
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Phase formation and photocatalytic properties of chalcostibite and tetrahedrite thin films derived from copper and antimony xanthates†
Marco
Sigl
 a,
Melissa
Egger
a,
Daniel
Knez
b,
Stephen
Nagaraju Myakala
c,
Connor M. J.
Marshall
d,
Joe
Kaye
d,
Ali
Salehi-Reyhani
e,
Heinz
Amenitsch
f,
Alexey
Cherevan
c,
Dominik
Eder
c,
Gregor
Trimmel
a,
Saif A.
Haque
d and
Thomas
Rath
*a
a,
Melissa
Egger
a,
Daniel
Knez
b,
Stephen
Nagaraju Myakala
c,
Connor M. J.
Marshall
d,
Joe
Kaye
d,
Ali
Salehi-Reyhani
e,
Heinz
Amenitsch
f,
Alexey
Cherevan
c,
Dominik
Eder
c,
Gregor
Trimmel
a,
Saif A.
Haque
d and
Thomas
Rath
*a
aInstitute for Chemistry and Technology of Materials, NAWI Graz, Graz University of Technology, Stremayrgasse 9, 8010 Graz, Austria. E-mail: thomas.rath@tugraz.at
bInstitute of Electron Microscopy and Nanoanalysis, Graz University of Technology, Steyrergasse 17, 8010 Graz, Austria
cInstitute of Materials Chemistry, TU Wien, Getreidemarkt 9, 1060 Vienna, Austria
dDepartment of Chemistry, Molecular Sciences Research Hub, Imperial College London, White City Campus, 82 Wood Lane, W12 0BZ London, UK
eDepartment of Surgery and Cancer, Imperial College London, London, W12 0HS, UK
fInstitute of Inorganic Chemistry, NAWI Graz, Graz University of Technology, Stremayrgasse 9, 8010 Graz, Austria
First published on 14th May 2025
Abstract
Metal sulfides receive great interest as solar absorber materials for photocatalysis and solar cells. Among them, copper antimony sulfide is a promising ternary metal sulfide. Copper antimony sulfide possesses four accessible phases, which have great potential in solar energy conversion and photocatalysis due to their energy levels and optical properties. However, the synthesis often requires high temperatures and prolonged reaction times. While the different phases are already well characterized, insights into their formation processes from specific precursors, which can enable targeted optimization of material properties, still remain largely unexplored. In this study, we investigated thin films of the two phases chalcostibite (CuSbS2) and tetrahedrite (Cu12Sb4S13), prepared from metal xanthate precursors. We used temperature-dependent grazing incidence X-ray scattering to analyze their thermal conversion process and crystal growth in detail. Furthermore, we evaluated their photocatalytic performance, revealing a good specific catalytic activity of 52 μmol g−1 h−1 for chalcostibite in methylene blue degradation. Additionally, tetrahedrite demonstrated high co-catalytic performance for hydrogen evolution in combination with mesoporous titania, achieving a specific activity exceeding 2.5 mmol g−1 h−1. The findings of this study provide valuable insights into the controlled synthesis of copper antimony sulfides and highlight their potential in solar-driven catalytic applications.
Introduction
Photocatalysis is getting increased attention in the continuous shift towards more environmentally friendly energy supply. It provides the possibility to synthesize solar fuels such as hydrogen, or decompose pollutants, e.g. in waste water.1–6 A photocatalyst with strong absorption in the visible spectrum should efficiently drive redox reactions under sunlight. However, its energy levels must be carefully aligned with the requirements of the target reaction. In this context, metal chalcogenides offer promising advantages, including highly tunable light absorption, controllable band gaps and energy levels.Especially ternary metal chalcogenides like ZnIn2S4,7–9 Cu3BiS3,10,11 CuInS2,12–14 CdZnS215,16 and AgBiS217,18 are promising as they show high absorption coefficients and low bandgaps. Another less explored but highly interesting ternary metal sulfide is copper antimony sulfide. It exists in four different phases: CuSbS2 (chalcostibite – orthorhombic), Cu12Sb4S13 (tetrahedrite – cubic), Cu3SbS3 (skinnerite – monoclinic) and Cu3SbS4 (fematinite – tetragonal), all with bandgaps between 1.1 eV to 1.8 eV19–22 and absorption coefficients over 105 cm−1. There are various reports on the suitability of copper antimony sulfides as solar absorber materials, investigating them in solar cells23,24 as well as in photocatalytic systems.25–27
The direct synthesis of all four phases from their elemental components (Cu, Sb, S) is very energy demanding, with high vacuum and multistage heating to 600 and 1000 °C required for a combined time of over 3 days.20 A faster approach reported by Rabhi et al. uses a solid solution of the elemental components in thermal evaporation, requiring 875 °C for 50 h.28 Thermal evaporation with significantly lower temperatures around 350 °C was achieved via a sequential stacked evaporation of Cu and Sb by Colambara et al., where they used an electrodeposited alloy precursor.29 The approach of Yang et al. is significantly less energy demanding: two stock solutions containing copper/sulfur and antimony/sulfur in hydrazine are used.30 This method for thin film preparation requires only 350 °C. However, hydrazine is a highly toxic and generally dangerous chemical.
Compared to these synthetic methods, metal sulfides can be accessed via a low temperature, solution-based preparation based on metal xanthates.10,11,14,17,31–38 The xanthates act as single source precursors providing both the metal and sulfur source, while also generating only volatile decomposition products beside the metal sulfide. In the case of copper antimony sulfide, the thermal conversion of the precursor film is completed after 15 minutes at 300 °C. Furthermore, the formation of either the chalcostibite or tetrahedrite can be easily controlled via adjusting the copper xanthate to antimony xanthate ratio in the precursor solution.19
In this work, we used the xanthate method to prepare chalcostibite and tetrahedrite thin films in a fast and reproducible manner. We used temperature dependent X-ray scattering measurements to get a deep understanding of the formation of the sulfides as well as their crystallization behavior. We investigated the activity of the thin films towards photocatalytic degradation of the synthetic pollutants Rhodamine B (RhB) and methylene blue (MB), further expanding towards light driven hydrogen evolution. Moreover, we looked at the reactive oxygen species (ROS) formation to gain a better understanding of the underlying mechanisms.
Results and discussion
Synthesis and thin film preparation
For the preparation of copper antimony sulfide films, the precursors copper(I) O-2,2-dimethylpentan-3-yl dithiocarbonate (CuXaC7) and antimony(III) O-propan-2-yl dithiocarbonate (SbXaC3i) were synthesized according to previous studies in the literature.19,39 To obtain thin films of chalcostibite and tetrahedrite, the precursors were dissolved in chlorobenzene resulting in individual solutions with concentrations of 0.25 mmol mL−1. These solutions were subsequently mixed in the respective volume ratios (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 for chalcostibite (CuSbS2); 3:1 for tetrahedrite (Cu12Sb4S13)), spin coated and thermally converted (300 °C) to the brown–grey chalcostibite and tetrahedrite films (thickness: approx. 80 nm) as depicted in Fig. 1a. As the decomposition properties of the xanthates significantly influence their thermal conversion to the sulfides, we recorded TGA curves of the individual xanthate precursors and their mixtures (Fig. 1b). They show that the antimony xanthate decomposes at much lower temperature than the copper xanthate, with a 5% mass loss at 152 °C compared to 183 °C for the copper xanthate. The mixtures of the xanthates show differences in the decomposition steps according to the copper to antimony xanthate ratios, with an additional slight shift to higher temperatures for the finished conversion in the 3:1 mixture, as summarized in Table 1. The X-ray diffraction patterns given in Fig. 1c confirm the formation of the desired phases for the two different precursor mixtures: the characteristic reflections from the orthorhombic chalcostibite (Fig. 1d) are well visible in the red curve at 24.5 (004), 28.4 (111) and 29.9° 2θ (013). Similarly, the blue curve (3:1 Cu:Sb) shows the characteristic reflections of the cubic tetrahedrite at 24.4 (220), 29.9 (222), 32.4 (321) and 34.7° 2θ (400). In addition, the two compounds develop from the respective precursor mixtures without any secondary phases detectable in the corresponding diffractograms.
:1 for chalcostibite (CuSbS2); 3:1 for tetrahedrite (Cu12Sb4S13)), spin coated and thermally converted (300 °C) to the brown–grey chalcostibite and tetrahedrite films (thickness: approx. 80 nm) as depicted in Fig. 1a. As the decomposition properties of the xanthates significantly influence their thermal conversion to the sulfides, we recorded TGA curves of the individual xanthate precursors and their mixtures (Fig. 1b). They show that the antimony xanthate decomposes at much lower temperature than the copper xanthate, with a 5% mass loss at 152 °C compared to 183 °C for the copper xanthate. The mixtures of the xanthates show differences in the decomposition steps according to the copper to antimony xanthate ratios, with an additional slight shift to higher temperatures for the finished conversion in the 3:1 mixture, as summarized in Table 1. The X-ray diffraction patterns given in Fig. 1c confirm the formation of the desired phases for the two different precursor mixtures: the characteristic reflections from the orthorhombic chalcostibite (Fig. 1d) are well visible in the red curve at 24.5 (004), 28.4 (111) and 29.9° 2θ (013). Similarly, the blue curve (3:1 Cu:Sb) shows the characteristic reflections of the cubic tetrahedrite at 24.4 (220), 29.9 (222), 32.4 (321) and 34.7° 2θ (400). In addition, the two compounds develop from the respective precursor mixtures without any secondary phases detectable in the corresponding diffractograms.
 | ||
| Fig. 1 (a) Scheme describing the preparation of the respective copper antimony sulfide films from the xanthate precursors, (b) TGA curves of the individual xanthate precursors and 1:1 and 3:1 (mol:mol) copper:antimony xanthate mixtures, (c) X-ray diffractograms of chalcostibite (red) and tetrahedrite (blue), prepared from 1:1 and 3:1 mixtures of copper and antimony xanthates, respectively, and annealed at 300 °C for 15 min with the reference patterns ICSD 14619 (cubic – blue) and ICSD 85133 (orthorhombic – red), (d) crystal structures of both phases. | ||
| Theoretical mass loss/% | Experimental mass loss/% | Temperature at 5% mass loss/°C | |
|---|---|---|---|
| CuXaC7 (→Cu2S) | 67.8 | 68.8 | 183 |
| SbXaC3i (→Sb2S3) | 67.8 | 67.6 | 152 |
| Cu:Sb = 1:1 (→CuSbS2) |
68.1 | 67.9 | 157 |
| Cu:Sb = 3:1 (→Cu12Sb4S13) |
67.8 | 68.2 | 159 |
Optical and electrical properties
For the optical characterization, we prepared thicker films of both copper antimony sulfide phases via drop casting instead of spin coating. The resulting films appeared darker and opaque compared to the grey–brown semitransparent thin films prepared via spin coating. We calculated the absorbance from the transmission and diffuse reflectance spectra. The absorbance and the diffuse reflectance spectra are shown in Fig. 2a. As seen in the absorbance spectra, the absorption onset of chalcostibite is significantly redshifted, compared to the tetrahedrite films. In the diffuse reflectance spectra, we see a continuous increase of the reflectance with a maximum slightly above 800 nm. The subsequent decrease is caused by the absorption of the material, as higher absorption typically counteracts the diffuse reflection.40 In the case of tetrahedrite, the maximum is less pronounced and blue shifted. | ||
| Fig. 2 (a) Absorbance and diffuse reflectance spectra of chalcostibite and tetrahedrite films on glass; (b) cyclic voltammograms of chalcostibite (red) and tetrahedrite (blue) films with ferrocene (grey) as external standard. | ||
The optical bandgaps were determined from diffuse reflectance spectra of the films via the Kubelka–Munk model and Tauc-plots as described in the literature.20,41 The equations are given in the ESI† (eqn (S1)–(S3)) and the plots derived from the Kubelka–Munk method are depicted in Fig. S3 (ESI†). Similarly to a previous study,19 we see an indirect band gap in the chalcostibite at lower energies at 1.42 eV but a stronger direct optical band gap at 1.53 eV. This duality in the bandgap has been reported in the literature, with the lowest transition being the indirect one and the direct one at slightly higher energy. This leads the material to behave optically more similar to a direct bandgap material.42
For the tetrahedrite we obtained a direct optical band gap of 1.78 eV, matching previous reports as well (Fig. 2a and Fig. S3, ESI†).19,22 Having a comparably low bandgap implies the potential for efficient utilization of a large part of the visible spectrum, making them promising candidates as solar absorbers. In very thin films, we observed a more gradual, less steep onset of the absorption, which is depicted in Fig. S4, in the ESI.†
The energy levels of the valence and conduction bands (VB, CB) in these materials were investigated by cyclic voltammetry. We used ferrocene as external standard and from the oxidation onset, we obtained the VB energy to be −6.0 and −5.9 eV for chalcostibite and tetrahedrite, respectively (Fig. 2b and Table 2). As the films partly dissolved during the CV experiments, we could not determine the CB energy via cyclic voltammetry. To determine the CB energies, we used the VB energies and the respective optical bandgaps of the films, resulting in −4.6 and −4.2 eV, respectively. These data together with the corresponding values with respect to NHE are summarized in Table 2.
| E opt/eV | VBel*/eV | CBel+opt*/eV | VBel/V (vs. NHE) | CBel+opt/V (vs. NHE) | |
|---|---|---|---|---|---|
| Chalcostibite | 1.55 | −6.0 | −4.6 | 1.29 | −0.26 |
| Tetrahedrite | 1.78 | −5.9 | −4.2 | −1.20 | −0.58 |
| Fc/Fc+ | — | −5.39 | — | 0.64 | — |
Temperature dependent X-ray scattering investigations
For a thorough understanding of the formation of the metal sulfides, we characterized the thermal conversion of the metal xanthate precursor films to the metal sulfide films with temperature dependent grazing incidence X-ray scattering experiments using synchrotron radiation. Simultaneous small-angle (SAXS) and wide-angle X-ray scattering (WAXS) in a grazing incidence (GI) condition reveals valuable information about the decomposition of the individual precursors, their interaction and the formation of the respective copper antimony sulfide phases.GISAXS measurements are very sensitive to structural changes within a film. Thus, we can observe the formation of particles during the heating process as well as preferred orientations within the films over the investigated temperature range from room temperature to 330 °C. Corresponding to the decomposition of the antimony xanthate at approx. 150 °C (observed in thermogravimetric analysis, Fig. 1b), an increased scattering intensity and the formation of an ordered mesophase is observed in the in-plane cuts (see Fig. 3a and b). Such an ordered mesophase in the GISAXS pattern has been already found in other metal xanthate derived thin films such as ZnS or ZnIn2S4.34,44 The ordered mesophase is indicated by a peak at approximately q = 1 and 2 nm−1 at 169 °C for the chalcostibite and tetrahedrite, respectively. It consists of decomposition products of the antimony xanthate, arranged in an ordered manner and is very likely responsible for the formation of meso- and microporosity in the metal sulfide films. These ordered decomposition products typically remain in the sample until a solid metal sulfide film has formed, after which they evaporate at a specific temperature, leaving behind well-defined pores.12,34 As the copper xanthate has a much higher decomposition onset, the mesophase in these samples is only formed by the antimony xanthate. In the 1:1 ratio, this signal is stronger and at a lower q value of 0.94 nm−1 (Fig. 3a). In contrast, corresponding to the lower antimony content in the 3:1 ratio, the mesophase signal is weaker and at higher q values of 1.56 nm−1, although it appears at the same temperature (Fig. 3b). The 2D GISAXS images (Fig. 3d) show the strong difference in intensity and scattering vector corresponding to this mesophase for both mixtures.
 | ||
| Fig. 3 In-plane GISAXS scattering curves at selected temperatures during the heating runs of (a) chalcostibite and (b) tetrahedrite thin films; (c) in-plane correlation length derived from the GISAXS data in a q range of q = 0.08 to q = 3.27 nm−1, and (d) 2D GISAXS images (left chalcostibite, right tetrahedrite) at 169 °C (top) and at 330 °C (bottom). | ||
When looking at the GIWAXS data (Fig. 4a and b), which were recorded simultaneously to the GISAXS patters, we observe the formation of amorphous antimony sulfide seeds at 150 °C (broad peak around 30–32° 2θ, see below). Additionally, we assume that the copper xanthate with its large ligands can act as a matrix hindering the growth of the antimony sulfide seeds. The capping limits the cluster sizes that can form in this time during the heating run, shifting the signal to higher q values (see Fig. 3b) compared to the chalcostibite sample. Furthermore, in the chalcostibite sample, we see a steep decrease of the GISAXS signal in the lower q region, while the tetrahedrite sample shows a plateau like Guinier regime45 corresponding to a size of around 30 nm. From these differences in the shapes of the GISAXS curves particularly at higher temperatures, it is evident that in the chalcostibite film much larger structures than in the tetrahedrite films are formed.
 | ||
| Fig. 4 GIWAXS data recorded during heating runs (vertically shifted for better visibility) of (a) chalcostibite thin films with an orthorhombic pattern (reference ICSD 85133) and (b) tetrahedrite thin films with a cubic pattern (reference ICSD 14619) – significant temperatures marked in black; (c) integrated intensities of the GIWAXS data from 27.5 to 31.3° 2θ (dotted lines, representing the amorphous antimony sulfide phase in the range below approx. 220 °C) as well as the integrated intensities of the 213 reflection of chalcostibite (integrated from 41.8 to 44.0°) and the 400 reflection of tetrahedrite (33.6 to 35.7° 2θ). | ||
To gain a deeper qualitative understanding of the structural evolution within the films and the particle formation, we analyzed the in-plane correlation length extracted from the GISAXS data (q = 0.08–3.27 nm−1), as shown in Fig. 3c. Up to 169 °C, an increase in the correlation length is observed in both cases, which can be attributed to the decomposition of the antimony xanthate and the formation of an amorphous antimony sulfide phase. This is consistent with the thermal decomposition of antimony xanthate at lower temperatures compared to the copper xanthate, as indicated by the TGA analysis (Fig. 1b). Moreover, the sharp increase in correlation length between 169 °C and 181 °C is very likely due to the evaporation of the remaining chlorobenzene solvent and decomposition products of the antimony xanthate. This is further supported by the enhanced scattering intensity observed in the GISAXS curves and the concurrent decrease of the mesophase signal (Fig. 3a and b), indicating significant structural reorganization during this temperature range.
Looking further at the correlation length, the chalcostibite mixture shows a maximum at 275 °C with an apparent decrease afterwards. This decrease is most likely due to a shift to larger structures beyond the accessible resolution. In addition, this is supported by the appearance of the curves in Fig. 3a, as the scattering intensity seemingly decreases at higher temperatures in the chalcostibite, but not in the tetrahedrite (Fig. 3b). The curves of the chalcostibite sample suggest that one phase (the amorphous antimony sulfide phase) is consumed by the larger ternary phase. We can see this in the transformation of the curves between 256 and 330 °C in Fig. 3a, where at 300 °C we still see a small shoulder at q = 0.2 nm−1 as a last remainder of the smaller binary phase.
This difference can be explained by the relatively higher copper xanthate content in the tetrahedrite sample, which might inhibit the cluster growth more strongly. The 2D images corresponding to these temperatures are depicted in the ESI,† Fig. S5. Additionally, at higher temperatures, in case of the tetrahedrite, we can observe a tendency towards a near-range order in the out of plane direction (Fig. 3d), which was also found in similar materials like ZnIn2S4 and CuInS2.12,44 This near-range order may be caused by pores growing perpendicular to the substrate.
To study the formation of both antimony sulfide phases and the nanocrystal growth, we combined the GISAXS data with simultaneously measured GIWAXS patterns. Here, during the heating, we can see the initial formation of two broad peaks at 29.9° and 49.7° 2θ (Fig. 4a and b) at temperatures below 200 °C, which we assign to an amorphous antimony sulfide phase due to its precursors’ earlier decomposition (Fig. 1b). Fig. 4c depicts the integrated intensity over the heating run between 27.5 to 31.3° 2θ (dotted lines) and the integrated intensity of reflections exclusively present in one of the phases (and no overlapping with the antimony sulfide phase). For the chalcostibite sample, we chose the 213 reflection (integration range: 41.8 to 44.0° 2θ) and for tetrahedrite the 400 reflection (integration range: 33.6 to 35.7° 2θ), respectively. The correlation length from the GISAXS data and the integrated intensity from the GIWAXS data correlate very well, with the initial increase originating from removal of the solvent and the formation of the antimony sulfide phase.
Comparing the different integrated ranges, it can be clearly seen that the ternary sulfide phases form only after the copper xanthate is fully decomposed. The formation of crystalline chalcostibite starts only around 256 °C and a steep increase in scattering intensity suggests a fast crystal growth. In contrast, the tetrahedrite formation starts at much lower temperatures (around 219 °C), with a much slower growth rate. This is most likely associated with the significantly smaller seeds in the tetrahedrite sample and a lower crystallization energy of the cubic phase. It has to be noted that the overall integral in the region of the amorphous antimony sulfide (27.5 to 31.3° 2θ) increases in both cases as both phases exhibit their most intense reflections in this 2θ range.
Moreover, to obtain information about the stability of the chalcostibite and tetrahedrite thin films over time, which is essential for the usage of the films in specific applications, we investigated the stability of the copper antimony sulfide films in different shelf-life conditions. In particular, we monitored changes in the primary crystallite sizes, estimated with the Scherrer equation, over time in air, in N2 atmosphere at room temperature, and in N2 atmosphere at 65 °C. In both materials, the primary crystallite size increases within the first 11 days. While only a minor increase is observed in the chalcostibite sample, the tetrahedrite films reveal a crystal growth from 33 nm to over 55 nm in the first 11 days. After this initial growth, the crystallite size in both materials remained relatively constant as depicted in Fig. S7 (ESI†), with the minor variations most likely being due to the individual measurements and peak fitting. Apart from the initial narrowing of the peaks within the first 11 days, which can be attributed to crystal growth, no further changes in the XRD patterns of either phase were observed under any of the three testing conditions.
Microstructure and porosity
Based on our previous studies on metal sulfide films prepared from metal xanthate precursors, we expected to find the materials highly porous.34,44 The formation of a mesophase observed in the time resolved GISAXS data supports this expectation. Therefore, we investigated the materials further with scanning transmission electron microscopy (STEM), instead of N2 adsorption measurements as this method is not well suited to investigate thin films.The STEM measurements revealed a highly porous material. The overview images (Fig. 5a and d) reveal that the crystals in the chalcostibite sample are much larger and interconnected, while the tetrahedrite sample mainly consists of spherical nanocrystals with sizes of 17 ± 8 nm. This is slightly smaller, however, still comparable with the distances between two scattering centers (30 nm), estimated from the GISAXS curves, considering this distance includes gaps between crystals, and also with the primary crystallite size of around 33 nm estimated via the Scherrer equation. The chalcostibite sample shows mesopores of 19 ± 6 nm, while the pores of the tetrahedrite sample are with a size of 4 ± 2 nm distinctly smaller (Fig. 5). Compared to other studied metal sulfides prepared with the xanthate method, the pores of chalcostibite are significantly larger and less frequent.34,44 The elemental maps based on energy-dispersive X-ray spectroscopy (EDX) measurements show the homogeneous distribution of copper, antimony and sulfur over the material (Fig. 5c and f), with occasionally occurring copper rich phases in the tetrahedrite.
 | ||
| Fig. 5 STEM high-angle annular dark-field (HAADF) micrographs of (a) and (b) chalcostibite and (d) and (e) tetrahedrite at two magnifications and the corresponding elemental maps from EDX measurements (c: chalcostibite; f: tetrahedrite; yellow: Cu, red: Sb, blue: S, mixed color: overlay of element distributions). | ||
For more information on the porosity, we fitted the GISAXS data of the in-plane line-cuts using a fitting function with a Schultz sphere distribution for the form factor and sticky hard sphere for the structure factor. Details on the fitting function, parameters and fitted curves are given in the ESI,† Fig. S6 and Table S1. The fitted pores of tetrahedrite with 7.3 ± 3.0 nm match the ones obtained from the STEM investigations very well, considering the different measurement methods and large size distribution. Furthermore, we could obtain the volume fraction of the pores via this fit as 0.16. For the chalcostibite sample, this fit however, was not possible as the resolution was insufficient and we could only fit the Porod region at high q values (ESI,† Table S1).
Based on these insights from the GISAXS and GIWAXS data and with the additional information revealed from TEM investigations, we propose the formation process illustrated in Fig. 6. At approx. 150 °C, we observe the formation of an amorphous antimony sulfide phase as a common first step in both chalcostibite and tetrahedrite phases (indicated as red dots in the scheme in Fig. 6). The decomposition products of SbXaC3i, already present in the film at this temperature, are shown in black and the still intact CuXaC7 in yellow. At 169 °C, an ordered mesophase composed of the SbXaC3i decomposition products is clearly visible in both cases, although it is more pronounced in the chalcostibite sample. Additionally, in the tetrahedrite sample, the amount of CuXaC7 compared to the amorphous antimony sulfide seeds is three times higher and we assume that the CuXaC7 matrix can hinder the growth of the amorphous Sb2S3 clusters. Therefore, a higher number of relatively smaller sized antimony sulfide seeds are present in the film at the temperature corresponding to the complete thermal decomposition of CuXaC7. For example, at 219 °C we do not observe a crystalline copper antimony sulfide phase in the chalcostibite sample, while the tetrahedrite mixture shows a slow but constant growth of small tetrahedrite crystals, which is very likely associated with the smaller antimony sulfide seeds. In the chalcostibite sample, a fast crystal growth of the ternary chalcostibite phase is observed starting at 256 °C, while in the tetrahedrite sample, the slower crystal growth is continued. From 300 °C onwards, only minor changes are observed in the GIWAXS patterns indicating that the formation of the chalcostibite and tetrahedrite phases are completed. In addition, the TEM investigations reveal that the films are porous and the chalcostibite film consists of significantly larger crystallites, which are well connected, while the tetrahedrite sample is comprised of spherical particles with sizes around 17 nm. Furthermore, we observe much larger pores in the chalcostibite film and a relatively higher number of smaller pores in the tetrahedrite film.
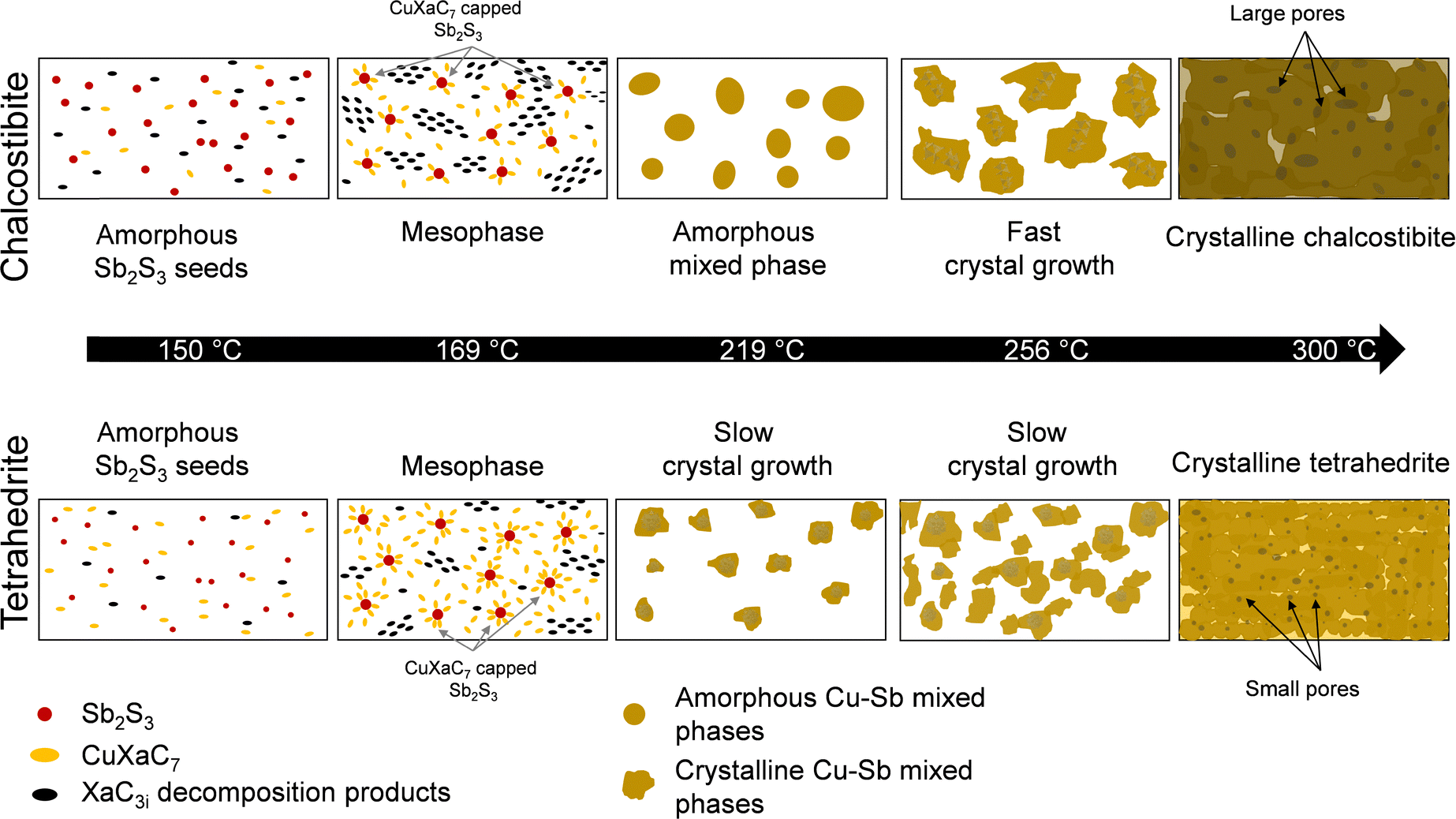 | ||
| Fig. 6 Schematic illustration of the formation of the chalcostibite and tetrahedrite films based on the time resolved X-ray scattering and TEM characterizations. | ||
Photocatalytic investigations
With the consideration of the optoelectronic properties, we employed the materials in dye degradation tests with the organic dyes methylene blue (MB) and Rhodamine B (RhB). These dyes are often applied as model compounds for photocatalytic wastewater treatment, enabling a good comparability of our experiments with literature data. Chalcostibite degrades about 52 μmol g−1 h−1of MB, while the tetrahedrite samples show a lower activity of about 31 μmol g−1 h−1 (Fig. 7). The activity towards MB degradation of these porous thin films is very well comparable with similar dispersed materials reported in the literature,2,46,47 with the advantage of the thin films being beneficial for recycling and simple separating the liquid from the catalyst. On the other hand, the activity of the pristine films towards the degradation of RhB was too low for quantification, so we used the xanthate precursors also to sensitize mesoporous titania films (1 μm). With only a 5 weight% loading, we observed a 12% increase in specific catalytic activity with chalcostibite compared to pristine mesoporous titania, while the tetrahedrite showed no significant difference to TiO2 (Fig. 7a and Table S2, ESI†). We can see a similar influence in the case of methylene blue, where the chalcostibite sensitized titania performed 60% better, while the tetrahedrite even slightly decreased the efficiency of the catalysis compared to pristine TiO2. In addition, to evaluate the photocatalytic performance over time, we exemplarily investigated the activity of chalcostibite films on glass over several cycles of methylene blue degradation and we observed a decrease in specific activity to 35% of its original activity in the 4th cycle (Fig. S9, ESI†). | ||
| Fig. 7 Photocatalytic activity towards the degradation of methylene blue (MB) and Rhodamine B (RhB) of the porous copper antimony sulfide films as well as from the metal sulfide sensitized mesoporous titania films (mp-TiO2). | ||
Previous studies have established superoxide (O2−) as one of the key species responsible for photocatalytic dye degradation when using semiconductor films. As such, the yield of O2− was determined for both pristine copper antimony sulfide (CAS) films and copper antimony sulfide sensitized TiO2 films. For the superoxide tests, we used dihydroethidium (DHE) as a molecular probe in methanol. DHE is known to react with superoxide to form the fluorescent products 2-hydroxyethidium (2-OH-E+) and ethidium (E+) which can be quantified with photoluminescence (PL) spectroscopy (see experimental for details).48,49
As shown in Fig. 8, both pristine CAS and CAS sensitized mp-TiO2 films show a significant ROS (O2−) formation when exposed to light and air. However, the trend in superoxide yield presented in Fig. 8 is different to that seen in the dye degradation studies depicted in Fig. 7. This suggests that the dye degradation may be initiated by direct electron transfer from the semiconductors CB to the dye, rather than being mediated via the ROS pathway. Additionally, differences in electron–hole recombination lifetime in the semiconductor, interfacial energetics and dye adsorption are also likely to influence the efficiency of the dye degradation. A full description of the parameters affecting the dye degradation is beyond the scope of this current study but will be considered in future work.
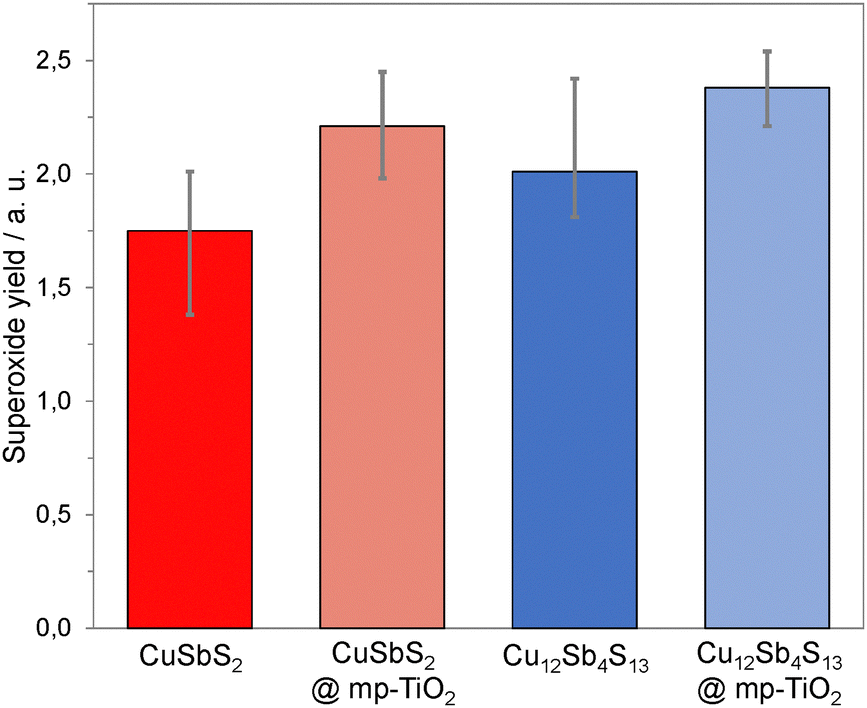 | ||
| Fig. 8 ROS formation over time using chalcostibite and tetrahedrite films as relative superoxide yield. | ||
For additional confirmation of the activity of the two materials, we investigated the films with EPR spectroscopy. We used DMPO (5,5′-dimethyl-1-pyrroline N-oxide) as a spin trap, showing EPR signals corresponding to a DMPO-OH˙ adduct50 in both cases after irradiation with 405 nm in air (Fig. S11, ESI†). However, the slight asymmetry observed in the EPR signals suggests the presence of a second radical species, most likely DMPO-OOH˙.51,52 Notably, no EPR signal was detected when the samples were saturated with nitrogen prior to irradiation, indicating that oxygen is essential for radical formation. Based on the literature, it is plausible that the initially formed DMPO–OOH˙ adduct undergoes conversion to DMPO–OH˙ within the time frame of our experimental setup.53–56 More details to the EPR measurements are given in Fig. S11 in the ESI.†
Based on the good photocatalytic performance of the CAS/mp-TiO2 composites for the degradation of organic dyes, we further investigated the activity of chalcostibite and tetrahedrite towards the hydrogen evolution reaction using methanol as a sacrificial agent (hole scavenger). From the CV measurements of the metal sulfide films (Fig. 2b) and the commonly found flatband potential of TiO2 reported in literature,57 we constructed a band diagram of the metal sulfides and the TiO2, depicted in Fig. 9a. This places the CB of chalcostibite roughly at the same level as that of TiO2 and the CB of tetrahedrite is positioned at a more negative potential on the NHE scale.
 | ||
| Fig. 9 (a) Diagram describing the energy levels of the copper antimony sulfide/mp-TiO2 samples used for hydrogen evolution with the CB of TiO2 taken from Nozik et al., measured as flatband potential,57 with the optical bandgap of mp-TiO2 obtained from diffuse reflectance spectroscopy (see ESI†); to account for kinetic overpotentials relevant in cyclic voltammetry used for the determination of the metal sulfide VB values and for a better comparability with the energy levels of TiO2, we added transparent grey boxes to the graph, which are upshifted by 0.1 V with the shifted values given in brackets; (b) hydrogen evolution of chalcostibite and tetrahedrite films on mp-TiO2 over time. | ||
When using the pristine copper antimony sulfides both showed no activity. However, when sensitizing mesoporous titania films with the copper antimony sulfides (5 weight%), we see a very strong activity of 2.5 mmol g−1 h−1 for the tetrahedrite sample, while the chalcostibite shows a lower, but still significant hydrogen formation of 0.75 mmol g−1 h−1, as outlined in Fig. 9b. This difference is most likely due to the energetically more favorable electron transfer from tetrahedrite to TiO2, compared to the chalcostibite/TiO2 heterojunction, where the conduction band energies of both materials are very close to each other. Comparing the hydrogen evolution performance to reports in the literature, the chalcostibite sensitized titania still compares well with rod like CuSbS2 and nanocrystalline CuSbS2.27 The tetrahedrite sensitized sample on the other hand competes very well with other highly active ternary sulfide materials and composites listed in Table S2 (ESI†).25,58–60
Conclusions
We prepared the two copper antimony sulfide phases chalcostibite and tetrahedrite with a facile solution-based method using xanthate precursors. Their optoelectronic properties were characterized with UV-vis spectroscopy and cyclic voltammetry, resulting in optical bandgaps of 1.55 and 1.78 eV for chalcostibite and tetrahedrite, respectively.In addition, we investigated the formation of the sulfides during the conversion with temperature dependent X-ray scattering experiments. In short, our observations reveal that both chalcostibite and tetrahedrite undergo an initial formation of an amorphous Sb2S3 phase at approx. 150 °C, followed by the development of an ordered mesophase at 169 °C. The presence of more CuXaC7 in the tetrahedrite sample appears to hinder the growth of amorphous Sb2S3 clusters, leading to smaller seeds and a slow gradual crystal growth, while the chalcostibite sample exhibits rapid crystal growth starting at 256 °C. Beyond 300 °C, no significant structural changes are observed. TEM analysis confirms a porous morphology, with chalcostibite forming larger, well-connected crystallites with larger pores and tetrahedrite consisting of approx. 17 nm spherical particles with a higher density of smaller pores.
As the optical characterizations suggest suitable properties for solar energy conversion applications and the high porosity is beneficial for heterogeneous catalysis, we successfully employed both materials in photocatalytic experiments. Chalcostibite performed very well in the oxidative photocatalytic dye degradation test using methylene blue showing a specific activity of 52 μmol g−1 h−1. Tetrahedrite shows a lower activity towards the dye degradation, although it shows a stronger formation of reactive oxygen species (O2−), which suggests that the dye degradation here is initiated by a direct electron transfer, rather than an O2− mediated pathway. On the other hand, tetrahedrite acts as a very efficient co-catalyst for titania in the reductive hydrogen evolution with a respectable HER rate level of over 2.5 mmol g−1 h−1. In particular, the very competitive performance of the tetrahedrite/mp-TiO2 sample, encourages us to refine the surface microstructure of the copper antimony sulfides and the heterojunction design to further increase the photocatalytic activity of these materials in future research.
Experimental section
The chemicals and solvents were purchased from commercial suppliers and used without further purification, unless explicitly specified otherwise: dichloromethane (99.8%, Fisher Scientific), hydrochloric acid (37%, Fisher Scientific), absolute ethanol (Sigma Aldrich, ≥99.8%), diethyl ether (Sigma Aldrich), Rhodamine B (Fluka). Copper(I) O-2,2-dimethylpentan-3-yl dithiocarbonate and antimony(III) O-propan-2-yl dithiocarbonate were synthesized according to previously published reports.19 We provide more details to the metal xanthate syntheses in the ESI.†Preparation of the mp-TiO2 films
Titania paste (30 NR-D, Greatcell Solar Materials) was mixed with terpineol (1:1.5 weight ratio) and stirred overnight. The glass substrates were cleaned first with water, then with 2-propanol in an ultrasonic bath. The cleaned substrates were treated with oxygen plasma (Femto plasma etcher from Diener electronic) for 3 min. The substrates were covered with the titania/terpineol mixture and spin coated (WS-650MZ-23NPPB) with 3000 rpm (ramp: 1000 rpm s−1) for 50 s. The layers were dried at 80 °C for 15 min and annealed at 500 °C for 1 h to obtain mesoporous mp-TiO2 films with a thickness of 1 μm.
Preparation of the CAS films
Glass substrates were cleaned with isopropanol and treated with an oxygen plasma for 3 min. For the preparation of the metal sulfide films, stock solutions of each xanthate precursor were prepared in chlorobenzene (0.25 mmol mL−1). To form chalcostibite, the copper and antimony precursor solutions were combined in a Cu:Sb ratio of 1:1 and for tetrahedrite in a Cu:Sb ratio of 3:1. The mixed precursor solution was then dropped onto the prepared glass substrates (50 μL cm−2) and spin coated with 1500 rpm (ramp: 1500 rpm s−1) for 30 s to obtain thin films with thicknesses around 80 nm. The same procedure was performed with mp-TiO2 coated glass substrates. The films were annealed at 300 °C for 15 minutes.
Characterization methods
sin(2θ/2)). The data have been corrected for fluctuation of the primary intensity and have been converted to in-plane cuts with SAXSDOG.62 The data have been analyzed with integrated quantities like integrated intensity and correlation length.63
 | (1) |
The specific photocatalytic activity was calculated by dividing the difference of dye molecules (μmol) before and after the photocatalytic experiment (the self-degradation of the dyes was measured simultaneously and subtracted from this difference) by the mass of the thin films (g) and time (h). We used the total mass of the deposited semiconductors for the calculation of the specific activity of the photocatalysts. In case of the CAS/mp-TiO2 films, the mass of both CAS and mp-TiO2 was used.
 | (2) |
Author contributions
M. S.: conceptualization, data curation, formal analysis, synthesis and investigation, characterization methodology, project administration, visualization, writing – original draft, writing – review & editing; M. E.: conceptualization, data curation, investigation, writing – review & editing; D. K.: investigation, data curation, review & editing; S. M.: conceptualization, investigation, data curation, review & editing; C. M. J. M.: investigation, data curation, review & editing; J. K.: investigation, data curation, review & editing; A. S.-R.: funding acquisition, supervision, writing – review & editing; H. A.: conceptualization, funding acquisition, data curation & investigation (GISAXS/GIWAXS), writing – review & editing; A. C.: conceptualization, data curation, review & editing; D. E.: conceptualization, data curation, resources, review & editing; G. T.: resources, supervision, funding acquisition, writing – review & editing; S. A. H.: conceptualization, data curation, supervision, review & editing; T. R.: conceptualization, funding acquisition, project administration, supervision, writing – original draft, writing – review & editing.Data availability
All evaluated data is available in the manuscript. The data supporting this article are included as part of the ESI.† Additional information/data can be provided upon reasonable request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge Graz University of Technology for financing this work through the Lead Project Porous Materials@Work for Sustainability (LP-03). Additionally, the authors thank the CERIC-ERIC Consortium for the access to experimental facilities of the Austrian SAXS beamline at Elettra Sincrotrone Trieste as well as financial support. SAH and JK are grateful for financial support provided by the EPSRC Centre for Doctoral Training in Next Generation Synthesis & Reaction Technology [‘rEaCt’, EP/S023232/1]. Furthermore, the authors thank Dmytro Neshchadin and Georg Gescheidt for conducting and evaluating EPR measurements of the investigated thin films.References
- T. Yan, Q. Yang, R. Feng, X. Ren, Y. Zhao, M. Sun, L. Yan and Q. Wei, Front. Environ. Sci. Eng., 2022, 16, 131 CrossRef CAS.
- H. M. El Sharkawy, A. M. Shawky, R. Elshypany and H. Selim, Sci. Rep., 2023, 13, 8845 Search PubMed.
- M. Khodamorady and K. Bahrami, Sci. Rep., 2023, 13, 2177 Search PubMed.
- M. Batool and M. N. Zafar, Mater. Adv., 2025, 6, 224–240 RSC.
- A. B. Siddique, M. A. Shaheen, S. Shafeeq, A. Abbas, Y. Zaman, M. Z. Ishaque and M. Aslam, Mater. Adv., 2025, 6, 1330–1344 Search PubMed.
- J. Su, J. Zhang, S. Chai, M. Anpo, Y. Fang and X. Wang, Nano Mater. Sci., 2024 DOI:10.1016/j.nanoms.2024.08.006.
- M. Tang, W. Yin, F. Zhang, X. Liu and L. Wang, Front. Chem., 2022, 10, 959414 Search PubMed.
- T. Zhang, T. Wang, F. Meng, M. Yang and S. Kawi, J. Mater. Chem. C, 2022, 10, 5400–5424 Search PubMed.
- M. H. Rahman, Y. Sun and A. Mannodi-Kanakkithodi, Mater. Adv., 2024, 5, 8673–8683 RSC.
- T. Rath, J. M. Marin-Beloqui, X. Bai, A.-C. Knall, M. Sigl, F. G. Warchomicka, T. Griesser, H. Amenitsch and S. A. Haque, ACS Appl. Mater. Interfaces, 2023, 15, 41624–41633 Search PubMed.
- C. Yan, E. Gu, F. Liu, Y. Lai, J. Li and Y. Liu, Nanoscale, 2013, 5, 1789–1792 Search PubMed.
- E. Vakalopoulou, T. Rath, F. G. Warchomicka, F. Carraro, P. Falcaro, H. Amenitsch and G. Trimmel, Mater. Adv., 2022, 3, 2884–2895 Search PubMed.
- W. Y. Lim, M. Hong and G. W. Ho, Dalton Trans., 2016, 45, 552–560 RSC.
- A. Fischereder, T. Rath, W. Haas, H. Amenitsch, D. Schenk, A. Zankel, R. Saf, F. Hofer and G. Trimmel, ACS Appl. Mater. Interfaces, 2012, 4, 382–390 CrossRef CAS PubMed.
- B. Li, W. Wang, J. Zhao, Z. Wang, B. Su, Y. Hou, Z. Ding, W.-J. Ong and S. Wang, J. Mater. Chem. A, 2021, 9, 10270–10276 RSC.
- B. Li, W. Guo, X. F. Lu, Y. Hou, Z. Ding and S. Wang, Mater. Rep. Energy, 2023, 3, 100230 CAS.
- E. Vakalopoulou, D. Knez, M. Sigl, G. Kothleitner, G. Trimmel and T. Rath, ChemNanoMat, 2023, 9, e202200414 CrossRef CAS.
- B. Pejova, D. Nesheva, Z. Aneva and A. Petrova, J. Phys. Chem. C, 2011, 115, 37–46 CrossRef CAS.
- T. Rath, A. J. MacLachlan, M. D. Brown and S. A. Haque, J. Mater. Chem. A, 2015, 3, 24155–24162 RSC.
- O. Abbassi, A. Rabhi and M. Kanzari, Optik, 2024, 300, 171641 CrossRef CAS.
- K. Ramasamy, H. Sims, W. H. Butler and A. Gupta, Chem. Mater., 2014, 26, 2891–2899 CrossRef CAS.
- J. van Embden, K. Latham, N. W. Duffy and Y. Tachibana, J. Am. Chem. Soc., 2013, 135, 11562–11571 CrossRef CAS PubMed.
- R. Zhou, X. Liu, S. Zhang, L. Liu, L. Wan, H. Guo, X. Yang, Z. Cheng, L. Hu, H. Niu and X. Mao, Mater. Sci. Semicond. Process., 2021, 124, 105613 CrossRef CAS.
- S. Chinnaiyah, R. Naik and R. Ramesh Babu, J. Mater. Sci.: Mater. Electron., 2024, 35, 1015 CrossRef CAS.
- W. Wang, Q. Sheng, G. Zhi, Y. Zhao, R. Qu, L. Sun and S. Zhang, Appl. Surf. Sci., 2023, 639, 158251 CrossRef CAS.
- Y. Bai, Z. H. J. Khoo, R. I. Made, H. Xie, C. Y. J. Lim, A. D. Handoko, V. Chellappan, J. J. Cheng, F. Wei, Y.-F. Lim and K. Hippalgaonkar, Adv. Mater., 2024, 36, e2304269 CrossRef PubMed.
- A. Sarilmaz, E. Genc, E. Aslan, A. Ozen, G. Yanalak, F. Ozel and I. H. Patir, J. Photochem. Photobiol., A, 2020, 400, 112706 Search PubMed.
- A. Rabhi, M. Kanzari and B. Rezig, Thin Solid Films, 2009, 517, 2477–2480 CrossRef CAS.
- D. Colombara, L. M. Peter, K. D. Rogers, J. D. Painter and S. Roncallo, Thin Solid Films, 2011, 519, 7438–7443 CrossRef CAS.
- B. Yang, L. Wang, J. Han, Y. Zhou, H. Song, S. Chen, J. Zhong, L. Lv, D. Niu and J. Tang, Chem. Mater., 2014, 26, 3135–3143 CrossRef CAS.
- M. Al-Shakban, P. D. Matthews and P. O'Brien, Chem. Commun., 2017, 53, 10058–10061 RSC.
- P. D. McNaughter, S. A. Saah, M. Akhtar, K. Abdulwahab, M. Azad Malik, J. Raftery, J. A. M. Awudza and P. O'Brien, Dalton Trans., 2016, 45, 16345 Search PubMed.
- N. Pradhan, B. Katz and S. Efrima, J. Phys. Chem. B, 2003, 107, 13843–13854 CrossRef CAS.
- E. Vakalopoulou, T. Rath, M. Kräuter, A. Torvisco, R. C. Fischer, B. Kunert, R. Resel, H. Schröttner, A. M. Coclite, H. Amenitsch and G. Trimmel, ACS Appl. Nano Mater., 2022, 5, 1508–1520 Search PubMed.
- L. Yu, X. He, B. Peng, W. Wang, G. Wan, X. Ma, S. Zeng and G. Zhang, Matter, 2023, 6, 1604–1621 Search PubMed.
- F. T. F. O'Mahony, U. B. Cappel, N. Tokmoldin, T. Lutz, R. Lindblad, H. Rensmo and S. A. Haque, Angew. Chem., Int. Ed., 2013, 52, 12047–12051 Search PubMed.
- L. X. Reynolds, T. Lutz, S. Dowland, A. MacLachlan, S. King and S. A. Haque, Nanoscale, 2012, 4, 1561–1564 RSC.
- T. Lutz, A. MacLachlan, A. Sudlow, J. Nelson, M. S. Hill, K. C. Molloy and S. A. Haque, Phys. Chem. Chem. Phys., 2012, 14, 16192–16196 RSC.
- T. Rath, M. Edler, W. Haas, A. Fischereder, S. Moscher, A. Schenk, R. Trattnig, M. Sezen, G. Mauthner, A. Pein, D. Meischler, K. Bartl, R. Saf, N. Bansal, S. A. Haque, F. Hofer, E. J. List and G. Trimmel, Adv. Energy Mater., 2011, 1, 1046–1050 CrossRef CAS.
- G. Kortüm, W. Braun and G. Herzog, Angew. Chem., Int. Ed. Engl., 1963, 2, 333–341 CrossRef.
- P. Makuła, M. Pacia and W. Macyk, J. Phys. Chem. Lett., 2018, 9, 6814–6817 CrossRef PubMed.
- T. Wada and T. Maeda, Phys. Status Solidi C, 2017, 14, 1600196 CrossRef.
- C. M. Cardona, W. Li, A. E. Kaifer, D. Stockdale and G. C. Bazan, Adv. Mater., 2011, 23, 2367–2371 CrossRef CAS PubMed.
- M. Sigl, M. Egger, F. Warchomicka, D. Knez, M. Dienstleder, H. Amenitsch, G. Trimmel and T. Rath, J. Mater. Chem. A, 2024, 12, 28965–28974 RSC.
- O. Glatter, J. Appl. Crystallogr., 1977, 10, 415–421 Search PubMed.
- J. Cao, D. Zhang and H. Zhang, Mater. Res. Bull., 2024, 179, 112990 CrossRef CAS.
- R. A. El-Gendy, H. M. El-Bery, M. Farrag and D. M. Fouad, Sci. Rep., 2023, 13, 7994 CrossRef CAS PubMed.
- N. Aristidou, C. Eames, I. Sanchez-Molina, X. Bu, J. Kosco, M. S. Islam and S. A. Haque, Nat. Commun., 2017, 8, 15218 CrossRef PubMed.
- N. Aristidou, I. Sanchez-Molina, T. Chotchuangchutchaval, M. Brown, L. Martinez, T. Rath and S. A. Haque, Angew. Chem., Int. Ed., 2015, 54, 8208–8212 CrossRef CAS PubMed.
- B. Kalyanaraman, E. Perezreyes and R. P. Mason, Biochim. Biophys. Acta, 1980, 630, 119–130 CrossRef CAS PubMed.
- E. Finkelstein, G. M. Rosen and E. J. Rauckman, J. Am. Chem. Soc., 1980, 102, 4994–4999 CrossRef CAS.
- E. Finkelstein, G. M. Rosen and E. J. Rauckman, Arch. Biochem. Biophys., 1980, 200, 1–16 CrossRef CAS PubMed.
- S. Fuloria, V. Subramaniyan, S. Karupiah, U. Kumari, K. Sathasivam, D. U. Meenakshi, Y. S. Wu, M. Sekar, N. Chitranshi, R. Malviya, K. Sudhakar, S. Bajaj and N. K. Fuloria, Antioxidants, 2021, 10, 128 CrossRef CAS PubMed.
- F. Parrino, S. Livraghi, E. Giamello, R. Ceccato and L. Palmisano, ACS Catal., 2020, 10, 7922–7931 CrossRef CAS.
- T. Shoji, L. Li, Y. Abe, M. Ogata, Y. Ishimoto, R. Gonda, T. Mashino, M. Mochizuki, M. Uemoto and N. Miyata, Anal. Sci., 2007, 23, 219–221 CrossRef PubMed.
- D. T. Sawyer, M. J. Gibian, M. M. Morrison and E. T. Seo, J. Am. Chem. Soc., 1978, 100, 627–628 CrossRef CAS.
- A. J. Nozik and R. Memming, J. Phys. Chem., 1996, 100, 13061–13078 CrossRef CAS.
- J. Zhao, X. Yan, N. Zhao, X. Li, B. Lu, X. Zhang and H. Yu, RSC Adv., 2018, 8, 4979–4986 RSC.
- P. D. Tran, L. Xi, S. K. Batabyal, L. H. Wong, J. Barber and J. S. C. Loo, Phys. Chem. Chem. Phys., 2012, 14, 11596–11599 RSC.
- J. Yu, B. Yang and B. Cheng, Nanoscale, 2012, 4, 2670–2677 RSC.
- H. Amenitsch, M. Rappolt, M. Kriechbaum, H. Mio, P. Laggner and S. Bernstorff, J. Synchrotron Radiat., 1998, 5, 506–508 CrossRef CAS PubMed.
- M. Burian, C. Meisenbichler, D. Naumenko and H. Amenitsch, J. Appl. Crystallogr., 2022, 55, 677–685 CrossRef CAS PubMed.
- O. Glatter and O. Kratky, Small Angle X-Ray Scattering. General Theory by Porod, Academic Press INC Ltd, 1982, ch. 2 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ma00212e |
| This journal is © The Royal Society of Chemistry 2025 |