
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceApplication of magnetic materials bearing Brønsted acid sites – based on the modification of amorphous carbon with ionic liquids as catalysts for synthesis of dihydropyrimidinone derivatives via the Biginelli reaction†
Thai-Phien Huynh
Dang‡
ab,
Thach Ngoc
Pham‡
ab,
Phuong Hoang
Tran
 ab and
Hai Truong
Nguyen
*ab
ab and
Hai Truong
Nguyen
*ab
aDepartment of Organic Chemistry, Faculty of Chemistry, University of Science, Ho Chi Minh City, Vietnam. E-mail: ngthai@hcmus.edu.vn; Tel: +84-908-108-824
bVietnam National University, Ho Chi Minh City, Vietnam
First published on 17th April 2025
Abstract
Biomass is a widely available and renewable natural resource derived from plant and animal materials. In addition to its role as an energy source, it is increasingly recognized as a valuable raw material for synthesizing solid catalysts. These catalysts play a crucial role in various organic synthesis reactions, offering a sustainable and eco-friendly alternative to conventional catalyst materials. In this study, by modifying amorphous carbon derived from rice husks with ionic liquid, followed by sulfonation and magnetization with magnetite, we successfully synthesized a solid catalyst Fe3O4@AmC/KSO3H (KS71), which is a magnetic material bearing Brønsted acid sites, for synthesizing dihydropyrimidinone derivatives via the Biginelli reaction. The synthesized catalyst can be easily separated, recovered, and reused without generating or releasing by-products while still providing high efficiency. The dihydropyrimidinone derivatives synthesized via the Biginelli reaction showed remarkable yields with our synthesized catalyst. With a short synthesis time, good yield, and especially being environmentally friendly, the catalyst that we offer is a great substance for reactions taking place during chemical synthesis. The results from analytical instruments such as FT-IR, EDX, ICP-MS, TGA, SEM, VSM, and 1H-NMR, 13C-NMR spectra have shown that we have successfully synthesized the solid catalyst and dihydropyrimidinone derivatives with good yields, along with ensuring safety according to green chemistry criteria.
1. Introduction
At present, common inorganic acids such as HCl and H2SO4 have been widely used as catalysts for many processes to produce renewable fuels (e.g., biodiesel) and fine chemicals (e.g., furfural and sugars).1 Furthermore, approximately 60% of the world's sulfuric acid production is utilized in fertilizer production and wastewater treatment. Owing to its low production cost, sulfuric acid has become increasingly prevalent and indispensable in numerous industries.2 However, several drawbacks, including strong corrosiveness, the costly and inefficient separation from homogeneous reaction mixtures, and the generation of significant amounts of waste, have limited the application of liquid acids in many fields.2 To replace such non-reusable liquid acid catalysts, the green catalysis approach has promoted the utilization of reusable solid strong acids.3 In recent years, acid-functionalized carbon materials have garnered significant attention in the field of chemistry. Notably, these materials can be readily obtained or synthesized from biomass sources. Given their abundance, affordability, and the inherent stability of acidic functional groups (such as carboxylic (–COOH) and phenolic (–OH) groups), these materials have garnered considerable interest.4The choice of amorphous carbon as the substrate material is primarily driven by its ease of preparation via carbonization of readily available natural precursors. Studies on rice husks have revealed a relatively stable carbon matrix with a porous structure, making it amenable to modification with ionic liquids. Unlike the time-consuming and chemically intensive process of obtaining graphene oxide (GO),4 amorphous carbon can be easily obtained through carbonization at low concentrations and short reaction times, resulting in a safer process. Carbon materials bearing –SO3H groups, also known as “sulfonated carbon”, represent an advanced class of solid Brønsted acids. These materials have been recognized as potent solid acid catalysts that can replace traditional liquid catalysts like H2SO4 in various fields, including catalysis, electrochemistry, water treatment, CO2 capture, and energy storage. Their chemical stability, high mechanical and thermal properties, tunable porous structure, and surface chemistry contribute to their versatility.2 Sulfonated carbons can be prepared by reacting carbon materials with concentrated H2SO4, sulfuric acid vapor, gaseous SO3, chlorosulfonic acid, p-toluenesulfonic acid, or certain aryldiazonium compounds containing –SO3H groups.2 Alternatively, sulfonated carbons can be synthesized by carbonizing or partially carbonizing macromolecules containing –SO3H acid groups through phenol–formaldehyde condensation reactions. Through C–C or C–S bonds, we can directly manipulate the structure, surface chemistry, and stability of the amorphous carbon substrate.
On the other hand, the issues of separation and recovery of solid catalysts are problems that need to be addressed. Compared to liquid acid catalysts, separation always poses difficulties. Therefore, in recent years, magnetic separation methods have been developed to replace conventional methods. The separation of magnetic solid acid catalysts has been proven to be quick, easy, convenient and efficient when using an external magnet. This new type of technique allows for the implementation of a process in an economical and technical manner with minimal loss of solid catalyst.5 Magnetic solid acid catalysts are solid acid catalysts that include Fe3O4 particles in their core or on their surface. In contrast to traditional solid acids, magnetic solid acids can be more readily isolated when subjected to an external magnetic field, particularly in viscous or solid reaction mixtures.6
The synthesized material KS71 has been characterized using FT-IR, ICP-MS, EDX, TGA, VSM, and SEM, confirming the successful synthesis of a layered material with porous structures and the incorporation of desired functional groups and metals. The high efficiency, short reaction time, availability of natural raw materials for KS71 synthesis, low cost, and ease of recovery and separation make KS71 an excellent candidate for green chemistry applications.
In recent years, chemists have concentrated on creating novel methods that minimize risks to human health and the environment, resulting in alterations to various synthetic processes. Multi-component reactions have gained prominence over multi-step reactions owing to their economic efficiency, energy and time conservation, and convergence in organic synthesis.7–11 The synthesis of dihydropyrimidinones is a prime example.
In the present investigation, we utilized our catalyst to perform the Biginelli reaction, which is a three-component reaction that involves aldehyde, β-ketoester, and either urea or thiourea. This reaction is a one-step reaction that consists of the synthesis of dihydropyrimidinones (DHPMs). Dihydropyrimidinones were first introduced by the Italian chemist Pietro Biginelli et al. in 1893.12 DHPM derivatives have also been synthesized through various methods, such as the four-component condensation of aliphatic or aromatic amines, diketones, aromatic aldehydes, and urea or thiourea catalyzed by p-toluenesulfonic acid in dichloromethane at room temperature, which was introduced by Shaabni et al. in 2010.13
DHPM derivatives exhibit diverse therapeutic applications in pharmaceuticals through various substitution patterns. It is hypothesized that the presence of the nitrogenous base of DHPM is one of the driving forces behind their activity, which is found in DNA and RNA, including nucleobases like thymine (T), uracil (U), and cytosine (C). DHPM and its derivatives serve as building blocks for nucleic acids, vitamins, amino acids, and others, contributing to proper growth and physiological function (Fig. 1 and 2).14
 | ||
| Fig. 1 Derivatives of dihydropyrimidinone.14 | ||
 | ||
| Fig. 2 FT–IR spectra of BMSA (black), KH–Si (red) and KS71 (blue). | ||
Additionally, DHPMs are known for their remarkable biological properties, including anticancer,15,16 antifungal,17 anti-inflammatory,18 antibacterial,19 antiviral,20 antidiabetic,21 antithyroid,22 antimuscarinic,23 hypolipidemic activities,20 contraceptive agents, calcium channel blockers, α-adrenergic antagonists, and hypertension medications, fatty acid transporter inhibitors, and mitotic kinesin inhibition. Biological studies have revealed that substituting different groups on the ring can lead to varying activities.15,24 Research conducted in the field of biology has demonstrated that the substitution of various groups on the ring leads to the occurrence of distinct activities. Numerous medications, including 5-fluorouracil, idoxuridine, methythiouracil, emivirin, and aminophylline, which include 3,4-dihydropyrimidine-2(1H)-one, have been found and have proven to be beneficial in the treatment or prevention of a wide range of disorders. Antibacterial, antihypertensive, anti-HIV, and anticancer properties have been investigated for DHPM and their derivatives, which are versatile compounds that have been investigated for the treatment of a variety of disorders.25–28 On the other hand, acid-catalyzed reactions are associated with a number of significant downsides, including poorer yields that range from 26–60% as a result.12,29 Moreover, environmental concerns arise from the use of strong acid catalysts. Consequently, numerous new catalysts have been synthesized to replace conventional acid catalysts, aiming for higher yields, shorter reaction times, and environmental friendliness (Schemes 1–3).
 | ||
| Scheme 1 Condensation reaction.12–14 | ||
 | ||
| Scheme 2 Synthesis process of KS71 catalyst. | ||
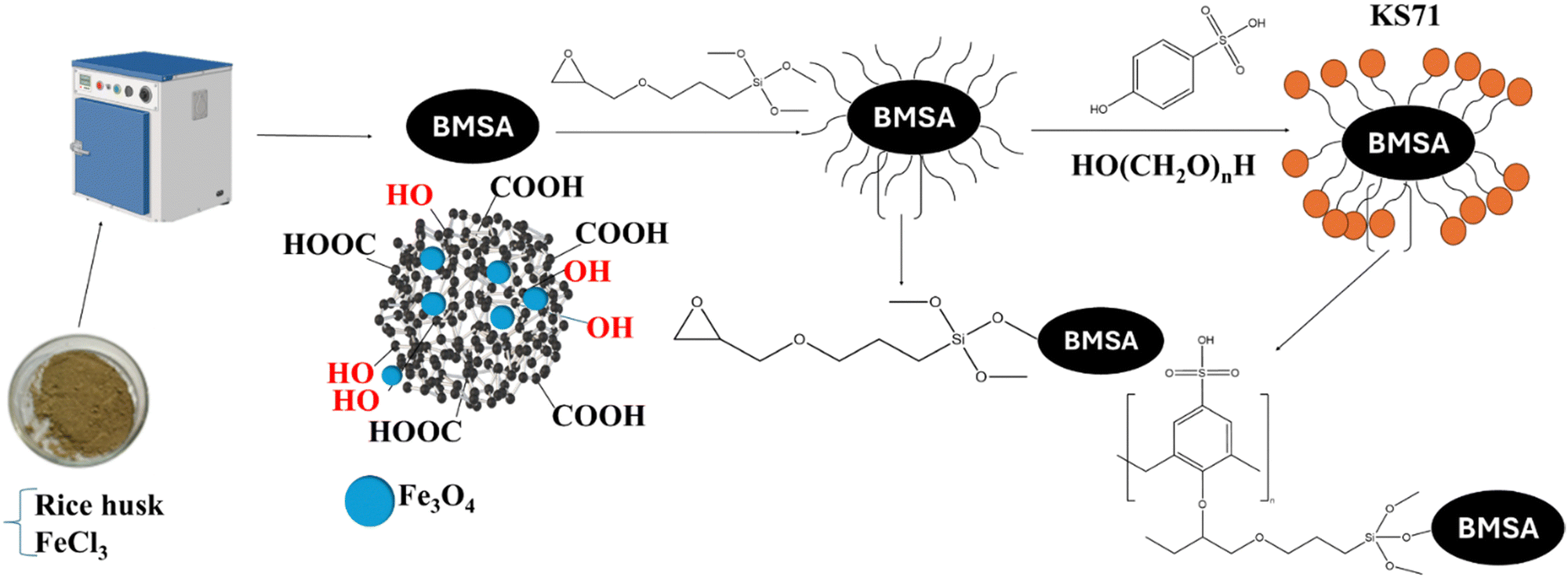 | ||
| Scheme 3 Structural changes in the synthesis process of catalysts KS71. | ||
The reaction was initially catalyzed by HCl in ethanol and subsequently explored using various homogeneous and heterogeneous catalysts. Currently, heterogeneous catalysts are of particular interest and have been synthesized for efficient separation and recovery, including polystyrene sulfonic acid,30 polystyrene-poly(ethylene glycol) resin supported sulfonic acid,31 mesoporous silica MCM-41,32 silica sulfuric acid,33 sulfonated carbon material, and silica gel supported L-pyrrolidine-2-carboxylic acid-4-hydrogen sulfate.34 Other heterogeneous catalysts bearing Brønsted or Lewis acid sites, such as Mag@Morph-AIL,15 [DABCO](SO3H)2CuCl4,7 and iron(III)trifluoroacetate [Fe(CF3CO2)3],35 has been effectively synthesized and applied to the Biginelli reaction, affording benefits such as quick reaction durations, high yields, and facile separation and recovery of the heterogeneous catalyst. These advantages have been achieved by the successful application of these compounds, thus minimizing environmental impact and meeting the criteria of green chemistry. However, the origins of these catalysts primarily involve metals, polymers, and organic frameworks. Therefore, the magnetic solid acid catalyst KS71 is a promising choice for the Biginelli reaction. It offers good yields (reached 52%), excellent separability and recoverability, no harmful byproducts, and is derived from natural materials, fully satisfying the criteria of green chemistry.
2. Experimental work
2.1. Materials
Rice husk (Long An Province, Viet Nam), 3-glycidyloxypropyltrimethoxysilane (98%), acid p-phenolsulfonic (85%), paraformaldehyde (95%), benzaldehyde (99.0%), 2-chlorobenzaldehyde (99.0%), 4-chlorobenzaldehyde (97%), 4-bromobenzaldehyde (99.0%), 4-fluorobenzaldehyde (98.5%), 4-methylbenzaldehyde (97%), 4-methoxybenzaldeyde (98.5%), 4-hydroxybenzaldehyde (98%), 4-hydroxy-3-methoxybenzaldehyde (99%), 4-(dimethylamino)benzaldehyde (98%), cinnamaldehyde (99%), cyclohexanecarbaldehyde (97%), ethyl acetoacetate (99%), urea (99%) were obtained from Sigma-Aldrich, ethanol (99.5%), cyclohexane (99.7%), ethyl acetate (99.5%), n-hexane (99.5%) were obtained from Chemsol, iron(III)chloride were obtained from China, Xilong 99%, thiourea (99.0%), TLC (silica gel 60 F254) were obtained from Merck.2.2. Techniques for analysis
After successful synthesis of the catalyst and DHPM along with its derivatives, we conducted Fourier-transform infrared spectroscopy (FT-IR) analysis on a JASCO FT/IR-6600 spectrometer in the range of 600–4000 cm−1; scanning electron microscopy (SEM) on a JSM-IT200, JEOL, Japan; elemental analysis (EDX) equipped with SEM on a JSM-IT200, JEOL, Japan; thermogravimetric analysis (TGA) using a TA Q500 system; vibrating sample magnetometer (VSM) on a PPMS600 system from Quantum Design, and inductively coupled plasma-mass spectrometry (ICP-MS). The 1H and 13C NMR spectra were recorded on Bruker Advance 500 instruments using DMSO-d6 as solvent and solvent peaks or TMS as internal standards.2.3. Synthesis of Fe3O4@AmC/K-SO3H (KS71)
A mixture of 0.5 L of 0.01 M FeCl3 and 5 g rice husk was added to a 1.0 L glass beaker. The amalgamation was agitated at 400 rpm at ambient temperature for 5 hours. Thereafter, it was heated to 100 °C until complete evaporation of the water occurred. The composite was desiccated at 100 °C for 12 hours, resulting in Fe-impregnated biomass.The Fe-loaded biomass was then carbonized at 400 °C in a furnace under a nitrogen atmosphere for 4 hours to form magnetic amorphous carbon (BMSA). A 50 mL round-bottom flask containing cyclohexane was filled with 1.2 g of BMSA and then 0.1 g of 3-glycidyloxypropyltrimethoxysilane (KH560). At 25 °C, the mixture was agitated for four hours at 400 rpm. After that, 16 mmol of paraformaldehyde and 8 mmol of p-phenolsulfonic acid were added, and the mixture refluxed in ethanol for 2 hours. Following the completion of the reaction, ethanol (10 mL × 3) was used to wash the result once the liquid had cooled to room temperature. In order to obtain KS71, the material was finally dried for 12 hours at 60 °C.
Determination of acidity of materials according to research by Thien-Hang T.N.:36
Determine the total acid concentration (–OH, –COOH, –SO3H) by adding NaOH (1 N) to the 3 stages of BMSA, HK–Si, KS71 catalysts and stirring at room temperature for 24 hours. The mixture after stirring was centrifuged and the clear aqueous solution was titrated with 0.1 N HCl with phenolphthalein as an indicator.
Determination of –SO3H functional groups: the content of the –SO3H group was determined by adding NaCl (1 N) was added to the KS71 catalyst and stirred at room temperature for 24 hours. The mixture after stirring was centrifuged to separate the aqueous solution, then titrated with NaOH (0.01 N) with phenolphthalein as an indicator.
2.4. Synthesis of DHPM and its derivatives
A 5 mL Biohall screw-cap tube was filled with a mixture that contained 10 mg of KS71, 1 mmol of aldehyde, 1 mmol of ethyl acetoacetate (EAA), and 1 mmol of urea/thiourea. In a solvent-free setting, the reaction was conducted for 2.5 hours at 90 °C and 300 rpm of stirring speed. Following the completion of the reaction, the catalyst was filtered and cleaned with EtOH (5 mL × 3) to get rid of any remaining contaminants. The reaction mixture was then allowed to settle to room temperature. The catalyst was then recovered by filtration after the reaction mixture was dissolved with ethyl acetate. By recrystallizing the crude product from ethyl acetate (10–15 mL), it was refined. The reaction was monitored by thin-layer chromatography (TLC) using a solvent system of n-hexane![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ethyl acetate (v:v = 5:5). The product was a white solid and its structure and purity were confirmed by 1H-NMR, 13C-NMR, and melting point.
:ethyl acetate = 5:5); m.p. = 206–209 °C [Lit. = 205–209 °C].15,371H-NMR (500 MHz, DMSO-d6): δ = 1.09 (t, J = 5.0 Hz, 3H), 2.24 (s, 3H), 3.98 (q, J = 5.0 Hz, 2H), 5.14 (d, J = 5.0 Hz, 1H), 7.25–7.22 (m, 3H), 7.33–7.30 (m, 2H), 7.72 (s, 1H), 9.18 (s, 1H) ppm. 13C-NMR (125 MHz, DMSO-d6): δ = 14.5, 18.2, 54.4, 59.7, 99.7, 126.7, 127.7, 128.9, 145.3, 148.8, 152.6, 165.8 ppm.
:ethyl acetate = 5:5); m.p. = 223–224 °C [Lit. = 217–218 °C].381H-NMR (500 MHz, DMSO-d6): δ = 1.09 (t, J = 10.0 Hz, 3H), 2.25 (s, 3H), 3.98 (q, J = 5.0 Hz, 2H), 5.14 (d, J = 5.0 Hz, 1H), 7.25 (d, J = 8.5 Hz, 2H), 7.39 (d, J = 8.5 Hz, 2H), 7.76 (s, 1H), 9.24 (s, 1H) ppm. 13C-NMR (125 MHz, DMSO-d6): δ = 14.6, 18.3, 53.9, 59.7, 99.3, 128.7, 128.8, 132.3, 144.3, 149.2, 152.4, 165.7 ppm.
:ethyl acetate = 5:5); m.p. = 228–230 °C [Lit. = 228–230 °C].71H-NMR (500 MHz, DMSO-d6): δ = 1.10 (t, J = 5.0 Hz, 3H), 2.25 (s, 3H), 3.98 (q, J = 5.0 Hz, 2H), 5.12 (d, J = 5.0 Hz, 1H), 7.19 (d, J = 8.5 Hz, 2H), 7.53 (d, J = 8.5 Hz, 2H), 7.78 (s, 1H), 9.26 (s, 1H) ppm. 13C-NMR (125 MHz, DMSO-d6): δ = 14.6, 18.3, 53.9, 59.7, 99.2, 120.8, 129.0, 131.8, 144.7, 149.2, 152.4, 165.7 ppm.
:ethyl acetate = 5:5); m.p. = 188–191 °C [Lit. = not found].371H-NMR (500 MHz, DMSO-d6): δ = 1.09 (t, J = 5.0 Hz, 3H), 2.25 (s, 3H), 4.00–3.96 (m, 2H), 5.15 (d, J = 5.0 Hz, 1H), 7.15 (t, J = 9.0 Hz, 2H), 7.26 (m, 2H), 7.74 (s, 1H), 9.22 (s, 1H) ppm. 13C-NMR (125 MHz, DMSO-d6): δ =14.5, 18.3, 53.8, 59.7, 99.6, 115.6 (J = 21.3 Hz), 128.7 (J = 8.8 Hz), 141.6 (J = 3.8 Hz), 141.6, 149.0, 152.4, 161.8 (J = 241.3 Hz), 165.7 ppm.
:ethyl acetate = 5:5); m.p. = 216–218 °C [Lit. = 216–217 °C].381H-NMR (500 MHz, DMSO-d6): δ = 1.10 (t, J = 5.0 Hz, 3H), 2.24 (s, 3H), 2.26 (s, 3H), 3.98 (q, J = 10.0 Hz, 2H), 5.10 (d, J = 5.0 Hz, 1H), 7.12 (s, 4H), 7.68 (s, 1H), 9.15 (s, 1H) ppm. 13C-NMR (125 MHz, DMSO-d6): δ =14.6, 18.2, 21.1, 54.1, 59.6, 99.9, 126.6, 129.4, 136.8, 142.4, 148.6, 152.6, 165.8 ppm.
:ethyl acetate = 5:5); m.p. = 204–206 °C [Lit. = 200–202 °C].71H-NMR (500 MHz, DMSO-d6): δ = 1.10 (t, J = 10.0 Hz, 3H), 2.24 (s, 3H), 3.72 (s, 3H), 3.98 (q, J = 10.0 Hz, 2H), 5.09 (d, J = 5.0 Hz, 1H), 6.87 (d, J = 8.5 Hz, 2H), 7.14 (d, J = 8.5 Hz, 2H), 7.66 (s, 1H), 9.15 (s, 1H) ppm. 13C-NMR (125 MHz, DMSO-d6): δ = 14.6, 18.2, 53.8, 55.5, 59.6, 100.0, 114.2, 127.9, 137.5, 148.5, 152.6, 158.9, 165.9 ppm.
:ethyl acetate = 5:5); m.p. = 223–224 °C [Lit. = 220–222 °C].71H-NMR (500 MHz, DMSO-d6): δ = 0.99 (t, J = 10.0 Hz, 3H), 2.30 (s, 3H), 3.89 (q, J = 5.0 Hz, 2H), 5.63 (d, J = 5.0 Hz, 1H), 7.28–7.25 (m, 1H), 7.34–7.29 (m, 2H), 7.40 (d, J = 7.5 Hz, 1H), 7.69 (s, 1H), 9.26 (s, 1H) ppm. 13C-NMR (125 MHz, DMSO-d6): δ = 14.4, 18.1, 52.0, 59.5, 98.4, 128.2, 129.3, 129.5, 129.8, 132.2, 142.2, 149.8, 151.8, 165.4 ppm.
:ethyl acetate = 5:5); m.p. = 226–228 °C [Lit. = 258–261 °C].151H-NMR (500 MHz, DMSO-d6): δ = 1.11 (t, J = 10.0 Hz, 3H), 2.23 (s, 3H), 2.85 (s, 6H), 4.01–3.94 (m, 2H), 5.03 (d, J = 5.0 Hz, 1H), 6.65 (d, J = 9.0 Hz, 2H), 7.03 (d, J = 9.0 Hz, 2H), 7.60 (s, 1H), 9.10 (s, 1H) ppm. 13C-NMR (125 MHz, DMSO-d6): δ = 14.6, 18.2, 40.7, 53.8, 59.6, 100.3, 112.7, 127.4, 133.1, 148.0, 150.2, 152.8, 166.0 ppm.
:ethyl acetate = 5:5); m.p. = 225–226 °C [Lit. = 225–226 °C]381H-NMR (500 MHz, DMSO-d6): δ = 1.10 (t, J = 5.0 Hz, 3H), 2.23 (s, 3H), 3.97 (q, J = 5.0 Hz, 2H), 5.04 (d, J = 5.0 Hz, 1H), 6.68 (d, J = 8.5 Hz, 2H), 7.02 (d, J = 8.5 Hz, 2H), 7.62 (s, 1H), 9.12 (s, 1H), 9.34 (s, 1H) ppm. 13C-NMR (125 MHz, DMSO-d6): δ = 14.6, 18.2, 53.9, 59.6, 100.2, 115.4, 127.9, 135.9, 148.2, 152.6, 157.0, 165.9 ppm.
:ethyl acetate = 5:5); m.p. = 215–217 °C [Lit. = 236–237 °C].391H-NMR (500 MHz, DMSO-d6): δ = 1.11 (t, J = 10.0 Hz, 3H), 2.23 (s, 3H), 3.72 (s, 3H), 3.99 (q, J = 5.0 Hz, 2H), 5.06 (d, J = 5.0 Hz, 1H), 6.61 (dd, J = 8.0 Hz, 1.5 Hz, 2H), 6.70 (d, J = 8.0 Hz, 1H), 6.80 (d, J = 1.5 Hz, 1H), 7.64 (s, 1H), 8.92 (s, 1H), 9.13 (s, 1H) ppm. 13C-NMR (125 MHz, DMSO-d6): δ = 14.6, 18.2, 54.0, 56.0, 59.6, 100.0, 111.3, 115.7, 118.7, 136.4, 146.2, 147.7, 148.4, 152.7, 165.9 ppm.
:ethyl acetate = 5:5); m.p. = 254–256 °C [Lit. = 228–230 °C].401H-NMR (500 MHz, DMSO-d6): δ = 1.20 (t, J = 10.0 Hz, 3H), 2.20 (s, 3H), 4.15–4.03 (m, 2H), 4.73 (t, J = 3.5 Hz, 1H), 6.20 (dd, J = 16.0 Hz, 6.5 Hz, 1H), 6.36 (d, J = 16.0 Hz, 1H), 7.24 (t, J = 7.0 Hz, 1H), 7.31 (t, J = 7.5 Hz, 2H), 7.40 (d, J = 7.5 Hz, 2H), 7.57 (s, 1H), 9.17 (s, 1H) ppm. 13C-NMR (125 MHz, DMSO-d6): δ = 14.7, 18.2, 52.4, 59.7, 98.2, 126.8, 128.1, 128.6, 129.2, 130.4, 136.7, 149.0, 153.1, 165.7 ppm.
:ethyl acetate = 5:5); m.p. = 209–210 °C [Lit. = 233–234 °C].381H-NMR (500 MHz, DMSO-d6): δ = 0.89–0.81 (m, 1H), 1.13–1.01 (m, 4H), 1.18 (t, J = 5.0 Hz, 3H), 1.39–1.28 (m, 2H), 1.69–1.58 (m, 4H), 2.16 (s, 3H), 3.92 (t, J = 5.0 Hz, 1H), 4.11–4.00 (m, 2H), 7.27 (s, 1H), 8.87 (s, 1H) ppm. 13C-NMR (125 MHz, DMSO-d6): δ = 14.7, 18.2, 26.1, 26.3, 26.5, 26.7, 29.0, 45.3, 55.4, 59.5, 98.5, 148.9, 153.7, 160.1, 166.3 ppm.
:ethyl acetate = 5:5); m.p. = 209–211 °C [Lit. = 209–210 °C].381H-NMR (500 MHz, DMSO-d6): δ = 1.10 (t, J = 10.0 Hz, 3H), 2.29 (s, 3H), 4.01 (q, J = 5.0 Hz, 2H), 5.18 (d, J = 5.0 Hz, 1H), 7.22 (d, J = 7.5 Hz, 2H), 7.27 (t, J =7.5 Hz, 1H), 7.35 (t, J = 7.5 Hz, 2H), 9.65 (s, 1H), 10.34 (s, 1H) ppm. 13C-NMR (125 MHz, DMSO-d6): δ = 14.5, 17.6, 54.5, 60.1, 101.2, 126.8, 128.2, 129.0, 143.9, 145.5, 165.6, 174.7 ppm.
:ethyl acetate = 5:5); m.p. = 220–221 °C [Lit. = 192–193 °C].381H-NMR (500 MHz, DMSO-d6): δ = 1.10 (t, J = 10.0 Hz, 3H), 2.26 (s, 3H), 3.99 (q, J = 6.5 Hz, 2H), 5.15 (d, J = 5.0 Hz, 1H), 7.25 (d, J = 8.5 Hz, 2H), 7.40 (d, J = 8.5 Hz, 2H), 7.77 (s, 1H), 9.25 (s, 1H) ppm. 13C-NMR (125 MHz, DMSO-d6): δ = 14.5, 18.3, 53.9, 59.7, 99.3, 128.7, 128.9, 132.3, 144.3, 149.2, 152.4, 165.7 ppm.
:ethyl acetate (v:v = 5:5). The product was a white solid and its structure and purity were confirmed by 1H-NMR, 13C-NMR, and melting point.
:ethyl acetate = 5:5); m.p. = 206–209 °C [Lit. = 205–209 °C].15,371H-NMR (500 MHz, DMSO-d6): δ = 1.09 (t, J = 5.0 Hz, 3H), 2.24 (s, 3H), 3.98 (q, J = 5.0 Hz, 2H), 5.14 (d, J = 5.0 Hz, 1H), 7.25–7.22 (m, 3H), 7.33–7.30 (m, 2H), 7.72 (s, 1H), 9.18 (s, 1H) ppm. 13C-NMR (125 MHz, DMSO-d6): δ = 14.5, 18.2, 54.4, 59.7, 99.7, 126.7, 127.7, 128.9, 145.3, 148.8, 152.6, 165.8 ppm.
:ethyl acetate = 5:5); m.p. = 223–224 °C [Lit. = 217–218 °C].381H-NMR (500 MHz, DMSO-d6): δ = 1.09 (t, J = 10.0 Hz, 3H), 2.25 (s, 3H), 3.98 (q, J = 5.0 Hz, 2H), 5.14 (d, J = 5.0 Hz, 1H), 7.25 (d, J = 8.5 Hz, 2H), 7.39 (d, J = 8.5 Hz, 2H), 7.76 (s, 1H), 9.24 (s, 1H) ppm. 13C-NMR (125 MHz, DMSO-d6): δ = 14.6, 18.3, 53.9, 59.7, 99.3, 128.7, 128.8, 132.3, 144.3, 149.2, 152.4, 165.7 ppm.
:ethyl acetate = 5:5); m.p. = 228–230 °C [Lit. = 228–230 °C].71H-NMR (500 MHz, DMSO-d6): δ = 1.10 (t, J = 5.0 Hz, 3H), 2.25 (s, 3H), 3.98 (q, J = 5.0 Hz, 2H), 5.12 (d, J = 5.0 Hz, 1H), 7.19 (d, J = 8.5 Hz, 2H), 7.53 (d, J = 8.5 Hz, 2H), 7.78 (s, 1H), 9.26 (s, 1H) ppm. 13C-NMR (125 MHz, DMSO-d6): δ = 14.6, 18.3, 53.9, 59.7, 99.2, 120.8, 129.0, 131.8, 144.7, 149.2, 152.4, 165.7 ppm.
:ethyl acetate = 5:5); m.p. = 188–191 °C [Lit. = not found].371H-NMR (500 MHz, DMSO-d6): δ = 1.09 (t, J = 5.0 Hz, 3H), 2.25 (s, 3H), 4.00–3.96 (m, 2H), 5.15 (d, J = 5.0 Hz, 1H), 7.15 (t, J = 9.0 Hz, 2H), 7.26 (m, 2H), 7.74 (s, 1H), 9.22 (s, 1H) ppm. 13C-NMR (125 MHz, DMSO-d6): δ =14.5, 18.3, 53.8, 59.7, 99.6, 115.6 (J = 21.3 Hz), 128.7 (J = 8.8 Hz), 141.6 (J = 3.8 Hz), 141.6, 149.0, 152.4, 161.8 (J = 241.3 Hz), 165.7 ppm.
:ethyl acetate = 5:5); m.p. = 216–218 °C [Lit. = 216–217 °C].381H-NMR (500 MHz, DMSO-d6): δ = 1.10 (t, J = 5.0 Hz, 3H), 2.24 (s, 3H), 2.26 (s, 3H), 3.98 (q, J = 10.0 Hz, 2H), 5.10 (d, J = 5.0 Hz, 1H), 7.12 (s, 4H), 7.68 (s, 1H), 9.15 (s, 1H) ppm. 13C-NMR (125 MHz, DMSO-d6): δ =14.6, 18.2, 21.1, 54.1, 59.6, 99.9, 126.6, 129.4, 136.8, 142.4, 148.6, 152.6, 165.8 ppm.
:ethyl acetate = 5:5); m.p. = 204–206 °C [Lit. = 200–202 °C].71H-NMR (500 MHz, DMSO-d6): δ = 1.10 (t, J = 10.0 Hz, 3H), 2.24 (s, 3H), 3.72 (s, 3H), 3.98 (q, J = 10.0 Hz, 2H), 5.09 (d, J = 5.0 Hz, 1H), 6.87 (d, J = 8.5 Hz, 2H), 7.14 (d, J = 8.5 Hz, 2H), 7.66 (s, 1H), 9.15 (s, 1H) ppm. 13C-NMR (125 MHz, DMSO-d6): δ = 14.6, 18.2, 53.8, 55.5, 59.6, 100.0, 114.2, 127.9, 137.5, 148.5, 152.6, 158.9, 165.9 ppm.
:ethyl acetate = 5:5); m.p. = 223–224 °C [Lit. = 220–222 °C].71H-NMR (500 MHz, DMSO-d6): δ = 0.99 (t, J = 10.0 Hz, 3H), 2.30 (s, 3H), 3.89 (q, J = 5.0 Hz, 2H), 5.63 (d, J = 5.0 Hz, 1H), 7.28–7.25 (m, 1H), 7.34–7.29 (m, 2H), 7.40 (d, J = 7.5 Hz, 1H), 7.69 (s, 1H), 9.26 (s, 1H) ppm. 13C-NMR (125 MHz, DMSO-d6): δ = 14.4, 18.1, 52.0, 59.5, 98.4, 128.2, 129.3, 129.5, 129.8, 132.2, 142.2, 149.8, 151.8, 165.4 ppm.
:ethyl acetate = 5:5); m.p. = 226–228 °C [Lit. = 258–261 °C].151H-NMR (500 MHz, DMSO-d6): δ = 1.11 (t, J = 10.0 Hz, 3H), 2.23 (s, 3H), 2.85 (s, 6H), 4.01–3.94 (m, 2H), 5.03 (d, J = 5.0 Hz, 1H), 6.65 (d, J = 9.0 Hz, 2H), 7.03 (d, J = 9.0 Hz, 2H), 7.60 (s, 1H), 9.10 (s, 1H) ppm. 13C-NMR (125 MHz, DMSO-d6): δ = 14.6, 18.2, 40.7, 53.8, 59.6, 100.3, 112.7, 127.4, 133.1, 148.0, 150.2, 152.8, 166.0 ppm.
:ethyl acetate = 5:5); m.p. = 225–226 °C [Lit. = 225–226 °C]381H-NMR (500 MHz, DMSO-d6): δ = 1.10 (t, J = 5.0 Hz, 3H), 2.23 (s, 3H), 3.97 (q, J = 5.0 Hz, 2H), 5.04 (d, J = 5.0 Hz, 1H), 6.68 (d, J = 8.5 Hz, 2H), 7.02 (d, J = 8.5 Hz, 2H), 7.62 (s, 1H), 9.12 (s, 1H), 9.34 (s, 1H) ppm. 13C-NMR (125 MHz, DMSO-d6): δ = 14.6, 18.2, 53.9, 59.6, 100.2, 115.4, 127.9, 135.9, 148.2, 152.6, 157.0, 165.9 ppm.
:ethyl acetate = 5:5); m.p. = 215–217 °C [Lit. = 236–237 °C].391H-NMR (500 MHz, DMSO-d6): δ = 1.11 (t, J = 10.0 Hz, 3H), 2.23 (s, 3H), 3.72 (s, 3H), 3.99 (q, J = 5.0 Hz, 2H), 5.06 (d, J = 5.0 Hz, 1H), 6.61 (dd, J = 8.0 Hz, 1.5 Hz, 2H), 6.70 (d, J = 8.0 Hz, 1H), 6.80 (d, J = 1.5 Hz, 1H), 7.64 (s, 1H), 8.92 (s, 1H), 9.13 (s, 1H) ppm. 13C-NMR (125 MHz, DMSO-d6): δ = 14.6, 18.2, 54.0, 56.0, 59.6, 100.0, 111.3, 115.7, 118.7, 136.4, 146.2, 147.7, 148.4, 152.7, 165.9 ppm.
:ethyl acetate = 5:5); m.p. = 254–256 °C [Lit. = 228–230 °C].401H-NMR (500 MHz, DMSO-d6): δ = 1.20 (t, J = 10.0 Hz, 3H), 2.20 (s, 3H), 4.15–4.03 (m, 2H), 4.73 (t, J = 3.5 Hz, 1H), 6.20 (dd, J = 16.0 Hz, 6.5 Hz, 1H), 6.36 (d, J = 16.0 Hz, 1H), 7.24 (t, J = 7.0 Hz, 1H), 7.31 (t, J = 7.5 Hz, 2H), 7.40 (d, J = 7.5 Hz, 2H), 7.57 (s, 1H), 9.17 (s, 1H) ppm. 13C-NMR (125 MHz, DMSO-d6): δ = 14.7, 18.2, 52.4, 59.7, 98.2, 126.8, 128.1, 128.6, 129.2, 130.4, 136.7, 149.0, 153.1, 165.7 ppm.
:ethyl acetate = 5:5); m.p. = 209–210 °C [Lit. = 233–234 °C].381H-NMR (500 MHz, DMSO-d6): δ = 0.89–0.81 (m, 1H), 1.13–1.01 (m, 4H), 1.18 (t, J = 5.0 Hz, 3H), 1.39–1.28 (m, 2H), 1.69–1.58 (m, 4H), 2.16 (s, 3H), 3.92 (t, J = 5.0 Hz, 1H), 4.11–4.00 (m, 2H), 7.27 (s, 1H), 8.87 (s, 1H) ppm. 13C-NMR (125 MHz, DMSO-d6): δ = 14.7, 18.2, 26.1, 26.3, 26.5, 26.7, 29.0, 45.3, 55.4, 59.5, 98.5, 148.9, 153.7, 160.1, 166.3 ppm.
:ethyl acetate = 5:5); m.p. = 209–211 °C [Lit. = 209–210 °C].381H-NMR (500 MHz, DMSO-d6): δ = 1.10 (t, J = 10.0 Hz, 3H), 2.29 (s, 3H), 4.01 (q, J = 5.0 Hz, 2H), 5.18 (d, J = 5.0 Hz, 1H), 7.22 (d, J = 7.5 Hz, 2H), 7.27 (t, J =7.5 Hz, 1H), 7.35 (t, J = 7.5 Hz, 2H), 9.65 (s, 1H), 10.34 (s, 1H) ppm. 13C-NMR (125 MHz, DMSO-d6): δ = 14.5, 17.6, 54.5, 60.1, 101.2, 126.8, 128.2, 129.0, 143.9, 145.5, 165.6, 174.7 ppm.
:ethyl acetate = 5:5); m.p. = 220–221 °C [Lit. = 192–193 °C].381H-NMR (500 MHz, DMSO-d6): δ = 1.10 (t, J = 10.0 Hz, 3H), 2.26 (s, 3H), 3.99 (q, J = 6.5 Hz, 2H), 5.15 (d, J = 5.0 Hz, 1H), 7.25 (d, J = 8.5 Hz, 2H), 7.40 (d, J = 8.5 Hz, 2H), 7.77 (s, 1H), 9.25 (s, 1H) ppm. 13C-NMR (125 MHz, DMSO-d6): δ = 14.5, 18.3, 53.9, 59.7, 99.3, 128.7, 128.9, 132.3, 144.3, 149.2, 152.4, 165.7 ppm.
2.5. Synthesis of Fe3O4@SiO2@AmC/KSO3H
The synthesis procedure was similar to KS71 and started from BMSA. Prepare a mixture of 25 mL EtOH, 1 mL H2O, 1 mL NH3 (aq.), 150 μL TEOS in a 100 mL flask, heat at 40 °C in a water bath and stir for 5 min. Then 0.3 g of BMSA was added to the above mixture and stirred at 40 °C for 12 h. After stirring, the mixture was washed with distilled water and dried overnight at 50 °C to obtain 0.27 g of BMSA-SiO2. The obtained 0.27 g of BMSA-SiO2 was added to 20 mL cyclohexane and 0.044 g of 3-glycidyloxypropyltrimethoxysilane (KH560). The mixture was stirred at room temperature for 4 h. Then, 1.8 mmol of p-phenolsulfonic acid and 3.6 mmol of paraformaldehyde were added to the mixture and stirred for 2 h in ethanol. The mixture was then cooled and washed twice with ethanol. The final solid product was dried at 60 °C for 12 h to obtain KS71–SiO2.2.6. Procedures for recycling KS71
A synthesis reaction of compound 1a was performed, followed by catalyst recovery and efficiency calculation to investigate the catalyst stability over multiple reactions. The reaction was conducted under solvent-free conditions at 90 °C for 2.5 hours with a stirring rate of 400 rpm, using benzaldehyde (1 mmol), ethyl acetoacetate (1 mmol), urea (1 mmol), and 5 mg of KS71 catalyst. After 2.5 hours, the mixture was cooled to room temperature, and the post-reaction mixture was washed with EtOH and filtered to collect the catalyst on filter paper. The catalyst was washed repeatedly with ethyl acetate. The recovered catalyst was subjected to 4 reaction cycles and the yield was calculated for each cycle.2.7. Leaching test
The experiment involved 6 mmol of benzaldehyde, 6 mmol of ethyl acetoacetate, 6 mmol of urea, and 60 mg of KS71 for 2 hours and 30 minutes at 90 °C under solvent-free conditions. After reacting for 1 hour and 15 minutes, ethanol (9 mL) was added to the mixture and divided into three equal parts:-Part 1: the reaction continued for the remaining 1 hour and 15 minutes. After the reaction was complete, the product was filtered, washed with ethanol, and recrystallized in ethyl acetate. The recovered catalyst was analyzed by ICP-MS to determine the sulfur (S) content.
-Part 2: the reaction mixture was filtered to recover the catalyst, and the filtrate was completely evaporated to remove ethanol. The residue was then subjected to a solvent-free reaction for 1 hour and 15 minutes, followed by washing and recrystallization in ethyl acetate. The filtrate after washing was analyzed by ICP-MS.
-Part 3: the sample was completely evaporated to remove ethanol and then subjected to a solvent-free reaction for 1 hour and 15 minutes. The mixture was then filtered, washed with ethanol, and recrystallized in ethyl acetate. The recovered catalyst was analyzed by ICP-MS to determine the sulfur (S) content.
3. Results and discussion
3.1. Catalyst characterization
The results of the Fourier-transform infrared spectroscopy (FT-IR) analysis for identifying functional groups showed that we had successfully attached functional groups to the amorphous carbon substrate originating from rice husk biomass.On the surface of the catalyst, the presence of hydroxyl groups and water molecules that have been adsorbed may be seen in the BMSA spectrum, namely in the bands that occur at 3412 and 1602 cm−1. There is a possibility that the stretching vibration of Si–OH and the broadband operating at 3412 cm−1, which represents hydrogen bonding between water molecules and hydroxyl groups with metals and non-metals, may overlap with the vibration of FeO(OH) (because in the Fe3O4 crystal lattice, hydroxyl groups are linked to each other by hydrogen bonds on the iron oxide surface), resulting in overlapping vibrations. The intensity of the vibrations at 3412 and 1602 cm−1 decreases upon the addition of KH560 to BMSA, indicating the hydrophobic nature of KH–Si. BMSA has been effectively grafted onto the surface of KH560 through a silylation reaction between the Si–OH groups that are present on the surface and the Si(OCH3)3 group that is present in KH560. These results show that the grafting technique was successful. Additionally, the intensity of the signals at 2927 and 2875 cm−1 in KS71 is enhanced due to the stretching vibration of C–H, and the stretching of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds is also observed at 1508 and 1433 cm−1, indicating that the sulfonic group (–SO3H) has been successfully immobilized on the carbon framework of KS71. It is important to observe that some bands, including the asymmetric OSO stretch at 1220 and 1167 cm−1, the symmetric OSO stretch at 1115 cm−1, and the C–S stretch at 1038 cm−1, coincide with the prominent Si–O–Si peak at 1094 cm−1.41 The vibration of Fe–O at 567 and 695 cm−1 proves that the catalyst remains magnetic after acid treatment.42
C bonds is also observed at 1508 and 1433 cm−1, indicating that the sulfonic group (–SO3H) has been successfully immobilized on the carbon framework of KS71. It is important to observe that some bands, including the asymmetric OSO stretch at 1220 and 1167 cm−1, the symmetric OSO stretch at 1115 cm−1, and the C–S stretch at 1038 cm−1, coincide with the prominent Si–O–Si peak at 1094 cm−1.41 The vibration of Fe–O at 567 and 695 cm−1 proves that the catalyst remains magnetic after acid treatment.42
The results of Fourier-transform infrared spectroscopy (FT-IR) analysis of KS71–SiO2 were shown in Fig. S1.1 (ESI†). The bands of the KS71–SiO2 catalyst also appeared like the KS71 catalyst. The bands at 3309 and 1640 cm−1 showed the adsorption of water on the catalyst surface, however, the signal intensity of these bands was weaker than that of the KS71 catalyst, possibly because the SiO2 layer on the surface limited the adsorption of water on the catalyst, as well as the O–H groups of the amorphous carbon matrix were affected. The difference lies in the bands 1080 and 1207 cm−1, which are two bands representing Si–O–Si and SO3H. The 1080 cm−1 band shows a higher intensity of Si–O–Si than its own band in the KS71 catalyst, even partially covering the signal of the 1207 cm−1 band of SO3H. This indicates that the SiO2 coating on the catalyst was successful. In addition, other bands such as 794 and 690 cm−1 represent the deformation vibration of Si–O–Si and the fingerprint region below 600 cm−1 of Fe–O.
Using inductively coupled plasma mass spectrometry (ICP-MS), we examined the sulfur content in the catalyst to verify the successful attachment of the acidic –SO3H groups. In the analysis of the magnetic amorphous carbon sample (BMSA), we were unable to detect any sulfur content. However, in the catalyst sample that we successfully synthesized, KS71, we did detect sulfur content. This indicates that we have successfully attached the –SO3H group to the amorphous carbon substrate, ensuring the catalyst's acidity. The three stages of the catalytic synthesis process, namely BMSA, KH–Si, and KS71, were measured by ICP-OES with the weighed masses for sample preparation before measurement being 13.3, 17.7, and 15.2 mg, respectively, to determine the Fe content in the catalyst samples of the three synthesis stages, The results are recorded as 76210.55, 76766.14, and 76512.09 mg kg−1, respectively (Table S1, ESI†). In addition, the ICP-MS measurement results for the sulfur element were calculated and presented for the first and last two stages of the catalytic synthesis process, namely BMSA and KS71, as shown in Table 1. In our work, we included the synthesis of a magnetic catalyst to facilitate the efficient recovery and reuse of the catalyst post-reaction. Furthermore, we evaluated the sulfur concentration on the catalyst surface, a primary catalytic site. The study revealed that 1 gram of catalyst had 18.7 mg of sulfur. According to the preparation process of KS71 catalyst was described in Fig. 3 (the revised manuscript). On the surface of BMSA were carried –SO3H groups which acted as the acid sites of the catalyst. Based on the results of ICP-MS analysis, it showed that BMSA material did not contain sulfur, however, after the synthesis process was completed, the sulfur content of KS71 material is recorded as 0.58748 mmol g−1.
 | ||
| Fig. 3 Scanning electron microscopy (SEM) images of BMSA, KH–Si, and KS71. | ||
Scanning electron microscopy (SEM) was utilized to characterize the catalyst's morphology. SEM produces high-resolution images at significant magnifications by scanning the sample surface, enabling the visualization of the material's structure.
SEM images revealed that our synthesized material, KS71, comprised carbon derived from the magnetization of rice husk-based amorphous carbon and subsequent sulfonation (Fig. 3). The material exhibited amorphous carbon particles with sizes ranging from 6–10 μm, possessing a porous structure with numerous pores. Additionally, small particles, presumably Fe3O4, were readily observed within the porous structure of the pyrolyzed biomass.
Energy-dispersive X-ray spectroscopy (EDX) was employed to analyze the material's elemental composition by recording the characteristic X-rays emitted upon interaction with incident radiation (Fig. 4). The resulting X-ray spectra provided information on the elements present in the sample and their relative proportions. By analyzing three samples involved in the synthesis of KS71, namely BMSA, KH–Si, and KS71, we could demonstrate the presence and variation of elements in each sample. The results from all three samples confirmed the successful incorporation of desired functional groups and metals. The presence of Fe in BMSA indicated successful magnetization of the amorphous carbon substrate. The results for KH–Si and KS71 (the final product) confirmed the presence of sulfur and an increased oxygen content, indicating successful attachment of the –SO3H group. Furthermore, the material retained its magnetic properties after sulfonation, as evidenced by the presence of Fe.
 | ||
| Fig. 4 EDX analysis of BMSA, KH–Si, and KS71. | ||
Fig. 5 presents the XRD patterns of three stages involved in the formation of the KS71 catalyst: BMSA, KH–Si, and KS71. The XRD patterns are combined with two reference XRD patterns of amorphous carbon and Fe3O4. The patterns of all three stages exhibit peaks in the 2θ range of 20–30°. The detection of these diffraction peaks can be attributed to the reflection of the (002) and (110) planes of the amorphous carbon structure.43 The intensity of the peaks representing the presence of the amorphous carbon structure is low in BMSA and gradually increases towards KS71 in the 2θ ranges of 40–60° and 70–80°. Additionally, distinct peaks appear in the 2θ range of 40–80°, indicating a match with the peaks of crystalline Fe3O4 (JCPDS 75-0033). The detection of crystalline Fe3O4 by XRD confirms the presence of Fe3O4 crystalline particles in the pores of the amorphous carbon structure. The modification of the amorphous carbon matrix using ionic liquid has led to more pronounced peaks in KS71 compared to BMSA.
 | ||
| Fig. 5 XRD patterns of BMSA, KH–Si, and KS71. | ||
Thermogravimetric analysis (TGA) is a valuable tool for assessing the thermal stability of materials and providing insights into their composition (Fig. 6). The temperature at which weight loss occurs is crucial for understanding a material's behavior under harsh conditions. The Fig. 5 presents the thermogravimetric decomposition curve of our synthesized catalyst, KS71. It can be observed that KS71 exhibits a high decomposition temperature (approximately 500 °C), indicating its excellent thermal stability. The catalyst undergoes three stages of decomposition: from room temperature (tR) to 95 °C, corresponding to the loss of water from the porous structure; from 100 °C to 300 °C, a relatively stable region with the loss of some fats, phenolics, resins, and simple sugars; and above 300 °C, continuous weight loss due to the decomposition of amorphous carbon and Fe3O4, with cellulose and lignin almost completely decomposing by 450 °C. After heating at 500 °C, approximately 20% of the sample mass remains. This is due to the fact that the material was heated in a nitrogen atmosphere and had been preloaded with Fe. The Fe3O4 particles on the biomass surface and the presence of SiO2 within the biomass did not reach the necessary temperature for decomposition at 500 °C.44
 | ||
| Fig. 6 Thermogravimetric analysis (TGA) curves for KS71. | ||
Magnetic hysteresis is a prominent characteristic of ferromagnetic materials. It refers to the irreversible relationship between magnetization and demagnetization processes, resulting from the material's ability to retain magnetization. When exposed to a strong magnetic field, these materials exhibit a magnetic moment that persists even after the external field is removed, behaving like permanent magnets. This property allows for the manipulation of the material's magnetic properties using external magnetic fields. To determine the magnetic properties of our catalyst, KS71, we measured its magnetic hysteresis using a vibrating sample magnetometer (VSM). The results, shown in the Fig. 7, indicate that the BMSA sample, obtained by carbonizing biomass impregnated with Fe at 400 °C, exhibited significant magnetic properties. After sulfonation, the resulting KS71 sample retained its magnetic properties, demonstrating that the catalyst possesses both acidic and magnetic characteristics. This facilitates the straightforward separation of the catalyst from the reaction mixture through the use of an external magnetic field.
 | ||
| Fig. 7 Magnetization curves of BMSA and KS71 at room temperature. | ||
The total titration results (–SO3H, –OH, –COOH…) with a repeatability of 3 times gave the results of the three stages of catalyst synthesis of 0.22, 0.12, 0.15 mmol g−1 for BMSA, KH–Si, KS71 respectively (ESI†). The titration results of the –SO3H group alone, the acid concentration was titrated with a repeatability of 3 times to 0.07 mmol g−1. Through the titration results of the total acid concentration, the acid concentration decreased when going from the BMSA stage to KH–Si. The reason may be that when modifying the surface of BMSA, some acid functional groups reacted, making the release of protons more difficult, so the acid concentration decreased. Acidification with para-phenolsulfonic acid increased the acid concentration, proving that the acidification was successful.
3.2. Catalytic activity of KS71 for DHPMs synthesis
Multi-component reactions have gained significant prominence in modern organic synthesis due to their numerous advantages, including economic viability, energy efficiency, time savings, and convergence. Building upon the well-established Biginelli reaction, we have successfully employed our custom-synthesized catalyst, KS71, for the synthesis of DHPMs. This one-pot, three-component reaction involves the condensation of aldehyde, β-ketoester, and urea or thiourea (Scheme 4). The initial reaction was carried out with HCl in ethanol (EtOH) as a catalyst, now we use our catalyst KS71 as a catalyst for the reaction involving the mixture mentioned above. In our study, we selected benzaldehyde, ethyl acetoacetate, and urea as model substrates and conducted the reaction under solvent-free conditions at 90 °C for 2.5 hours using KS71 as the catalyst. The successful synthesis of the desired DHPM product was confirmed by 1H-NMR and 13C-NMR spectroscopic analysis. | ||
| Scheme 4 DHPM synthesis reaction in the presence of KS71 catalyst. | ||
The initial overall yield when conducting the reaction was 52%. We continued to investigate the reaction conditions, including reaction temperature, reaction time, suitable solvents for the reaction, the required amount of catalyst, and various types of Brønsted acid-supported catalysts (Table 2). By investigating with increasing reaction temperatures from 30 °C to 110 °C (entries 1–6, Table 2), it can be seen that the yield increased from 0% at 30 °C (entry 1, Table 2) and 50 °C (entry 2, Table 2) to 52% at 90 °C (entry 4, Table 2). Under conditions below 50 °C, there may not be enough heat for the substrates to melt, so the reaction is difficult to occur below 50 °C. However, at temperatures greater than 90 °C, the yield gradually decreased, possibly due to the concentration of the substances at temperatures greater than 90 °C, leading to a significant decrease in the contact area, making the reaction more difficult. Therefore, it can be said that 90 °C is the optimal temperature for the synthesis of DHPM with the KS71 catalyst.
| Entry | Catalysts | Amount of catalyst (mg) | Temp. (°C) | Time (h) | Solvents | V solvent (mL) | Yieldb (%) |
|---|---|---|---|---|---|---|---|
| a Reaction conditions: benzaldehyde (1 mmol), ethyl acetoacetate (1 mmol), urea (1 mmol), 90 °C, 2.5 hours, solvent-free. b Isolated yield through crystallization in ethyl acetate (EA) (10–15 mL). c cc-pTSA = the carbon material was acidified with p-toluenesulfonic acid (pTSA); BG-TsOH; BG-H2SO4 = carbon-based solid acids; BMSA-H2SO4 = solid acid derived from biomass acidified with H2SO4 acid. | |||||||
| 1 | KS71 | 5 | 30 | 2.5 | — | — | 0 |
| 2 | KS71 | 5 | 50 | 2.5 | — | — | 0 |
| 3 | KS71 | 5 | 70 | 2.5 | — | — | 26 |
| 4 | KS71 | 5 | 90 | 2.5 | — | — | 52 |
| 5 | KS71 | 5 | 100 | 2.5 | — | — | 29 |
| 6 | KS71 | 5 | 110 | 2.5 | — | — | 25 |
| 7 | KS71 | 5 | 90 | 1 | — | — | 28 |
| 8 | KS71 | 5 | 90 | 2 | — | — | 41 |
| 9 | KS71 | 5 | 90 | 2.25 | — | — | 43 |
| 10 | KS71 | 5 | 90 | 2.5 | — | — | 52 |
| 11 | KS71 | 5 | 90 | 2.75 | — | — | 32 |
| 12 | KS71 | 5 | 90 | 3 | — | — | 30 |
| 13 | KS71 | 5 | 90 | 2.5 | Cyclohexane | 5 | 35 |
| 14 | KS71 | 5 | 90 | 2.5 | Toluene | 5 | 34 |
| 15 | KS71 | 5 | 90 | 2.5 | Ethanol | 5 | 22 |
| 16 | KS71 | 5 | 90 | 2.5 | n-Butanol | 5 | 34 |
| 17 | KS71 | 5 | 90 | 2.5 | Tetrahydrofuran | 5 | 31 |
| 18 | KS71 | 5 | 90 | 2.5 | Acetone | 5 | 33 |
| 19 | KS71 | 5 | 90 | 2.5 | Dimethylfomamide | 5 | 0 |
| 20 | KS71 | 5 | 90 | 2.5 | H2O | 5 | 0 |
| 21 | KS71 | 5 | 90 | 2.5 | — | — | 52 |
| 22 | KS71 | 0 | 90 | 2.5 | — | — | 22 |
| 23 | KS71 | 1 | 90 | 2.5 | — | — | 28 |
| 24 | KS71 | 5 | 90 | 2.5 | — | — | 52 |
| 25 | KS71 | 10 | 90 | 2.5 | — | — | 52 |
| 26 | BMSA | 5 | 90 | 2.5 | — | — | 29 |
| 27 | HK–Si | 5 | 90 | 2.5 | — | — | 28 |
| 28 | KS71 | 5 | 90 | 2.5 | — | — | 52 |
| 29 | cc-pTSAc | 5 | 90 | 2.5 | — | — | 26 |
| 30 | BG-TsOHc | 5 | 90 | 2.5 | — | — | 30 |
| 31 | BG-H2SO4c | 5 | 90 | 2.5 | — | — | 29 |
| 32 | BMSA-H2SO4c | 5 | 90 | 2.5 | — | — | 32 |
| 33 | HCl | 5 | 90 | 2.5 | — | — | 0 |
| 34 | H2SO4 | 5 | 90 | 2.5 | — | — | 0 |
Based on the optimal temperature conditions, we proceeded to investigate the reaction time in the range of 1–3 h (entries 7–12, Table 2) to learn about the effect of reaction time on the yield. Referring to the survey table, it can be seen that the yield increased gradually from 1 h to 2.5 h; however, the yield decreased when the time was longer than 2.5 h to 3 h, showing that the yield of the reaction is optimal when the reaction is carried out in 2.5 h (entry 10, Table 2).
Initially, the reaction was carried out in a solvent-free environment. However, the investigation of various solvents is significant (entries 13–20, Table 2). The influence of solvents on the reaction is considerable, as shown by the yields of non-polar solvents such as 35% for cyclohexane (entry 13, Table 2), 34% for toluene (entry 14, Table 2). Protic polar solvents also showed significant yields, such as 22% for ethanol (entry 15, Table 2), 34% for n-butanol (entry 16, Table 2), as well as aprotic polar solvents like acetone (entry 18, Table 2) at 33%, and tetrahydrofuran (THF) (entry 17, Table 2) at 31%. On the other hand, when investigating H2O and dimethylformamide (DMF) as polar solvents, we found the absence of the desired product. Additionally, under solvent-free conditions, the highest yield obtained was 52% (entry 21, Table 2). Under solvent-free conditions, it can be said that the synthesis of DHPM in the presence of the KS71 catalyst is a mild reaction when carried out in a solvent-free environment, causing less harm to the environment.
The yield of the reaction can be maximized depending on the amount of catalyst. The catalyst amount was investigated in the range of 0–10 mg (entries 22–25, Table 2). The results showed that the best yield was 52% when using 5 mg (entry 24, Table 2) or 10 mg (entry 25, Table 2) of catalyst. Additionally, the investigation of catalysts with origins similar to the catalyst we are using, KS71 (entries 26–35, Table 2), i.e., catalysts also derived from biomass like KS71, was conducted. Furthermore, HCl and H2SO4 were also investigated as inorganic Brønsted acid catalysts to clearly show the effect of different types of catalysts on the DHPM synthesis reaction. Using two catalysts from the first and second stages of the synthesis of KS71, which are magnetic porous carbon (BMSA) (entry 26, Table 2) and silanized magnetic porous carbon (KH–Si) (entry 27, Table 2), showed yields of 29% and 28%, respectively. Using KS71 gave us the best yield of 52% (entry 28, Table 2). Other biomass-based catalysts such as cc-pTSA (entry 29, Table 2), BG-TsOH (entry 30, Table 2), BG-H2SO4 (entry 31, Table 2), BMSA-H2SO4 (entry 32, Table 2) also did not yield very high results, ranging from 26% to 29%. The use of two Brønsted acids, HCl and H2SO4, did not produce satisfactory results, as the desired product was not obtained. Finally, the synthesized KS71 catalyst showed the most potential for DHPM synthesis based on the Biginelli reaction among similar biomass-derived catalysts like KS71.
Besides, the application of KS71–SiO2 catalyst is also applied to the Biginelli reaction. In addition, the application of KS71–SiO2 catalyst was also applied to the Biginelli reaction. The application of the new catalyst to the Biginelli reaction was also under the same conditions as the KS71 catalyst with 1 mmol (0.10612 g) benzaldehyde, 1 mmol (0.13014 g) ethylacetoacetate, 1 mmol (0.06006 g) urea at 90 °C for 2.5 h with a stirring speed of 400 rpm and no solvent. The product after the reaction was treated as when using the KS71 catalyst. The product yield of the Biginelli reaction using the KS71–SiO2 catalyst was 22.3%, which was lower than when using the KS71 catalyst. The reason may be that the SiO2 coating may reduce some of the active sites of the catalyst needed to catalyze the reaction.
3.3. The proposed mechanism for DHPM synthesis using KS71 as a catalyst
The synthesis mechanism of DHPM and its derivatives using KS71 catalyst is depicted in Scheme 5. Initially, the catalyst KS71 activates the carbonyl group, and under the nucleophilic attack of urea/thiourea combined with water elimination, an intermediate (A) bearing an imine group is formed. Ethyl acetoacetate is also activated by the catalyst KS71, followed by a nucleophilic attack on the carbon of the imine group to form intermediate (B). The remaining lone pair of electrons on the –NH2 group in urea/thiourea interacts with the carbon of the carbonyl group, which is activated by the KS71 catalyst, leading to ring closure and water elimination, resulting in the final product (C). | ||
| Scheme 5 The proposed mechanism for the synthesis DHPM using KS71 as a catalyst. | ||
3.4. The reusability of the KS71 catalyst in the synthesis of DHPM
The stability of KS71 catalyst in the Biginelli reaction (1 mmol of benzaldehyde, 1 mmol of ethyl acetoacetate, 1 of mmol urea) at 90 °C, 2.5 h, with a stirring speed of 400 rpm, and solvent-free conditions was investigated through four reaction runs. The statistical data on product yield is illustrated in Fig. 8. Accordingly, the product yield obtained from the reaction decreased slightly but insignificantly, with yield deviations ranging from 1–3%. The magnetic nature of the catalyst has enabled excellent catalyst separation, facilitating the process of conducting catalytic reactions. The insignificant decrease in product yield over successive reactions demonstrates the remarkable stability of the KS71 catalyst. The crystallized products from each reaction run were examined and found to have the exact structure as the initially synthesized product, further confirming that KS71 is a catalyst that is both easily separable and recoverable, and highly efficient over multiple reaction cycles. | ||
| Fig. 8 Reusability of the KS71. | ||
After four times of recovery and reuse, FT-IR spectra was used to test the stability of the catalyst, the results are shown in the Fig. 9.
 | ||
| Fig. 9 FT-IR spectra of fresh KS71 (blue) and recovered KS71 (orange). | ||
The overlapping vibrations of the –OH group at 3242 cm−1 and the additional asymmetric –NH stretching at 3116 cm−1 are primarily due to the porous carbon structure of the material, allowing the DHPM product of the Biginelli reaction and residual reactants to enter the pores. This is more evident at 3116 and 1294 cm−1, corresponding to the stretching vibrations of –NH and –CN, respectively. The signals of the product and reactants are also clearly observed at 1708 cm−1 for the CO stretching vibration (mainly due to residual unreacted urea and a portion of the DHPM product), and additional signals at 1645 and 1463 cm−1 for CC and CN, respectively. The catalyst retains its acidity after multiple reactions, as evidenced by the vibrations at 1221 cm−1 for SOS (in the –SO3H group) and at 1031 cm−1 for C–S (Fig. 9).
The magnetism of the catalyst is still retained, as shown by the vibrations at 696 and 509 cm−1. Considering the magnetism of the recovered catalyst after four uses, the vibrating sample magnetometer (VSM) results show that the catalyst remains magnetic after four recovery and reuse cycles, thus allowing for easy separation from the reaction mixture using an external magnetic field (Fig. 10).
 | ||
| Fig. 10 Magnetization curves of KS71 (red) and recovered KS71 (blue) at room temperature. | ||
After the reaction, the KS71 catalyst was recovered and analyzed by XRD pattern to evaluate the transformation of Fe3O4 into Fe2O3. Fig. S1.4 (ESI†) shows the results of the XRD pattern of the recovered KS71 catalyst. The diffraction peaks of the 002 and 110 lattice planes of amorphous carbon have decreased in intensity compared to the KS71 catalyst and the diffraction peaks of Fe2O3 have been found and clearly shown in the 2θ range from 30–70° (JCPDS 39-1346). In addition, we could not detect the peaks of the Fe3O4 lattice plane. However, from the XRD pattern, we can conclude that there has been a transformation of the existing form from Fe3O4 to Fe2O3 and this transformation may occur more if the recovered KS71 catalyst is used in subsequent reactions.
The KS71–SiO2 catalyst after the reaction was also recovered to be used for the next reactions to evaluate the durability of the KS71–SiO2 catalyst through many reactions compared to KS71. The reaction recovery efficiency of KS71–SiO2 was shown in Fig. S1.2 (ESI†). The product yield gradually decreased with reaction times and after three reactions, the yield decreased significantly, which can be seen that the operating cycle of the KS71–SiO2 catalyst was lower than that of KS71. The FT-IR analysis results of the KS71–SiO2 catalyst are also shown in Fig. S1.1 (ESI†), showing that the 1088 cm−1 bands of Si–O–Si were reduced after the reaction and the 1230 cm−1 bands of OSO or 1450 and 1365 cm−1 or 1597 cm−1 bands of CO appeared more clearly.
3.5. Leaching test
A leaching test was carried out in order to shed light on the catalyst's involvement, and it was conducted within the confines of the optimal reaction conditions (Fig. 11). | ||
| Fig. 11 Leaching test procedure: (1) reaction stopped; (2) reaction without KS71; (3) continuous reaction with KS71. | ||
With a reaction time of 1 hour and 15 minutes, we proceeded to divide the reaction mixture into three equal parts. For the first part, the product yield was 37.7% after catalyst removal and crystallization in ethyl acetate. The second part underwent catalyst removal and was allowed to react for the remaining 1 hour and 15 minutes, yielding a product yield of 3.1%. The remaining part was allowed to react for the remaining 1 hour and 15 minutes, resulting in a product yield of 50.4%. The product yields from the three parts indicate the significant role of the KS71 catalyst in the DHPM synthesis reaction.
The results of ICP-MS analysis of the three leaching test samples to determine the Fe (mg kg−1) and S (mg kg−1) content are shown in Table 3. The amount of Fe in sample LT2 (entry 2, Table 3) is very small, meaning that the filtrate after the reaction does not contain catalyst or metal fragments, and we have successfully separated the catalyst from the post-reaction mixture. This suggests that our catalyst can be easily separated. The amount of Fe in both samples LT1 (entry 1, Table 3) and LT3 (entry 3, Table 3) is high, indicating that our catalyst still contains a significant amount of Fe to continue acting as a magnetic solid catalyst in the second half of the reaction or can be easily separated and recovered. Similarly to the amount of Fe, the amount of S in sample LT2 (entry 2, Table 3) is also very small, and the amount of S in samples LT1 (entry 1, Table 3) and LT3 (entry 3, Table 3) is very high, although we cannot determine it accurately due to the different amounts of samples used for measurement. The ICP-MS results clearly demonstrate the importance of the KS71 catalyst in the DHPM synthesis reaction and the excellent separation and recovery capabilities of KS71.
| No. | Sample | Fe density (mg kg−1) | S density (mg kg−1) |
|---|---|---|---|
| Condition of reaction: benzaldehyde (6 mmol), ethyl acetoacetate (6 mmol), urea (6 mmol), KS71 (30 mg), 2.5 h, 90 °C, solvent-free. | |||
| 1 | LT1 | 23319.18 |
<970 |
| 2 | LT2 | <2 | <36 |
| 3 | LT3 | 14784.61 |
<1400 |
3.6. Synthesis of DHPM derivatives
The establishment of optimal conditions aims to achieve the best yield in the synthesis of DHPM derivatives (Scheme 6). The synthesis of DHPM derivatives using aryl aldehydes, ethyl acetoacetate, and urea/thiourea was carried out to investigate the influence of substituents on the reaction. Substituents such as electron-donating groups (4-OH, 4-OMe) and electron-withdrawing groups (4-Cl, 4-Br) were sequentially attached to the aromatic ring of the aldehyde to synthesize DHPM derivatives. The synthesized DHPM derivatives, along with their corresponding yields, are presented in the attached Scheme 6. | ||
| Scheme 6 DHPM derivatives synthesis substrate scope. Reaction conditions: aldehydes (1 mmol), ethyl acetoacetate (1 mmol), urea/thiourea (1 mmol), KS71 (5 mg), under solvent-free conditions. | ||
When performing reactions with aldehydes containing substituents on the aromatic ring, a significant decrease in the yield of 2a–12a (Scheme 6, compounds 2a–12a) was observed compared to the initially synthesized 1a (Scheme 6, compound 1a) without any substituents. The primary cause is attributed to the steric effect, which hinders the interaction between the catalyst and the functional group, thereby making the reaction less likely to occur and resulting in lower yields compared to benzaldehyde. Electron-donating groups such as –Cl and –Br, when attached to the benzene-ring, reduce the carbonyl activity and introduce a steric effect, especially at the ortho position, making it more difficult for the catalyst to access the reactive functional group, leading to a sharp decrease in product formation. Electron-withdrawing groups such as –OCH3 and –OH, when attached to the benzene-ring, also decrease the carbonyl activity and introduce a steric effect from the substituents, making it more difficult for the catalyst to access the reactive functional group, leading to a significant decrease in product formation. In addition to modifying the substituents on the aromatic ring of the aldehyde, we conducted reactions with thiourea and synthesized thiourea derivatives 13a (Scheme 6, compound 13a) and 14a (Scheme 6, compound 14a). However, the use of thiourea did not yield satisfactory results. Thin-layer chromatography (TLC) analysis using a n-hexane:ethyl acetate (v/v = 5:5) eluent system consistently showed a single spot, and the synthesized products were white solids with melting points matching those reported in the literature, indicating successful synthesis of these derivatives. Furthermore, nuclear magnetic resonance (NMR) spectroscopy, specifically 1H-NMR and 13C-NMR, was used to accurately characterize these compounds.
3.7. A comparison between this work and prior works
The reports on compound 1a, along with the types of catalysts used, are presented in Table 4. We selected compound 1a due to its highest yield among the derivatives we synthesized. Compared to previously published reports in Table 4, our KS71 catalyst did not yield the highest performance but was also not significantly lower. KS71 offers a shorter reaction time and a moderate temperature under solvent-free conditions compared to catalysts that require longer reaction times, higher temperatures, and the use of various solvents. Additionally, KS71 has demonstrated comparable stability to previously reported catalysts. Derived from natural sources and operating under favorable reaction conditions, especially solvent-free conditions, which is a current trend in green chemistry, along with its stable performance, makes KS71 a reliable catalyst for the Biginelli reaction.| Entry | Catalyst and reaction conditions | Temp. (°C) | Time (min) | Solvents | Yield (%) | Ref. |
|---|---|---|---|---|---|---|
| 1 | β-CD (0.5 mol%) | Reflux | 480 | H2O | 20 | 45 |
| 2 | H2SO4/C (100% w/w) | Reflux | 60 | CH3CN | 30 | 46 |
| 3 | Ca–Al–CO3 hydrotalcite | 80 | 35 | Solvent-free | 69 | 47 |
| 4 | PS-AFDPAT (5 mol%) | Reflux | 300 | EtOH | 90 | 48 |
| 5 | PSBIL (50 mg) | 110 | 1080 | EtOH | 92 | 12 |
| 6 | KS71 (5 mg) | 90 | 150 | Solvent-free | 52 | This work |
4. Conclusion
In conclusion, our study indicates that the solid catalyst KS71, which we synthesized, exhibits high effectiveness in the Biginelli reaction for DHPM synthesis. With the initial raw material being biomass, our catalyst is completely green, environmentally friendly, and easy to recover and separate. The efficiency of the Biginelli reaction to synthesize DHPM is promising (reached 52%), the reaction time is not too long, and it is carried out in a solvent-free environment. From the tested results, we confirm that we have successfully synthesized KS71 and DHPM derivatives.Author contributions
Thai-Phien Huynh Dang: investigation, methodology, resources, formal analysis, validation, data curation, writing – original draft; Thach Ngoc Pham: investigation, methodology, resources, formal analysis; Phuong Hoang Tran: methodology, resources, formal analysis, validation, data curation, writing – review & editing; Hai Truong Nguyen: methodology, resources, formal analysis, validation, data curation, writing – review & editing, supervision.Data availability
The authors confirm that the data supporting the findings of this study are available within the article. Raw data that support the findings of this study are available from the corresponding author, upon reasonable request.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This research is funded by University of Science, VNU-HCM under grant number T2024-114.References
- C.-H. Zhou, X. Xia, C.-X. Lin, D.-S. Tong and J. Beltramini, Chem. Soc. Rev., 2011, 40, 5588–5617 RSC.
- L. J. Konwar, P. Mäki-Arvela and J.-P. Mikkola, Chem. Rev., 2019, 119, 11576–11630 CrossRef CAS.
- P. Anastas and N. Eghbali, Chem. Soc. Rev., 2010, 39, 301–312 RSC.
- N. T. Tinh, L. M. Bao, D. D. Nhan, P. P. Bao, T. M. Hoang, N. T. H. Nam, N. M. Dat, N. N. Khoa, N. T. Hanh, Q. T. T. Huong, T. T. Danh and H. H. Nguyen, Renewable Energy, 2024, 232, 121039 CrossRef CAS.
- S. Alaei, M. Haghighi, J. Toghiani and B. Rahmani Vahid, Ind. Crops Prod., 2018, 117, 322–332 CrossRef CAS.
- C. S. Gill, B. A. Price and C. W. Jones, J. Catal., 2007, 251, 145–152 CrossRef CAS.
- N. Fekrat, M. Mazloumi and F. Shirini, RSC Adv., 2023, 13, 35659–35671 RSC.
- M.-N. Chen, L.-P. Mo, Z.-S. Cui and Z.-H. Zhang, Curr. Opin. Green Sustainable Chem., 2019, 15, 27–37 CrossRef.
- R. Afshari and A. Shaabani, ACS Comb. Sci., 2018, 20, 499–528 CrossRef CAS PubMed.
- Z. Zhang, Y. You and C. Hong, Macromol. Rapid Commun., 2018, 39, 1800362 CrossRef PubMed.
- C. Lamberth, Bioorg. Med. Chem., 2020, 28, 115471 CrossRef CAS PubMed.
- A. G. Khiratkar, P. N. Muskawar and P. R. Bhagat, RSC Adv., 2016, 6, 105087–105093 RSC.
- A. Shaabani, M. Seyyedhamzeh, A. Maleki and F. Hajishaabanha, Tetrahedron, 2010, 66, 4040–4042 CrossRef CAS.
- S. M. Sondhi, R. N. Goyal, A. M. Lahoti, N. Singh, R. Shukla and R. Raghubir, Bioorg. Med. Chem., 2005, 13, 3185–3195 CrossRef CAS PubMed.
- H. S. Oboudatian, H. Naeimi and M. Moradian, RSC Adv., 2021, 11, 7271–7279 RSC.
- K. De, S. Chandra, B. Sarkar, S. Ganguly and M. Misra, J. Radioanal. Nucl. Chem., 2010, 283, 621–628 CrossRef CAS.
- I. M. T. C. Crevel, M. C. Alonso and R. A. Cross, Curr. Biol., 2004, 14, R411–R412 CrossRef CAS.
- Sung H. Choi and D. McCollum, Curr. Biol., 2012, 22, 225–230 CrossRef CAS PubMed.
- J. C. Cochran and S. P. Gilbert, Biochem., 2005, 44, 16633–16648 CrossRef CAS.
- L. Duan, T.-Q. Wang, W. Bian, W. Liu, Y. Sun and B.-S. Yang, Spectrochim. Acta, Part A, 2015, 137, 1086–1091 CrossRef CAS.
- S. DeBonis, J.-P. Simorre, I. Crevel, L. Lebeau, D. A. Skoufias, A. Blangy, C. Ebel, P. Gans, R. Cross, D. D. Hackney, R. H. Wade and F. Kozielski, Biochemistry, 2003, 42, 338–349 CrossRef CAS PubMed.
- K. Drosopoulos, C. Tang, W. C. H. Chao and S. Linardopoulos, Nat. Commun., 2014, 5, 3686 CrossRef CAS.
- C. Joel Funk, A. S. Davis, J. A. Hopkins and K. M. Middleton, Anal. Biochem., 2004, 329, 68–76 CrossRef.
- T. Peters, H. Lindenmaier, W. E. Haefeli and J. Weiss, Pharmacology, 2006, 372, 291–299 CAS.
- B. N. Naidu, M. E. Sorenson, M. Patel, Y. Ueda, J. Banville, F. Beaulieu, S. Bollini, I. B. Dicker, H. Higley, Z. Lin, L. Pajor, D. D. Parker, B. J. Terry, M. Zheng, A. Martel, N. A. Meanwell, M. Krystal and M. A. Walker, Bioorg. Med. Chem. Lett., 2015, 25, 717–720 CrossRef CAS PubMed.
- P. Skehan, R. Storeng, D. Scudiero, A. Monks, J. McMahon, D. Vistica, J. T. Warren, H. Bokesch, S. Kenney and M. R. Boyd, J. Natl. Cancer Inst., 1990, 82, 1107–1112 CrossRef CAS PubMed.
- R. K. Yadlapalli, O. P. Chourasia, K. Vemuri, M. Sritharan and R. S. Perali, Bioorg. Med. Chem. Lett., 2012, 22, 2708–2711 CrossRef CAS.
- B. S. Holla, B. S. Rao, B. K. Sarojini and P. M. Akberali, Eur. J. Med. Chem., 2004, 39, 777–783 CrossRef CAS PubMed.
- B. Ahmed, R. A. Khan, Habibullah and M. Keshari, Tetrahedron Lett., 2009, 50(24), 2889–2892 CrossRef CAS.
- V. Polshettiwar and R. S. Varma, Tetrahedron Lett., 2007, 48, 7343–7346 CrossRef CAS.
- Z.-J. Quan, Y.-X. Da, Z. Zhang and X.-C. Wang, Catal. Commun., 2009, 10, 1146–1148 CrossRef CAS.
- H. Murata, H. Ishitani and M. Iwamoto, Org. Biomol. Chem., 2010, 8, 1202–1211 RSC.
- S. Rostamnia and K. Lamei, Chin. Chem. Lett., 2012, 23, 930–932 CrossRef CAS.
- A. Ghorbani-Choghamarani and P. Zamani, Chin. Chem. Lett., 2013, 24, 804–808 CrossRef CAS.
- H. Adibi, H. A. Samimi and M. Beygzadeh, Catal. Commun., 2007, 8, 2119–2124 CrossRef CAS.
- T.-H. T. Nguyen, H. B. Phan, T. H. Nguyen, K. N. Tran, L. H. T. Nguyen, T. L. H. Doan and P. H. Tran, RSC Adv., 2023, 13, 7257–7266 RSC.
- S. Farooq, F. A. Alharthi, A. Alsalme, A. Hussain, B. A. Dar, A. Hamid and S. Koul, RSC Adv., 2020, 10, 42221–42234 RSC.
- B.-J. Yao, W.-X. Wu, L.-G. Ding and Y.-B. Dong, J. Org. Chem., 2021, 86, 3024–3032 CrossRef CAS.
- M. Wang, Z.-C. Wang, Z.-L. Sun and H. Jiang, Transition Met. Chem., 2005, 30, 792–796 CrossRef CAS.
- Ş. Gülten, J. Heterocycl. Chem., 2017, 54, 1252–1260 CrossRef.
- R. Zhou, Y. Jiang, H. Zhao, B. Ye, L. Wang and Z. Hou, Fuel, 2021, 291, 120207 CrossRef CAS.
- H. Guo, Y. Lian, L. Yan, X. Qi and R. L. Smith, Green Chem., 2013, 15, 2167–2174 RSC.
- R. Krishna, A. N. Jones and B. J. Marsden, Radiat. Phys. Chem., 2015, 107, 121–127 CrossRef CAS.
- L. Hu, X. Tang, Z. Wu, L. Lin, J. Xu, N. Xu and B. Dai, Chem. Eng. J., 2015, 263, 299–308 CrossRef CAS.
- N. A. Liberto, S. de Paiva Silva, Â. de Fátima and S. A. Fernandes, Tetrahedron, 2013, 69, 8245–8249 CrossRef CAS.
- K. A. Dilmaghani, B. Zeynizadeh and H. Parasajam, Phosphorus, Sulfur Silicon Relat. Elem., 2012, 187, 544–553 CrossRef CAS.
- J. Lal, M. Sharma, S. Gupta, P. Parashar, P. Sahu and D. D. Agarwal, J. Mol. Catal. A: Chem., 2012, 352, 31–37 CrossRef CAS.
- M. Lei, D.-D. Wu, H.-G. Wei and Y.-G. Wang, Synth. Commun., 2009, 39, 475–483 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ma00045a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |