
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProton insertion chemistry in a phenazine-based cathode for aqueous Zn-organic batteries†
Yutian
Xiang‡
,
Xinran
Li‡
,
Chaoyi
Qiu
,
Wenhui
Yang
,
Lei
Liu
,
Haoxiang
Yu
,
Liyuan
Zhang
 ,
Lei
Yan
* and
Jie
Shu
*
,
Lei
Yan
* and
Jie
Shu
*
School of Materials Science and Chemical Engineering, Ningbo University, Ningbo, Zhejiang 315211, China. E-mail: yanlei@nbu.edu.cn; shujie@nbu.edu.cn
First published on 8th January 2025
Abstract
Highly active and stable cathodes play a crucial role in aqueous Zn-organic batteries, enabling them to achieve high capacity, rapid redox kinetics, and an extended lifespan. However, currently reported electrode materials for Zn-organic batteries face challenges such as low capacity and inadequate cycling stability. In this contribution, aiming to overcome the challenges above, we develop a new Zn-organic battery. In this battery, saturated ZnSO4 served as an electrolyte and its cathode is based on dipyrido [3,2-a:2′,3′-c] phenazine (DPPZ). Theoretical calculations and ex situ analyses demonstrate that the Zn//DPPZ batteries mainly undergo an H+ uptake/removal process with a highly reversible structural evolution of DPPZ. As a result, the aqueous Zn//DPPZ full cell exhibits a remarkable capacity of 94 mA h g−1 at a mass-loading of 2 mg cm−2 (achieved at 0.5 A g−1), and rapid kinetics. Moreover, the cell possesses remarkable cycling durability such that at a mass-loading of 2 mg cm−2, the cell owns a long lifespan of 8000 cycles with a current density of 5 A g−1, and even at a high mass-loading of 8 mg cm−2, it can still endure 600 cycles with a current density of 0.5 A g−1. These findings pave the way for the development of advanced organic electrodes.
Introduction
Aqueous zinc-ion batteries (ZIBs) offer great potential as future energy storage solutions, primarily due to the advantages associated with using a Zn anode. These advantages include a high specific capacity of 820 mA h g−1, a low redox potential (−0.76 V vs. standard hydrogen electrode), cost efficiency, environmental sustainability, and enhanced safety.1–8 Inorganic materials such as Prussian blue, manganese substances, and vanadium oxides have been extensively studied as cathodes for Zn batteries.9–15 However, their practical application is often hindered by sustainability, dynamics, and stability issues. In particular, repeated intercalation of Zn2+ can lead to significant structural degradation of these materials.16–25Organic electrode materials are considered as an environmentally friendly and sustainable alternative for rechargeable Zn batteries. One of the key advantages of organic electrodes is their ability to possess suitable redox potentials for aqueous electrolytes, which can be easily adjusted through molecular design.26–30 Additionally, the spacious internal structure of organic materials allows for excellent cycling stability by effectively accommodating the volume changes caused by the insertion of hydrated Zn2+/H+ ions. Recently, carbonyl (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) and azine (CN) compounds have shown promise due to their n-type redox characteristics. These compounds store charge through coordination reactions between Zn2+/H+ ions and CO/CN groups.31–36 However, organic electrodes for Zn batteries are still in the early stages of development and face limitations such as low capacity and poor cyclability. In particular, small molecular electrode materials, while advantageous for mass production and practical applications due to their simplicity and relatively low cost, often exhibit high solubility in electrolyte.37–43 This leads to the undesired “shuttling” of active materials and rapid capacity decay in batteries. Therefore, the development of highly redox-active organic materials remains a significant challenge but is vital for the advancement of Zn batteries.
O) and azine (CN) compounds have shown promise due to their n-type redox characteristics. These compounds store charge through coordination reactions between Zn2+/H+ ions and CO/CN groups.31–36 However, organic electrodes for Zn batteries are still in the early stages of development and face limitations such as low capacity and poor cyclability. In particular, small molecular electrode materials, while advantageous for mass production and practical applications due to their simplicity and relatively low cost, often exhibit high solubility in electrolyte.37–43 This leads to the undesired “shuttling” of active materials and rapid capacity decay in batteries. Therefore, the development of highly redox-active organic materials remains a significant challenge but is vital for the advancement of Zn batteries.
In this study, a novel Zn-organic battery is presented which operates with a mild aqueous electrolyte, saturated ZnSO4. The cathode consists of dipyrido [3,2-a:2′,3′-c] phenazine (DPPZ), while Zn foil serves as the anode. The battery's function is intimately linked to the coordination reaction occurring in the cathode and the zinc oxidation/reduction process happening on the anode. DPPZ is classified as a π-conjugated aromatic compound that contains CN groups. These CN groups can mainly form coordination bonds with H+ ions. As a result, the Zn//DPPZ cells demonstrate a high capacity of 94 mA h g−1 at 0.5 A g−1, along with exceptional cycling performance for 8000 repetitive discharge–charge cycles at 5 A g−1. This signifies that even with a high mass-loading of DPPZ (8 mg cm−2) in the cathode, a respectable cycle life of 600 cycles can be achieved at 0.5 A g−1. Additionally, through a combination of theoretical calculation and ex situ characterizations, the charge storage mechanism of the DPPZ organic electrode has been comprehensively elucidated.
Results and discussion
In this work, we assemble an aqueous Zn-organic battery, which involves a dipyrido [3,2-a:2′,3′-c] phenazine (DPPZ) cathode, a Zn anode and a mild aqueous electrolyte (i.e., saturated ZnSO4). The component of the organic cathode is non-toxic, showing an environment-friendly nature. The pH value of the saturated ZnSO4 solution is 3.1, which implies the presence of many H+ ions. Therefore, the H+ insertion is taking place in the DPPZ electrode by the coordination reaction in the discharge process (Scheme 1). The charge process reverses discharge.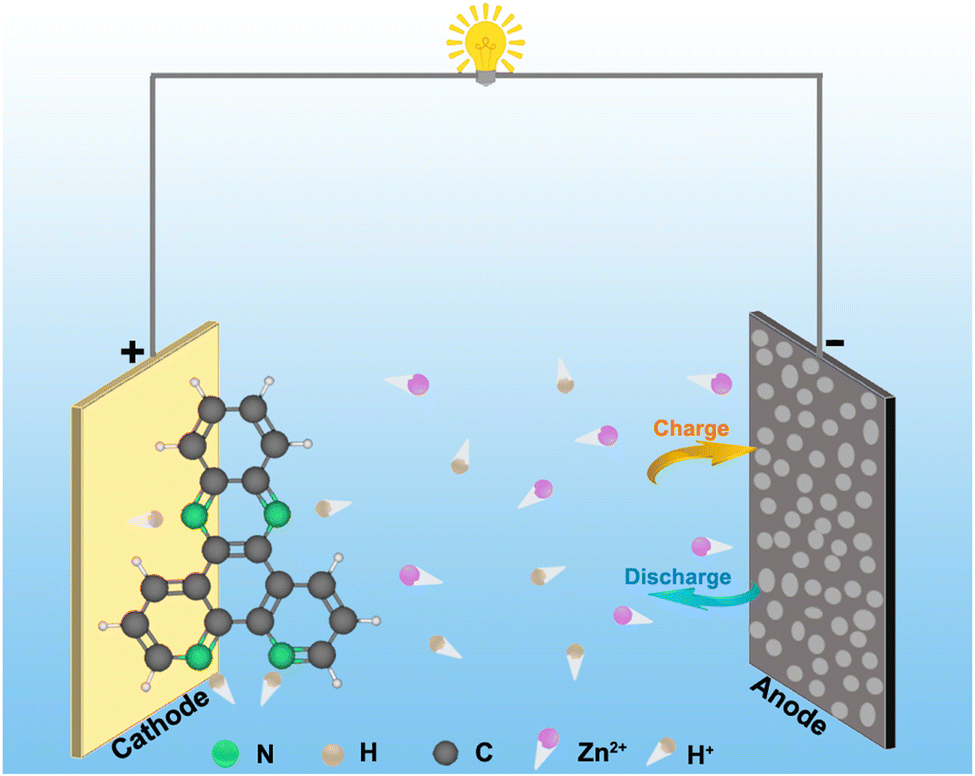 | ||
| Scheme 1 Schematic illustration of the aqueous Zn//DPPZ cell. | ||
The morphology of the DPPZ sample was examined using scanning electron microscopy (SEM). Additionally, we performed various characterization techniques to confirm the chemical composition of DPPZ, including Fourier transform infrared (FTIR) spectroscopy, nuclear magnetic resonance (NMR) spectroscopy, and X-ray diffraction (XRD) analysis. Fig. S1 and S2 (ESI†) show the SEM images of DPPZ, displaying a nanowire morphology with an average diameter of 200 nm. Fig. 1a exhibits the FTIR spectroscopy of DPPZ. The peaks located at 1134 cm−1, 1485 cm−1 and 1574 cm−1, are assigned to the stretching vibration of C–N, CC and CN bonds, respectively. Furthermore, the C–H in- plane and out-of-plane bending vibration in the aromatic ring corresponds to the peaks appearing at 1071 and 744 cm−1, respectively. The 1H NMR spectrum (Fig. 1b) shows five distinct peaks, corresponding to the five types of hydrogen atoms present in DPPZ. In addition, the 13C NMR spectrum (Fig. 1c) shows nine peaks, which is consistent with the nine types of C atoms in the DPPZ structure. The purity of the DPPZ sample was also confirmed by the X-ray diffraction (XRD) pattern, as shown in Fig. 1d.
 | ||
| Fig. 1 (a) FT-IR spectrum of DPPZ. (b) 1H NMR spectrum of DPPZ. (c) 13C NMR spectrum of DPPZ. (d) The XRD pattern of DPPZ. | ||
The electrochemical performance of the DPPZ electrode was assessed using a Zn-organic coin cell (i.e., Zn//DPPZ) with a saturated ZnSO4 aqueous electrolyte. Fig. 2a illustrates the cyclic voltammetry (CV) profiles of the Zn//DPPZ cells at scan rates ranging from 0.2 mV s−1 to 0.6 mV s−1 within a voltage window of 0.2 V to 1.6 V. The CV curves display one pair of redox peaks at 0.53/0.69 V, indicating a one-step redox reaction. As the scan rate increases, the cathodic peaks shift to higher potentials, while the anodic peaks shift to lower potentials. By analyzing the relationship between scan rate (v) and peak current (i), it is found that the calculated values of b for peaks 1 and 2 are 0.74 and 0.85, respectively (Fig. 2b). These values suggest a surface capacitive-dominant charge storage nature, indicating fast kinetics. Additionally, the capacitive contribution increases with increasing scan rate, ultimately reaching 80% at a scan rate of 0.6 mV s−1 (Fig. 2c). Due to its fast reaction kinetics, the Zn//DPPZ cell delivers a high capacity of 94 mA h g−1 with a current density of 0.5 A g−1 and maintains specific capacities of 89, 83, and 77 mA h g−1, respectively, as the current density increases (1.0, 2.0, and 5 A g−1) (Fig. 2d). Even when the current density increases to 10 A g−1, a respectable specific capacity of 70 mA h g−1 can still be obtained, verifying its excellent rate performance. Fig. 2d shows the data at different current densities, which is used to calculate the energy density and power density of the battery. As a result, the cells possess a high energy density of 58 W h kg−1 at a power density of 700 W kg−1 (Fig. S3, ESI†). The cycling stability measurement (Fig. 2e) reveals a specific capacity of 60 mA h g−1 and an ultra-stable durability with only 23% decay after 8000 cycles at 5 A g−1, demonstrating preeminent cycling performance.
 | ||
| Fig. 2 (a) CV curves of DPPZ at different scan rates. (b) The linear fit of the peak current density with scan rate. (c) The capacitive contributions at various scan rates. (d) Rate performance measured in the current range of 0.5–10 A g−1. (e) Cycling performance at the current density of 5 A g−1. | ||
Density functional theory (DFT) calculations were performed to explore the interactions of Zn2+ and H+ with the active sites in the DPPZ molecule. Firstly, we calculated the frontier molecular orbitals for DPPZ binding Zn2+ and/or H+, including the lowest unoccupied molecular orbital (LUMO) and the highest occupied molecular orbital (HOMO). As shown in Fig. 3a, the energies of the LUMO and HOMO for DPPZ binding only H+ (i.e., DPPZ–H4) were calculated to be −0.0066 and −0.1639 eV, respectively. For DPPZ binding two H+ and one Zn2+ (i.e., DPPZ–ZnH2), the energies of the LUMO and HOMO were calculated to be −0.0001 and −0.1725 eV, respectively. Thus, it is indicated that the storage of H+ in DPPZ is more favorable, because its energy gap (ΔE) of DPPZ–H4 between the HOMO and LUMO is 0.1573 eV, which is lower than that of DPPZ–ZnH2 (0.1724 eV). This can be also reflected by the calculated binding free energy (ΔGb). As shown in Fig. 3b, ΔGb of DPPZ–H4 is −50.46 eV, which is lower than the ΔGb for DPPZ–ZnH2 (−44.87 eV). This result suggests that DPPZ mainly tending to store H+ during the discharge process.
 | ||
| Fig. 3 (a) Optimized structures and calculated frontier molecular orbital energies of DPPZ–H4 and DPPZ–ZnH2 molecules. (b) Calculated MESP distributions on the van der Waals surface of DPPZ–H4 and DPPZ–ZnH2 molecules. | ||
The charge storage mechanism of the Zn//DPPZ cell was further investigated using ex situ characterizations at different discharge–charge states. Demonstrated in Fig. 4a is the ex situ FT-IR spectra collected to explore the intrinsic electrochemical activity of DPPZ, such as the active sites and reaction process of the cathode material. During the process of discharging, the peak intensity of the CN bond at 1560 cm−1 gradually declines, revealing that the CN bond is the redox-active site in DPPZ. Simultaneously, the peak intensities of the C–N (1109 cm−1) and N–H (3072 cm−1) bonds increase, suggesting that the CN bond in DPPZ is reduced to a C–N bond and H+ ions in the electrolyte are stored to form N–H by the coordination reaction. In the process of charging, the peak intensities of these functional groups (i.e., CN, C–N and N–H) gradually return to their initial states, manifesting that these structural changes of DPPZ resulting in electrochemical activity are reversible. The electrochemical reaction mechanism of the DPPZ electrode was further confirmed by ex situ XPS spectra. Fig. 4b–d shows the recorded XPS spectra of N 1s at different discharged/charged states. After discharge from the initial state to 0.2 V, it is observed that the characteristic peak of the C–N bond appears at 399.3 eV, while the characteristic peak of the CN bond at 398 eV disappears. This phenomenon is attributed to the coordination reaction between the CN bonds and H+ ions. Upon recharging to 1.6 V (i.e., returning from 0.2 V to 1.6 V), the CN peak reappears and the C–N peak disappears, indicating the release of H+ ions. This result indicates that the DPPZ electrode is mainly responsible for the storage of H+ ions, which is consistent with the findings of the DFT results. In addition, to explore whether Zn2+ ions are involved in storage, we compared the galvanostatic charge/discharge behaviors of DPPZ electrodes in a non-aqueous electrolyte containing Zn2+ (i.e., 0.5 M Zn(OTF)2 in N,N-dimethylformamide (DMF) and a saturated ZnSO4 electrolyte. As shown in Fig. S4 (ESI†), DPPZ electrodes exhibit significantly reduced capacity (59 mA h g−1) in 0.5 M Zn(OTF)2/DMF electrolyte compared to that (188.9 mA h g−1) in saturated ZnSO4 electrolyte, indicating that the capacity contribution of Zn2+ storage in the DPPZ electrodes is around 31%. This result reveals that the H+ storage dominates the overall reaction.
 | ||
| Fig. 4 (a) The ex situ FTIR spectra of DPPZ electrode at the marked points in the discharge/charge curve. (b)–(d) The ex situ N 1s XPS spectra of the DPPZ electrode at different states. | ||
Indeed, it is essential to the practical application of batteries to achieve a high gravimetric capacity in a high mass-loading electrode. To address this, we fabricated DPPZ electrodes with mass-loading ranging from 2 to 8 mg cm−2. The charge/discharge profiles of the DPPZ cathode with different kinds of mass-loading at an applied current density of 0.5 A g−1 are displayed in Fig. 5a. As Fig. 5a depicts, the Zn//DPPZ cell with a mass-loading of 2 mg cm−2 delivers a capacity of 94 mA h g−1. Even when its mass-loading reaches 8 mg cm−2, the cell still exhibits a capacity of 85.5 mA h g−1. The rate performance of the Zn//DPPZ cell was also investigated with different mass-loading, and the results are depicted in Fig. 5b and Fig. S5 and S6 (ESI†). Fig. 5b demonstrates excellent rate ability of the Zn//DPPZ cell at mass-loading of 2, 5 and 8 mg cm−2 in the cathode. As described in Fig. 5b, when the mass-loading is at 5 mg cm−2 and under a current density of 10 A g−1, the cell retains a reversible capacity of 50 mA h g−1, which corresponds to 59% of the capacity achieved at a current density of 0.5 A g−1. Even at a higher mass-loading of 8 mg cm−2, with a current density of 5 A g−1, the reversible capacity of the cell reaches 40.2 mA h g−1, which corresponds to 47% of the capacity achieved at a current density of 0.5 A g−1. Based on the rate capability data presented in Fig. 5b, the energy and power densities of the Zn//DPPZ cell are computed and are illustrated in a Ragone plot (Fig. S7, ESI†). The plot shows that at a mass-loading of 5 mg cm−2, the Zn//DPPZ cell features a prominent energy density of 54 W h kg−1, and even at a higher mass-loading of 8 mg cm−2, it still maintains a decent energy density of 52 W h kg−1. Furthermore, we evaluated the cycling stability of the Zn//DPPZ cell at a high mass-loading. Fig. 5c demonstrates that the cell exhibits good cycling stability at a mass-loading of 8 mg cm−2. As shown in Fig. 5c, it can cycle for more than 600 cycles at a current density of 0.5 A g−1 and there is only 24% attenuation of its initial capacity.
 | ||
| Fig. 5 (a) The discharge–charge curves of the Zn//DPPZ cell with different active mass-loading. (b) Rate capability of the Zn//DPPZ cell with various active mass-loadings. (c) Cycling performance at 0.5 A g−1 with mass-loading of 8 mg cm−2. | ||
Conclusion
In summary, we have investigated the potential of a novel organic material, dipyrido [3,2-a:2′,3′-c] phenazine (DPPZ), as a cathode material for aqueous Zn batteries. Through theoretical calculations and various ex situ characterization techniques, we have comprehensively analyzed the charge storage behavior in DPPZ. Our findings indicate that the CN groups within DPPZ are mainly the active site for H+ storage. The high reaction kinetics of the DPPZ cathode contribute to the exceptional performance of Zn//DPPZ full cells, which exhibit a notable discharge capacity of 94 mA h g−1 at a current density of 0.5 A g−1 with a mass-loading of 2 mg cm−2. Furthermore, these cells display excellent rate capability and extended cycling stability, maintaining a high-capacity retention ratio of 78% after 8000 cycles at a current density of 5 A g−1. Additionally, the mass-loading coming to 8 mg cm−2 in the DPPZ cathode, the Zn//DPPZ cell achieves a remarkable capacity of 85.5 mA h g−1 at a current density of 0.5 A g−1 and exhibits excellent cycling performance with over 600 cycles. Overall, our study introduces DPPZ as an up-and-coming organic cathode material for zinc batteries and extends the way for the exploitation of high-performance organic cathodes in the future.
Author contributions
Yutian Xiang: conceptualization, methodology, software, data processing, writing – original draft preparation. Xinran Li: methodology, validation, writing – review & editing. Chaoyi Qiu, Wenhui Yang, Lei Liu, Haoxiang Yu, Liyuan Zhang: visualization, investigation. Lei Yan and Jie Shu: validation, supervision, writing – review & editing.Data availability
The authors declare that all the relevant data are available within the paper and its ESI† file or from the corresponding author on reasonable request.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could influence the work reported in this paper.Acknowledgements
This work is supported by the National Natural Science Foundation of China (22209082), the Natural Science Foundation of Zhejiang Province (LY23B030005), and the Natural Science Foundation of Ningbo (2023J098).References
- B. Y. Tang, L. T. Shan, S. Q. Liang and J. Zhou, Energy Environ. Sci., 2019, 12, 3288–3304 RSC.
- J. N. Hao, B. Li, X. L. Li, X. H. Zeng, S. L. Zhang, F. H. Yang, S. L. Liu, D. Li, C. Wu and Z. P. Guo, Adv. Mater., 2020, 32, 2003021 CrossRef CAS PubMed.
- H. Z. Zhang, Y. B. Fang, F. Yang, X. Q. Liu and X. H. Lu, Energy Environ. Sci., 2020, 13, 2515–2523 RSC.
- Z. W. Tie, L. J. Liu, S. Z. Deng, D. B. Zhao and Z. Q. Niu, Angew. Chem., Int. Ed., 2020, 59, 4920–4924 CrossRef CAS PubMed.
- W. Sun, F. Wang, S. Y. Hou, C. Y. Yang, X. L. Fan, Z. H. Ma, T. Gao, F. D. Han, R. Z. Hu, M. Zhu and C. S. Wang, J. Am. Chem. Soc., 2017, 139, 9775–9778 CrossRef CAS PubMed.
- F. N. Mo, G. J. Liang, Q. Q. Meng, Z. X. Liu, H. F. Li, J. Fan and C. Y. Zhi, Energy Environ. Sci., 2019, 12, 706 RSC.
- H. F. Liang, Z. Cao, F. W. Ming, W. L. Zhang, D. H. Anjum, Y. Cui, L. Cavallo and H. N. Alshareef, Nano Lett., 2019, 19, 3199 CrossRef CAS PubMed.
- N. Zhang, F. Cheng, J. Liu, L. Wang, X. Long, X. Liu, F. Li and J. Chen, Nat. Commun., 2017, 8, 405 CrossRef PubMed.
- Y. Zhao, Y. X. Huang, F. Wu, R. J. Chen and L. Li, Adv. Mater., 2021, 33, 2106469 CrossRef CAS PubMed.
- Z. Wang, J. H. Huang, Z. W. Guo, X. L. Dong, Y. Liu, Y. G. Wang and Y. Y. Xia, Joule, 2019, 3, 1289–1300 CrossRef CAS.
- J. H. Huang, Z. Wang, M. Y. Hou, X. L. Dong, Y. Liu, Y. G. Wang and Y. Y. Xia, Nat. Commun., 2018, 9, 2906 CrossRef PubMed.
- Y. Ma, M. Xu, R. Liu, H. H. Xiao, Y. Liu, X. Wang, Y. Y. Huang and G. H. Yuan, Energy Storage Mater., 2022, 48, 212–222 CrossRef.
- H. L. Pan, Y. Y. Shao, P. F. Yan, Y. W. Cheng, K. S. Han, Z. M. Nie, C. M. Wang, J. H. Yang, X. L. Li, P. Bhattacharya, K. T. Mueller and J. Liu, Nat. Energy, 2016, 1, 16039 CrossRef CAS.
- Y. X. Liao, H. C. Chen, C. Yang, R. Liu, Z. W. Peng, H. J. Cao and K. K. Wang, Energy Storage Mater., 2022, 44, 508–516 CrossRef.
- F. Wan and Z. Q. Niu, Angew. Chem., Int. Ed., 2019, 58, 16358–16367 CrossRef CAS PubMed.
- F. W. Ming, H. F. Liang, Y. J. Lei, S. Kandambeth, M. Eddaoudi and H. N. Alshareef, ACS Energy Lett., 2021, 6, 2982 CrossRef CAS.
- L. Y. Zhang, L. Chen, X. F. Zhou and Z. P. Liu, Adv. Energy Mater., 2015, 5, 1400930 CrossRef.
- Y. X. Zeng, X. F. Lu, S. L. Zhang, D. Y. Luan, S. Li and X. W. Lou, Angew. Chem., Int. Ed., 2021, 60, 22189–22194 CrossRef CAS PubMed.
- P. Hu, T. Zhu, X. P. Wang, X. F. Zhou, X. J. Wei, X. H. Yao, W. Luo, C. W. Shi, K. A. Owusu, L. Zhou and L. Q. Mai, Nano Energy, 2019, 58, 492–498 CrossRef CAS.
- M. H. Alfaruqi, V. Mathew, J. Gim, S. Kim, J. Song, J. P. Baboo, S. H. Choi and J. Kim, Chem. Mater., 2015, 27, 3609–3620 CrossRef CAS.
- M. Li, Z. L. Li, X. P. Wang, J. S. Meng, X. Liu, B. K. Wu, C. H. Han and L. Q. Mai, Energy Environ. Sci., 2021, 14, 3796–3839 RSC.
- X. Y. Wu, Y. T. Qi, J. J. Hong, Z. F. Li, A. S. Hernandez and X. L. Ji, Angew. Chem., Int. Ed., 2017, 56, 13026–13030 CrossRef CAS PubMed.
- Y. Zhang, F. Wan, S. Huang, S. Wang, Z. Q. Niu and J. Chen, Nat. Commun., 2020, 11, 2199 CrossRef CAS PubMed.
- Y. Zhao, Y. N. Wang, Z. M. Zhao, J. W. Zhao, T. Xin, N. Wang and J. Z. Liu, Energy Storage Mater., 2020, 28, 64–72 CrossRef.
- S. Chen, J. Zheng, L. Yu, X. Ren, M. Engelhard, C. Niu, H. Lee, W. Xu, J. Xiao, J. Liu and J. Zhang, Joule, 2018, 2, 1548–1558 CrossRef CAS.
- Z. Chen, Y. Tang, X. Du, B. Chen, G. Lu, X. Han, Y. Zhang, W. Yang, P. Han, J. Zhao and G. Cui, Angew. Chem., Int. Ed., 2020, 59, 21769–21777 CrossRef CAS PubMed.
- N. Liu, X. Wu, Y. Zhang, Y. Yin, C. Sun, Y. Mao, L. Fan and N. Zhang, Adv. Sci., 2020, 7, 200146 Search PubMed.
- S. K. M. Nalluri, Z. Liu, Y. Wu, K. R. Hermann, A. Samanta, D. J. Kim, M. D. Krzyaniak, M. R. Wasielewski and J. F. Stoddart, J. Am. Chem. Soc., 2016, 138, 5968–5977 CrossRef CAS PubMed.
- M. Song, H. Tan, D. Chao and H. J. Fan, Adv. Funct. Mater., 2018, 28, 1802564 CrossRef.
- S. Gu, S. Wu, L. Cao, M. Li, N. Qin, J. Zhu, Z. Wang, Y. Li, Z. Li, J. Chen and Z. Lu, J. Am. Chem. Soc., 2019, 141, 9623–9628 CrossRef CAS PubMed.
- Z. Guo, Y. Ma, X. Dong, J. Huang, Y. Wang and Y. Xia, Angew. Chem., Int. Ed., 2018, 130, 11911–11915 CrossRef.
- Y. Luo, F. Zheng, L. Liu, H. Meng, J. Shi and F. Li, ChemSusChem, 2020, 13, 2239–2244 CrossRef CAS PubMed.
- D. Kundu, P. Oberholzer, C. Glaros, A. Bouzid, E. Tervoort, A. Pasquarello and M. Niederberger, Chem. Mater., 2018, 30, 3874–3881 CrossRef CAS.
- Y. Chen, J. Li, Q. Zhu, K. Fan, Y. Cao, G. Zhang, C. Zhang, Y. Gao, J. Zou, T. Zhai and C. Wang, Angew. Chem., Int. Ed., 2022, 61, e202116289 CrossRef CAS PubMed.
- Q. Dou, N. Wu, H. Yuan, K. H. Shin, Y. Tang, D. Mitlin and H. S. Park, Chem. Soc. Rev., 2021, 50, 6734–6789 RSC.
- Y. Liang, Y. Jing, S. Gheytani, K. Y. Lee, P. Liu, A. Facchetti and Y. Yao, Nat. Mater., 2017, 16, 841 CrossRef CAS PubMed.
- U. Mittal, F. Colasuonno, A. Rawal, M. Lessio and D. Kundu, Energy Storage Mater., 2022, 46, 129–137 CrossRef.
- L. Yan, Q. Zhu, Y. Qi, J. Xu, Y. Peng, J. Shu, J. Ma and Y. G. Wang, Angew. Chem., Int. Ed., 2022, 61, e202211107 CrossRef CAS PubMed.
- Z. Y. Song, L. Miao, H. Duan, L. Ruhlmann, Y. K. Lv, D. Z. Zhu, L. C. Li, L. H. Gan and M. X. Liu, Angew. Chem., Int. Ed., 2022, 61, e202208821 CrossRef CAS PubMed.
- L. Yan, Y. Zhang, Z. Ni, Y. Zhang, J. Xu, T. Kong, J. Huang, W. Li, J. Ma and Y. G. Wang, J. Am. Chem. Soc., 2021, 143, 15369–15377 CrossRef CAS PubMed.
- H. L. Cui, D. C. Zhang, Z. X. Wu, J. X. Zhu, P. Li, Y. Hou, R. Zhang, X. Q. Wang, X. Jin, S. C. Bai and C. Y. Zhi, Energy Environ. Sci., 2024, 17, 114–122 RSC.
- Y. L. Lin, H. L. Cui, C. Liu, R. Li, S. P. Wang, G. M. Qu, Z. Q. Wei, Y. H. Yang, Y. X. Wang, Z. J. Tang, H. F. Li, H. Y. Zhang, C. Y. Zhi and H. M. Lv, Angew. Chem., 2023, 135, e202218745 CrossRef.
- H. L. Cui, L. T. Ma, Z. D. Huang, Z. Chen and C. Y. Zhi, Smart Mater., 2022, 4, 33–49 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ma01128g |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |