
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSorafenib nanocrystals enhance bioavailability and overcome multidrug resistance in liver cancer cells: an approach based on P-gp inhibition strategy
Mohamed
Nasr
 *ab,
Sameh
Saber
c,
Heba I.
Elagamy
b,
Soha M.
El-Masry
d,
Haydy
Asad
e,
Ahmed A. E.
Mourad
f,
Ahmed Gaafar Ahmed
Gaafar
f and
Shaimaa K.
Mostafa
b
*ab,
Sameh
Saber
c,
Heba I.
Elagamy
b,
Soha M.
El-Masry
d,
Haydy
Asad
e,
Ahmed A. E.
Mourad
f,
Ahmed Gaafar Ahmed
Gaafar
f and
Shaimaa K.
Mostafa
b
aDepartment of Pharmaceutics and Industrial Pharmacy, Faculty of Pharmacy, Helwan University, Cairo 11790, Egypt. E-mail: m2nasr@yahoo.com
bDepartment of Pharmaceutics, Faculty of Pharmacy, Delta University for Science and Technology, Gamasa 11152, Egypt
cDepartment of Pharmacology, Faculty of Pharmacy, Delta University for Science and Technology, Gamasa 11152, Egypt
dDepartment of Pharmaceutics, Faculty of Pharmacy, Damanhour University, Damanhour 22511, Egypt
eDrug analysis and chromatography research lab, Faculty of Pharmacy, Delta University for Science and Technology, Gamasa 11152, Egypt
fDepartment of Pharmacology and Toxicology, Faculty of Pharmacy, Port Said University, Port Said 42511, Egypt
First published on 8th April 2025
Abstract
This research aimed to improve the oral bioavailability of Sorafenib (SF) and overcome multidrug resistance in hepatocellular carcinoma cells based on P-glycoprotein (P-gp) inhibition strategy. Four nanocrystal formulations (F1–F4) were developed using Labrasol® (LB) or Gelucire® (GL) as stabilizers as well as P-gp inhibitors. The prepared SF nanocrystal (SF-NC) formulations were characterized in vitro and in vivo. The results of in vitro studies showed that LB-based nanocrystals (F2) prepared using 0.02% LB, significantly reduced the crystal size and improved the aqueous saturation solubility of SF compared to the GL-based nanocrystals. Crystal morphology analysis of SF-NC (F2) revealed a uniform arrangement of nanosized crystals with significantly smaller particle size compared to plain SF. Furthermore, in vitro cytotoxicity studies showed that LB had no significant effects on cell viability of MDR-HepG2 and SF-resistant Huh-7 cells and can be considered safe in the in vivo environment at concentrations more than 10 times its corresponding concentration in SF-NC. However, LB-stabilized SF-NC significantly reduced IC50 values in MDR-HepG2 and SF-resistant Huh-7 cells compared to plain SF. In vivo absorption studies revealed that SF-NC significantly increased the rate and extent of absorption with a 1.27-fold increase in relative bioavailability. The developed SF-NC stabilized by LB as a P-gp inhibitor is expected to be a promising approach to improve oral bioavailability and restore SF's activity against multidrug-resistant hepatocellular carcinoma cells.
Introduction
Sorafenib tosylate (SF) is a multi-kinase inhibitor and is currently the only drug approved by the FDA for the treatment of advanced liver cancer.1–3 It can facilitate apoptosis, attenuate angiogenesis, suppress tumor cell proliferation, and improve overall survival by approximately three months compared to a placebo. However, SF's clinical efficacy is hindered by the limited oral absorption from the gastrointestinal tract, which is primarily due to the poor aqueous solubility and its classification as a P-gp substrate.4,5P-gp is only overexpressed in certain kinds of cancers and carries a lot of anticancer medications, which makes the malignancies resistant to those medications.6 The decrease in antitumor drug intracellular concentration is due to drug efflux by P-glycoprotein (P-gp) which is considered a key factor contributing to the development of MDR in cancer cells.7 Previous reports indicated that the therapeutic anticancer efficacy of SF relies on various efflux transporters, such as P-glycoprotein (P-gp),8–11 multidrug resistance-associated proteins 2 and 3 (MRP2 and 3), breast cancer resistance protein (BCRP), organic anion transporting polypeptide 1B1/3 (OATP1B1/3), and organic cation transporter-1 (OCT1). These efflux transporters play a key role in cancer chemoresistance8,12–15 due to the efflux extrusion of either SF itself or its metabolites, which show chemical characteristics typical of P-gp and MRP2 substrates.16–18 Accordingly, a potential reduction of its intracellular concentration and therapeutic efficacy could be expected.15 Previous reports indicated that P-gp was involved in the transport of SF, and coadministration with P-gp inhibitors like verapamil10 or morphine9 could increase SF absorption in either Caco-2 cell model or rats.
Several formulation approaches have been applied to overcome SF's poor water solubility to improve its bioavailability, such as cyclodextrin-based inclusion complex,19 solid self-nano-emulsifying drug delivery systems,20 liquid nanocrystals,21 solid dispersion,22 mesoporous solid dispersion,23 in addition to nanoparticle-based drug delivery systems, including liposomes,24,25 polymeric nanoparticles,26–28 polyethylene glycol nanoparticles,29,30 nanostructured lipid carriers,31,32 silica nanoparticles,33,34 and solid lipid nanoparticles.1,35–37
Nanocrystals or solid micelles are commonly employed as a formulation strategy to enhance the solubility of poorly water-soluble drugs by increasing the surface area available for dissolution and subsequently enhancing the bioavailability.38 Drug nanocrystals consist of solid drug particles surrounded by a stabilizer layer, preventing individual particles' aggregation. Various stabilizers have been utilized in the stabilization of nanocrystals such as polymers like hydroxypropyl methylcellulose, polyvinylpyrrolidone K30, and Pluronic® as well as amphiphilic surfactants like polysorbates,39 Labrasol,40 and sodium lauryl sulfate.41–43 Amphiphilic surfactants enhance the solubility of nanocrystals via better wetting and solubilizing effects. In addition to their solubilization effect, some stabilizers like LB, and Gelucire 44/14 (GL) have been found to possess inhibitory activity against efflux transporters, particularly P-gp.44–46
This study aimed to develop a novel SF-nanocrystals (SF-NC) by using LB or GL as stabilizers and P-gp inhibitors. The newly developed SF-NC will be in vitro and in vivo evaluated. In vitro characterizations of SF-NC include crystal characteristics, saturation aqueous solubility, dissolution, and cytotoxicity in terms of IC50 against resistant cancer cells compared to plain SF crystals. In vivo, the influence of SF-NC on oral absorption in rats will be evaluated. The developed SF-NC formulation is expected to be a promising approach to improve SF oral bioavailability and potentiate its activity against resistant cancer cells.
Materials and methods
Materials
Sorafenib tosylate was purchased from Cayman Chemical (Ann Arbor, MI, USA). Gelucire® 44/14 and Labrasol® (caprylocaproyl polyoxyl-8 glycerides) were kindly provided as a gift by Gattefosse, Saint-Priest Cedex, France. Acetonitrile (HPLC grade) was obtained from BDH, Poole, England. We bought methanol, phosphoric acid, sodium dihydrogen orthophosphate dihydrate (pharmaceutical quality), and hydrochloric acid (analytical grade) from El Nasr Chemical Company in Cairo, Egypt.Methods
Characterization of SF-NC
The establishment of SF-resistant Huh-7 cell line was conducted according to the method described by Verslype, van Malenstein.54 In summary, Huh-7 cells were cultured for 21 days straight in a medium containing 5 μM SF. Every three days, the medium containing SF was changed, and the cells were divided when they achieved 80% confluency. The developed SF-resistant Huh-7 cells were maintained in DMEM supplemented with 10% fetal bovine serum, 100 U mL−1 penicillin G, and 100 μg mL−1 streptomycin and kept in the 37 °C humidified CO2 incubator. Previous reports indicated that resistance of hepatocellular carcinoma to SF was associated with increased expression of P-gp.55,56
Bioavailability study
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :45, v/v), pumped by a flow rate of 0.25 mL min−1. The detection wavelength was 265 nm. The validity of the method was examined regarding selectivity, linearity, precision, and accuracy according to ICH guidelines.60 SF plasma calibration curve was established by spiking 200 μL blank plasma with 20 μL of working standard solutions to obtain concentrations ranging from (1–1000 μg mL−1) prepared by dilutions of stock of SF solution (1000 μg mL−1) in HPLC methanol. Plasma samples (200 μL) were added to 20 μL Celecoxib solution (100 μg mL−1 in methanol, as an internal standard). To precipitate plasma proteins, 400 μL of acetonitrile was added. The resultant mixture was centrifuged (Tabletop Centrifuge, PLC-03, Gemmy Industrial Corporation, Taiwan) at 4000 rpm for 15 min and finally, the supernatant was filtered through 0.22 μm Millipore filter and 20 μL was injected into HPLC.
:45, v/v), pumped by a flow rate of 0.25 mL min−1. The detection wavelength was 265 nm. The validity of the method was examined regarding selectivity, linearity, precision, and accuracy according to ICH guidelines.60 SF plasma calibration curve was established by spiking 200 μL blank plasma with 20 μL of working standard solutions to obtain concentrations ranging from (1–1000 μg mL−1) prepared by dilutions of stock of SF solution (1000 μg mL−1) in HPLC methanol. Plasma samples (200 μL) were added to 20 μL Celecoxib solution (100 μg mL−1 in methanol, as an internal standard). To precipitate plasma proteins, 400 μL of acetonitrile was added. The resultant mixture was centrifuged (Tabletop Centrifuge, PLC-03, Gemmy Industrial Corporation, Taiwan) at 4000 rpm for 15 min and finally, the supernatant was filtered through 0.22 μm Millipore filter and 20 μL was injected into HPLC.
HPLC method was validated regarding selectivity, linearity, precision, and accuracy according to ICH guidelines. HPLC chromatogram of blank plasma, plasma spiked with SF, and celecoxib (internal standard) showed selectivity of the method with no interferences with plasma components. From the SF calibration curve in plasma, the linearity was achieved over the concentration range of 1–1000 μg mL−1 with R2 = 0.9976. Precision was 2.8702 to 9.458% and accuracy was 96.83 to 103.3% while inter-day precision was 0.178 to 8.034% and accuracy was 94.65 to 108.4%.
Results and discussion
Characterization of SF-NC
| Stabilizer (concentration % w/v) | Particle size (nm ± SD, n = 3) | PDI | SF saturated aqueous solubility (μg mL−1 ± SD, n =3) | |
|---|---|---|---|---|
| Plain SF | — | 893.80 ± 15.66 | 0.937 | 0.01 ± 0.003 |
| F1 | LB (0.01) | 320.40 ± 8.86 | 0.445 | 2.00 ± 0.119 |
| F2 | LB (0.02) | 132.30 ± 5.57 | 0.641 | 4.45 ± 0.072 |
| F3 | GL (0.01) | 350.70 ± 9.69 | 0.668 | 1.69 ± 0.075 |
| F4 | GL (0.02) | 310.90 ± 7.61 | 0.603 | 1.66 ± 0.081 |
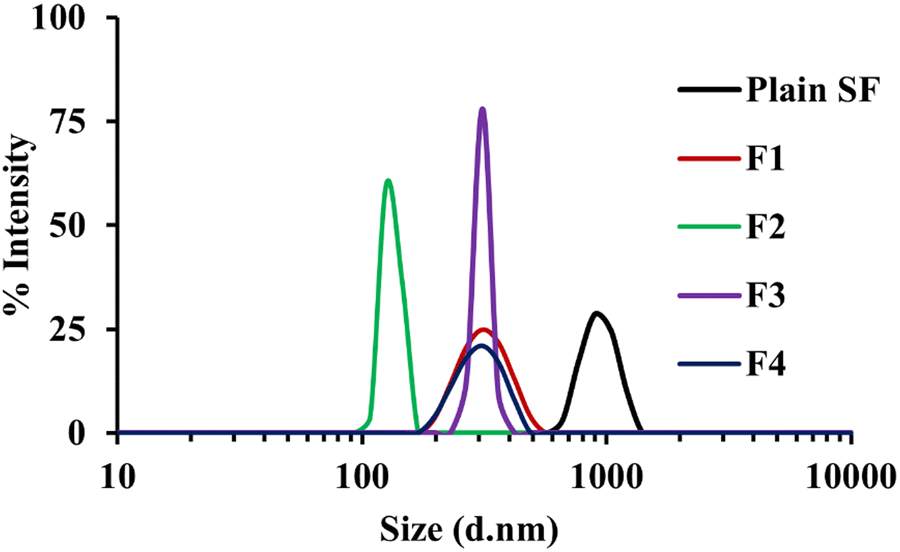 | ||
| Fig. 1 Particle size distribution of plain SF and SF-NC formulations (F1 and F2 stabilized by 0.01% and 0.02% w/v LB, respectively, F3 and F4 were stabilized by 0.01% and 0.02% w/v GL, respectively). | ||
The results of particle size analysis indicated that a significant reduction in crystal size was achieved by using LB compared to the size of nanocrystals prepared by GL. This might be explained based on the surface activity of LB that can prevent agglomeration and promote the formation of smaller, more uniformly distributed nanocrystals.61,62 On the other hand, GL, a combination of mono, di, triglycerides, and polyethylene glycol esters of fatty acids, stabilizes the formed crystals through the formation of gel phase or liquid crystals, which may limit particle size reduction.63,64 Accordingly, it is believed that the higher solubilizing and surfactant properties of LB lead to a more prominent reduction in particle size than GL. The mechanism behind the particle size reduction by stabilizers like LB and GL implicates their surfactant characteristics. They are known to develop self-emulsifying systems, which are isotropic mixtures that form oil in water emulsions under mild stirring.62 These systems can decrease the particle size of the drug effectively, thus improving its solubility and bioavailability.63
 | ||
| Fig. 2 SEM photographs of (A) plain SF and (B) F2 nanocrystals. | ||
 | ||
| Fig. 3 DSC thermogram of plain SF and SF-NC formulations (F2). | ||
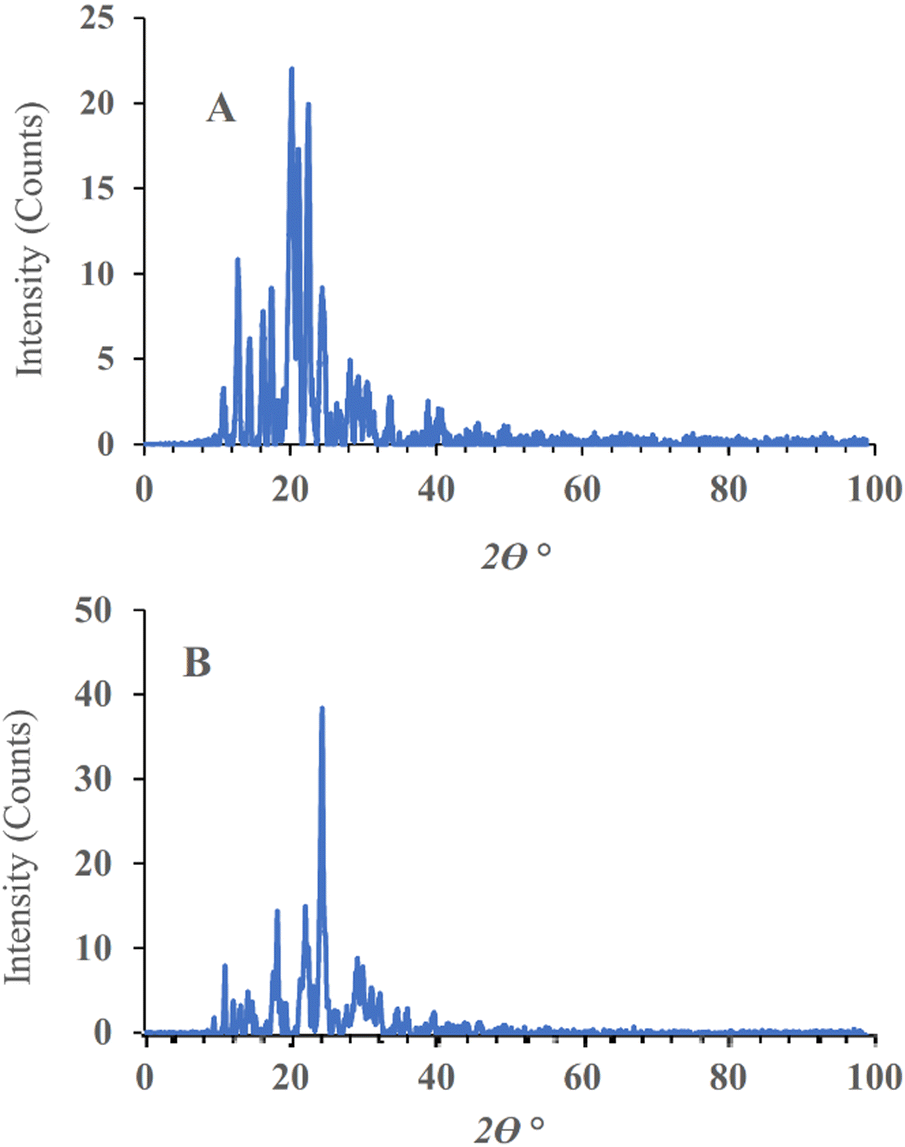 | ||
| Fig. 4 Powder XRD patterns of plain-SF (A), and SF-NC (B). | ||
 | ||
| Fig. 5 In vitro dissolution profiles of SF-NC (F2) and plain SF. | ||
The values of dissolution parameters of plain SF including the % drug dissolved after 10 and 120 min (Q10% and Q120%) were 25.22 ± 1.23 and 49.55 ± 1.44% compared to 90.12 ± 2.6 and 99.60 ± 1.55 in the case of F2 formulation, with dissolution efficiency % at 15 min (% DE15min) of 25.23 ± 1.65 and 61.9 ± 2.31 for plain SF and F2 respectively. It was obvious that dissolution parameters were significantly higher for F2 compared to plain SF. This enhancement resulted from the solubilization effects of LB which improved SF wettability and hydrophilicity in dissolution media due to its amphiphilic characters and the reduced particle size. This leads to increasing drug surface area available for dissolution medium and subsequently, the dissolution velocity increases, as verified by the Noyes–Whitney equation.65
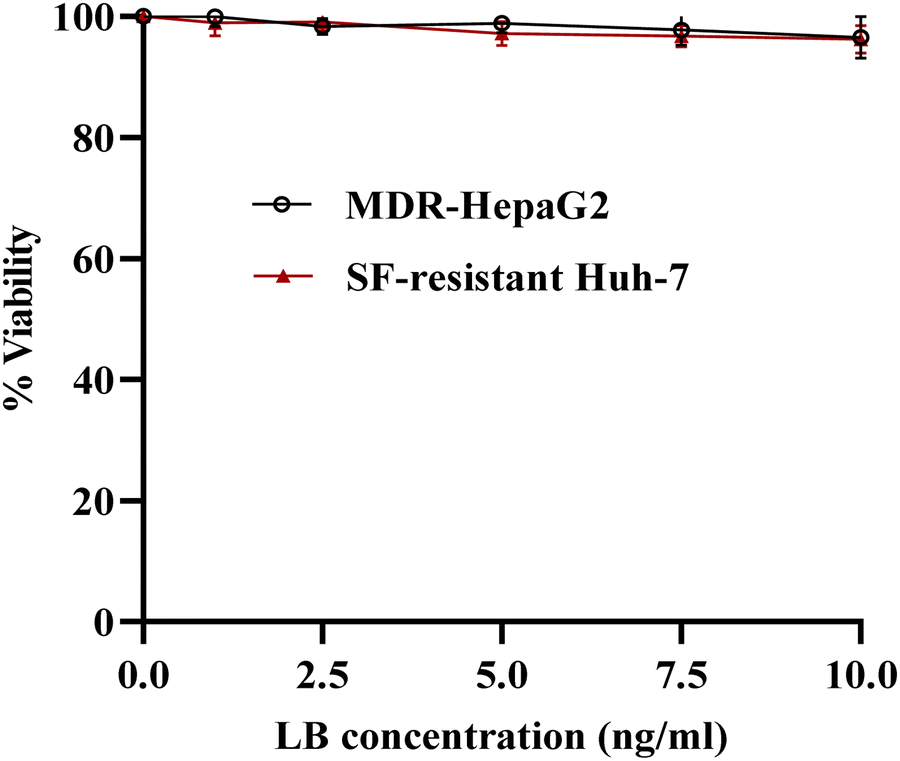 | ||
| Fig. 6 In vitro cytotoxicity of LB against MDR-HepaG2 and SF-resistant Huh-7 cells after 24 h of incubation. | ||
Fig. 7(A) and (B) illustrates the results of cell viability of MDR HepG2 and SF-resistant Huh-7 cells after exposure to either SF-plain crystals or SF-NC formulation (F2), as well as SF-plain in addition to 50 μM verapamil. Table 2 summarizes the calculated IC50 values for each treatment. The IC50 values for SF-plain crystals were 9.60 ± 1.07 μM for MDR HepG2 cells and 10.67 ± 1.30 μM for SF-resistant Huh7 cells. In contrast, the IC50 values were significantly reduced to 6.31 ± 0.71 μM and 7.06 ± 0.84 μM, respectively, when using SF-NC, which may be attributed to the presence of LB as a P-gp inhibitor on the surface of SF-NC.
 | ||
| Fig. 7 The cytotoxicity of SF-plain, SF-NC (F2), and SF-plain + 50 μM against MDR-HepG2 (A), and SF-resistant Huh-7 (B) cells after 24 h incubation. The measurements were taken using MTT cell viability assay. The data represent mean ± S.D. of three independent experiments. | ||
| Cancer cells | IC50 (Mean ± SD μM) | ||
|---|---|---|---|
| SF-plain crystals | SF-NC (F2) | SF-plain + verapamil | |
| The data represent the mean ± SD of three independent experiments. | |||
| MDR-HepG2 | 9.60 ± 1.07 | 6.31 ± 0.71 | 4.99 ± 0.53 |
| SF-resistant Huh-7 | 10.67 ± 1.30 | 7.06 ± 0.84 | 4.81 ± 0.66 |
Notably, when different concentrations of SF-plain crystals were combined with 50 μM verapamil, a remarkable decrease in IC50 values to 4.99 ± 0.53 and 4.81 ± 0.66 μM in the case of MDR HepG2 and SF-resistant Huh7 cells, respectively compared to either SF-plain crystals or SF-NC formulation. This reduction can be attributed to the presence of verapamil, which is known as a P-gp inhibitor capable of completely reversing the resistance caused by the P-gp efflux pump.57 Although the SF-NC formulation achieved a relatively higher IC50 value than the SF-plain + 50 μM verapamil, it still demonstrated significantly lower values than the SF-plain. The findings suggest that SF-NC successfully reduces the viability of MDR-HepG2 and SF-resistant Huh-7 cells, although its potency is much less compared to verapamil. Further investigations are needed to assess LB's potential in reducing MDR.
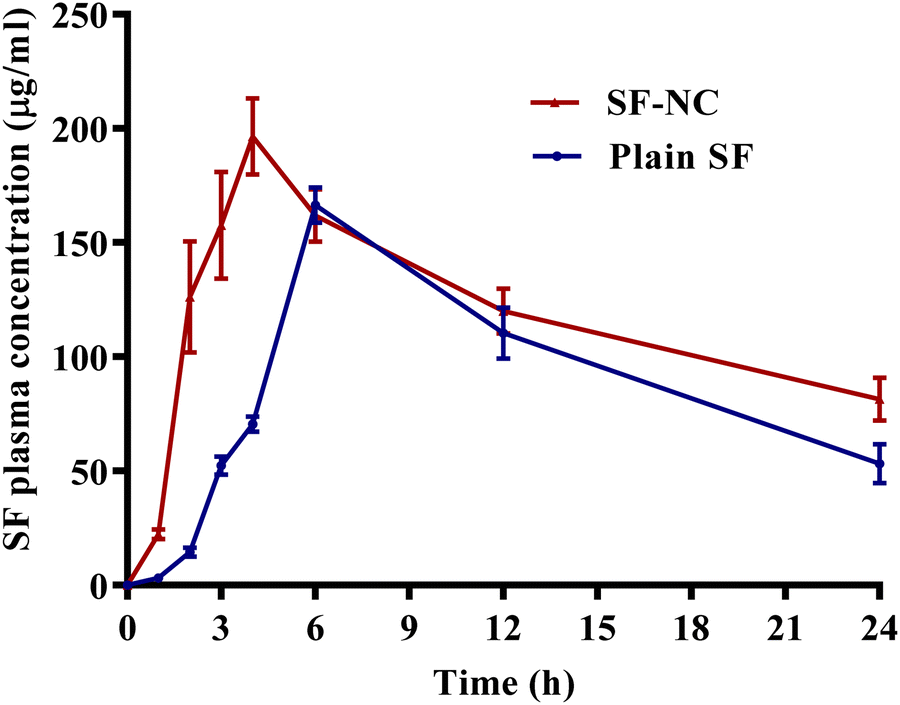 | ||
| Fig. 8 SF plasma concentration-time profiles (mean ± SD, n = 6) after administration of a single oral dose (50 mg kg−1) of SF-NC and plain SF aqueous suspensions to rats. | ||
| Bioavailability parameters | SF-NC | Plain SF |
|---|---|---|
| AUC0–24 (μg h mL−1) | 2949.40 ± 268.60 | 2320.90 ± 79.32 |
| C max (μg mL−1) | 196.40 ± 19.09 | 166.47 ± 6.52 |
| T max (h) | 4.00 | 6.00 |
| Relative bioavailability (%) | 127.08% |
Conclusion
In conclusion, the results of this study revealed that SF-NC formulation (F2) prepared by 0.02% LB, significantly improved the aqueous saturation solubility and dissolution of SF compared to plain SF. Moreover, in vitro cytotoxicity studies showed that LB had no significant effects on the cell viability of MDR-HepaG2 and SF-resistant Huh-7. However, SF-NC significantly reduces IC50 values compared to plain SF. In vivo absorption studies indicated that formulation of SF-NC, stabilized by LB as a P-gp inhibitor, remarkably increased both the rate and extent of oral bioavailability of FS in terms of Cmax and AUC0–24, as indicated by a 1.27-fold increase in the relative bioavailability. Overall, the developed SF-NC formulation utilizing LB as a stabilizer and P-gp inhibitor showed promising improvements in the physicochemical properties and oral bioavailability of SF and could be a reliable strategy for overcoming multidrug resistance in hepatocellular carcinoma cells.Author contributions
Mohamed Nasr: conceptualization, formal analysis, supervision, writing – reviewing and editing. Sameh Saber: conceptualization, supervision, and visualization. Heba I. Elagamy: methodology, writing – reviewing and editing. Soha M. El-Masry: investigation, methodology, writing and reviewing. Haydy Asad: methodology, writing, and reviewing. Ahmed A. E. M., methodology, Ahmed G. A. G. methodology, Shaimaa K. Mostafa: Methodology, material preparation, data collection, and analysis.Data availability
Data will be made available on request.Conflicts of interest
The authors declare that they have no conflicts of interest.Acknowledgements
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors. The authors are thankful to Gattefosse (France) for providing the gift samples of Gelucire® 44/14 and Labrasol®.References
- F.-H. Kong, Q.-F. Ye, X.-Y. Miao, X. Liu, S.-Q. Huang and L. Xiong, et al., Current status of sorafenib nanoparticle delivery systems in the treatment of hepatocellular carcinoma, Theranostics, 2021, 11(11), 5464 CrossRef CAS PubMed.
- O. Brunetti, A. Gnoni, A. Licchetta, V. Longo, A. Calabrese and A. Argentiero, et al., Predictive and prognostic factors in HCC patients treated with sorafenib, Medicina, 2019, 55(10), 707 CrossRef PubMed.
- M. Monajati, S. Tavakoli, S. S. Abolmaali, G. Yousefi and A. Tamaddon, Effect of PEGylation on assembly morphology and cellular uptake of poly ethyleneimine-cholesterol conjugates for delivery of sorafenib tosylate in hepatocellular carcinoma, BioImpacts, 2018, 8(4), 241 CrossRef CAS PubMed.
- X.-Q. Wang, J.-M. Fan, Y.-O. Liu, B. Zhao, Z.-R. Jia and Q. Zhang, Bioavailability and pharmacokinetics of sorafenib suspension, nanoparticles and nanomatrix for oral administration to rat, Int. J. Pharm., 2011, 419(1–2), 339–346 CrossRef CAS PubMed.
- S. Jiang, Y. Qin, S. Wu, S. Xu, K. Li and P. Yang, et al., Solubility correlation and thermodynamic analysis of sorafenib free base and sorafenib tosylate in monosolvents and binary solvent mixtures, J. Chem. Eng. Data, 2017, 62(1), 259–267 CrossRef CAS.
- A. K. Nanayakkara, C. A. Follit, G. Chen, N. S. Williams, P. D. Vogel and J. G. Wise, Targeted inhibitors of P-glycoprotein increase chemotherapeutic-induced mortality of multidrug resistant tumor cells, Sci. Rep., 2018, 8(1), 967 CrossRef PubMed.
- R. N. Montesinos, A. Béduneau, Y. Pellequer and A. Lamprecht, Delivery of P-glycoprotein substrates using chemosensitizers and nanotechnology for selective and efficient therapeutic outcomes, J. Controlled Release, 2012, 161(1), 50–61 CrossRef PubMed.
- J. S. Lagas, R. A. van Waterschoot, R. W. Sparidans, E. Wagenaar, J. H. Beijnen and A. H. Schinkel, Breast cancer resistance protein and P-glycoprotein limit sorafenib brain accumulation, Mol. Cancer Ther., 2010, 9(2), 319–326 CrossRef CAS PubMed.
- A. Karbownik, D. Szkutnik-Fiedler, T. Grabowski, A. Wolc, J. Stanisławiak-Rudowicz and R. Jaźwiec, et al., Pharmacokinetic drug interaction study of Sorafenib and morphine in rats, Pharmaceutics, 2021, 13(12), 2172 CrossRef CAS PubMed.
- X. Wang, X. Zhang, X. Huang, Y. Li, M. Wu and J. Liu, The drug–drug interaction of sorafenib mediated by P-glycoprotein and CYP3A4, Xenobiotica, 2016, 46(7), 651–658 CrossRef CAS PubMed.
- W. Tang, Z. Chen, W. Zhang, Y. Cheng, B. Zhang and F. Wu, et al., The mechanisms of sorafenib resistance in hepatocellular carcinoma: theoretical basis and therapeutic aspects, Signal Transduction Targeted Ther., 2020, 5(1), 87 CrossRef PubMed.
- M. Chen, C. Neul, E. Schaeffeler, F. Frisch, S. Winter and M. Schwab, et al., Sorafenib activity and disposition in liver cancer does not depend on organic cation transporter 1, Clin. Pharm. Ther., 2020, 107(1), 227–237 CrossRef CAS PubMed.
- A. Vasilyeva, S. Durmus, L. Li, E. Wagenaar, S. Hu and A. A. Gibson, et al., Hepatocellular shuttling and recirculation of sorafenib-glucuronide is dependent on Abcc2, Abcc3, and Oatp1a/1b, Cancer Res., 2015, 75(13), 2729–2736 CrossRef CAS PubMed.
- Y. Shibayama, K. Nakano, H. Maeda, M. Taguchi, R. Ikeda and M. Sugawara, et al., Multidrug resistance protein 2 implicates anticancer drug-resistance to sorafenib, Biol. Pharm. Bull., 2011, 34(3), 433–435 CrossRef CAS PubMed.
- J. P. Rigalli, N. Ciriaci, A. Arias, M. P. Ceballos, S. S. M. Villanueva and M. G. Luquita, et al., Regulation of multidrug resistance proteins by genistein in a hepatocarcinoma cell line: impact on sorafenib cytotoxicity, PLoS One, 2015, 10(3), e0119502 CrossRef PubMed.
- C. D. Klaassen and L. M. Aleksunes, Xenobiotic, bile acid, and cholesterol transporters: function and regulation, Pharmacol. Rev., 2010, 62(1), 1–96 CrossRef CAS PubMed.
- S. Ghassabian, T. Rawling, F. Zhou, M. R. Doddareddy, B. N. Tattam and D. E. Hibbs, et al., Role of human CYP3A4 in the biotransformation of sorafenib to its major oxidized metabolites, Biochem. Pharm., 2012, 84(2), 215–223 CAS.
- P. H. Cui, T. Rawling, T. B. Gillani, K. Bourget, X.-S. Wang and F. Zhou, et al., Anti-proliferative actions of N′-desmethylsorafenib in human breast cancer cells, Biochem. Pharm., 2013, 86(3), 419–427 CAS.
- A. Aman, S. Ali, P. Mahalapbutr, K. Krusong, P. Wolschann and T. Rungrotmongkol, Enhancing solubility and stability of sorafenib through cyclodextrin-based inclusion complexation: in silico and in vitro studies, RSC Adv., 2023, 13(39), 27244–27254 CAS.
- C. Lim, D. Lee, M. Kim, S. Lee, Y. Shin and J. D. Ramsey, et al., Development of a sorafenib-loaded solid self-nanoemulsifying drug delivery system: Formulation optimization and characterization of enhanced properties, J. Drug Delivery Sci. Technol., 2023, 82, 104374 CAS.
- N. Diddi, S. Kumar, S. Pavani and P. Neelima, Formulation and Evaluation of Liquid Nanocrystals of Sorafenib Tosylate, Global J. Pharm. Pharm. Sci., 2019, 7(4), 137–142 Search PubMed.
- D. H. Truong, T. H. Tran, T. Ramasamy, J. Y. Choi, H.-G. Choi and C. S. Yong, et al., Preparation and characterization of solid dispersion using a novel amphiphilic copolymer to enhance dissolution and oral bioavailability of sorafenib, Powder Technol., 2015, 283, 260–265 CAS.
- S. Xingjie, Y. Zhankuan, Y. Chaofeng and L. Chuanjun, The preparation and characterization of sorafenib solid dispersion, J. Pharm. Practice Service, 2016, 34(4), 320–342 Search PubMed.
- S. Yang, B. Zhang, X. Gong, T. Wang, Y. Liu and N. Zhang, In vivo biodistribution, biocompatibility, and efficacy of sorafenib-loaded lipid-based nanosuspensions evaluated experimentally in cancer, Int. J. Nanomed., 2016, 2329–2343 CAS.
- J. Zhang, T. Wang, S. Mu, L. D. Olerile, X. Yu and N. Zhang, Biomacromolecule/lipid hybrid nanoparticles for controlled delivery of sorafenib in targeting hepatocellular carcinoma therapy, Nanomedicine, 2017, 12(8), 911–925 CrossRef CAS PubMed.
- E. F. Craparo, C. Sardo, R. Serio, M. G. Zizzo, M. L. Bondì and G. Giammona, et al., Galactosylated polymeric carriers for liver targeting of sorafenib, Int. J. Pharm., 2014, 466(1–2), 172–180 CrossRef CAS PubMed.
- G. Babos, E. Biró, M. Meiczinger and T. Feczkó, Dual drug delivery of sorafenib and doxorubicin from PLGA and PEG-PLGA polymeric nanoparticles, Polymers, 2018, 10(8), 895 CrossRef PubMed.
- J. Varshosaz, F. Raghami, M. Rostami and A. Jahanian, PEGylated trimethylchitosan emulsomes conjugated to octreotide for targeted delivery of sorafenib to hepatocellular carcinoma cells of HepG2, J. Liposome Res., 2019, 29(4), 383–398 CrossRef PubMed.
- L. Zheng, X. Huang, X. Lin, W. Lin, F. Yang and T. Chen, Thermosensitive hydrogels for sustained-release of sorafenib and selenium nanoparticles for localized synergistic chemoradiotherapy, Biomaterials, 2019, 216, 119220 CrossRef CAS PubMed.
- Y. Wang, H. Yu, D. Zhang, G. Wang, W. Song and Y. Liu, et al., Co-administration of combretastatin A4 nanoparticles and sorafenib for systemic therapy of hepatocellular carcinoma, Acta Biomater., 2019, 92, 229–240 CrossRef CAS PubMed.
- M. L. Bondì, C. Botto, E. Amore, M. R. Emma, G. Augello and E. F. Craparo, et al., Lipid nanocarriers containing sorafenib inhibit colonies formation in human hepatocarcinoma cells, Int. J. Pharm., 2015, 493(1–2), 75–85 CrossRef PubMed.
- J.-Y. Wang, Y.-Q. Song, J. Peng and H.-L. Luo, Nanostructured lipid carriers delivering Sorafenib to enhance immunotherapy induced by doxorubicin for effective esophagus cancer therapy, ACS Omega, 2020, 5(36), 22840–22846 CrossRef CAS PubMed.
- H. Tang, D. Chen, C. Li, C. Zheng, X. Wu and Y. Zhang, et al., Dual GSH-exhausting sorafenib loaded manganese-silica nanodrugs for inducing the ferroptosis of hepatocellular carcinoma cells, Int. J. Pharm., 2019, 572, 118782 CrossRef CAS PubMed.
- J. Ye, R. Zhang, W. Chai and X. Du, Low-density lipoprotein decorated silica nanoparticles co-delivering sorafenib and doxorubicin for effective treatment of hepatocellular carcinoma, Drug Delivery, 2018, 25(1), 2007–2014 CrossRef CAS PubMed.
- A. Grillone, E. R. Riva, A. Mondini, C. Forte, L. Calucci and C. Innocenti, et al., Active targeting of sorafenib: preparation, characterization, and in vitro testing of drug-loaded magnetic solid lipid nanoparticles, Adv. Healthcare Mater., 2015, 4(11), 1681–1690 CrossRef CAS PubMed.
- S. Benizri, L. Ferey, B. Alies, N. Mebarek, G. Vacher and A. Appavoo, et al., Nucleoside-lipid-based nanocarriers for sorafenib delivery, Nanoscale Res. Lett., 2018, 13, 1–8 CrossRef CAS PubMed.
- L. Tunki, H. Kulhari, L. N. Vadithe, M. Kuncha, S. Bhargava and D. Pooja, et al., Modulating the site-specific oral delivery of sorafenib using sugar-grafted nanoparticles for hepatocellular carcinoma treatment, Eur. J. Pharm. Sci., 2019, 137, 104978 CrossRef CAS PubMed.
- Y. Lu, Y. Li and W. Wu, Injected nanocrystals for targeted drug delivery, Acta Pharm. Sin. B, 2016, 6(2), 106–113 CrossRef PubMed.
- A. Tuomela, P. Liu, J. Puranen, S. Rönkkö, T. Laaksonen and G. Kalesnykas, et al., Brinzolamide nanocrystal formulations for ophthalmic delivery: reduction of elevated intraocular pressure in vivo, Int. J. Pharm., 2014, 467(1–2), 34–41 CrossRef CAS PubMed.
- M. Kurakula, A. El-Helw, T. R. Sobahi and M. Y. Abdelaal, Chitosan based atorvastatin nanocrystals: effect of cationic charge on particle size, formulation stability, and in vivo efficacy, Int. J. Nanomed., 2015, 321–334 CrossRef PubMed.
- P. Liu, X. Rong, J. Laru, B. van Veen, J. Kiesvaara and J. Hirvonen, et al., Nanosuspensions of poorly soluble drugs: preparation and development by wet milling, Int. J. Pharm., 2011, 411(1–2), 215–222 CrossRef CAS PubMed.
- H. Rahim, A. Sadiq, S. Khan, M. A. Khan, S. M. H. Shah and Z. Hussain, et al., Aceclofenac nanocrystals with enhanced in vitro, in vivo performance: formulation optimization, characterization, analgesic and acute toxicity studies, Drug Des., Dev. Ther., 2017, 2443–2452 CrossRef CAS PubMed.
- M. Nasr, Influence of microcrystal formulation on in vivo absorption of celecoxib in rats, AAPS PharmSciTech, 2013, 14, 719–726 CrossRef CAS PubMed.
- O. Dubray, V. Jannin, F. Demarne, Y. Pellequer, A. Lamprecht and A. Béduneau, In-vitro investigation regarding the effects of Gelucire® 44/14 and Labrasol® ALF on the secretory intestinal transport of P-gp substrates, Int. J. Pharm., 2016, 515(1–2), 293–299 CAS.
- Y. Lin, Q. Shen, H. Katsumi, N. Okada, T. Fujita and X. Jiang, et al., Effects of Labrasol and other pharmaceutical excipients on the intestinal transport and absorption of rhodamine 123, a P-glycoprotein substrate, in rats, Biol. Pharm. Bull., 2007, 30(7), 1301–1307 CrossRef CAS PubMed.
- K. Sachs-Barrable, A. Thamboo, S. D. Lee and K. M. Wasan, Lipid excipients Peceol and Gelucire 44/14 decrease P-glycoprotein mediated efflux of rhodamine 123 partially due to modifying P-glycoprotein protein expression within Caco-2 cells, J. Pharm. Pharm. Sci., 2007, 10(3), 319–331 CAS.
- V. R. Gupta, S. Mutalik, M. M. Patel and G. K. Jani, Spherical crystals of celecoxib to improve solubility, dissolution rate and micromeritic properties, Acta Pharm., 2007, 57(2), 173–184 CAS.
- A. Tan, S. Simovic, A. K. Davey, T. Rades and C. A. Prestidge, Silica-lipid hybrid (SLH) microcapsules: a novel oral delivery system for poorly soluble drugs, J. Controlled Release, 2009, 134(1), 62–70 CrossRef CAS PubMed.
- Y. Liu, C. Sun, Y. Hao, T. Jiang, L. Zheng and S. Wang, Mechanism of dissolution enhancement and bioavailability of poorly water soluble celecoxib by preparing stable amorphous nanoparticles, J. Pharm. Pharm. Sci., 2010, 13(4), 589–606 CAS.
- R. Panda and K. Kuotsu, Fabrication, characterization, and in vitro evaluation of pegylated glyceride labrasol® nanostructured lipid carrier composites of methotrexate: the pathway to effective cancer therapy, Asian J. Pharm. Clin. Res., 2019, 12(6), 229–237 CrossRef CAS.
- P. M.-K. Tang, D.-M. Zhang, N.-H. B. Xuan, S. K.-W. Tsui, M. M.-Y. Waye and S.-K. Kong, et al., Photodynamic therapy inhibits P-glycoprotein mediated multidrug resistance via JNK activation in human hepatocellular carcinoma using the photosensitizer pheophorbide a, Mol. Cancer, 2009, 8, 1–12 CrossRef PubMed.
- J. Y. Chan, A. C. Chu and K. P. Fung, Inhibition of P-glycoprotein expression and reversal of drug resistance of human hepatoma HepG2 cells by multidrug resistance gene (mdr1) antisense RNA, Life Sci., 2000, 67(17), 2117–2124 CrossRef CAS PubMed.
- J. Zhou, M. Liu, R. Aneja, R. Chandra, H. Lage and H. C. Joshi, Reversal of P-glycoprotein-mediated multidrug resistance in cancer cells by the c-Jun NH2-terminal kinase, Cancer Res., 2006, 66(1), 445–452 CrossRef CAS PubMed.
- C. Verslype, H. van Malenstein, J. Dekervel, P. Windmolders, L. Libbrecht and R. van Eijsden, et al., Resistance development after long-term sorafenib exposure in hepatocellular cancer cell lines and risk of rebound growth and epithelial to mesenchymal transition, J. Clin. Oncol., 2012, 30(4_suppl), 216 Search PubMed.
- J. Li, B. Duan, Y. Guo, R. Zhou, J. Sun and B. Bie, et al., Baicalein sensitizes hepatocellular carcinoma cells to 5-FU and Epirubicin by activating apoptosis and ameliorating P-glycoprotein activity, Biomed. Pharmacother., 2018, 98, 806–812 CrossRef CAS PubMed.
- J. Dong, B. Zhai, W. Sun, F. Hu, H. Cheng and J. Xu, Activation of phosphatidylinositol 3-kinase/AKT/snail signaling pathway contributes to epithelial-mesenchymal transition-induced multi-drug resistance to sorafenib in hepatocellular carcinoma cells, PLoS One, 2017, 12(9), e0185088 CrossRef PubMed.
- M. Huang and G. Liu, The study of innate drug resistance of human hepatocellular carcinoma Bel7402 cell line, Cancer Lett., 1998, 135(1), 97–105 CrossRef PubMed.
- P. W. Shueng, H. W. Chan, W. C. Lin, D. Y. Kuo and H. Y. Chuang, Orlistat Resensitizes Sorafenib-Resistance in Hepatocellular Carcinoma Cells through Modulating Metabolism, Int. J. Mol. Sci., 2022, 23(12), 6501 CrossRef CAS PubMed.
- C.-T. Ting, Y.-Y. Cheng and T.-H. Tsai, Preclinical Pharmacokinetic Interaction and Histopathological Analyses of Hedyotis diffusa on Sorafenib in Rats, ACS Omega, 2021, 6(4), 3060–3067 CrossRef CAS PubMed.
- Guideline IHT, Validation of analytical procedures: text and methodology. Q2 (R1), ICH Harmonised Tripartite, 2005, 1(20), 05 Search PubMed.
- S. Fernandez, V. Jannin, S. Chevrier, Y. Chavant, F. Demarne and F. Carrière, In vitro digestion of the self-emulsifying lipid excipient Labrasol® by gastrointestinal lipases and influence of its colloidal structure on lipolysis rate, Pharm. Res., 2013, 30, 3077–3087 CrossRef CAS PubMed.
- S. Rani, R. Rana, G. K. Saraogi, V. Kumar and U. Gupta, Self-emulsifying oral lipid drug delivery systems: advances and challenges, AAPS PharmSciTech, 2019, 20, 1–12 CrossRef PubMed.
- V. R. Kallakunta, B. B. Eedara, R. Jukanti, R. K. Ajmeera and S. Bandari, A Gelucire 44/14 and labrasol based solid self emulsifying drug delivery system: formulation and evaluation, J. Pharm. Invest., 2013, 43, 185–196 CrossRef CAS.
- A. Karataş, N. Yüksel and T. Baykara, Improved solubility and dissolution rate of piroxicam using gelucire 44/14 and labrasol, Il Farmaco, 2005, 60(9), 777–782 CrossRef PubMed.
- A. A. Noyes and W. R. Whitney, The rate of solution of solid substances in their own solutions, J. Am. Chem. Soc., 1897, 19(12), 930–934 CrossRef.
- P. Á. Spósito, A. L. Mazzeti, K. C. M. P. de Castro, P. F. Mendes, J. A. Urbina and M. T. Bahia, et al., Higher oral efficacy of ravuconazole in self-nanoemulsifying systems in shorter treatment in experimental chagas disease, Exp. Parasitol., 2021, 228, 108142 CrossRef PubMed.
- M. V. Varma, Y. Ashokraj, C. S. Dey and R. Panchagnula, P-glycoprotein inhibitors and their screening: a perspective from bioavailability enhancement, Pharmacol. Res., 2003, 48(4), 347–359 CrossRef CAS PubMed.
- Y. Kono, I. Kawahara, K. Shinozaki, I. Nomura, H. Marutani and A. Yamamoto, et al., Characterization of P-glycoprotein inhibitors for evaluating the effect of P-glycoprotein on the intestinal absorption of drugs, Pharmaceutics, 2021, 13(3), 388 CrossRef CAS PubMed.
- J. Van Asperen, O. Van Tellingen, A. Sparreboom, A. Schinkel, P. Borst and W. Nooijen, et al., Enhanced oral bioavailability of paclitaxel in mice treated with the P-glycoprotein blocker SDZ PSC 833, Br. J. Cancer, 1997, 76(9), 1181–1183 CrossRef CAS PubMed.
- R. P. Keller, H. J. Altermatt, P. Donatsch, H. Zihlmann, J. A. Laissue and P. C. Hiestand, Pharmacologic interactions between the resistance-modifying cyclosporine sdz psc 833 and etoposide (VP 16–213) enhance In Vivo cytostatic activity and toxicity, Int. J. Cancer, 1992, 51(3), 433–438 CrossRef CAS PubMed.
- J.-O. Kwak, S. H. Lee, G. S. Lee, M. S. Kim, Y.-G. Ahn and J. H. Lee, et al., Selective inhibition of MDR1 (ABCB1) by HM30181 increases oral bioavailability and therapeutic efficacy of paclitaxel, Eur. J. Pharmacol., 2010, 627(1–3), 92–98 CrossRef CAS PubMed.
- J. Hendrikx, J. Lagas, E. Wagenaar, H. Rosing, J. Schellens and J. Beijnen, et al., Oral co-administration of elacridar and ritonavir enhances plasma levels of oral paclitaxel and docetaxel without affecting relative brain accumulation, Br. J. Cancer, 2014, 110(11), 2669–2676 CrossRef CAS PubMed.
- M. V. Varma and R. Panchagnula, Enhanced oral paclitaxel absorption with vitamin E-TPGS: effect on solubility and permeability in vitro, in situ and in vivo, Eur. J. Pharm. Sci., 2005, 25(4–5), 445–453 CrossRef CAS PubMed.
- Y. Lin, Q. Shen, H. Katsumi, N. Okada, T. Fujita and X. Jiang, et al., Effects of Labrasol and other pharmaceutical excipients on the intestinal transport and absorption of rhodamine 123, a P-glycoprotein substrate, in rats, Biol. Pharm. Bull., 2007, 30(7), 1301–1307 CrossRef CAS PubMed.
- K. Koga, S. Kawashima and M. Murakami, In vitro and in situ evidence for the contribution of Labrasol® and Gelucire 44/14 on transport of cephalexin and cefoperazone by rat intestine, Eur. J. Pharm. Biopharm., 2002, 54(3), 311–318 CrossRef CAS PubMed.
- S. Eaimtrakarn, Y. Rama Prasad, T. Ohno, T. Konishi, Y. Yoshikawa and N. Shibata, et al., Absorption enhancing effect of labrasol on the intestinal absorption of insulin in rats, J. Drug Targeting, 2002, 10(3), 255–260 CrossRef CAS PubMed.
- Z. Hu, R. Tawa, T. Konishi, N. Shibata and K. Takada, A novel emulsifier, Labrasol, enhances gastrointestinal absorption of gentamicin, Life Sci., 2001, 69(24), 2899–2910 CrossRef CAS PubMed.
- Y. R. Prasad, S. Puthli, S. Eaimtrakarn, M. Ishida, Y. Yoshikawa and N. Shibata, et al., Enhanced intestinal absorption of vancomycin with Labrasol and D-α-tocopheryl PEG 1000 succinate in rats, Int. J. Pharm., 2003, 250(1), 181–190 CrossRef CAS PubMed.
- X. Sha, G. Yan, Y. Wu, J. Li and X. Fang, Effect of self-microemulsifying drug delivery systems containing Labrasol on tight junctions in Caco-2 cells, Eur. J. Pharm. Sci., 2005, 24(5), 477–486 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2025 |