
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBiocompatible PMAO-coated Gd2O3/Fe3O4 composite nanoparticles as an effective T1–T2 dual-mode contrast agent for magnetic resonance imaging†
Le T. T.
Tam
 ab,
Nguyen T. N.
Linh
c,
Le T.
Tam
d,
Duong V.
Thiet
e,
Pham H.
Nam
f,
Nguyen T. H.
Hoa
c,
Le A.
Tuan
g,
Ngo T.
Dung
b and
Le T.
Lu
*ab
ab,
Nguyen T. N.
Linh
c,
Le T.
Tam
d,
Duong V.
Thiet
e,
Pham H.
Nam
f,
Nguyen T. H.
Hoa
c,
Le A.
Tuan
g,
Ngo T.
Dung
b and
Le T.
Lu
*ab
aGraduate University of Science and Technology, Vietnam Academy of Science and Technology, 18 Hoang Quoc Viet, Hanoi, Vietnam. E-mail: ltlu@itt.vast.vn
bInstitute for Tropical Technology, Vietnam Academy of Science and Technology, 18 Hoang Quoc Viet, Hanoi, Vietnam
cThai Nguyen University of Sciences, Tan Thinh Ward, Thai Nguyen City 25000, Thai Nguyen, Vietnam
dVinh University, 182 Le Duan, Vinh City, Vietnam
eSchool of Mechanical and Automotive Engineering, Hanoi University of Industry, Hanoi 100000, Vietnam
fInstitute of Materials Science, Vietnam Academy of Science and Technology, 18 Hoang Quoc Viet, Hanoi, Vietnam
gPhenikaa University, Nguyen Thanh Binh Street, Yen Nghia Ward, Ha Dong District, Hanoi 12116, Vietnam
First published on 9th January 2025
Abstract
In this study, we developed novel Gd2O3/Fe3O4 composite nanoparticles (GFO CNPs) via a simple one-step thermal decomposition of Fe(III) actylacetonate and Gd(III) acetate in octadecene solvent, with oleic acid (OA) and oleylamine (OM) serving as surfactants. The effects of the Gd/Fe molar ratio on the formation, size, and T1, T2 relaxation efficiency of the NPs were investigated. The as-synthesised GFO CNPs were characterised using various analytical techniques such as TEM, HRTEM, XRD, UV-Vis, VSM, EDX and XPS. The results showed that GFO CNPs synthesised at a Gd/Fe ratio of 7/3 (GFO-7/3 CNPs) had an average size of about 10 nm, exhibited monodispersity with a narrow size distribution, and demonstrated superparamagnetic properties at room temperature. Additionally, surface modification of the NPs with PMAO improved their dispersibility and stability in aqueous media. Cytotoxicity tests confirmed the biocompatibility of the PMAO encapsulated GFO CNPs. In vitro relaxivity studies showed high r1 and r2 values of 18.20 and 94.75 mM−1 s−1, respectively, with an r2/r1 ratio of 5.21, outperforming commercial products such as Feraheme, Resovist, Feridex, and Dotarem. These findings suggest that GFO CNPs provide a promising nanoplatform for non-invasive T1–T2 dual-modality MRI diagnosis.
Introduction
Magnetic resonance imaging (MRI) is one of the modern non-invasive diagnostic techniques widely used in clinical practice. This technique utilizes non-ionizing safe radiation, providing high spatial resolution and almost infinite tissue penetration depth, which enhances the ability to distinguish between normal and pathological tissues.1–3 However, the primary limitation of MRI lies in its relatively low sensitivity, especially when distinguishing between tissues with similar properties. To address this challenge, contrast agents are often employed to enhance signal intensity differences, making structures or abnormalities more visible.4–6In clinical practice, gadolium (Gd) complexes are most commonly used as MRI contrast agents. They are effective for T1-weighted imaging by brightening areas of interest through shortening of the longitudinal relaxation time (T1).7,8 However, most clinically relevant T1 contrast agents are small molecules with limitations, including low longitudinal relaxivity (with r1 usually ≤ 10 mM−1 s−1), short circulation time in the body, lack of specificity to tumors and the requirement for high dosages.9,10 Additionally, Gd complex-based contrast agents pose a high risk for patients with kidney or liver disease.11,12 Recently, iron oxide NPs (IONs), such as Fe3O4 and γ-Fe2O3, have attracted a great deal of attention as T2-weighted contrast agents due to their high magnetic properties, biocompatibility and low toxicity.13,14 However, T2-weighted dark signals generated by IONs are susceptible to disturbances caused by signals originating from calcification, bleeding, or metal deposits, and these artifacts can impact the quality of background imaging.15,16
Each type of contrast agent has its own strengths and limitations, leading to increased interest in developing multimodal imaging agents for more precise diagnostic information. T1–T2 dual-modal contrast agents, which combine the benefits of both positive and negative contrasts, could enhance diagnosis by providing sharper anatomical details in MR images. Recently, significant efforts have been made to synthesise T1–T2 dual modal CAs for MRI by incorporating gadolinium species with iron species into a single nanosystem such as Gd chelate conjugating to the surface of IONs,17,18 Gd-doped IONs,19,20 and Gd-compound coatings on magnetic cores.21–23 For example, Yang et al. developed a T1–T2 dual-mode MRI contrast agent by utilising gadolinium complex-coupled SiO2@Fe3O4 core–shell NPs, which exhibited excellent MRI contrast capabilities.24 Similarly, Mustafina and colleagues fabricated a core–shell structure by incorporating ultra-small iron oxides as the core and Gd(III) complexes doped into the silica nanoparticles as the exterior layer, achieving high r1 and r2 relaxivity values.25 Sun et al. designed a yolk-like nanostructure with an ION core and a mesoporous Gd2O3 shell, separated by an interstitial hollow space. This structure effectively reduces the disturbance of the magnetic core with the T1 image, thereby significantly enhancing the T1–T2 contrast effect.26 Miao and co-workers reported a highly efficient T1–T2 dual-mode MRI contrast agent using ultra-small gadolinium oxide-decorated magnetic iron oxide nanocrystals (GMIOs).27 Zhang and colleagues developed a mesoporous IO/GdO nanocomposite as a T1–T2 dual-modal MRI contrast agent through a hydrothermal synthetic method enhancing the attenuation of T1 and T2 relaxation times.28
However, the aforementioned nanostructures encountered different problems. Previous studies have shown that the effectiveness of T1 contrast agents depends on their contact area with water molecules, while T2 agents rely on the distance between the agent and water molecules. Therefore, increasing the contact area between T1 agents and water protons and minimizing the distance between T2 agents and water molecules can optimize the T1–T2 dual imaging performance. In the case of Gd compounds located outside and directly in contact with a magnetic core, the magnetic field generated by a magnetic core can perturb the spin–lattice relaxation process of the paramagnetic T1 contrast agent, which can lead to a decrease in T1 contrast ability.24–26,29 Therefore, gadolinium centers need to be spatially separated from the magnetic core to reduce local magnetic disturbance. In contrast, Gd ions inside superparamagnetic IONs (SIONs), such as Gd-doped SIONs, exhibit parallel spin alignment with the magnetic field generated by the SIONs, which could effectively prevent magnetic disturbance, while significantly enhancing local magnetic field intensity and inhomogeneity, thus improving both T1 and T2 imaging performance.30 Despite progress in developing T1–T2 dual-modal CAs based on gadolinium and iron compounds, challenges remain, such as a low contact area of gadolinium with water-proton, inappropriate distance between Fe3O4 and water molecule, or undesirable disturbance of gadolinium by Fe3O4, which compromise their dual-contrast capabilities.25,31 Therefore, further research is essential to strike a balance between the saturated magnetism and superparamagnetism of magnetic NPs.
In this work, Gd2O3/Fe3O4 composite nanoparticles with a unique structure were synthesized using a one-step thermal decomposition process to develop a novel T1–T2 dual-modality contrast agent. By utilizing a high Gd/Fe precursor ratio, the resulting GFO CNP structure demonstrated partial integration of Gd3+ ions into the Fe3O4 lattice, along with the deposition of ultrasmall Gd2O3 NPs on their surface. This not only enhanced the longitudinal T1 relaxation effect but also optimized the transverse T2 relaxation effect. The as-prepared GFO CNPs were systematically characterized to determine the optimal composition for dual-mode MRI applications. Among the obtained GFO samples, GFO CNPs synthesised at a Gd/Fe ratio of 7/3 demonstrated the most promising balance of properties, showing both high T1 and T2 relaxivity values and excellent biocompatibility. Furthermore, these CNPs exhibited uniform size distribution and good aqueous stability, making them well-suited for potential clinical applications. The results suggest that GFO CNPs could serve as an effective dual-modality MRI contrast agent, overcoming the limitations associated with conventional single-mode agents.
Experimental
Chemicals
Gadolinium(III) acetate hydrate (Gd(Ac)3, 99.9%), iron(III) acetylacetonate 97% (Fe(acac)3), oleic acid (OA, 90%), oleyamine (OM, 70%), 1-octadecene (ODE, 90%), poly(maleic anhydride-alt-1-octadecene) (PMAO), chloroform, absolute ethanol and hexane were purchased from Sigma-Aldrich, Ltd and used as received without further purification.Synthesis of GFO CNPs
The GFO CNPs were synthesized by a thermal decomposition method following our previously reported work with some modifications.32,33 In a typical process, a mixture of 10 mmol precursor containing gadolinium(III) acetate and iron(III) acetylacetonate at different molar ratios was added to a 100 mL round-bottomed three-neck flask containing a pre-mixed solution of OA/OM (molar ratio of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) and 40 mL of octadecene solvent. The reaction mixture was stirred at 100 °C for 1 h until the precursors completely dissolved. Subsequently, the reaction mixture was heated to 200 °C and kept at this temperature for 3 h, followed by a further increase to 320 °C and maintained for 2 h. Throughout the process, a continuous flow of nitrogen gas was maintained. After the reaction, the solution was cooled to room temperature, and the GFO CNPs were collected by centrifugation and washed four times with absolute ethanol. Finally, GFO CNPs were resuspended in chloroform at a concentration of 10 mg mL−1.
:1) and 40 mL of octadecene solvent. The reaction mixture was stirred at 100 °C for 1 h until the precursors completely dissolved. Subsequently, the reaction mixture was heated to 200 °C and kept at this temperature for 3 h, followed by a further increase to 320 °C and maintained for 2 h. Throughout the process, a continuous flow of nitrogen gas was maintained. After the reaction, the solution was cooled to room temperature, and the GFO CNPs were collected by centrifugation and washed four times with absolute ethanol. Finally, GFO CNPs were resuspended in chloroform at a concentration of 10 mg mL−1.
Surface modification of GFO CNPs
To achieve a stable dispersion of GFO CNPs in aqueous medium, we encapsulated the CNPs with the poly(maleic anhydride-alt-1-octadecene) (PMAO) polymer using our previously reported procedure.32,33 In a typical experiment, 10 mL of GFO CNPs in chloroform (10 mg mL−1) were mixed with 10 mL of PMAO polymer in chloroform (0.2 g mL−1). The mixture was continuously stirred at room temperature until all the chloroform evaporated. Next, a diluted NaOH solution was added, and the mixture was sonicated for 30 min. The resulting GFO@PMAO solution was collected by centrifugation to eliminate excess free PMAO and NaOH, and redispersed in 10 mL of distilled water. The synthesis and surface modification procedure of GFO CNPs with PMAO is presented in Scheme 1.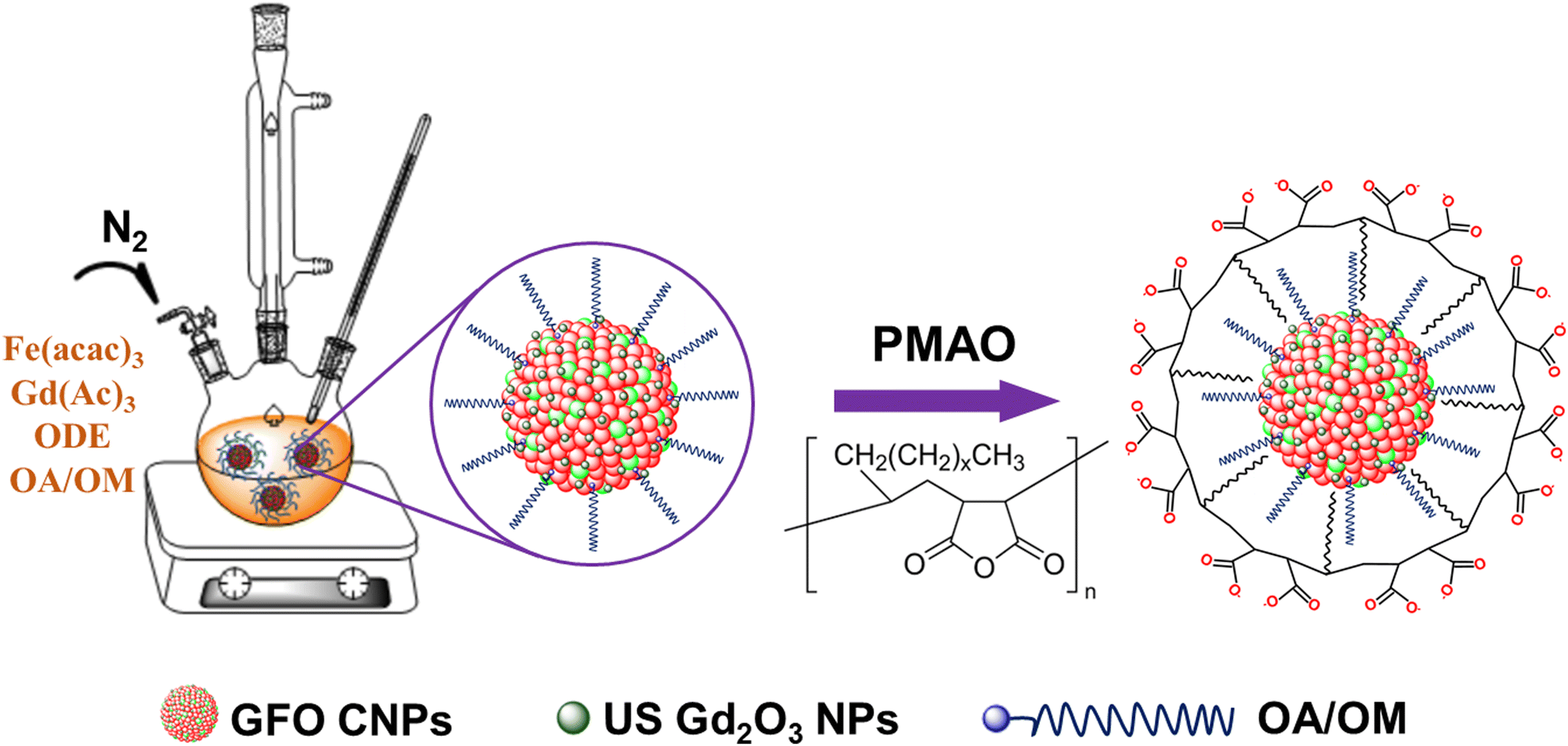 | ||
| Scheme 1 Illustration of the synthesis and surface modification process of GFO CNPs. | ||
Characterisation
The morphology of GFO CNPs and the selective area electron diffraction (SAED) pattern were studied by transmission electron microscopy (TEM, JEM1010-JEOL) and HRTEM. The mean size and the standard deviation were evaluated from the electron micrographs by counting around 300 particles. The crystal structure and phase composition of GFO CNPs were determined by X-ray diffraction analysis. The distribution of the elements within the GFO samples was determined by energy dispersive X-ray spectroscopy (EDX) analysis using Hitachi S-4800 FESEM equipment. The magnetic properties of the samples were studied using a vibrating sample magnetometer (VSM, DMS 800, Quantum Design, Inc.). The saturation magnetization (Ms) was measured at room temperature with the magnetic field ranging from −11 to 11 kOe. X-ray photoelectron spectroscopy measurements (XPS, Thermo Fisher) were carried out using monochromatic Al Kα radiation (hm = 1486.6 eV). The UV-Vis spectra of the samples were recorded on a Jasco V-670 spectrometer. FT-IR spectroscopy (Nicolet 6700) and TGA (Nezsch, Germany) analyses were used to characterise the surface structure of GFO CNPs. Dynamic light scattering (DLS) and zeta potential measurement were used to investigate the distribution and stability of GFO CNPs in aqueous medium. The concentration of Fe and Gd in the GFO samples was measured by inductively coupled plasma mass spectroscopy (ICP-MS) (Agilent Technologies, Japan).Cytotoxicity assay
The cytotoxicity of GFO CNPs was assessed by the methyl thiazolyl tetrazolium (MTT) assay using Vero cells (Kidney, African green monkey) as model cells. Firstly, Vero cells with a density of 1.5 × 105 were seeded in a 96-well plate and further incubated in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin solution at 37 °C for 24 hours in the incubator containing 5% CO2. The culture medium was then replaced with fresh medium containing GFO CNPs at various concentrations of 8, 16, 32, 64 and 128 μg mL−1 and then incubated for 48 hours. The cell viability was determined by adding 20 μL of MTT to each well and incubating for 4 h at 37 °C. The crystalline formazan metabolite formed within the cells was dissolved by adding dimethyl sulfoxide (DMSO, Sigma-Aldrich) to each well, and the optical density was measured at 540 nm using a Tecan Spark device (Männedorf, Switzerland).MRI relaxivity measurement
For MRI phantom study, GFO solutions with concentrations ranging from 0.0625 to 0.8 mM were used for the MRI test. To evaluate the properties of the dual-mode contrast agents, T1-weighted (T1W) and T2-weighted (T2W) magnetic resonance images were obtained using a clinical 1.5 T MRI scanner (Siemens Magnetom, Germany). The T1W images were obtained using a turbo spin-echo sequence with the following parameters: the repetition time (TR) varied from 100 to 1000 ms, with a constant echo time (TE) value of 12 ms, the field of view (FOV) was set to 190 × 190 mm2, and the matrix size was 256 × 192. For the T2W images, a multi-spin echo sequence was applied with a TR = 3000 and TE ranging from 24 to 106 ms. The FOV was set to 230 × 230 mm2, the matrix size set to 320 × 240 and the slice thickness was 1.6 mm. The relaxivity r1 and r2 values were calculated by determining the slope of the relaxation time versus the total metal concentration in mM, respectively.Results and discussion
Characterisation of GFO CNPs
In this work, GFO CNPs were synthesised through a one-step thermal decomposition process using a precursor mixture of Fe(acac)3 and Gd(Ac)3 in octadecene solvent containing oleic acid (OA) and oleylamine (OM) as surfactants. The mixture was thermally decomposed at 320 °C for 2 h to form GFO CNPs. In this procedure, we investigated the effect of the Gd/Fe precursor molar ratio on the formation of the GFO CNPs by varying the Gd/Fe molar ratios to 7/3, 8/2, 9/1, and sole Gd, while maintaining the reaction time, temperature, and surfactant constant. Fig. S1 (ESI†) shows TEM images and photographs of the obtained GFO solutions (insets of Fig. S1a–d, ESI†) synthesized at different Gd/Fe precursor ratios. The TEM images confirm that the resulting GFO CNPs are spherical. It can be seen that the mean size of the GFO CNPs decreases as the Gd/Fe molar ratio increases. This trend is possible due to the higher binding energy required to form gadolinium–oxygen bonds compared to ferric–oxygen bonds, so the incorporation of Gd3+ ions slow down the crystal growth of the GFO CNPs.23Photographs for the GFO CNP solutions reveal that the solution color turned lighter progressively when the molar ratio of Gd/Fe in the precursors increases. The Gd and Fe contents in the GFO CNPs, as determined by inductively coupled plasma mass spectrometry (ICP-MS), are shown in Table S1 (ESI†). The obtained molar ratios of Gd/Fe for GFO-7/3, GFO-8/2 and GFO-9/1 were 1.237, 2.093 and 3.751, respectively. Notably, the Gd/Fe molar ratio in the GFO CNPs is slightly lower than that of the precursors, suggesting that Gd(III) has a lower reaction efficiency compared to Fe(III) under the same conditions. In the current study, GFO-7/3 CNPs were selected for further study.
From the TEM image and size distribution histogram, it can be observed that the GFO CNPs have a relatively uniform spherical shape with a mean size of 10.1 ± 0.7 nm. In addition, the high-resolution TEM (HRTEM) image (Fig. 1c) reveals that the interplanar spacing distance within the CNP is 2.46 Å, which corresponds to the (222) crystal plane in the cubic structure of Fe3O4.34 However, this value is slightly larger than that expected for pure Fe3O4, likely due to the substitution of bigger Gd3+ ions (0.94 Å) into the position of smaller Fe3+ ions (0.67 Å), resulting in lattice expansion and distortion.
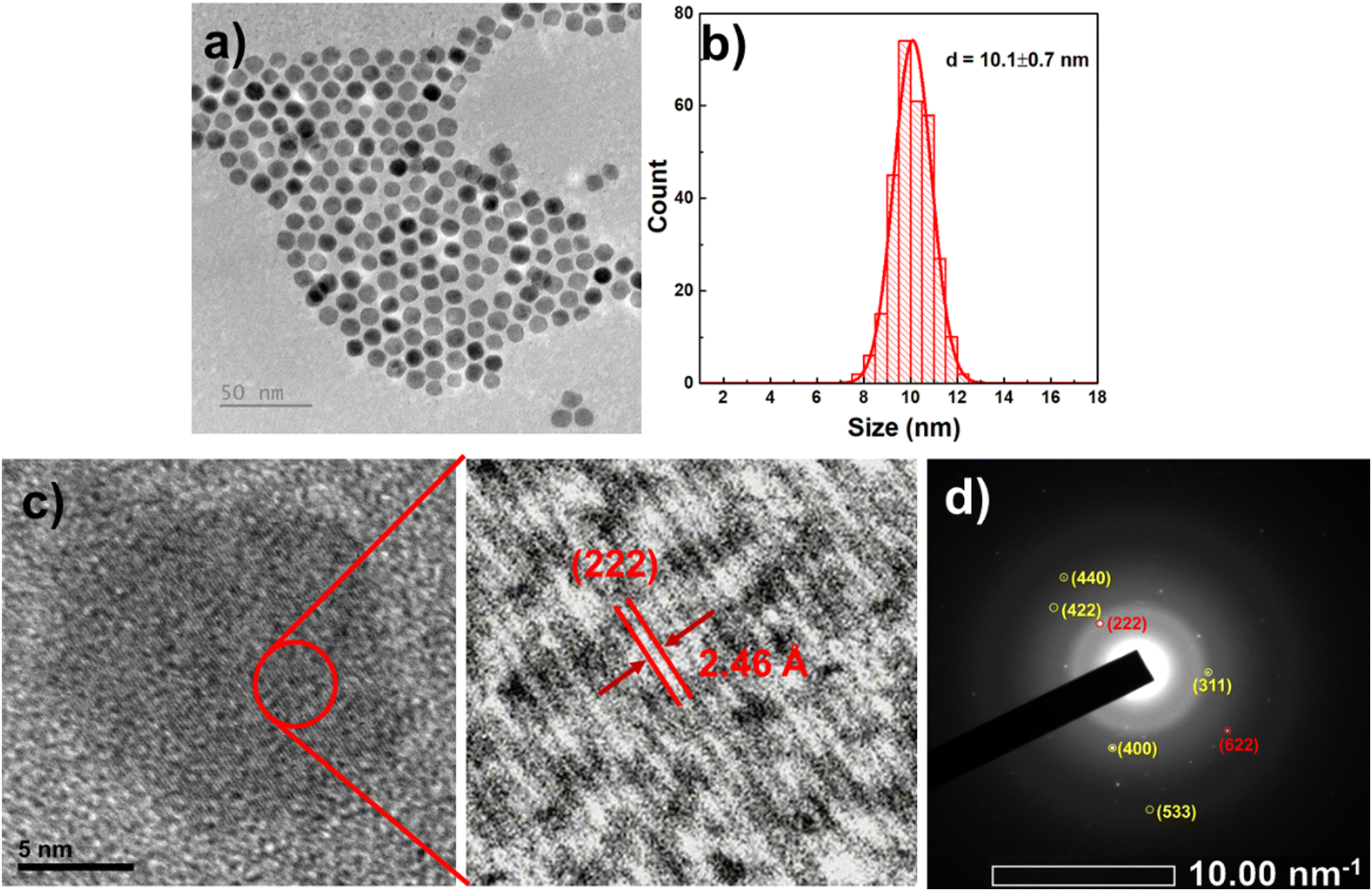 | ||
| Fig. 1 TEM image (a), histogram of size distribution (b), HRTEM images (c) and SAED pattern (d) of the GFO-7/3 CNPs. | ||
To further confirm the phases present, selected area electron diffraction (SAED) analysis was conducted, as shown in Fig. 1d. The obtained SAED pattern revealed concentric rings with spots, indicating that the as-synthesized GFO is polycrystalline in nature. Interplanar spacings, calculated from the diffraction pattern and summarized in Table S2 (ESI†), are consistent with those reported for cubic phase Fe3O4 (JCPDS, card 19-0629) and Gd2O3 (PDF Card No. 2106881). The diffraction rings correspond to the Bragg reflections of (311), (400), (422), (440), and (533) planes of Fe3O4 (highlighted in yellow) and the (222) and (622) planes of Gd2O3 (highlighted in red).22,35,36 Notably, the calculated d-spacing values for Fe3O4 are slightly larger than those reported in the JCPDS card, suggesting a minor structural distortion of the Fe3O4 lattice possibly due to the incorporation of Gd3+.13,20,37,38 These findings confirm the coexistence of Fe3O4 and Gd2O3 phases and indicate partial Gd doping within the Fe3O4 lattice.
The crystal structure of the synthesized GFO-7/3 CNPs was analysed using X-ray diffraction (XRD). As illustrated in Fig. S2a (ESI†), the XRD pattern of the CNPs reveals peak broadening across the spectrum with relatively low diffraction peak intensity. A broad peak centered in the 2θ range of approximately 25° to 40° exhibited the mixed diffraction peaks assigned to the characteristic (220), (311) and (222) diffraction peaks of Fe3O4 and the (222) and (440) diffraction peaks of Gd2O3 NPs. The lack of sharp and well-defined peaks can be attributed to the presence of very small (1–2 nm) or highly amorphous Gd2O3 clusters on the surface of Fe3O4 NPs resulting from the excess of free Gd3+ ions. This incorporation or clustering could lead to the lattice distortion within the Fe3O4 NPs. The evidence for the presence of very small Gd2O3 NPs or clusters is provided by UV-Vis spectroscopy (Fig. S2b, ESI†), which shows a weak absorption peak around 270 nm, consistent with previous studies on Gd2O3.39,40
The magnetic properties of GFO-7/3 CNPs were evaluated at room temperature (300 K), as shown in Fig. S2c (ESI†). The remanence and coercivity were found to be very low, suggesting that the GFO-7/3 CNPs exhibit superparamagnetic behavior. The field-dependent magnetisation (M/H) curves of the GFO-7/3 CNPs showed a moderate saturation magnetization (Ms) of approximately 10.3 emu g−1, which is significantly lower than that of Fe3O4 NPs.41 The reduction in saturation magnetization is likely due to the decreased iron oxide content in the NPs. Additionally, when Gd3+ ions are introduced into the spinel structure of Fe3O4, they preferentially occupy octahedral B sites rather than tetrahedral A sites due to their larger size, disrupting the normal magnetic interactions between Fe3+ ions at these sites.20 At higher Gd/Fe molar ratios, the magnetic properties of the CNPs begin to resemble those of cubic Gd2O3 due to the formation of Gd–O–Gd bonds instead of Gd–O–Fe bonds, which further disrupts the magnetic structure and reduces the overall magnetization of the particles.13
To verify the composition and distribution of elements in the GFO-7/3 CNPs, the energy-dispersive X-ray element mapping was performed (Fig. 2). The EDS spectrum shows that iron, gadolinium and oxygen were the main elements present in the CNPs. No other impurity elements can be detected except carbon element, which stems from OA and OLA molecules. EDS mapping indicates that iron and gadolinium distribute uniformly in the scanned area. The molar ratio of Gd/Fe for GFO-7/3 CNPs is approximately 1.237, determined by inductively coupled plasma mass spectroscopy (ICP-MS).
 | ||
| Fig. 2 SEM image (a), EDX spectrum (b) and elemental mapping of GFO-7/3 CNPs (c)–(e). | ||
The chemical composition and oxidation states of the different chemical species in the GFO-7/3 CNPs were further analyzed using X-ray photoelectron spectroscopy (XPS), with the results presented in Fig. 3. The XPS survey spectrum (Fig. 3a) confirms the presence of C, N, O, Fe, and Gd elements within the binding energy range of 0 to 1300 eV. Fig. 3b shows the high-resolution XPS spectrum of Fe 2p peaks, where Fe 2p3/2 and Fe 2p1/2 peaks are observed at binding energies of approximately 710 eV and 723.5 eV, respectively. This suggests the presence of both Fe(II) and Fe(III) oxidation states in the NPs. The gap between these two peaks is about 13.5 eV. Further deconvolution of these peaks revealed five main peaks at 708, 710, 712, 722.9, and 725.1 eV, along with a satellite peak at 718.3 eV. The peaks at 708, 710, 722.9, and 725.1 eV correspond to the Fe–O bond of the Fe2+ ion, while the one at 712 eV is attributed to the Fe–O bond of the Fe3+ ion in Fe3O4. The satellite peak at 718.3 eV confirms the presence of Fe3+ species.
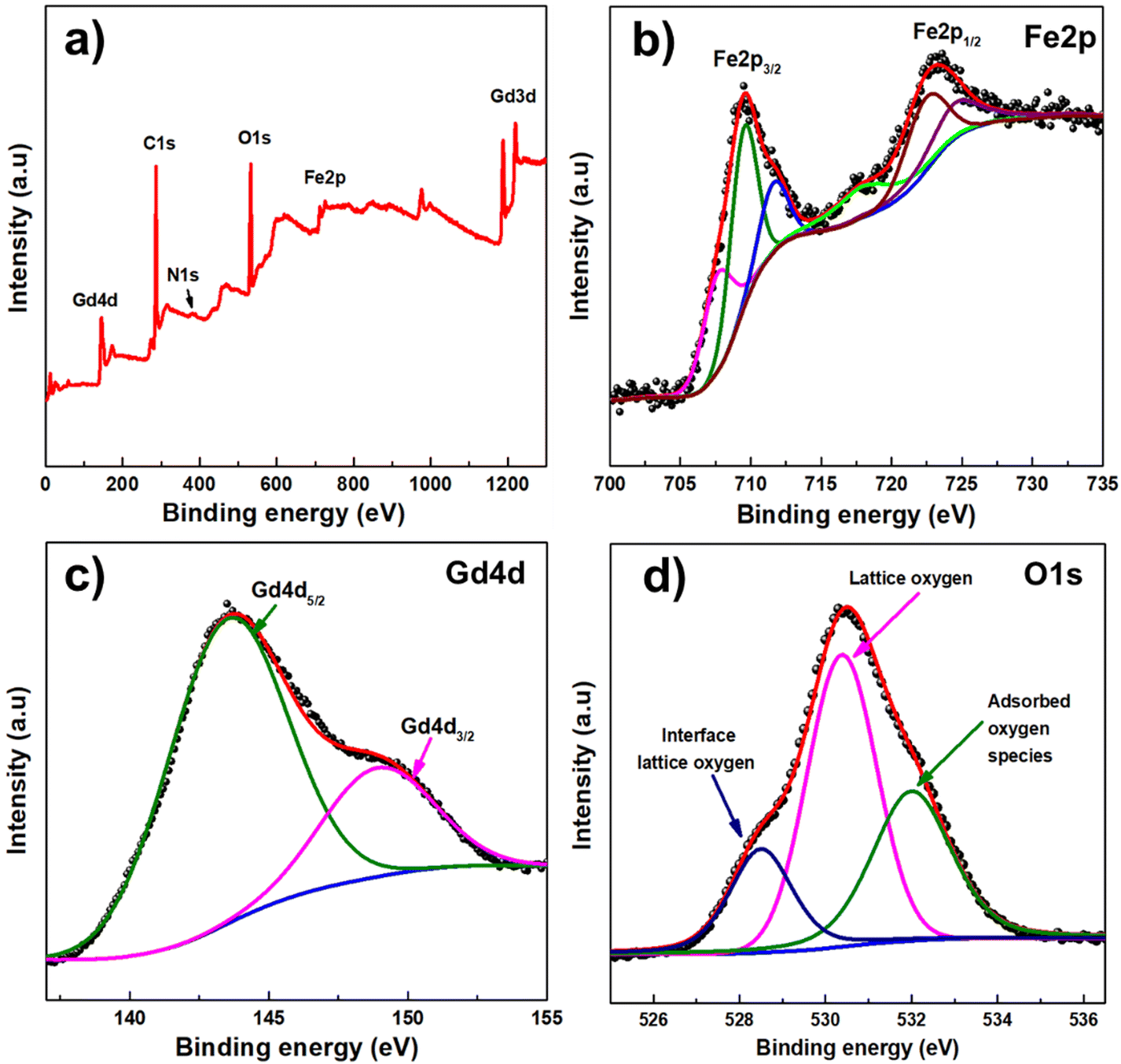 | ||
| Fig. 3 (a) Scan survey XPS spectrum of GFO-7/3 CNPs and high-resolution XPS spectra of (b) Fe 2p, (c) Gd 4d and (d) O 1s. | ||
As seen in Fig. 3c, the Gd 4d spectrum displays Gd 4d5/2 and Gd 4d3/2 peaks around 143.6 eV and 149.1 eV, confirming the presence of Gd(III). Fig. 3d presents the high-resolution XPS spectrum of O 1s, which features three peaks at binding energies of 528.5, 530.4, and 532 eV. The main peak at 530.4 eV is attributed to lattice oxygen in metal–oxygen bonds, while the peak at 528.5 eV corresponds to interface lattice oxygen. The O 1s peak at 532 eV is assigned to oxygen in absorbed OH− and H2O species on the surface.
In this study, the GFO CNPs synthesised in organic solvents were initially coated with surfactants of OA and OM, resulting in hydrophobic NPs. For biomedical applications, it is crucial for NPs to be dispersible and stable in aqueous medium. To achieve this, the synthesised NPs were surface modified by coating them with the amphiphilic polymer poly(maleic anhydride-alt-1-octadecene) (PMAO). Fig. 4a shows the FTIR spectra of the GFO CNPs before and after encapsulation with PMAO. All samples exhibit the absorption peaks at 2854 and 2923 cm−1, corresponding to the stretching vibrations of C–H bonds in the hydrocarbon chains. A broad band in the range of 3300 cm−1 to 3600 cm−1 is attributed to the –OH stretching. In the FTIR spectrum of GFO-7/3@OA/OM, the characteristic absorption peaks for the –COOH and –NH2 groups from oleic acid and oleylamine have disappeared. Instead, the appearance of peaks at 1546 cm−1 and 1423 cm−1 correspond to the asymmetric stretching vibrations of the COO− group and the bending vibration of C–H bonds, respectively, indicating the formation of an acid–base complex between OA and OM on the GFO CNPs’ surface.
 | ||
| Fig. 4 (a) FT-IR spectra, (b) TGA scans of GFO CNPs before and after encapsulation with PMAO, (c) DLS pattern and (d) zeta potential of the GFO@PMAO CNPs. | ||
Additionally, two intense absorption bands located at 547 and 434 cm−1 are assigned to the Gd–O stretching vibration of the Gd2O3 and Fe–O vibration of Fe3O4, respectively. For the GFO@PMAO sample, the FT-IR spectrum shows the disappearance of the characteristic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (1781 cm−1 and 1862 cm−1) and C–O (1083 cm−1 and 925 cm−1) vibrations in the anhydride groups, and the appearance of a broadening peak at 1712 cm−1 corresponding to the CO stretching vibration of carboxylic acid, which indicates the formation of carboxylate groups due to the hydrolysis of PMAO's anhydride ring.
O (1781 cm−1 and 1862 cm−1) and C–O (1083 cm−1 and 925 cm−1) vibrations in the anhydride groups, and the appearance of a broadening peak at 1712 cm−1 corresponding to the CO stretching vibration of carboxylic acid, which indicates the formation of carboxylate groups due to the hydrolysis of PMAO's anhydride ring.
TGA was performed to quantify the amount of PMAO polymer bound to the surface of GFO CNPs. As shown in Fig. 4b, for the sample before encapsulating (OA/OM coated GFO CNPs), we recorded a weight loss of about 25.8% in the temperature range of 100 to 700 °C due to the evaporation of physical adsorbents (such as water or solvents) and decomposition of OA and OM. After modification with PMAO, a large weight loss of about 63.9% was observed, which is attributed to the evaporation of water adsorbed on the particle surface during the storage of the sample and the decomposition of OA/OM and PMAO. These results in the current study are similar with our previous works using PMAO as a surface-capping agent for NPs.32,33,41
The colloidal stability of the GFO@PMAO is essential for their biomedical applications. To evaluate this stability, we measured the hydrodynamic diameter and zeta potential of the GFO@PMAO CNPs using a dynamic light scattering (DLS) technique. As shown in Fig. 4c and d, the hydrodynamic diameter and the zeta potentials of the NPs are 58.7 nm and –63.4 mV, respectively. To further assess colloidal stability, the hydrodynamic diameter and zeta potential of GFO@PMAO CNPs were monitored over time and their stability under various acidic, basic saline conditions were analyzed. The results, as shown in Fig. 5, indicate that both the hydrodynamic diameter and zeta potential of the CNPs remained relatively constant over two months. In addition, the CNPs demonstrate their stability across a pH range of 2 to 13 and in salt solutions up to 0.45 M. The results demonstrate that GFO@PMAO CNPs exhibit excellent colloidal stability, which is highly beneficial for the further biological applications.
 | ||
| Fig. 5 (a) Hydrodynamic diameters and (b) zeta potential of the GFO@PMAO CNPs over time, and (c) and (d) the photographs of the GFO@PMAO CNPs at different pH values and NaCl concentrations. | ||
Cytotoxicity assay
Toxicity is an important factor to consider for the clinical use of magnetic NPs as contrast agents. To evaluate the suitability of GFO@PMAO CNPs, we conducted an MTT assay to assess their cytotoxicity on Vero cells. As shown in Fig. 6, there was no significant change in cell morphology across the GFO@PMAO CNP concentration range of 8 to 128 μg mL−1, indicating that the GFO@PMAO CNPs do not cause any morphological alterations in the cells, even at high concentrations. Additionally, cell viability remained above 80% after 48 hours of incubation with NP concentrations up to 128 μg mL−1, suggesting low cytotoxicity. This is likely due to the PMAO coating, which prevents direct interaction between the inorganic core and the cells. Moreover, this result is comparable to that observed with Gd2O3@PMAO nanoplates at a lower concentration (100 μg mL−1), as reported in our previous study.33 These findings suggest that the incorporation of Fe into the NP composition improved the biocompatibility of GFO CNPs compared to pure Gd2O3 nanoplates, thus enhancing their potential for biomedical applications. | ||
| Fig. 6 MTT assay of Vero cell viability after incubation with different concentrations of GFO@PMAO CNPs for 48 hours. | ||
In vitro MR imaging
To quantitatively analyze the relaxivities of the GFO@PMAO CNPs, we measured the T1 and T2 relaxation times utilizing a 1.5 T clinical MRI scanner. The longitudinal relaxivity (r1) and transverse relaxivity (r2) of the CNPs were determined from the slopes of the plots of 1/T1 and 1/T2 as a function of metal concentrations, respectively. As shown in Fig. 7a and b, a gradual brightening of the T1-weighted phantom images and a darkening of the T2-weighted phantom images were observed with increasing concentration of the CNPs. The determined r1 and r2 values were 18.2 mM−1 s−1 and 94.75 mM−1 s−1, respectively. A moderate r2/r1 ratio of 5.21 within a range of 5–10 suggests that GFO@PMAO CNPs are advantageous for T1–T2 dual-mode MR imaging.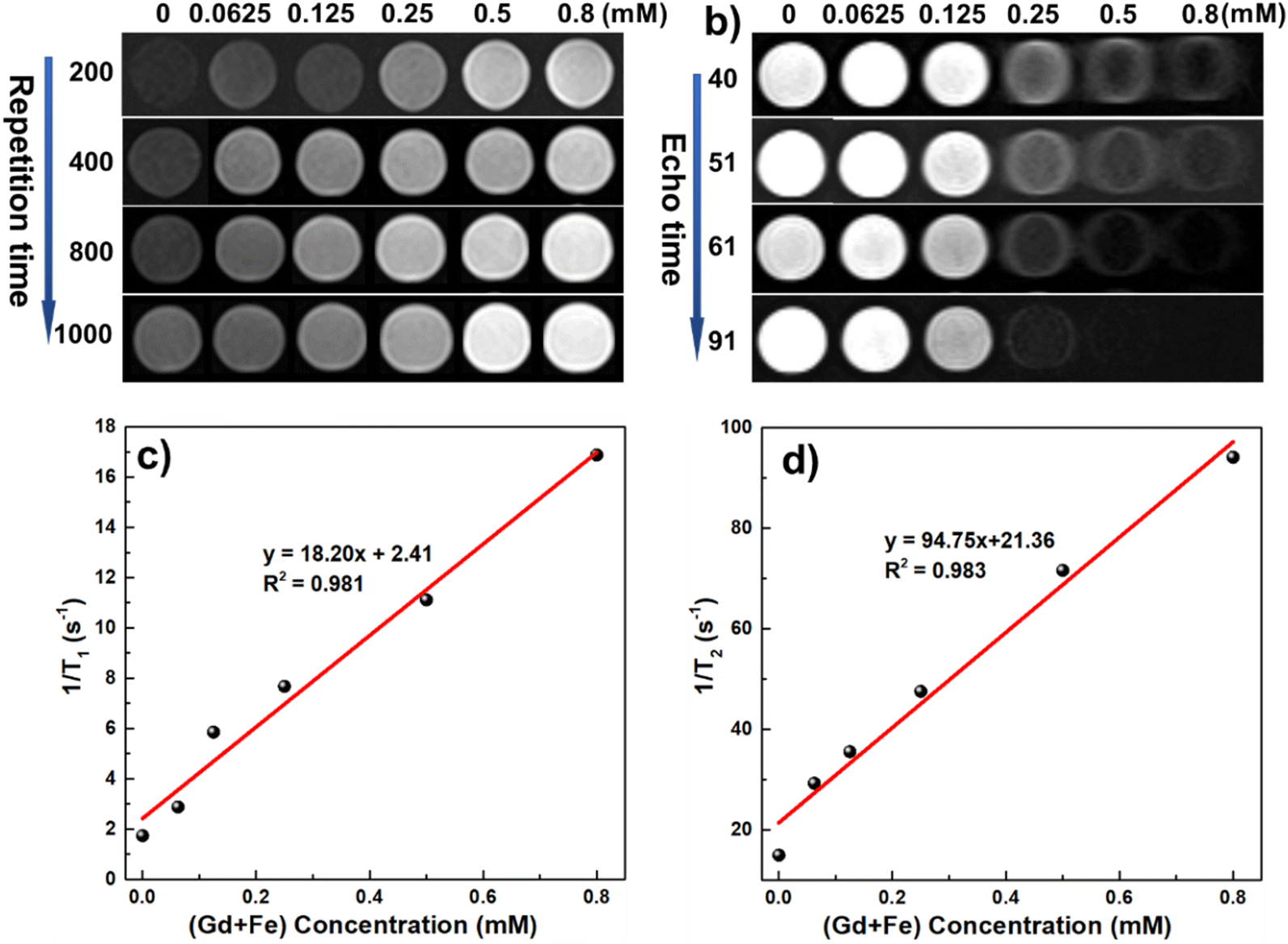 | ||
| Fig. 7 T1–T2 dual-modal contrast effect: T1-weighted (a) and T2-weighted (b) MR images of GFO@PMAO CNPs; longitudinal relaxation (c) and transverse relaxation (d) of GFO@PMAO as a function of (Gd + Fe) concentration. | ||
Notably, both r1 and r2 values of the GFO@PMAO CNP are higher than those of several previously reported Fe- and Gd-containing contrast agents.13,42 The r1 value of GFO@PMAO CNPs is approximately four times larger than that of commercial T1 contrast agents (Dotarem, Magnevist, Gadovist) while the r2 value is still comparable to that of commercial T2 contrast agents (Ferumoxytol).43,44 These high relaxivity values are possible due to the synergistic effect of the iron and gadolinium elements within the GFO CNPs. The presence of Gd, both incorporated into the Fe3O4 lattice and as Gd2O3 on the particle surface, plays a crucial role in enhancing the relaxivity properties. Gd3+ ions in the Fe3O4 lattice ensure that the spin order of Gd3+ has the identical direction with the magnetisation of the iron oxide NPs under an external magnetic field, which significantly increases the local magnetic field of the T2 contrast material, and further enhances the T2 contrast effect. Meanwhile, the presence of a large number of Gd3+ ions in the ultrasmall Gd2O3 structure on the surface of the GFO CNPs provides a high surface-to-volume ratio, enabling direct interaction with water molecules. This configuration contributes to the T1 contrast effect by promoting efficient water exchange. Additionally, the partial T1 shortening effect from the iron oxide nanoparticles further enhances the overall T1 contrast performance. This unique dual-modality behavior demonstrates the potential of GFO@PMAO CNPs to serve as effective contrast agents for both T1 and T2 imaging applications.
To systematically evaluate the MRI performance of GFO CNPs with varying compositions, the longitudinal and transverse relaxivity of the GFO@PMAO CNPs at different Gd/Fe ratios were also measured. The r1 and r2 values for these Gd/Fe ratios are also summarized in Table S1 (ESI†) and demonstrated in Fig. S3 (ESI†). It can be found that the r1 value increased slightly from 18.20 to 20.45 mM−1 s−1 while the r2 value decreased significantly from 94.75 to 54.60 mM−1 s−1 as the Gd/Fe ratio increased from 7:3 to 9:1. Consequently, the r2/r1 ratio of the samples decreased from 5.21 to 2.69, indicating that the GFO@PMAO CNPs are more suitable as T1 contrast agents with a higher Gd/Fe ratio. This phenomenom can be explained by the existence of a large amount of effective paramagnetic Gd ions in the GFO CNP composition, which enhances a local magnetic field. This enhancement improves the relaxivity of Gd ions, and influences the positive contrast effects of the NPs. At higher Gd/Fe ratios, the magnetisation value of the GFO CNPs significantly decreases, primarily due to the paramagnetic nature of Gd and also because of the significant reduction in the NP size. The decrease in magnetism lead to a reduction in the r2 value.
Conclusion
In summary, Gd2O3/Fe3O4 composite nanoparticles with high dispersibility, colloidal stability and low toxicity were successfully synthesised by a simple procedure. The Gd and Fe contents in the NPs could be adjusted by varying the Gd/Fe ratios. The results demonstrated that PMAO encapsulated GFO CNPs were well dispersed in aqueous solution, colloidally stable and biocompatible. The r1 and r2 relaxivities of the GFO@PMAO CNPs with various Gd/Fe ratios were measured. The high values of both r1 and r2 relaxivities are possibly attributed to the synergistic effect of the Gd and Fe elements in the NP composition. While further in vivo experiments are necessary, these findings suggest that GFO CNPs show potential as a promising candidate for an effective T1–T2 dual-modality MRI contrast agent.Author contributions
Le T. T. Tam and Le T. Lu conceptualized the study, designed the methodology and wrote the manuscript. Nguyen T. N. Linh and Nguyen T. H. Hoa conducted the toxicity tests and collected the associated data, while Le T. Tam performed the MRI measurements. Duong V. Thiet and Pham H. Nam contributed to the laboratory work, conducted experiments, and were involved in data collection and analysis. Le A. Tuan assisted in editing and revising the manuscript. Ngo T. Dung and Le T. Lu acquired funding and supervised the overall project. All authors reviewed and approved the final manuscript.Data availability
The data supporting this article have been included in the main text and as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was financially supported by the Development program in the Field of Physics for the 2021-2025 period, through the Ministry of Science and Technology of Vietnam (Grant number: ĐTĐL.CN.16/23). Le Thi Thanh Tam was funded by the Master, PhD Scholarship Programme of Vingroup Innovation Foundation (code: VINIF.2023.TS.104).References
- E. J. Werner, A. Datta, C. J. Jocher and K. N. Raymond, Angew. Chem., Int. Ed., 2008, 47, 8568–8580 CrossRef CAS PubMed.
- H. B. Na, I. C. Song and T. Hyeon, Adv. Mater., 2009, 21, 2133–2148 CrossRef CAS.
- H. Xing, S. Zhang, W. Bu, X. Zheng, L. Wang, Q. Xiao, D. Ni, J. Zhang, L. Zhou, W. Peng, K. Zhao, Y. Hua and J. Shi, Adv. Mater., 2014, 26, 3867–3872 CrossRef CAS.
- J. Wahsner, E. M. Gale, A. Rodríguez-Rodríguez and P. Caravan, Chem. Rev., 2019, 119, 957–1057 CrossRef CAS.
- J. Lin, P. Xin, L. An, Y. Xu, C. Tao, Q. Tian, Z. Zhou, B. Hu and S. Yang, Chem. Commun., 2019, 55, 478–481 RSC.
- M. Qin, Y. Peng, M. Xu, H. Yan, Y. Cheng and X. Zhang, Beilstein J. Nanotechnol., 2020, 11, 1000–1009 CrossRef CAS.
- T. J. Clough, L. Jiang, K.-L. Wong and N. J. Long, Nat. Commun., 2019, 10, 1420 CrossRef PubMed.
- E. Gallo, C. Diaferia, E. D. Gregorio, G. Morelli, E. Gianolio and A. Accardo, Pharmaceuticals, 2020, 13, 19 CrossRef CAS.
- M. Ahren, L. Selegard, A. Klasson, F. Soderlind, N. Abrikossova, C. Skoglund, T. Bengtsson, M. Engstrom, P. O. Kall and K. Uvdal, Langmuir, 2010, 26, 5753–5762 CrossRef CAS PubMed.
- B. H. Kim, N. Lee, H. Kim, K. An, Y. I. Park, Y. Choi, K. Shin, Y. Lee, S. G. Kwon, H. B. Na, J. G. Park, T. Y. Ahn, Y. W. Kim, W. K. Moon, S. H. Choi and T. Hyeon, J. Am. Chem. Soc., 2011, 133, 12624–12631 CrossRef CAS PubMed.
- M. J. Cho, R. Sethi, J. S. A. Narayanan, S. S. Lee, D. N. Benoit, N. Taheri, P. Decuzzi and V. L. Colvin, Nanoscale, 2014, 6, 13637 RSC.
- L. Prodi, E. Rampazzo, F. Rastrelli, A. Speghini and N. Zaccheroni, Chem. Soc. Rev., 2015, 44, 4922–4952 RSC.
- G. Zhang, R. Du, L. Zhang, D. Cai, X. Sun, Y. Zhou, J. Zhou, J. Qian, K. Zhong, K. Zheng, D. Kaigler, W. Liu, X. Zhang, D. Zou and Z. Wu, Adv. Funct. Mater., 2015, 25, 6101–6111 CrossRef CAS.
- Y. Hu, S. Mignani, J.-P. Majoral, M. Shen and X. Shi, Chem. Soc. Rev., 2018, 47, 1874–1900 RSC.
- J. Chen, W. J. Zhang, Z. Guo, H. B. Wang, D. D. Wang, J. J. Zhou and Q. W. Chen, ACS Appl. Mater. Interfaces, 2015, 7, 5373 CrossRef CAS.
- A. Charidimou, G. Boulouis, S. M. Greenberg and A. Viswanathan, Neurology, 2019, 93, e2192–202 Search PubMed.
- S. Santra, S. D. Jativa, C. Kaittanis, G. Normand, J. Grimm and J. M. Perez, ACS Nano, 2012, 6, 7281–7294 CrossRef CAS PubMed.
- W. Zhao, H. Huang, Y. Sun, X. Zhang, Y. Li and J. Wang, RSC Adv., 2015, 5, 97675–97680 RSC.
- G. Singh, B. H. McDonagh, S. Hak, D. Peddis, S. Bandyopadhyay, I. Sandvig, A. Sandvig and W. R. Glomm, J. Mater. Chem. B, 2017, 5, 418–422 RSC.
- H. Xiang, P. Dong, L. Pi, Z. Wang, T. Zhang, S. Zhang, C. Lu, Y. Pan, H. Yuan and H. Liang, J. Mater. Chem. B, 2020, 8, 1432–1444 RSC.
- M. Yang, L. Gao, K. Liu, C. Luo, Y. Wang, L. Yu, H. Peng and W. Zhang, Talanta, 2015, 131, 661–665 CrossRef CAS PubMed.
- Z. Zhou, C. Wu, H. Liu, X. Zhu, Z. Zhao, L. Wang, Y. Xu, H. Ai and J. Gao, ACS Nano, 2015, 9, 3012–3022 CrossRef CAS PubMed.
- F. Li, D. Zhi, Y. Luo, J. Zhang, X. Nan, Y. Zhang, W. Zhou, B. Qiu, L. Wen and G. Liang, Nanoscale, 2016, 8, 12826–12833 RSC.
- H. Yang, Y. M. Zhuang, Y. Sun, A. T. Dai, X. Y. Shi, D. M. Wu, F. Y. Li, H. Hu and S. P. Yang, Biomaterials, 2011, 32, 4584–4593 CrossRef CAS.
- S. Fedorenko, A. Stepanov, R. Zairov, O. Kaman, R. Amirov, I. Nizameev, K. Kholin, I. Ismaev, A. Voloshina, A. Sapunova, M. Kadirov and A. Mustafina, Colloids Surf., A, 2018, 559, 60–67 CrossRef CAS.
- X. Sun, R. Du, L. Zhang, G. Zhang, X. Zheng, J. Qian, X. Tian, J. Zhou, J. He, Y. Wang, Y. Wu, K. Zhong, D. Cai, D. Zou and Z. Wu, ACS Nano, 2017, 11, 7049–7059 CrossRef CAS.
- Y. Miao, P. Chen, M. Yan, J. Xiao, B. Hong, K. Zhou, G. Zhang, J. Qian and Z. Wu, Biomed. Mater., 2021, 16, 044104 CrossRef CAS PubMed.
- Q. Li, J. Xiao, C. Zhang, P. Wang, W. Li, Y. Wang, L. Ran, X. Qin, M. Yan, J. Qian, Z. Wu, G. Tian, P. Wei and G. Zhang, Mater. Today Adv., 2022, 16, 100278 CrossRef CAS.
- M. H. Kim, H. Y. Son, G. Y. Kim, K. Park, Y. M. Huh and S. Haam, Biomaterials, 2016, 101, 121–130 CrossRef CAS PubMed.
- Y. Si, G. Zhang, D. Wang, C. Zhang, C. Yang, G. Bai, J. Qian, Q. Chen, Z. Zhang, Z. Wu, Y. Xu and D. Zou, Chem. Eng. J., 2019, 360, 289–298 CrossRef CAS.
- J. Qin, G. Liang, Y. Feng, B. Feng, G. Wang, N. Wu, Y. Zhao and J. Wei, Nanoscale, 2020, 12, 6096–6103 RSC.
- N. T. Dung, N. V. Long, L. T. T. Tam, P. H. Nam, L. D. Tung, N. X. Phuc, L. T. Lu and N. T. K. Thanh, Nanoscale, 2017, 9, 8952–8961 RSC.
- N. T. T. Khue, L. T. T. Tam, N. T. Dung, L. T. Tam, N. X. Chung, N. T. N. Linh, N. D. Vinh, B. M. Quy and L. T. Lu, ChemistrySelect, 2022, 7, e202202062 CrossRef.
- M. I. Dar and S. A. Shivashankar, RSC Adv., 2014, 4, 4105–4113 RSC.
- Z. Zhou, D. Huang, J. Bao, Q. Chen, G. Liu, Z. Chen, X. Chen and J. Gao, Adv. Mater., 2012, 24(46), 6223–6228 CrossRef CAS PubMed.
- R. Fu, M. Ou, C. Yang, Y. Hu and H. Yin, Nanotechnology, 2020, 31, 395705 CrossRef CAS.
- K. P. Hazarika, C. Borgohain and J. P. Borah, ACS Omega, 2024, 9, 6696–6708 CrossRef CAS PubMed.
- C. Guida, A. Chappaz, A. Poulain, J.-M. Grenèche, A. Gloter, N. Menguy, N. Findling and L. Charlet, ACS Nanosci. Au, 2024, 4(5), 322–326 CrossRef CAS PubMed.
- S. A. Khan, S. Gambhir and A. Ahmad, Beilstein J. Nanotechnol., 2014, 5, 249–257 CrossRef PubMed.
- A. Khumaeni, S. Avicenna and I. Nurhasanah, J. Mater. Sci.: Mater. Electron., 2024, 35, 1422 CrossRef CAS.
- N. T. N. Linh, N. T. Dung, L. T. T. Tam, L. T. Tam, N. P. Hung, N. D. Vinh, N. T. Ha, P. H. Nam, L. V. Thanh, N. V. Dong, L. G. Nam, N. V. Dang, N. X. Phuc, L. D. Tung, N. T. K. Thanh and L. T. Lu, New J. Chem., 2023, 47, 4052–4067 RSC.
- K. Wang, L. An, Q. Tian, J. Lin and S. Yang, RSC Adv., 2018, 8, 26764–26770 RSC.
- M. Rohrer, H. Bauer, J. Mintorovitch, M. Requardt and H. J. Weinmann, Invest. Radiol., 2005, 40, 715–724 CrossRef PubMed.
- H. Wei, O. T. Bruns, M. G. Kaul, E. C. Hansen, M. Barch, A. Wiśniowska, O. Chen, Y. Chen, N. Li, S. Okada, J. M. Cordero, M. Heine, C. T. Farrar, D. M. Montana, G. Adam, H. Ittrich, A. Jasanoff, P. Nielsen and M. G. Bawendi, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 2325–2330 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ma01067a |
| This journal is © The Royal Society of Chemistry 2025 |