction coefficient of P3HT, but also because of the opacity of PCL, stemming from its semi-crystallinity. The specific shape and lack of transmission of these samples make the study of their optical properties challenging. This is the reason why the UV-vis spectra of the samples are collected in diffuse reflectance mode instead of conventional transmission. Despite this selected mode, the sample containing 1 wt% of P3HT still led to a saturated signal, so the ratio of P3HT was further reduced to 0.4 wt% to observe the P3HT characteristic bands. Fig. 7 shows the UV-vis diffusion spectra of the PCL–P3HT sample printed using the 140 °C_700 kPa_4 mm s−1 parameters, along with the spectrum of P3HT in solution and that of a drop-cast P3HT film. P3HT solubilized in chloroform exhibits an orange color with a single band around 460 nm. Upon solvent evaporation, the P3HT color transitions from orange to dark purple, indicating aggregation. The processing method plays a significant role in influencing the aggregation of P3HT, which in turn impacts its charge mobility properties.39 The ratio of the 580 and 610 nm bands observed in the UV-vis spectra of the P3HT drop cast film, corresponding to the A0-1 and A0-0 transitions, respectively, can be used to assess the type of interactions leading to the P3HT aggregation. The spectrum of the 3D printed samples is similar to that of the P3HT drop cast film, showing the typical vibronic bands of P3HT in aggregate form, i.e. the 520, 580 and 610 nm bands. The stronger absorption observed at wavelengths shorter than 520 nm is attributed to unaggregated molecules. The band at 520 nm acts as a boundary band, representing a mixture of contributions from aggregates and disordered P3HT chains, while the bands at 580 and 610 nm are linked to interchain coupling and ordered aggregates.42,43 This result indicates that the aggregation can be controlled during the formulation process, prior to the printing, as it is not dramatically perturbed by the HME processing. | ||
| Fig. 7 Transmission UV-visible spectra of a P3HT solution in CHCl3 and drop cast P3HT film combined with the diffuse reflectance UV-vis spectrum of PCL–P3HT-0.4 digital snake printed at 140 °C. | ||
Another experiment was conducted to assess if the aggregation of P3HT could be controlled via one of the printing parameters, i.e. temperature. For instance, conducting the printing at high temperature using the 250 °C_150 kPa_4 mm s−1 set of parameters for the PCL–P3HT-0.4 blend led to the disappearance of the vibronic bands of the P3HT. This result indicates that after the melting of the P3HT crystals at 250 °C during the HME, the P3HT could not come back to its initial aggregated form: the P3HT chains are mainly non aggregated which is represented by the band at 520 nm in the UV-visible spectrum of the sample (Fig. S11, ESI†).44 In this specific case, P3HT struggles to reorganize and aggregate within the crystallizing PCL matrix. P3HT remains in a disorganized form which has less charge mobility than its intermolecular aggregate counterparts.45 In addition, the printing of the sample at 250 °C was done in less than 10 min. According to the isothermal TGA at 250 °C in Fig. 2, it corresponds to a decomposition lower than 5 wt%, which is acceptable. While we have shown that the resulting microstructure of the formulations differs when being printed below and above the melting temperature of P3HT, printing beyond the decomposition temperature of the thermoplastic matrix is not recommended.
The microstructure of the 3D printed samples, and more specifically phase separation between the components of the blend, can also be studied using atomic force microscopy (AFM). However, AFM alone is agnostic to chemical composition. While it provides high-resolution topographical mapping, it does not inherently distinguish chemical differences between phases. In contrast, AFM-IR, combining atomic force microscopy with infrared spectroscopy, enables the detection of chemical composition through the identification of functional groups. This combination makes AFM-IR particularly useful for studying phase separation,46 as it provides both spatially resolved topographical data and chemical information, allowing for a more comprehensive understanding of the material's microstructure and the distribution of components like P3HT within the PCL matrix. Before analyzing the samples with AFM-IR, FTIR was performed on pristine PCL compressed film and pristine P3HT drop-cast film to identify analyzable and distinguishable bands for both polymers (Fig. S12, ESI†). Indeed, a band for each polymer of the blend that does not overlap with the bands of the other is necessary to observe the distribution of each component within the sample. P3HT does not exhibit many bands that are distinguishable from those of PCL in the 1900–900 cm−1 range. The only relatively intense band that does not overlap with the PCL is the band at 1508 cm−1 which is assigned to asymmetric thiophene ring stretching vibration.47 On the other hand, PCL shows a band at 1728 cm−1 which is assigned to the stretching vibration of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O carbonyl group. The high intensity of this band allows for a good contrast in the IR chemical mapping. Of note, the IR resolution of AFM-IR is determined by the probe of the AFM, allowing it to provide more detailed information compared to conventional FTIR. Notably, the carbonyl vibration band observed at 1728 cm−1 in conventional FTIR appears as a single band, whereas AFM-IR spectra reveal a splitting into two distinct bands: one at 1724 cm−1, attributed to crystalline PCL, and another at 1732 cm−1, associated with amorphous PCL. Typically, the band at 1732 cm−1 exhibits greater intensity than the band at 1724 cm−1, so all the IR chemical mappings are realized at this wavenumber.
O carbonyl group. The high intensity of this band allows for a good contrast in the IR chemical mapping. Of note, the IR resolution of AFM-IR is determined by the probe of the AFM, allowing it to provide more detailed information compared to conventional FTIR. Notably, the carbonyl vibration band observed at 1728 cm−1 in conventional FTIR appears as a single band, whereas AFM-IR spectra reveal a splitting into two distinct bands: one at 1724 cm−1, attributed to crystalline PCL, and another at 1732 cm−1, associated with amorphous PCL. Typically, the band at 1732 cm−1 exhibits greater intensity than the band at 1724 cm−1, so all the IR chemical mappings are realized at this wavenumber.
Fig. 8 consists of three distinct panels for the PCL–P3HT-1 material prepared using the 120 °C_700 kPa_4 mm s−1 parameters for both the surface of the filament and its cross-section. The left panel shows a high-resolution topographical image showing the surface morphology of the sample. It provides detailed information about the height variations and surface topology. The center panel provides infrared (IR) chemical mapping focused on the characteristic absorption band at 1732 cm−1, corresponding to the carbonyl group in PCL. This mapping highlights the chemical composition and distribution of PCL within the sample. Colors ranging from red to yellow represent a PCL-rich domain, while the color blue indicates a PCL-poor domain. The right panel corresponds to the IR spectra collected on specific spots on the sample, highlighting PCL-rich domain (green spectrum and spot) and PCL-poor domain (blue spectrum and spot).
 | ||
| Fig. 8 AFM height images and chemical mapping at 1732 cm−1 for PCL–P3HT-1 (printed at 120 °C and 700 kPa) on the filament surface or its cross-section. Blue and green dots on these images represent the location where the FTIR spectra are taken. | ||
Together, these results provide a comprehensive analysis of the physical and chemical characteristics of the PCL–P3HT-1 sample. Of note, for the pristine PCL in Fig. S13 (ESI†), the IR chemical mappings depend on its surface topography. In this context, the colors ranging from red to yellow corresponds to domains with a greater height, which leads to a relatively higher thermal expansion: the yellow zone is the highest, followed by the red zone, and with the blue zone being the lowest. A uniform response is expected for pristine PCL when 1732 cm−1 is used for IR chemical mapping, but due to the variations in surface heights, it seems that there are different domains. However, at this scale, the PCL is considered to be uniform,48 the height images correlate well with the IR mapping, showing that greater heights correspond to a stronger thermal response of the PCL. This trend is no longer true as the P3HT ratio increases: it is no longer simply a height effect that dictates the IR mapping images, it is rather the phase separation that causes PCL rich and poor domains. This is particularly evident with PCL–P3HT-10, where there is no longer a clear correlation between the height image and the IR mapping, and where the characteristic band of P3HT at 1508 cm−1 becomes noticeable in Fig. S14 (ESI†). The domain differences observed in PCL–P3HT-1 in Fig. 8 are likely a result of the combined influence of the presence of P3HT and height variations. The 1508 cm−1 band of P3HT lacks sufficient intensity to be detected in the IR spectra of PCL–P3HT-1 and PCL–P3HT-4. However, in a drop-cast sample of a PCL–P3HT-50 blend prepared using our formulation method (Fig. S15, ESI†), the 1508 cm−1 band is visible. Multiple IR spectra collected randomly across different spots on the image exhibit similar band intensities, indicating that P3HT is well distributed in the system despite phase separation, which is in agreement with the observation in fluorescence microscopy.
Overall, these analyses confirmed that the PCL–P3HT resulting materials undergo phase separation at the sub-micron scale and that the aggregated state of P3HT is largely preserved during the HME process. In the context of charge mobility in OFET devices for example, we can hypothesize that these phenomena would be beneficial. The sub-micron scale phase separation would help to minimize discontinuous charge transport pathways with insulating domains. Additionally, the preservation of the P3HT aggregation promotes effective π–π stacking interactions, which contributes to the charge mobility in a device.49 Additional methods, such as Time-of-Flight Secondary Ion Mass Spectrometry (ToF-SIMS),50 and grazing incidence wide-angle X-ray scattering (GIWAXS)51,52 could be used to further our understanding of the phase separation and molecular organization, respectively, of the resulting materials. This behavior makes possible the printing of an organic semiconductor, P3HT, without the use of solvent and in a non-molten state, up to 10 wt% using a 400 μm nozzle, upon its blending with PCL acting as a thermoplastic matrix.
Conclusions
The study conducted on the PCL–P3HT model system enabled an in-depth exploration of the behavior of blends composed of a thermoplastic matrix and a π-conjugated compound in the context of hot-melt extrusion. Thermal and rheological characterizations were used to identify the lower and upper temperature limits for hot-melt extrusion, referred to as the printability window. Rheological studies within this temperature range revealed that PCL–P3HT blends exhibit composite-like behavior, attributed to the P3HT remaining in its aggregated state. Additionally, the blends demonstrated shear thinning and thixotropic properties, which are crucial for ensuring printability. Print fidelity tests further established a correlation between the uniformity and roundness parameters of the extruded architectures and the thixotropic behavior of the blends. UV-visible spectroscopy, combined with optical and fluorescence microscopy, provided insights into the microstructure of the blends, particularly the size and distribution of P3HT aggregates within the PCL matrix. These analyses confirmed a phase separation between PCL and P3HT at the sub-micron scale and demonstrated that the aggregated state of P3HT is largely preserved during the hot-melt extrusion process. These findings contribute to advance the field of the additive manufacturing of π-conjugated compound by highlighting that the phase separation and aggregation state of a π-conjugated compound, induced herein during the preparation of the blend, can be retained upon additive manufacturing, provided that the thermoplastic matrix containing the π-conjugated compound can be extruded at lower temperatures than the melting temperature of the π-conjugated compound. This strategy can be used advantageously to transition from conventional to unconventional architectures while preserving their function, which is dictated by the molecular organization of the π-conjugated compound within the thermoplastic matrix. Overall, these findings represent a significant advancement in understanding the structure–processing–property relationships of semiconducting polymer blends in the context of additive manufacturing. They highlight that employing a suitable thermoplastic matrix for hot-melt extrusion is an effective strategy to enable the 3D printing of π-conjugated compounds.Experimental section
Materials
Highly regioregular poly(3-hexylthiophene) (P3HT) (97.3% RR, Mw ∼74 kg mol−1) was purchased from Ossila. Poly(ε-caprolactone) (PCL) (Mw ∼45 kg mol−1) was supplied by MilliporeSigma. Chloroform (CHCl3) was purchased from Fisher Chemical. All materials were used as received without further purification.PCL–P3HT blends
For the preparation of the PCL–P3HT-X blends, where X corresponds to the weight percent of P3HT blended in PCL (in the following example, X = 1 wt%), 1 g of PCL was dissolved in CHCl3 in a round-bottom flask and stirred at room temperature for 1 h. In a separate round bottom flask, 10 mg of P3HT were dissolved in CHCl3 and stirred at 50 °C for 20 min. The two solutions were blended in a round bottom flask and mixed for 10 min at room temperature. The solution was transferred to a Teflon Petri dish and heated on a hot plate at 90 °C for 2 h. The Petri dish was then transferred in a dynamic vacuum for 15 min and left under static vacuum for 48 h. The resulting film was cut into small pieces and stored in a desiccator until printing and characterization. These steps were repeated with the adjusted quantities of materials for PCL–P3HT blends containing 0.1, 0.4, 4 and 10 wt% of P3HT.Characterization
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Data availability
The data supporting this article have been included as part of this article and the ESI.†Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
JC thanks the Fonds de Recherche du Québec for a doctoral fellowship. AL acknowledges the Canada Research Chair program. The authors gratefully acknowledge financial support from the Natural Sciences and Engineering Research Council of Canada (NSERC), and from the Canada Foundation for Innovation (CFI).References
- E. K. Solak and E. Irmak, RSC Adv., 2023, 13, 12244–12269 RSC.
- G. Kumar and F. C. Chen, J. Phys. D: Appl. Phys., 2023, 56, 353001 CrossRef CAS.
- J. Y. Woo, M. H. Park, S. H. Jeong, Y. H. Kim, B. Kim, T. W. Lee and T. H. Han, Adv. Mater., 2023, 35, 2207454 CrossRef CAS PubMed.
- P. L. dos Santos, P. Stachelek, Y. Takeda and P. Pander, Mater. Chem. Front., 2024, 8, 1731–1766 RSC.
- H. Li, W. Shi, J. Song, H. J. Jang, J. Dailey, J. S. Yu and H. E. Katz, Chem. Rev., 2019, 119, 3–35 CrossRef CAS PubMed.
- Y. Niu, Z. Qin, Y. Zhang, C. Chen, S. Liu and H. Chen, Mater. Futures, 2023, 2, 042401 CrossRef CAS.
- U. Bielecka, P. Lutsyk, K. Janus, J. Sworakowski and W. Bartkowiak, Org. Electron., 2011, 12, 1768–1776 CrossRef CAS.
- S. H. Wang, L. Peng, H. B. Sun and W. Huang, J. Mater. Chem. C, 2022, 10, 12468–12486 RSC.
- Z. S. Wang, Q. E. Zhang, S. C. Long, Y. X. Luo, P. K. Yu, Z. B. Tan, J. Bai, B. H. Qu, Y. Yang, J. Shi, H. Zhou, Z. Y. Xiao, W. J. Hong and H. Bai, ACS Appl. Mater. Interfaces, 2018, 10, 10437–10444 CrossRef CAS PubMed.
- M. P. Motloung, T. G. Mofokeng and S. S. Ray, Materials, 2022, 15, 104 CrossRef CAS PubMed.
- T. M. Eggenhuisen, Y. Galagan, A. F. K. V. Biezemans, T. M. W. L. Slaats, W. P. Voorthuijzen, S. Kommeren, S. Shanmugam, J. P. Teunissen, A. Hadipour, W. J. H. Verhees, S. C. Veenstra, M. J. J. Coenen, J. Gilot, R. Andriessen and W. A. Groen, J. Mater. Chem. A, 2015, 3, 7255–7262 RSC.
- Y. M. Chen, L. J. Zhou, J. Wei, C. T. Mei, S. H. Jiang, M. Z. Pan and C. Y. Xu, J. Electron. Mater., 2019, 48, 3157–3168 CrossRef CAS.
- J. S. Sidhu, A. Misra and A. Bhardwaj, J. Mater. Sci: Mater. Electron, 2024, 35, 1958 CrossRef CAS.
- S. H. Park, R. T. Su, J. Jeong, S. Z. Guo, K. Y. Qiu, D. Joung, F. B. Meng and M. C. McAlpine, Adv. Mater., 2018, 30, 1803980 CrossRef PubMed.
- F. Gagnon, V. Tremblay, A. Soldera, M. U. Ocheje, S. Rondeau-Gagné, M. Leclerc and J. F. Morin, Mater. Adv., 2022, 3, 599–603 RSC.
- Y. Zhao, A. Gumyusenge, J. Z. He, G. Qu, W. W. McNutt, Y. Long, H. Y. Zhang, L. B. Huang, Y. Diao and J. G. Mei, Adv. Funct. Mater., 2018, 28, 1705584 CrossRef.
- A. Gumyusenge, X. K. Zhao, Y. Zhao and J. G. Mei, ACS Appl. Mater. Interfaces, 2018, 10, 4904–4909 CrossRef CAS PubMed.
- L. Y. Yu, E. Pavlica, R. P. Li, Y. F. Zhong, C. Silva, G. Bratina, C. Müller, A. Amassian and N. Stingelin, Adv. Mater., 2022, 34, 2103002 CrossRef CAS PubMed.
- N. A. Masarra, M. Batistella, J. C. Quantin, A. Regazzi, M. F. Pucci, R. El Hage and J. M. Lopez-Cuesta, Materials, 2022, 15, 762 CrossRef CAS PubMed.
- C. M. B. Ho, A. Mishra, P. T. P. Lin, S. H. Ng, W. Y. Yeong, Y. J. Kim and Y. J. Yoon, Macromol. Biosci., 2017, 17, 1600250 CrossRef PubMed.
- B. T. DiTullio, X. Kuang, A. Österholm, A. W. Lang, P. J. Kinlen, N. Stingelin, H. J. Qi and J. R. Reynolds, J. Mater. Chem. C, 2023, 11, 4404–4414 RSC.
- J. H. Kim, S. Lee, M. Wajahat, H. Jeong, W. S. Chang, H. J. Jeong, J. R. Yang, J. T. Kim and S. K. Seol, ACS Nano, 2016, 10, 8879–8887 CrossRef CAS PubMed.
- M. Wang, P. Baek, A. Akbarinejad, D. Barker and J. Travas-Sejdic, J. Mater. Chem. C, 2019, 7, 5534–5552 RSC.
- M. Gharibshahian, M. Salehi, N. Beheshtizadeh, M. Kamalabadi-Farahani, A. Atashi, M. S. Nourbakhsh and M. Alizadeh, Front. Bioeng. Biotech., 2023, 11, 1168504 CrossRef PubMed.
- G. M. Gratson, F. Garcia-Santamaria, V. Lousse, M. J. Xu, S. H. Fan, J. A. Lewis and P. V. Braun, Adv. Mater., 2006, 18, 461–465 CrossRef CAS.
- X. Wan, L. Luo, Y. Liu and J. Leng, Adv. Sci., 2020, 7, 2001000 CrossRef CAS PubMed.
- L. Li, Q. Lin, M. Tang, A. J. E. Duncan and C. Ke, Chem. – Eur. J., 2019, 25, 10768–10781 CrossRef CAS PubMed.
- G. Y. Zhang, S. Lee, E. Gutiérrez-Meza, C. Buckley, M. McBride, D. A. Valverde-Chávez, Y. H. Kwon, V. Savikhin, H. Xiong, T. J. Dunn, M. F. Toney, Z. B. Yuan, C. Silva and E. Reichmanis, Chem. Mater., 2019, 31, 6530–6539 CrossRef CAS.
- S. Goffri, C. Muller, N. Stingelin-Stutzmann, D. W. Breiby, C. P. Radano, J. W. Andreasen, R. Thompson, R. A. J. Janssen, M. M. Nielsen, P. Smith and H. Sirringhaus, Nat. Mater., 2006, 5, 950–956 CrossRef CAS PubMed.
- D. E. Dunstan, Sci. Rep., 2019, 9, 543 CrossRef PubMed.
- H. T. Lim, K. H. Ahn, J. S. Hong and K. Hyun, J. Rheol., 2013, 57, 767–789 CrossRef CAS.
- M. Baghgar, J. A. Labastide, F. Bokel, R. C. Hayward and M. D. Barnes, J. Phys. Chem. C, 2014, 118, 2229–2235 CrossRef CAS.
- N. E. Persson, P. H. Chu, M. McBride, M. Grover and E. Reichmanis, Acc. Chem. Res., 2017, 50, 932–942 CrossRef CAS PubMed.
- V. Carnicer, C. Alcázar, M. J. Orts, E. Sánchez and R. Moreno, Open. Ceram., 2021, 5, 100052 CrossRef CAS.
- A. Gebrekrstos, M. Sharma, G. Madras and S. Bose, Cryst. Growth Des., 2016, 16, 2937–2944 CrossRef CAS.
- M. Li, J. Hasjim, F. W. Xie, P. J. Halley and R. G. Gilbert, Starch-Starke, 2014, 66, 595–605 CrossRef CAS.
- L. Sardelli, M. Tunesi, F. Briatico-Vangosa and P. Petrini, Soft Matter, 2021, 17, 8105–8117 RSC.
- E. S. Ortega, A. Sanz-Garcia, A. Pernia-Espinoza and C. Escobedo-Lucea, Materials, 2019, 12, 613 CrossRef CAS PubMed.
- S. C. Wong, A. Baji and S. W. Leng, Polymer, 2008, 49, 4713–4722 CrossRef CAS.
- A. D. Scaccabarozzi and N. Stingelin, J. Mater. Chem. A, 2014, 2, 10818–10824 RSC.
- C. Müller, S. Goffri, D. W. Breiby, J. W. Andreasen, H. D. Chanzy, R. A. J. Janssen, M. M. Nielsen, C. P. Radano, H. Sirringhaus, P. Smith and N. Stingelin-Stutzmann, Adv. Funct. Mater., 2007, 17, 2674–2679 CrossRef.
- J. Peng and Y. C. Han, Giant, 2020, 4, 100039 CrossRef.
- P. J. Brown, D. S. Thomas, A. Köhler, J. S. Wilson, J. S. Kim, C. M. Ramsdale, H. Sirringhaus and R. H. Friend, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 67, 064203 CrossRef.
- J. Clark, C. Silva, R. H. Friend and F. C. Spano, Phys. Rev. Lett., 2007, 98, 206406 CrossRef PubMed.
- V. Coropceanu, J. Cornil, D. A. da Silva, Y. Olivier, R. Silbey and J. L. Brédas, Chem. Rev., 2007, 107, 926–952 CrossRef CAS PubMed.
- I. Jalan, L. K. E. Ericsson, E. Moons and J. van Stam, ACS Appl. Polym. Mater., 2024, 6(18), 11312–11319 CrossRef CAS.
- T. Jarosz, K. Kepska, P. Ledwon, M. Procek, W. Domagala and A. Stolarczyk, Polymers, 2019, 11, 205 CrossRef PubMed.
- P. N. Tri and R. E. Prud'homme, Crystallization and Segregation Behavior at the Submicrometer Scale of PCL/PEG Blends, Macromolecules, 2018, 51(18), 7266–7273 CrossRef.
- A. Khasbaatar, Z. Xu, J. H. Lee, G. Campillo-Alvarado, C. Hwang, B. N. Onusaitis and Y. Diao, Chem. Rev., 2023, 123, 8395–8487 CrossRef CAS PubMed.
- H. Mei, T. S. Laws, T. Terlier, R. Verduzco and G. E. Stein, J. Polym. Sci., 2022, 60, 1174–1198 CrossRef CAS.
- A. Nogales, E. Gutiérrez-Fernández, M. C. García-Gutiérrez, T. A. Ezquerra, E. Rebollar, I. Sics, M. Malfois, S. Gaidukovs, E. Gecis, K. Celms and G. Bakradze, Macromolecules, 2019, 52, 9715–9723 CrossRef CAS.
- F. Dou, Y. L. Ren, L. Y. Yu, B. Xu, M. Dyson, J. Martin, Z. P. Fei, J. H. Bannock, Y. W. Zhang, P. N. Stavrinou, M. Heeney, J. de Mello and X. P. Zhang, ACS Appl. Polym. Mater., 2025, 7, 2986–2996 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional thermal, rheological, microscopy and spectroscopic characterization results. See DOI: https://doi.org/10.1039/d5ma00326a |
| This journal is © The Royal Society of Chemistry 2025 |