
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEfficient advanced reduction with cobalt phthalocyanine-decorated N-doped mesoporous carbon†
Mengjiao
Xu
,
Kaizhi
Wang
,
Wendi
Guo
,
Zehui
Sun
,
Mugeng
Chen
,
Yongmei
Liu
 * and
Yong
Cao
*
* and
Yong
Cao
*
Shanghai Key Laboratory of Molecular Catalysis and Innovative Materials, State Key Laboratory of Porous Materials for Separation and Conversion, Department of Chemistry, Fudan University, Shanghai 200433, China. E-mail: ymliu@fudan.edu.cn; yongcao@fudan.edu.cn
First published on 11th March 2025
Abstract
Effective removal of environmental pollutants relies heavily on advanced reduction processes (ARPs), and developing effective and low cost catalysts is pivotal. Herein, it is demonstrated that cobalt phthalocyanine (CoPc) adsorbed on nitrogen-doped mesoporous carbon (NMC) can serve as an efficient catalyst for ARPs. The CoPc-NMC composite stands out for its remarkable ability to reduce hexavalent Cr(VI) using formic acid as the reducing agent. This exceptional performance is attributed to the distinct characteristics of the composite, including atomically dispersed CoPc and strong π–π interactions with the rich pyridinic N-doped carbon support. It also reveals the complex involvement of active species, such as electrons and hydrogen radicals, which clarifies the mechanism behind this catalytic process. Ultimately, this study not only sheds light on the factors that influence the catalytic activity of CoPc-NMC but also offers critical insights into developing more efficient strategies for Cr(VI) reduction in environmental applications.
Introduction
In response to the growing environmental concerns, advanced reduction processes (ARPs) have emerged as promising solutions for effectively addressing persistent and refractory pollutants. These approaches employ various reducing agents to generate highly reactive species capable of targeting a wide range of organic and inorganic contaminants.1–4 Formic acid (FA) has gained considerable attention in this context due to its cost-effectiveness, safety, recyclability, and environmentally friendly properties.5–9 Unlike alternative reductants, FA can directly yield hydrogen (H2) and carbon dioxide (CO2) through catalytic processes (HCOOH → H2 + CO2). The resulting CO2 can be efficiently converted back into FA, establishing a reversible hydrogen storage–release cycle that helps mitigate greenhouse gas emissions.10–12 The potent reactive hydrogen produced during FA dehydrogenation exhibits robust reducing capabilities, making it well-suited for remediating environmental contaminants, including organic compounds and heavy metals.13–17 Furthermore, FA-centric ARPs offer a viable alternative in situations where other reductants may prove unsuitable.18 Various strategies, such as the use of metallic catalysts, metallic compounds, ultrasonication, alkali treatment, and short-wavelength UV irradiation, activate FA to generate highly reactive reducing species like hydrogen radicals (H˙), or employ nonradical pathways to degrade recalcitrant pollutants.19–22 However, the practical implementation of these strategies is largely hindered by their high energy and chemical consumption, underscoring the imperative need for the development of more sustainable and environmentally friendly catalysts to address these challenges.Within the context of heterogeneous FA-ARPs, the pivotal step for effective catalytic reduction lies in the adsorption of pollutants onto catalyst surfaces. Expanding on this concept, the development of dual- or multi-sites materials, incorporating both adsorptive and catalytic domains, emerges as a critical strategy to selectively concentrate target pollutants.23,24 This design confines the degradation reaction within the catalyst framework, reducing interference from substances in the bulk solution. Notably, research has unveiled that adducts formed between FA and electro-conductive composite-based catalysts play a crucial role in facilitating pollutant breakdown through catalyst-mediated FA activation, involving a directed electron-transfer mechanism or the co-generation of reactive radical species.25,26 This mechanism operates in two stages: initially, FA forms a surface adduct upon interaction with the catalyst, followed by the transfer of electrons from the catalyst–FA adduct to adsorbed pollutants, initiating the reduction process. This approach implies that the migration of active hydrogen radical (H˙) species and electrons may both play important roles in facilitating the overall FA consumption process. Furthermore, the directed transport of H˙ and/or electrons from FA to contaminants on the composite catalyst may involve a potential synergy, either long-range or short-range, between distinct active sites.23 However, the source and spatial arrangement of this synergy remain unclear, presenting challenges to the advancement of FA-ARPs.
In recent times, single-atom catalysts (SACs), characterized by their isolated metal centers, have demonstrated remarkable catalytic efficiency across a diverse range of reactions.27–30 This efficacy is attributed to their unique electronic structures and efficient atom utilization. Nitrogen-doped porous carbon has emerged as an ideal substrate for SACs, offering substantial specific surface areas and abundant nitrogen content.31–33 This structure facilitates the effective adsorption of various pollutants, efficient electron conduction, and optimal material usage by concentrating on the active surface while minimizing the contribution of inactive interior regions.34 Concurrently, the dehydrogenative activation of FA, often involving FA deprotonation upon surface adsorption, followed by subsequent hydride transfer or elimination, is expected to benefit from Lewis basic nitrogen sites working in synergy with transition metal-based SACs (TM-SACs) embedded within a carbon framework.35–38 However, the simultaneous activation of FA and the remediation of harmful pollutants, as well as the underlying mechanisms involving SAC-based materials, remain largely unexplored. Moreover, the intricate manufacturing processes for TM-N-co-doped carbonaceous materials, typically involving direct pyrolysis of metal–organic framework precursors or associated composites, prove to be impractical for environmental applications.4 Consequently, there is a compelling need to explore cost-effective alternatives with uniform dual-functional active sites for FA activation.
The strategy presented herein is both convenient and effective, employing easily accessible cobalt phthalocyanine (CoPc) molecules anchored onto nitrogen-doped mesoporous carbon (NMC). These materials function as highly reactive and stable catalysts, efficiently converting hazardous hexavalent Cr(VI) into less toxic Cr(III) through FA activation. Given the prevalent contamination of industrial wastewater with Cr(VI) at concentrations ranging from 20 ppm to 100 ppm, there are significant health risks associated with its carcinogenic and mutagenic properties.39,40 By controlling the nitrogen speciation in NMC materials, it was able to make full use of the synergistic effect between surface-adsorbed CoPc and the pyridinic N-doped carbonaceous support to initiate the desired FA activation. This finding is substantiated by a comprehensive analysis of experimental data. Through capture experiments and electron paramagnetic resonance (EPR) tests, it is confirmed that the reduction of Cr(VI) is driven by a facile FA-consuming catalytic pathway involving coordinated electron transfer processes and H˙. The optimized cobalt single-atom molecular catalyst exhibited remarkable performance, achieving a high turnover frequency (TOF) value of up to 18.1 μmol s−1 g−1 for Cr(VI) degradation. Overall, this research provides valuable insights into a cobalt single-atom molecular catalyst featuring π–π stacking interactions, offering an efficient and sustainable approach for addressing aquatic inorganic pollutants stemming from post-industrial activities.41
Results and discussion
Synthesis and characterization
The initial investigation focused on exploring nitrogen-doped NMC materials with diverse N characteristics. Employing an impregnation process with DMF as the solvent for the molecular precursor enabled uniform dispersion of CoPc molecules onto the N-doped carbonaceous supports.42 Importantly, the unique mesostructure of the different pristine supports remained unchanged after the introduction of CoPc, a fact verified through HRTEM (Fig. 1a, b and Fig. S1, ESI†). Further examinations involving N2 sorption and ICP-OES confirmed the permanent nanoporosity of all the catalysts, with closely matched Co contents (Tables S1 and S2, ESI†). In the analysis of the electronic structure and coordination environment of the resulting complexes, our focus centered on comparing 0.4% CoPc-NMC900 with pristine CoPc and the as-prepared MNC900 material. Notably, when the CoPc content was below 1 wt%, distinct XRD diffraction peaks corresponding to CoPc crystals were absent (Fig. S2, ESI†), indicating the highly dispersed nature of CoPc within the support's surface layer. HAADF-STEM images with aberration correction (Fig. 1a and b) further validated the atomically dispersed form of active CoPc in 0.4% CoPc-NMC900, predominantly appearing as scattered bright spots. Elemental distribution analysis aligned with the results from XRD and HAADF-STEM, visually confirming the atomic dispersion of CoPc species. | ||
| Fig. 1 (a) and (b) HAADF-STEM images and (c)–(f) corresponding elemental mappings of Co, N, C and O for 0.4% CoPc-NMC900. | ||
To gain deeper insights into the nature of the supported CoPc species, a variety of characterization techniques were employed. EXAFS measurements conducted at the Co K-edge indicated that the coordination environment of the single-atomic Co center remained essentially consistent for both 0.4% CoPc-NMC900 and CoPc precursors (Fig. 2). The R-space spectrum fitting of 0.4% CoPc-NMC900 indicated that each Co atom coordinated with four N atoms (Fig. S3 and Table S3, ESI†). However, Co K-edge X-ray absorption near-edge structure (XANES) analysis revealed that the rising edge position for 0.4% CoPc-NMC900 fell between those of Co foil and the CoPc precursor (Fig. 2a). This positioning appears to indicate that the average oxidation state of cobalt in 0.4 wt% CoPc-NMC900 lays between Co0 and Co2+. This observation can be attributed to the extended delocalization of π electrons, a consequence of the π–π interactions between CoPc and NMC900. Furthermore, XPS analysis provided additional evidence for the formation of a charge-transfer complex between CoPc and NMC900. This was evident as the BE of Co 2p in 0.4 wt% CoPc-NMC900, compared to pure CoPc, shifted to a lower BE (Fig. 3a). This shift confirmed the alteration in the coordination environment of the Co atom in CoPc-NMC900, resulting in a change in the charge state of the Co species.43
 | ||
| Fig. 2 (a) Co K-edge XANES spectra and (b) FT-EXAFS spectra of the Co K-edge of samples. | ||
 | ||
| Fig. 3 XPS spectra of (a) Co 2p and (b) N 1s of materials. | ||
Furthermore, the peaks within the N 1s XPS of both 0.4 wt% CoPc-NMC900 and NMC900 can be deconvoluted as follows: oxidized N at 404.5 eV (15.2%) and 403.8 eV (14.6%), graphitic N at 402.3 eV (15.7%) and 402.1 eV (15.1%), pyrrolic N at 400.7 eV (36.1%) and 400.4 eV (35.5%), Co–N at 399.7 eV (1.1%, CoPc-NMC900), and pyridinic N at 398.6 eV (31.9%) and 398.3 eV (34.8%) (Fig. 3b), respectively.44,45 The analysis indicates that the content of pyridinic N species in NMC900 is approximately 34.8 at%, and these species serve as anchors for stabilizing CoPc. This is due to the stronger coordination affinity between pyridinic N and the central atom of CoPc compared to other N species like pyrrolic N and graphitic N.23 The weight ratio of pyridinic N (in NMC900) to anchor CoPc (0.4 wt% CoPc-NMC900) is calculated to be approximately 6.9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. Moreover, the close anchoring of CoPc on the surface of NMC900 results in a decrease in the content of pyridine N in CoPc-NMC900 compared to NMC900. The positive shift in BE for graphitic N, pyrrolic N, and pyridinic N in 0.4 wt% CoPc-NMC900 compared to NMC900 provides evidence of the electronic interaction between CoPc and NMC900. This phenomenon is consistent with the results from Co 2p XPS. This enhanced interaction can be regarded as a driving force that facilitates the even dispersion of CoPc molecules on the surface of NMC900.
:1. Moreover, the close anchoring of CoPc on the surface of NMC900 results in a decrease in the content of pyridine N in CoPc-NMC900 compared to NMC900. The positive shift in BE for graphitic N, pyrrolic N, and pyridinic N in 0.4 wt% CoPc-NMC900 compared to NMC900 provides evidence of the electronic interaction between CoPc and NMC900. This phenomenon is consistent with the results from Co 2p XPS. This enhanced interaction can be regarded as a driving force that facilitates the even dispersion of CoPc molecules on the surface of NMC900.
It is worth mentioning in this context that, in a separate experiment where conventional mineral supports such as TiO2 and C (see Table S2, entries 5 and 6, ESI†) were utilized, the formation of a charge-transfer complex induced by π–π interactions did not occur (Fig. S4, ESI†). This highlights the distinct advantages of employing N-doped carbonaceous supports when crafting electronically modified CoPc-based molecular catalysts. Also noteworthy is that, similar to the previously mentioned CoPc-NMC900 case, the utilization of N-doped carbon mesostructures other than NMC900 as supports yielded similar redshifts in the BE of N 1s across various CoPc-NMCT samples (where T denotes the pyrolysis temperature of the NMC support), as confirmed through XPS analysis (see Fig. S5a, ESI†). The shifts toward lower BEs followed the sequence of CoPc-NMC900 > CoPc-NMC1000 > CoPc-NMC800, suggesting a stronger interaction between exfoliated CoPc and NMC900 compared to NMC800 and NMC1000. This phenomenon can be attributed to the higher prevalence of pyridinic N on NMC900, which amplifies the π–π interactions between CoPc and the underlying supports. Conversely, CoPc and NMC900, obtained through a simple mixing approach of CoPc and NMC900, exhibited minimal shifts in the BE of N 1s (see Fig. S5b, ESI†). Evidently, the randomly dispersed CoPc nanostructures obtained via simple mixing lack the capacity to engage in π–π stacking interactions with NMC900.
Catalytic performance
To evaluate the catalytic performance of CoPc-NMC900, Cr(VI) solution was chosen as model wastewater. In the absence of a catalyst, the absorption intensity of a mixture containing Cr(VI) (50 ppm) and FA (4.3 mmol) remained virtually unchanged over a 20-minute period (Fig. 4a), indicating that FA alone could not efficiently reduce Cr(VI). Conversely, with the addition of CoPc-NMC900 into the solution, the absorption intensity decreased rapidly and disappeared within 20 minutes, accompanied by a color change from yellow to colorless in the reaction solution. This visually confirmed the successful conversion of Cr(VI) to Cr(III). The formation of Cr(III) in the colorless solution was further confirmed by the generation of a green solution upon the addition of excess NaOH (Fig. S6a, ESI†). Notably, when 50 mmol FA (nFA:nCr(VI) = 1040) was used, the reduction of Cr(VI) could be completed within just 3 minutes, demonstrating significantly higher efficiency compared to previous reports.22,25 A comparison of the catalytic reduction of Cr(VI) between CoPc-NMC900 and previously reported catalysts is presented in Table S4 (ESI†). The calculated turnover frequency (TOF) for the composite was 18.1 μmol s−1 g−1, surpassing that of other listed heterogeneous catalysts, even when using a lower molar ratio of FA:Cr(VI) = 90:1, highlighting the superior performance of CoPc-NMC900 in Cr(VI) reduction. It is worth noting that achieving an optimal ratio of CoPc to NMC900 is crucial for Cr(VI) reduction. As shown in Fig. S6b (ESI†), the catalytic performance of the composite exhibited an initial increase followed by a decrease with an increasing CoPc content within the CoPc-NMC900 composite. The best catalytic activity of CoPc-NMC900 with the 0.4 wt% Co content is likely due to the optimal balance between active site exposure and material dispersion, as shown in Fig. S7 (ESI†) by the XPS-determined surface Co/C ratio.
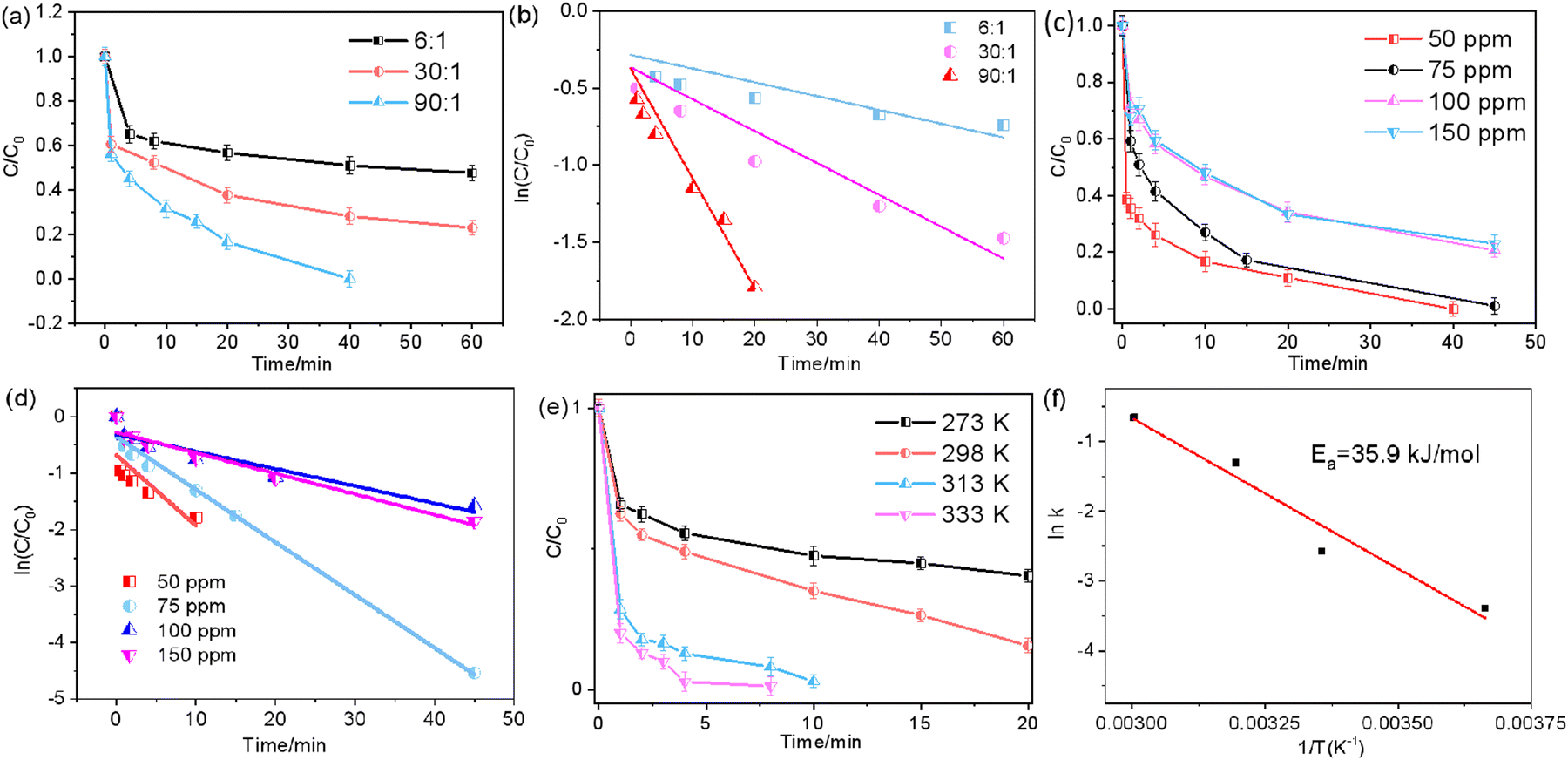 | ||
| Fig. 4 (a) Effect of the molar ratio of FA and Cr(VI) on the removal efficiency of CoPc-NMC900 and (b) corresponding plots of ln(Ct/C0) versus reaction time. (c) Effect of the concentration of Cr(VI) on the removal efficiency of CoPc-NMC900 and (d) corresponding plots of ln(Ct/C0) versus reaction time. (e) Effect of the reaction temperature on the removal efficiency of CoPc-NMC900 and (f) corresponding plots of Arrhenius. | ||
The reduction of Cr(VI) was found to be highly reliant on various reaction conditions, including the concentrations of FA, which illustrates the impact of different FA concentrations on Cr(VI) reduction. It is evident that the reaction rate increased significantly with higher concentrations of FA, as the increased FA concentration provided more reductant and H+ ions, which promoted the reduction of Cr(VI). From Fig. 4b, the reaction rate constant (k) was calculated to be 0.076 min−1 at a lower molar ratio of FA:Cr(VI) of 90:1, demonstrating that this cost-effective and readily available catalyst exhibits exceptional performance in Cr(VI) reduction using FA. In Fig. 4c, the efficiency of Cr(VI) reduction at different initial Cr(VI) concentrations, with a fixed FA:Cr(VI) ratio of 90:1, is depicted. It is evident that the reduction of Cr(VI) gradually decreases with increasing initial Cr(VI) concentration. Specifically, complete elimination of Cr(VI) requires 40 minutes at an initial concentration of 50 ppm, while an 80% reduction efficiency is achieved at an initial Cr(VI) concentration of 100 ppm for 45 min. Simultaneously, the corresponding k decreases from 0.125 to 0.03 min−1 (Fig. 4d). This reduction in the reaction rate may be attributed to the coverage of excess Cr ions on the available active sites provided by the catalyst. As shown in Fig. 4e, the rate of Cr(VI) reduction increased markedly with increasing reaction temperature, indicating that the reduction of Cr(VI) by FA is a temperature-sensitive reaction. The associated reaction rate increased from 0.059 to 0.115 min−1 as the reaction temperature increased from 0 to 45 °C, which can be attributed to the acceleration effect at higher temperatures, facilitating FA molecular collisions on the catalyst surface and leading to a faster release of reactive species. According to the Arrhenius equation, the activation energy (Ea) for Cr(VI) reduction catalyzed by CoPc-NMC900 was calculated to be 35.9 kJ mol−1 (Fig. 4f), which is lower than that observed for previously reported activation energies.22
Furthermore, CoPc-NMC900 demonstrates superior catalytic activity compared to the widely used heterogeneous inorganic single-atom Co–N–C catalyst (Co-CNZIF, see Table S2, entry 7, ESI†) derived from the controlled pyrolysis of the Zn/Co bimetallic ZIF-8.23 Despite having a similar C/N ratio, Co–N4 structure and larger BET surface area (1366 m2 g−1), the lower performance in Cr(VI) reduction observed for Co-CNZIF may be attributed to its poor conductivity, as confirmed in the following section. To discern the key factors governing the catalytic properties, a series of CoPc-NMCT catalysts were prepared. Among these, CoPc-NMC900 exhibited the highest catalytic activity (Fig. S8a, ESI†). Based on the correlation between the relative N content evolution and the corresponding catalytic performance, it becomes evident that pyridinic-N is the most influential nitrogen species (Fig. S8b, ESI†). To further validate the importance of the CoPc-NMC hybrid structure, CoPc deposited on different supports was investigated (see Table S2 and Fig. S9, ESI†). The results revealed significantly lower performance when using CoPc alone, possibly due to CoPc's poor hydrophilicity.44 Even when CoPc is combined with TiO2 or carbon support, it still exhibited considerably lower catalytic activity for the studied reaction. All together, these findings imply that beneficial π–π interaction between isolated CoPc and the pyridinic-N doped carbon matrix is essential to efficiently reduce Cr(VI).
Apart from the high activity displayed by the CoPc-NMC900 catalyst for FA-mediated Cr(VI) reduction, other aspects such as reusability and tolerance to coexisting ions are important for any practical application. Hence, as shown in Fig. S10a (ESI†), the activity remained largely unchanged after five cycles, demonstrating the excellent stability of the CoPc-NMC900 composite. Furthermore, the XPS of Co 2p for CoPc-NMC900 showed minimal changes after repeated catalytic reactions, confirming its robust catalytic stability (Fig. S11a, ESI†). Notably, the catalyst retains its crystalline structure (XRD, Fig. S11b, ESI†), morphological features (TEM, Fig. S11c and d, ESI†), and porous architecture (BET, Table S1, ESI†) following five consecutive reaction cycles. Remarkably, only trace amounts of Co species were detected in the Cr(VI) solution, and the CoPc loading (0.38%) showed minimal change after five cycles (Table S2, entry 3, ESI†). Therefore, the CoPc-NMC900 catalyst exhibited excellent activity and recyclability, underscoring its potential for practical applications. Additionally, the impact of typical coexisting ions, such as Na+, K+, and NH4+ cations, and Cl−, SO42−, and NO3− anions (0.05 M), on FA-mediated Cr(VI) reduction was investigated to evaluate the universal applicability of the CoPc-NMC900 composite (Fig. S10b, ESI†). The results indicated that the catalytic activity of the samples toward Cr(VI) reduction remained unaffected by the presence of interfering ions, highlighting its strong robustness and anti-interference ability. Furthermore, to evaluate the practical one-step conversion of Cr(VI) into Cr(III), a continuous-flow mode employing the CoPc-NMC900 catalyst was performed. In this setup, a 10 mL syringe was used to drive the mixed solution of Cr(VI) (10 mL, 50 ppm) and FA (1 M) through three pieces of the PES membrane (diameter: 2.5 cm) loaded with CoPc-NMC900 (total mass: 15 mg), as illustrated in Fig. S12a (ESI†). Remarkably, it exhibited a powerful capability for efficient Cr(VI) conversion even at a permeation flux of 1650 L m−2 h−1 (LMH), as evidenced by the rapid transformation of the effluent from yellow to colorless. UV-vis absorption spectroscopy analysis of the original and filtered solutions (Fig. S12b, ESI†) revealed a conversion efficiency above 99%, confirming the nearly complete reduction of Cr(VI) to Cr(III) through the continuous-flow mode.
Clarification of catalytic sites and the mechanism
In our investigation of the mechanistic aspects of CoPc-NMC900-catalyzed Cr(VI) reduction, the study was initiated by evaluating the role of H2 as a reductant in the absence of FA. Our findings, illustrated in Fig. S13a (ESI†), clearly demonstrated that H2 is not a feasible active species in the CoPc-NMC900/FA-Cr(VI) system, as efficient reduction of adsorbed Cr(VI) to Cr(III) did not occur. To gain further insights into the observed performance related to the assembly of CoPc and pyridinic-N-based catalytic pairs, a control study involving spiking experiments was conducted. The addition of piperidine, a known poison for transition-metal-based Lewis acid sites, significantly reduced the conversion of Cr(VI) from 95% to 50%, underscoring the indispensability of anchored CoPc for the observed activity of the CoPc-NMC900 material. Furthermore, to block the Py-N sites, CoPc-NMC900 was soaked in a 5 M aqueous H3PO4. Consequently, the conversion of Cr(VI) was notably suppressed to half of the original value (as shown in Fig. 5a). To validate this mechanism, we performed poisoning and regeneration tests. Treating CoPc-NMC900 with 5 M H3PO4 reduced the pyridinic N content (via XPS) and halved its catalytic activity (Fig. S14, ESI†). Upon regenerating the acid-treated sample with K2CO3, pyridinic N levels rebounded, and catalytic activity was nearly restored to its initial value (Fig. S15, ESI†). These findings collectively affirm that the catalytic pairs consisting of anchored CoPc and Py-N sites are indeed responsible for the exceptional activity in Cr(VI) reduction. To further figure out the reaction pathways involved in the reduction of Cr(VI) by FA, kinetic isotope effect (KIE) studies were conducted to determine the rate-determining step (refer to Table S5, ESI†). This aspect is rarely explored in other reports.13–17,45 When the substrate was allowed to react with HCOOD instead of FA, a KIE value of kHCOOH/kHCOOD = 1.52 was obtained, which was smaller than the values obtained when using DCOOH and DCOOD (1.68 and 1.98). These results, along with a KIE value of approximately 1.1 for 0.4% CoPc-NMC900 with pyridinic N sites blocked by H3PO4 (Table S6, ESI†), suggest that the rate-determining step in Cr(VI) reduction involves the dissociation of FA, which includes the cleavage of the C–H bond at active sites with CoPc-pyridinic N pairs.46 | ||
| Fig. 5 (a) Control reactions performed in the presence of various scavengers. (Reaction conditions: 50 mL of Cr(VI) (50 ppm), 4.3 mmol FA, 50 mg of the catalyst, and 25 °C.) (b) EPR spectra of the (A) FA-Cr(VI) system, (B) 0.4% CoPc-NMC900/FA-Cr(VI) system with DMPO for 1 min and (C) 0.4% CoPc-NMC900/FA-Cr(VI) system with DMPO for 3 min. | ||
To elucidate the active species that facilitate the reduction of Cr(VI), various scavengers were introduced into the reaction system. Specifically, AgNO3 was used, known for its ability to scavenge electrons (e−), and DMPO, effective in trapping hydrogen free radicals (H˙).22 The impact of these scavengers on the reaction's effectiveness is illustrated in Fig. 5a. Notably, the introduction of AgNO3 (10 mg) led to a significant decrease in the reaction's efficiency. This, together with the effectiveness in reducing Cr(VI) dropped from an initial 95% to just 52% when 5 mg of DMPO was added, indicating that both electrons and hydrogen radicals play crucial roles in this reaction process. To provide additional evidence for the generation of hydrogen radicals during the reaction, EPR spectroscopy was employed, using DMPO as a spin-trapping agent. The results, displayed in Fig. 5b, revealed a distinct EPR signal characterized by a unique pattern: a triplet signal followed by a doublet, specifically a 1:1:1 triplet of a 1:2:1 triplet. This pattern is the characteristic of a DMPO-H˙ adduct. This finding in the CoPc-NMC900/FA-Cr(VI) system conclusively demonstrates the formation of active hydrogen radicals during the reduction process, thus substantiating the integral role of these radicals in the reaction mechanism.
To better understand how Co-based SACs affect the activation of FA, a series of comparative tests focusing the sole dehydrogenation of FA were conducted. These tests were essential, as dehydrogenating aqueous FA at room temperature is impractical. Therefore, these experiments were performed at 110 °C, using propylene carbonate as a solvent. According to the results presented in Table S7 (ESI†), the CoPc-NMC900 catalyst demonstrated markedly higher activity in FA dehydrogenation compared to CoPc, CoPc-TiO2, and CoPc-C. It is noteworthy that when only NMC900 was used as the catalyst, there was no observable gas production. Interestingly, while Co-CNZIF showed a significantly higher TOF in sole FA dehydrogenation, its efficacy in reducing Cr(VI) via FA was significantly less than expected, especially compared to the results with CoPc-NMC900. Co-CNZIF, being a fully inorganic compound containing atomically dispersed CoN4 entities embedded in porous N-doped graphitic carbon, is created by pyrolyzing a cobalt-zinc-based bimetallic zeolitic imidazolate framework (Co-Zn-BMZIF-8).47,48 This contrast in performance suggests that there are other crucial factors that influence the efficiency of Cr(VI) reduction.
To gain deeper insights into the exceptional activity of CoPc-NMC900, analysis of its adsorption capacity was conducted under reaction conditions. It was observed that CoPc-NMC900 exhibited a substantial adsorption capacity for Cr(VI), adsorbing approximately 26% of the Cr(VI) present (Fig. S8, ESI†). This observation aligns with the established understanding that efficient Cr(VI) reduction relies on effective Cr(VI) adsorption. Notably, the point of zero charge for CoPc-NMC900, approximately 8, is higher than that of Co-CNZIF, which is around 7 (Fig. S13b, ESI†). This discrepancy indicates that, especially under acidic conditions, the highly positively charged surface of CoPc-NMC900 can offer stronger electrostatic attraction to Cr(VI) anions. To identify the specific surface sites responsible for Cr(VI) adsorption, XPS analysis on the catalyst surface after the catalytic Cr(VI) reduction was performed. The Cr 2p spectrum revealed a distinct peak at approximately 577.3 eV, corresponding to Cr(III). This result implies that Cr(VI) species were successfully anchored on the catalyst surface and subsequently reduced to Cr(III) (Fig. S16a, ESI†). Additionally, the high-resolution C 1s spectrum after the catalytic reaction displayed a decrease in the proportion of C–OH functional groups, declining from 32.2% to 23.8% (Fig. S16b, ESI†). This decrease suggests that some surface functional groups may serve as anchor sites for Cr(VI) species. To further investigate the factors contributing to the CoPc-NMC900's superior Cr(VI) reduction performance, electrochemical impedance spectroscopy (EIS) tests were conducted (Fig. S17, ESI†). It indicated that CoPc-NMC900 exhibited a smaller charge-transfer resistance (Rct) compared to NMC900 and Co-CNZIF. This finding suggests a higher charge-transfer rate for CoPc-NMC900, indicating its enhanced ability for electron transport. Collectively, these results suggest that CoPc-NMC900's superior performance in Cr(VI) reduction can be attributed to its enhanced adsorption and electron transport properties compared to Co-CNZIF.
Based on the above characterization analysis and experimental results, a plausible mechanism is formulated to explain the Cr(VI) reduction (Scheme 1). Initially, FA and Cr(VI) are adsorbed onto the surface of CoPc-NMC900 due to electrostatic attraction. Within this context, the adsorbed FA molecules undergo deprotonation, leading to the formation of bridging bidentate formate species through O–H dissociation at the Lewis basic sites (pyridinic-N). Subsequently, HCOO− undergoes decomposition via C–H breakage, which is facilitated by another catalytic active center (CoPc), situated on the N-decorated carbon surface. This decomposition process generates critical active H˙ species and CO2. It is noteworthy that the rate-limiting step for active H˙ generation is the C–H cleavage, and the kinetic barrier of this step is closely influenced by the adsorbed configuration of the formate intermediate. As a result, the adsorbed Cr(VI) can be efficiently reduced to Cr(III) through a H˙ transfer pathway when electron-donor acids are present. These mechanistic insights can be succinctly summarized using the following equations:
| HCOOHabs → HCOOabs− + H+ | (1) |
| HCOOabs− → CO2 + Habs− | (2) |
| Habs− → e− + H˙ | (3) |
| Cr(VI)abs + 3H˙ → Cr(III) + 3H+ | (4) |
| Cr2O72− + 14H+ + 6e− → 2Cr3+ + 7H2O | (5) |
 | ||
| Scheme 1 The possible catalytic mechanism of Cr(VI) reduction. | ||
Conclusions
In summary, this study presents a promising approach for advanced reduction processes by harnessing the potential of CoPc integrated with nitrogen-doped mesoporous carbon (NMC). The CoPc-NMC composite exhibits exceptional efficiency in reducing various pollutants, with a primary focus on Cr(VI) reduction using FA as the reducing agent. The outstanding performance of CoPc-NMC can be attributed to its distinctive characteristics, including the presence of atomically dispersed CoPc species, a high pyridinic nitrogen content, and robust π–π interactions between CoPc and the N-doped carbonaceous support. Furthermore, our investigation into the active species involved in the reaction, encompassing electrons and hydrogen radicals, underscores the intricate nature of the catalytic process. This research not only elucidates the pivotal factors governing CoPc-NMC's catalytic activity but also provides valuable insights into the mechanism of Cr(VI) reduction. These insights have significant implications for the design and optimization of catalysts for addressing environmental pollutant remediation challenges, with promising applications extending beyond Cr(VI) reduction.Author contributions
Mengjiao Xu: conceptualization, data curation, investigation, methodology, validation, and writing and editing; Kaizhi Wang: data curation, validation, and review and editing; Wendi Guo: validation and review and editing; Zehui Sun: validation and review and editing; Mugeng Chen: validation and review and editing; Yongmei Liu: data curation, validation, and review and editing; Yong Cao: funding acquisition, methodology, investigation, project administration, resources, supervision, and writing – review and editing.Data availability
The data supporting this article have been included as part of the ESI.† The other data are available from the corresponding author upon reasonable request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (22272032, 21972024, and 22088101), the National Key R&D Program of China (2022YFA1503804), and the Science & Technology Commission of Shanghai Municipality (19DZ2270100) and SINOPEC (X514005).Notes and references
- B. P. Vellanki, B. Batchelor and A. Abdel-Wahab, Environ. Eng. Sci., 2013, 30, 264–271 CrossRef CAS PubMed
.
- S. Y. Yang and D. Zheng, Prog. Chem., 2016, 28, 934–941 CAS
- A. G. Capodaglio, Appl. Sci., 2020, 10, 4549 CAS
- L. Oar-Arteta, T. Wezendonk, X. Sun, F. Kapteijn and J. Gascon, Mater. Chem. Front., 2017, 1, 1709–1745 CAS
- A. Boddien, D. Mellmann, F. Gärtner, R. Jackstell, H. Junge, P. J. Dyson, G. Laurenczy, R. Ludwig and M. Beller, Science, 2011, 333, 1733–1736 CrossRef CAS PubMed
- X. Liu, S. Li, Y. Liu and Y. Cao, Chin. J. Catal., 2015, 36, 1461–1475 CrossRef CAS
- Q. Y. Bi, X. L. Du, Y. M. Liu, Y. Cao, H. Y. He and K. N. Fan, J. Am. Chem. Soc., 2012, 134, 8926–8933 CAS
- Q. Y. Bi, J. D. Lin, Y. M. Liu, H. Y. He, F. Q. Huang and Y. Cao, J. Power Sources, 2016, 328, 463–471 CrossRef CAS
- T. Jiang, P. Lu, M. J. Xu, J. D. Lin, Y. M. Liu, Y. Cao and H. Y. He, J. Energy Chem., 2020, 49, 205–213 Search PubMed
- M. Yadav and Q. Xu, Energy Environ. Sci., 2012, 5, 9698–9725 CAS
- K. Sordakis, C. Tang, L. K. Vogt, H. Junge, P. J. Dyson, M. Beller and G. Laurenczy, Chem. Rev., 2018, 118, 372–433 CAS
- Q. Y. Bi, J. D. Lin, Y. M. Liu, X. L. Du, J. Q. Wang, H. Y. He and Y. Cao, Angew. Chem., Int. Ed., 2014, 53, 13583 CAS
- P. Veerakumar and K. C. Lin, Chemosphere, 2020, 253, 126750 CrossRef CAS
- Z. H. Farooqi, M. W. Akram, R. Begum, W. Wu and A. Irfan, J. Hazard. Mater., 2021, 402, 123535 CrossRef CAS PubMed
- M. A. Omole, I. O. K’Owino and O. A. Sadik, Appl. Catal., B, 2007, 76, 158–167 CrossRef CAS
- M. Celebi, M. Yurderi, A. Bulut, M. Kaya and M. Zahmakiran, Appl. Catal., B, 2016, 180, 53–64 CrossRef CAS
- C. Qin, G. Pan, Y. Zhang, F. Ding, J. Qu, X. Xu and X. Su, Catalysts, 2022, 12, 179 CrossRef CAS
- B. Jiang, Y. Gong, J. Gao, T. Sun, Y. Liu, N. Oturan and M. A. Oturan, J. Hazard. Mater., 2019, 365, 205–226 CrossRef CAS PubMed
- H. Karimi-Maleh, A. Ayati, S. Ghanbari, Y. Orooji, B. Tanhaei, F. Karimi, M. Alizadeh, J. Rouhi, L. Fu and M. Sillanpää, J. Mol. Liq., 2021, 329, 115062 CrossRef CAS
- M. M. Islam, A. A. Mohana, M. A. Rahman, M. Rahman, R. Naidu and M. M. Rahman, Toxics, 2023, 11, 252 CrossRef CAS PubMed
- G. Yan, Y. Gao, K. Xue, Y. Qi, Y. Fan, X. Tian, J. Wang, R. Zhao, P. Zhang, Y. Liu and J. Liu, Front. Environ. Sci., 2023, 11, 1131204 CrossRef
- J. Jiang, S. Yang, W. Wei, S. Tu, X. Zheng and L. Ai, Appl. Surf. Sci., 2023, 625, 157170 CrossRef CAS
- Q. Zhang, M. Peng, Z. Gao, W. Guo, Z. Sun, Y. Zhao, W. Zhou, M. Wang, B. Mei, X. L. Du, Z. Jiang, W. Sun, C. Liu, Y. Zhu, Y. M. Liu, H. Y. He, Z. H. Li, D. Ma and Y. Cao, J. Am. Chem. Soc., 2023, 145, 4166–4176 CrossRef CAS
- Y. Shi, B. Luo, R. Liu, R. Sang, D. Cui, H. Junge, Y. Du, T. Zhu, M. Beller and X. Li, Angew. Chem., Int. Ed., 2023, 62, e202313099 CrossRef CAS
- S. Li, L. Liu, Q. Zhao, C. He and W. Liu, Phys. Chem. Chem. Phys., 2018, 20, 3457–3464 RSC
- Z. Lv, S. Liu, Y. Liu, P. Liu, M. Fang, X. Tan, W. Xu, M. Kong and X. Wang, Sep. Purif. Technol., 2022, 295, 121289 CrossRef CAS
- L. Liu and A. Corma, Chem. Rev., 2018, 118, 4981–5079 CAS
- H. Yang, G. Li, G. Jiang, Z. Zhang and Z. Hao, Appl. Catal., B, 2023, 325, 122384 CrossRef CAS
- A. Wang, J. Li and T. Zhang, Nat. Rev. Chem., 2018, 2, 65–81 CrossRef CAS
- K. Yin, R. Wu, Y. Shang, D. Chen, Z. Wu, X. Wang, B. Gao and X. Xu, Appl. Catal., B, 2023, 329, 122558 CrossRef CAS
- Y. Zeng, E. Almatrafi, W. Xia, B. Song, W. Xiong, M. Cheng, Z. Wang, Y. Liang, G. Zeng and C. Zhou, Coord. Chem. Rev., 2023, 475, 214874 CrossRef CAS
- J. F. Sun, Q. Q. Xu, J. L. Qi, D. Zhou, H. Y. Zhu and J. Z. Yin, ACS Sustainable Chem. Eng., 2020, 8, 14630–14656 CrossRef CAS
- P. Xia, Z. Ye, L. Zhao, Q. Xue, S. Lanzalaco, Q. He, X. Qi and I. Sirés, Appl. Catal., B, 2023, 322, 122116 CrossRef CAS
- M. B. Gawande, P. Fornasiero and R. Zbořil, ACS Catal., 2020, 10, 2231–2259 CrossRef CAS
- J. F. Perez-Benito and C. Arias, Can. J. Chem., 1993, 71, 649 CrossRef CAS
- Q. Y. Bi, J. D. Lin, Y. M. Liu, H. Y. He, F. Q. Huang and Y. Cao, Angew. Chem., Int. Ed., 2016, 55, 11849 CrossRef CAS PubMed
- Y. Yao, J. Zhang, H. Chen, M. Yu, M. Gao, Y. Hu and S. Wang, Appl. Surf. Sci., 2018, 440, 421–431 CrossRef CAS
- Z. Lv, W. Chen, Y. Cai, K. Chen, K. Li, M. Fang, X. Tan and X. Wang, Appl. Surf. Sci., 2022, 575, 151748 CrossRef CAS
- Y. X. Zhang, M. J. Xu, H. Li, H. Ge and Z. F. Bian, Appl. Catal., B, 2018, 226, 213–219 CrossRef CAS
- R. F. Oliveira, K. G. P. Nunes, I. V. Jurado, I. C. B. Amador, D. C. Estumano and L. A. Féris, Environ. Technol. Innovation, 2020, 20, 101092 CrossRef CAS
- T. Li, F. Zhu, Y. Q. Gao, M. R. Iribagiza, G. Y. Hu and J. Guan, Environ. Sci.: Water Res. Technol., 2024, 10, 339–352 RSC
- S. Xie, Green Chem. Lett. Rev., 2024, 17, 2356614 CrossRef
- X. Yu, S. Lai, S. Xin, S. Chen, X. Zhang, X. She, T. Zhan, X. Zhao and D. Yang, Appl. Catal., B, 2021, 280, 119437 CrossRef CAS
- J. Su, C. B. Musgrave, Y. Song, L. Huang, Y. Liu, G. Li, Y. Xin, P. Xiong, M. M. J. Li, H. Wu, M. Zhu, H. M. Chen, J. Zhang, H. Shen, B. Z. Tang, M. Robert, W. A. Goddard and R. Ye, Nat. Catal., 2023, 6, 818–828 CrossRef CAS
- Z. Lv, X. Tan, C. Wang, A. Alsaedi, T. Hayat and C. Chen, Chem. Eng. J., 2020, 389, 123428 CrossRef CAS
- S. S. Li, L. Tao, F. Z. R. Wang, Y. M. Liu and Y. Cao, Adv. Synth. Catal., 2016, 358, 1410–1416 CrossRef CAS
- Y. Zhao, K. Z. Wang, Z. H. Sun, Q. Zhang, Z. J. Wang, Y. M. Liu, H. Y. He and Y. Cao, Green Chem., 2022, 24, 4095–4107 RSC
- A. Boontanom, M. Maddaloni, P. Suwanpinij, I. Vassalini and I. Alessandri, Environ. Sci.: Water Res. Technol., 2024, 10, 551–564 RSC
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ma01129e |
| This journal is © The Royal Society of Chemistry 2025 |