
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHalide-mediated electrochemical modification of copper phthalocyanine for humidity sensing applications†
Busarakham
Ngokpho
a,
Pattanaphong
Janphuang
b,
Supinya
Nijpanich
 b,
Narong
Chanlek
b,
Suttipong
Wannapaiboon
b,
Theeranun
Siritanon
a and
Kamonwad
Ngamchuea
*a
b,
Narong
Chanlek
b,
Suttipong
Wannapaiboon
b,
Theeranun
Siritanon
a and
Kamonwad
Ngamchuea
*a
aSchool of Chemistry, Institute of Science, Suranaree University of Technology, 111 University Avenue, Suranaree, Muang, Nakhon Ratchasima 30000, Thailand. E-mail: kamonwad@g.sut.ac.th; Tel: +66 0 44 224 637
bSynchrotron Light Research Institute (Public Organization), 111 University Avenue, Suranaree, Muang, Nakhon Ratchasima, 30000, Thailand
First published on 11th December 2024
Abstract
This work presents a one-step electrochemical halogenation and exfoliation of copper phthalocyanine (CuPc), producing thin-layered halogenated CuPc with tunable physicochemical properties. By incorporating halogen into the CuPc structure, the copper centers acquire an enhanced positive charge, which significantly improves their affinity for water molecules. Coupled with the strong water-interactive properties of polyvinylpyrrolidone, this halogenation-driven modification creates a synergistic effect, resulting in markedly increased sensitivity and responsiveness for humidity sensing applications. As a proof of concept, chlorinated CuPc integrated with polyvinylpyrrolidone on flexible interdigitated electrodes achieves high sensitivity (1.2 × 104%), excellent reproducibility (<5% RSD), and a broad dynamic range (11–94%RH), firmly establishing its potential as a high-performance humidity sensor.
1 Introduction
Copper(II) phthalocyanine (CuPc) is a pivotal member of the metal phthalocyanine family, renowned for its structural integrity derived from strong π–π interactions and a high electron count, which stabilize its various polymorphic forms.1 The positioning of a centrally coordinated copper(II) ion within the nitrogen-rich macrocyclic ligand imparts intrinsic optoelectronic properties, including favourable energy level alignments, narrow bandgaps, and high carrier mobilities.2,3 Such attributes, combined with its chemical versatility and thermal stability, have elevated CuPc from a historically significant blue pigment4 to a key functional material in the development of optoelectronic devices,5–7 thin-film transistors,8 and sensing platforms.9,10Despite CuPc's immense potential, conventional modification and integration methods often rely on physical or chemical exfoliation techniques that, while effective for a wide range of two-dimensional materials, frequently compromise its structural and compositional integrity, ultimately affecting performance in downstream applications.11 Mechanical exfoliation methods, such as adhesive tape cleavage or controlled shear under inert conditions, facilitate the isolation of pristine nanosheets but are inherently low-yield, labor-intensive, and unsuitable for large-scale applications.12 Ultrasonic exfoliation employs high-frequency acoustic waves in organic solvents or surfactant solutions to overcome interlayer van der Waals forces, yielding amorphous ultrathin nanosheets.13 However, this technique is highly dependent on the intrinsic defect density and disorder of the bulk material. Highly ordered, defect-deficient systems resist delamination due to robust interlayer π–π interactions, resulting in low exfoliation efficiencies and broad thickness distributions.13 Chemical strategies, such as liquid-phase exfoliation using strong acids, bases, or oxidative reagents, enhance interlayer spacing by disrupting chemical bonds or introducing functional groups. While these methods can increase yield, they often compromise the crystalline order and electronic properties of the material.14,15 Gas-phase approaches, including chemical vapor deposition, provide greater control over the formation of uniform thin films but demand precise environmental conditions and are challenging to scale, with the potential for contamination or unintended doping.16,17 These limitations highlight the need for scalable, defect-minimizing, and environmentally sustainable methods for CuPc processing.
Recent advancements have explored electrochemical methods, which offer precise control over modifications through voltage and electrolyte composition adjustments.18–20 While promising, these techniques often still necessitate direct contact with the material, which can introduce defects or lead to uneven modifications.19,20 Addressing these challenges, this study introduces a novel halide-based electrochemical technique that innovatively eliminates the need for direct electrical contact with the material being processed.21–24 By applying a potential difference across two electrodes immersed in a solution containing CuPc, this method induces the oxidation of halide ions into reactive oxidative species. These species interact with CuPc, facilitating halogenation and a consequent reduction in particle size, an effect attributed to the interaction between the electrolysis products of water and halides, such as oxygen, hydrogen, and halogen gases, and the CuPc structure. This interaction promotes the separation of layers, enhancing the material's physicochemical properties and suitability for advanced applications (refer to Scheme 1). This approach not only promotes efficient and sustainable manufacturing and material modification for processing CuPc and other similar 2D materials but also minimizes the use of hazardous solvents and generates fewer byproducts, reducing environmental impact.25,26 By optimizing the electrochemical parameters, we can tailor the properties of CuPc to meet specific application needs. For example, our work demonstrates how incorporating chlorine species can enhance CuPc's ability to interact with other molecules, such as water.
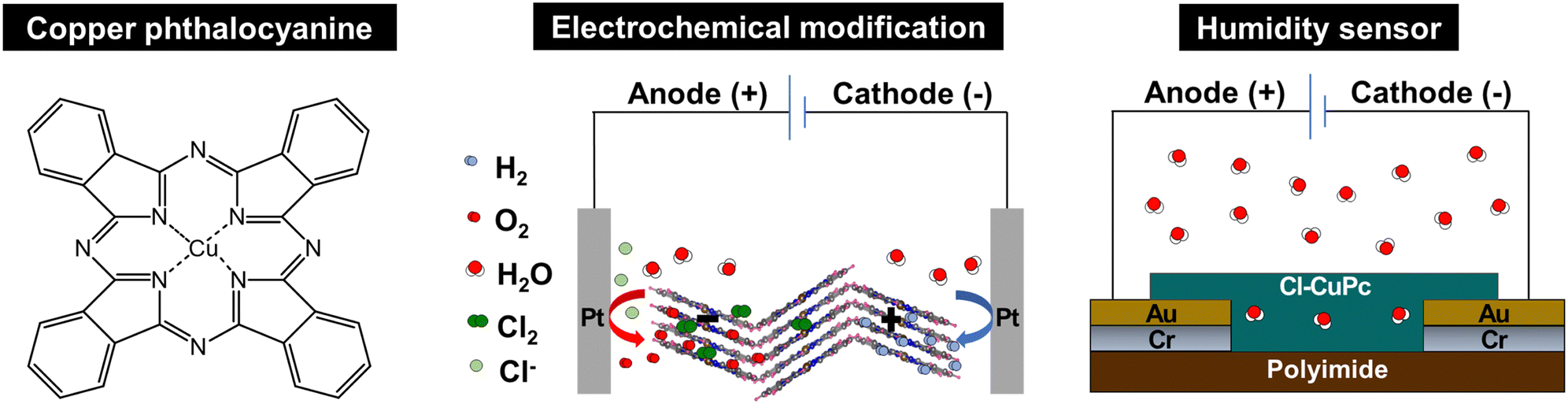 | ||
| Scheme 1 (Left) Chemical structure of copper phthalocyanine (CuPc). (middle) Halide-mediated electrochemical modifications of CuPc. (right) Application of the electrochemically modified CuPc in a humidity sensor. | ||
The application of the halogenated CuPc prepared via electrochemical synthesis proves particularly effective in humidity sensing applications, which are essential for environmental monitoring, industrial processing, consumer electronics, etc.27,28 Density functional theory calculations have identified potential adsorption configurations of water molecules on the CuPc surface, indicating molecular interactions crucial for sensor functionality.29 The electrochemically modified CuPc serves as a highly responsive and reliable medium for these sensors. Utilizing CuPc on flexible interdigitated electrodes further enhances the adaptability of these sensors. This flexibility ensures that the sensors can be integrated seamlessly into wearable technologies or applied to non-standard surfaces, providing practical alternatives to traditional rigid sensors.
2 Experimental
2.1 Chemical reagents
All reagents were of analytical grades and used as received without further purification: copper(II) phthalocyanine (α-form, >99.0%, TCI Chemicals), sodium chloride (NaCl, 99.8%, QRëC), sodium sulfate (Na2SO4, 99.5%, TCI Chemicals), lithium chloride (LiCl, 98%, Sigma Aldrich), lithium sulfate (Li2SO4, 98.0%, TCI Chemicals), polyvinylpyrrolidone (PVP, (C6H9NO)n, TCI Chemicals), magnesium chloride (MgCl2, 99.5%, QRëC), magnesium nitrate hexahydrate (Mg(NO3)2·6H2O, 99.5%, QRëC), potassium chloride (KCl, 99.0–100.5%, Sigma-Aldrich), potassium nitrate (KNO3, 99%, QRëC), and tetrabutylammonium hexafluorophosphate (TBAPF6, ≥99.0%, Sigma-Aldrich). Deionized water was obtained from the Elga Purelab Ultra water purification system (Elga Labwater, UK).2.2 Electrochemical modification and characterization of CuPc
First, 1.0 mM polyvinylpyrrolidone (PVP) was dispersed in a 0.50 M LiCl electrolyte solution and then sonicated for 3 minutes. After that, α-CuPc powder with a concentration of 0.50 mg mL−1 was added to the solution. The mixture was then sonicated at an ultrasonic power of 50 W and a frequency of 40 kHz (GT Sonic-D2, China) for 2 hours. Two Pt electrodes (10 × 10 × 0.2 mm3, 99.99% purity, Ilience) were immersed in the solution, with a 2.0 cm distance between the electrodes. A constant DC potential of 10 V was applied using a power supply (Agilent, 33220A, 20 Hz Function, Arbitrary Waveform Generator) for 90 minutes. Subsequently, the modified CuPc powder was separated from the electrolyte solution by centrifugation at 5000 rpm for 15 minutes (Hangzhou Miu Instruments CO., LTD, China), washed with deionized water, and dried at room temperature.The morphology of the CuPc samples was evaluated by a field-emission scanning electron microscope (Zeiss Auriga FESEM/FIB/EDX, 3.00 kV, Carl Zeiss, Oberkochen, Germany). The size distribution of the CuPc samples was measured using dynamic light scattering (Zetasizer-ZS, Malvern, United Kingdom) in a ZSU1002 capillary cell. The spectrophotometric measurements were performed in the spectral range of 200–900 nm using a double-beam UV-visible spectrophotometer (T80+, PG Instrument, United Kingdom). The functional groups of the CuPc samples were analyzed using a Fourier-transform infrared spectrometer (ATR-FTIR, Tensor 27, Bruker, Germany).
X-ray diffraction (XRD) analyses were conducted on the CuPc samples utilizing an X-ray diffractometer (Bruker, D2 PHASER, Germany). X-ray absorption spectroscopy (XAS) investigations, including X-ray absorption near-edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) analyses targeting the Cu K-edge, were carried out at Beamline 1.1W: Multiple X-ray Techniques, Synchrotron Light Research Institute (SLRI), Thailand. This was performed in combined transmission and fluorescent mode under ambient conditions, with simultaneous measurements of the samples in fluorescent mode and Cu foil as a standard reference in transmission mode for alignment of energy shifts during synchrotron operation. The data obtained underwent analysis using ATHENA software.30
X-ray photoelectron spectroscopy (XPS) analysis of the samples’ surface elemental composition and oxidation state was conducted at the SUT-NANOTEC-SLRI Joint Research Facility, Synchrotron Light Research Institute (SLRI), Thailand, utilizing the PHI5000 VersaProbe II instrument from ULVAC-PHI, Japan. A monochromatized Al-Kα X-ray source (hν = 1486.6 eV) was employed for sample excitation. The XPS spectra underwent fitting using the PHI MultiPak XPS software, employing a combination of Gaussian–Lorentzian lines and a Shirley-type background subtraction method. The survey spectra were acquired with an energy step of 1.000 eV and a pass energy of 117.4 eV. Conversely, the high-resolution spectra were obtained with an energy step of 0.05 eV and a pass energy of 46.95 eV. The C1s spectrum served as the peak reference, delineating the C–C/C–H binding energy at 284.8 eV.
2.3 Humidity sensor fabrication
The interdigitated electrode (IDE) is prepared through the following steps (Scheme 2). Initially, the polyimide (PI) substrate undergoes standard cleaning, which includes immersing it in acetone for 5 minutes, followed by a 5-minute immersion in isopropyl alcohol (IPA), rinsing it in deionized water for 5 minutes, and drying it with nitrogen gas for 10 seconds. Subsequently, dehydration is carried out at 80 °C for 2 hours. Next, photolithography is performed as follows by applying AZ 4620 coating onto the PI substrate, which is then spun at 500 rpm for 10 seconds. Subsequently, it is baked on a hotplate at 110 °C for 4 minutes, followed by cooling. Using a mask aligner, the substrate is post-exposed for 50 seconds, then developed using a solution of RO water, boric acid, and KOH (in a ratio of 300![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:4) for 10 minutes. After development, the substrate is baked at 80 °C for 60 minutes, and microscopic inspection is conducted for resolution and alignment. Regarding the sputtering process, it begins with plasma etching at 150 watts for 20 minutes, followed by the deposition of a chromium (Cr) adhesion layer using plasma at 150 watts for 10 minutes. Subsequently, in the evaporation process, plasma etching at 150 watts for 20 minutes precedes the evaporation of gold (Au) onto a 1.5 cm length wire at 100 mA for 20–30 nm. Finally, the lift-off process involves immersing the substrate in acetone for 3 minutes, followed by a 5-second immersion in IPA, rinsing it in oxygenated water for 60 seconds, and drying it with nitrogen gas. Finally, the sensitive layer consisting of the PVP-coated electrochemically modified CuPc (Cl-CuPc/PVP), prepared via electrochemical synthesis as described in Section 2.2, is dropcasted onto the interdigitated electrode and dried in an oven at 55 °C for 30 minutes.
:1:4) for 10 minutes. After development, the substrate is baked at 80 °C for 60 minutes, and microscopic inspection is conducted for resolution and alignment. Regarding the sputtering process, it begins with plasma etching at 150 watts for 20 minutes, followed by the deposition of a chromium (Cr) adhesion layer using plasma at 150 watts for 10 minutes. Subsequently, in the evaporation process, plasma etching at 150 watts for 20 minutes precedes the evaporation of gold (Au) onto a 1.5 cm length wire at 100 mA for 20–30 nm. Finally, the lift-off process involves immersing the substrate in acetone for 3 minutes, followed by a 5-second immersion in IPA, rinsing it in oxygenated water for 60 seconds, and drying it with nitrogen gas. Finally, the sensitive layer consisting of the PVP-coated electrochemically modified CuPc (Cl-CuPc/PVP), prepared via electrochemical synthesis as described in Section 2.2, is dropcasted onto the interdigitated electrode and dried in an oven at 55 °C for 30 minutes.
 | ||
| Scheme 2 Schematic diagram showing the fabrication process of Au/Cr interdigitated electrodes. | ||
2.4 Humidity measurement
The humidity-sensing capabilities of the sensors were assessed across various levels of relative humidity (%RH) using saturated aqueous salt solutions: LiCl, MgCl2, Mg(NO3)2, NaCl, KCl, and KNO3, corresponding to 11%, 33%, 54%, 75%, 84%, and 94% RH, respectively (refer to Fig. S1 in the ESI†). These evaluations were conducted at room temperature, with humidity and temperature control maintained using a Bentech GM1361 Temperature and Humidity Meter (Bentech, China). Linear sweep voltammetry (current–voltage or I–V analysis) was conducted via a PalmSens4 potentiostat (PalmSens, Netherlands) within a potential range of 0–1 V. The humidity response (S) of the sensor is defined by eqn (1): | (1) |
3 Results and discussion
A halide-mediated electrochemical approach was first developed for the modification of copper phthalocyanine (CuPc), followed by an extensive characterization of its altered physicochemical properties. Upon establishing the successful modification, the electrochemically functionalized CuPc was integrated into a flexible resistive humidity sensor and subjected to performance evaluation.3.1 Physicochemical properties of the electrochemically modified CuPc
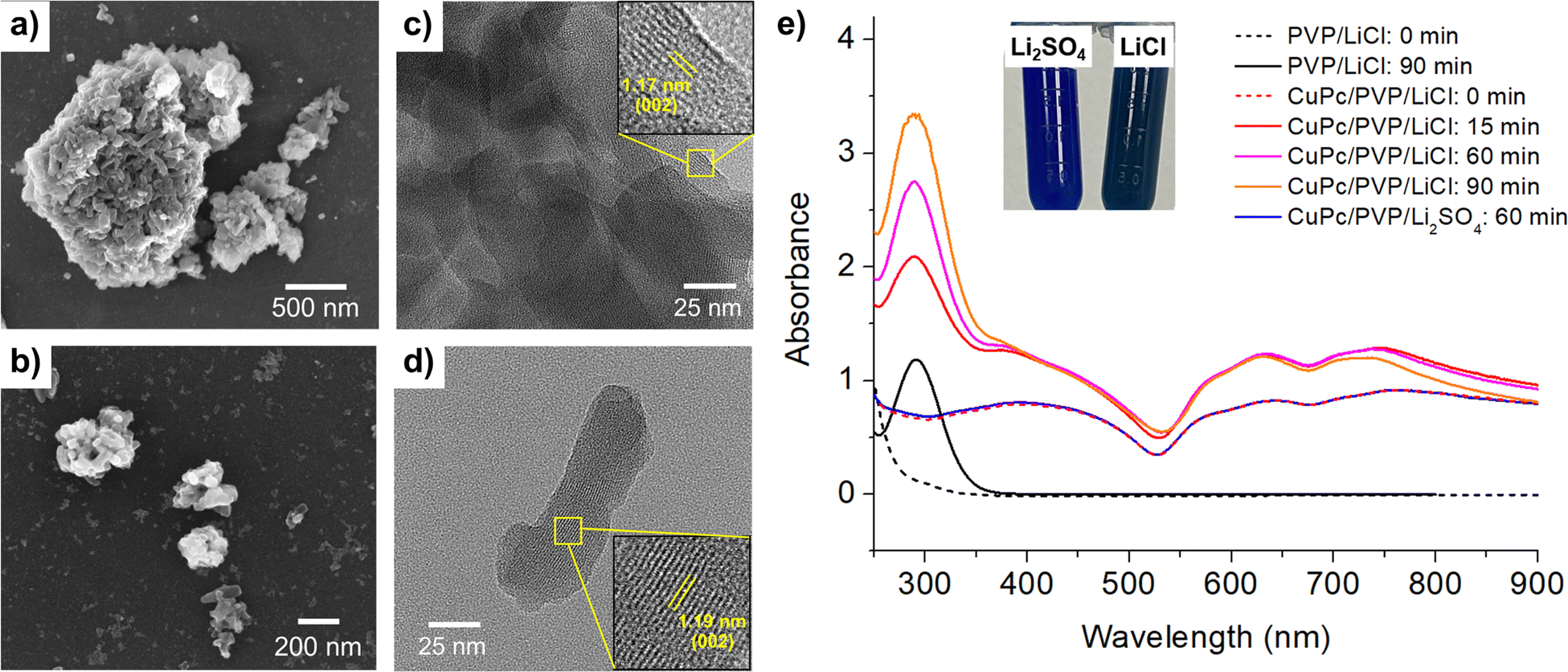 | ||
| Fig. 1 SEM and TEM images, and UV-vis spectra of CuPc: (a) SEM image before modification. (b) SEM image after modification in 0.50 M LiCl and 1.0 mM PVP at 10 V for 90 minutes. (c) TEM image before modification. (d) TEM image after modification under the same conditions as in (b). (e) UV-vis absorption spectra of CuPc suspensions before and after modification in either 0.50 M LiCl or 0.50 M Li2SO4, with 1.0 mM PVP at 10 V. Inset: Visual comparison of CuPc suspensions after modification in LiCl vs. Li2SO4. | ||
The transmission electron microscope (TEM) images shown in Fig. 1(c) and (d) further elucidate the structural changes before and after modification. Fig. 1(c) depicts the unmodified material, characterized by densely stacked and thick particles. A lattice d-spacing of 1.17 nm, corresponding to the (002) plane of the CuPc structure, is observed.34 Post-modification, Fig. 1(d) reveals a significant reduction in particle thickness, demonstrating the effectiveness of the electrochemical modification process in weakening interlayer bonding and resulting in a thinner, more dispersed structure. Additionally, the interplanar spacing corresponding to (002) plane slightly increases to 1.19 nm, indicating an expansion in lattice parameters following the modification.
Dynamic light scattering (DLS) measurements confirms a substantial reduction in the size of CuPc particles following the electrochemical modification. Before modification, the CuPc exhibits a hydrodynamic size of 1872 ± 53 nm, indicating the presence of large, aggregated particles in the solution. After subjecting the CuPc to the electrochemical modification process in 0.50 M LiCl at 10 V for 90 minutes, the DLS measurements shows a significant reduction in hydrodynamic size to 890 ± 15 nm. This notable decrease in particle size aligns well with the TEM observations, indicating the breakdown of larger CuPc aggregates into smaller, more discrete particles.
Fig. 1(e) displays the UV-vis spectrum of CuPc, where the Q-band, ranging from 600 to 750 nm, is observed. This band is attributed to π–π* transitions, influenced by the molecular orbitals within the aromatic 18 π-electron system and overlapping orbitals on the central metal.35,36 Post-modification in sulfate electrolyte (Li2SO4), the UV-vis spectra of CuPc remained unchanged, consistent with the persistent blue color.
However, following the modification of CuPc in a chloride electrolyte (LiCl), a new absorption band, Soret band, appeared at approximately 292 nm.37 This peak also coincided with the PVP absorption band which appeared post-modification (Fig. 1(e), black line).38 The results suggest that the application of potentials and the presence of oxidizing chlorine species39 such as Cl2, HClO and ClO− generated in the modification process not only change the morphology of CuPc, but also cause chemical changes to both CuPc and PVP.
The observed color changes can be attributed to subtle alterations in the electronic structure resulting from halide incorporation. Under electrochemical conditions in chloride-containing electrolytes, the formation of Cu–Cl and C–Cl bonds introduces slight shifts in electron density and the distribution of molecular orbitals within the CuPc framework. These changes affect the energies and intensities of the π–π* transitions in the UV-vis spectrum, thereby altering the perceived color. In contrast, no such halogenation occurs in sulfate electrolytes, preserving the original electronic structure and the characteristic blue hue of CuPc. Likewise, bromide electrolytes create a distinct halogen environment that modifies the electronic states differently, yielding unique color variations. More detailed evidence on the specific structural and electronic modifications responsible for these color changes is provided in subsequent sections.
 | ||
| Fig. 2 (a) XRD patterns of CuPc before modification (black line) and after modification (red line) in 0.50 M LiCl for 90 minutes. (b) XAS spectra at the CuPc K-edge of CuPc before modification (black line) and after modification (red line) in 0.50 M LiCl for 90 minutes. Insets: Top: Derivative μ(E) plots of the XANES and EXAFS spectra; bottom left: k-space plots of the EXAFS spectra; bottom right: R-space plots of the EXAFS spectra (generated from Fourier transformation of k-space from 3 to 9.5 Å−1). | ||
The X-ray absorption spectra (Fig. 2(b)) reveal notable insights into the structural and electronic changes of CuPc at the Cu K-edge before and after undergoing a 90-minute modification in a LiCl electrolyte. The main plot, representing the normalized absorption coefficients (μ(E)), shows that the peak positions in the X-ray absorption near edge structure (XANES) region are relatively consistent before and after modification, suggesting that the copper centers largely retain their oxidation state and electronic configuration.40,41 However, slight variations in peak intensity and shape hint at subtle changes in the local electronic structure or coordination environment.42 In the extended X-ray absorption fine structure (EXAFS) region, as detailed in the insets, there are indications of changes in local structure: the derivative plot (top inset) shows a reduction in peak sharpness and intensity post-modification, while the EXAFS oscillations (bottom-left inset) and their Fourier transforms (bottom-right inset) suggest minor adjustments in bond distances and possibly coordination numbers. This could potentially be attributed to the interactions at the Cu–Cl center and the C–Cl binding within the structure, as discussed along with the XPS results in the next section.
The X-ray photoelectron spectroscopy (XPS) results presented in Fig. 3 provide insightful observations into the chemical and electronic transformations of CuPc upon modification in a 0.50 M LiCl solution, particularly highlighting modifications to the chemical environment of the copper centers. The Cu 2p region presents a doublet peak corresponding to Cu 2p3/2 and Cu 2p1/2, which exhibits a shift from 935.7 eV and 955.4 eV in the unmodified state to 933.5 eV and 953.4 eV after modification,43 with additional peaks at 935.3 eV and 955.0 eV, suggesting the formation of Cu–Cl bonds and electron donation from chlorine atoms. These shifts in the Cu 2p peaks, while indicative of changes in the local electronic environment, do not necessarily reflect a change in the formal oxidation state of copper, as verified by the stable peak positions in the XANES region.
 | ||
| Fig. 3 XPS spectra of CuPc before and after modification in 0.50 M LiCl at 10 V for 90 minutes, showing peaks for (a) Cu 2p, (b) Cl 2p, (c) C 1s, (d) O 1s, and (e) N 1s. | ||
In the Cl 2p high-resolution spectra, the emergence of new peaks post-modification highlights the active role of chlorine in forming diverse coordination complexes within the CuPc and PVP structures, likely leading to the formation of Cu−Cl and C−Cl bonds. Shifts in the C 1s, O 1s, and N 1s high-resolution spectra also indicate substantial modifications in the carbon framework, oxygen functionalities, and nitrogen environments, details of which are elaborated in Section S4 of the ESI.† These spectral changes are consistent with the establishment of Cu−Cl and C−Cl bonds subsequent to modification, suggesting that the modification process triggers both surface modifications and deeper structural reconfigurations within the CuPc molecule. Further examination of the XPS spectra following modification in a bromide electrolyte (LiBr), as detailed in Fig. S10 (ESI†), reveals the presence of Cu−Br and C−Br bonds, indicating a similar transformation as observed with chloride electrolytes. This underscores the significant roles of halides in facilitating these modification processes. Additional XPS data are available in Fig. S7–S10, ESI.†
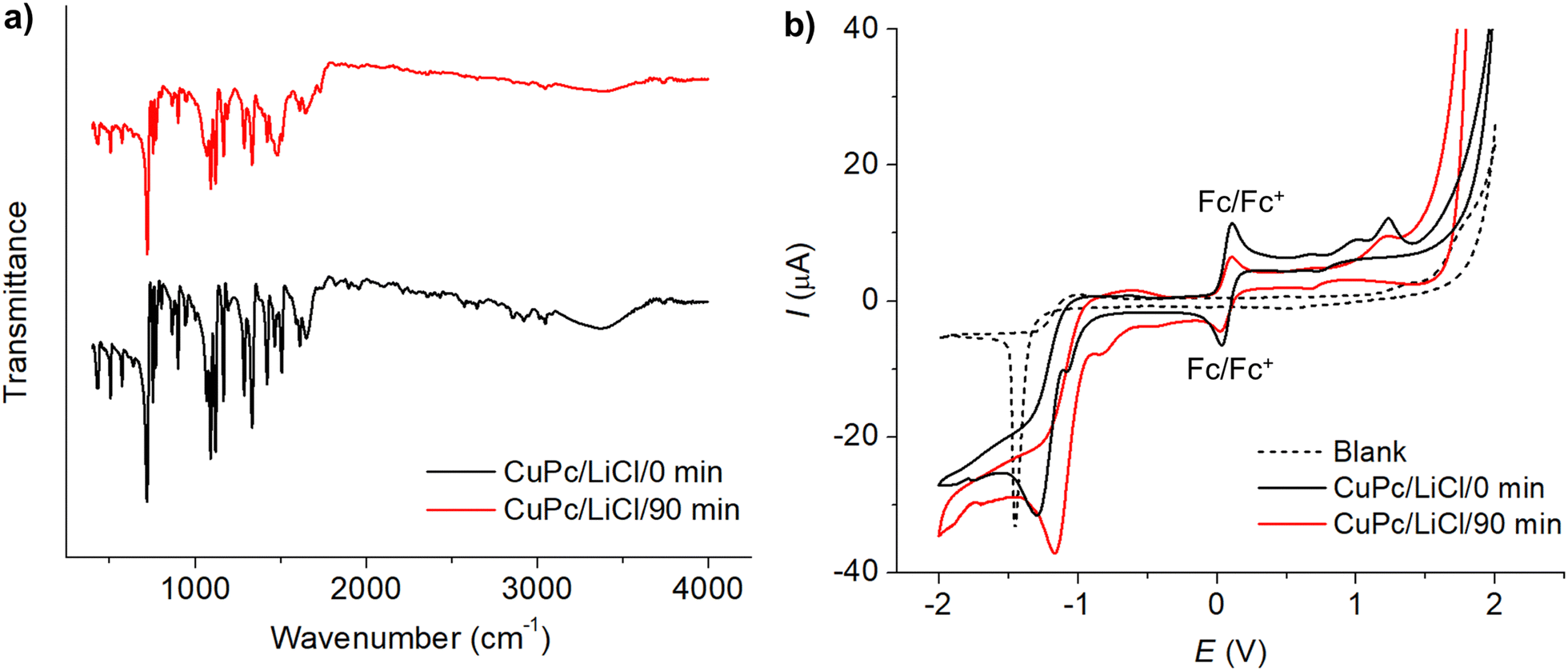 | ||
| Fig. 4 (a) FTIR spectra of CuPc before modification (black line) and after modification (red line) in 0.50 M LiCl for 90 minutes. (b) Cyclic voltammograms of 1.0 mM CuPc before modification (black line) and after modification (red line) in the presence of 1.0 mM ferrocene and 0.10 M TBAPF6 in acetonitrile at a scan rate of 50 mV s−1. | ||
The voltammograms of CuPc before and after the modification process are depicted in Fig. 4(b). Prior to modification, the cyclic voltammetry of CuPc exhibited three distinct oxidation peaks at 0.669 V, 1.008 V, and 1.233 V, and two reduction peaks at −1.082 V and −1.294 V. Post-modification, the voltammogram shows two oxidation peaks at 0.694 V and 1.23 V, and reduction peaks at −0.831 V and −1.175 V. Notably, the reduction peaks observed at very negative potentials of −1.175 V (before modification) and −1.294 V (after modification) showed characteristics similar to the blank electrolyte response at −1.453 V, suggesting that these peaks might not directly relate to CuPc's intrinsic electrochemical properties but rather to reactions such as proton reduction occurring at the electrode interface.
The frontier molecular orbital energies were subsequently determined based on the redox potentials. The energy of the highest occupied molecular orbital (HOMO) was derived from the first oxidation peak according to eqn (2):44
| EHOMO = −4.8 + [E1/2,Fc − Eox] | (2) |
The first reduction wave was employed to determine the energy of the lowest unoccupied molecular orbital (LUMO), according to eqn (3):44
| ELUMO = −4.8 + [E1/2,Fc − Ered] | (3) |
The energy gap (ΔE) was then calculated from the difference between HOMO and LUMO energies. The results are summarized in Table 1.
| E ox (V) | E red (V) | HOMO (eV) | LUMO (eV) | ΔE (eV) | |
|---|---|---|---|---|---|
| Before modification | 0.669 | −1.082 | −5.406 | −3.655 | 1.751 |
| After modification | 0.694 | −0.831 | −5.448 | −3.906 | 1.542 |
The changes in oxidation and reduction potentials are indicative of modifications in CuPc's electronic environment, likely due to the interaction with chlorine species during the modification process. XPS and XAS results corroborate this, showing shifts in the Cu 2p peaks and the appearance of Cl 2p peaks, which signify the formation of Cu−Cl bonds. This interaction contributes to the stabilization of copper, influencing the electron density around copper centers. Consequently, this affects the HOMO and LUMO energy levels of CuPc, where the HOMO level deepened slightly from −5.406 eV to −5.448 eV and the LUMO shifted from −3.655 eV to −3.906 eV, narrowing the energy gap from 1.751 eV to 1.542 eV. These modifications in the frontier molecular orbitals, as evidenced by shifts in energy levels post-modification, reflect an enhanced ability of CuPc to facilitate electron transfer.
3.2 Application of the modified CuPc in humidity sensor
This section employs the modified CuPc in the development of a flexible humidity sensor using an interdigitated electrode (IDE) composed of gold and chromium on a polyimide substrate (Fig. 5(a)). The functionalized CuPc, polyvinylpyrrolidone-coated chlorinated copper phthalocyanine (Cl–CuPc/PVP), prepared as described in Section 2.2, acts as the humidity-sensitive material and is coated onto the IDE, as illustrated in Fig. 5(b). The sensor's flexibility is demonstrated in Fig. 5(c), which shows that it can bend up to 84 degrees without significant changes in the I–V characteristics (within ±5% experimental error). Fig. 5(d) presents an optical microscope image of the fabricated IDE, which incorporates 60 interleaved bands, with an electrode length of 12.00 ± 0.01 mm, an electrode width of 79 ± 1 μm, and a gap width of 57 ± 3 μm. | ||
| Fig. 5 (a) A bare Au/Cr interdigitated electrode (Au/Cr-IDE). (b) Optical microscope image of the Au/Cr-IDE. (b) Cl-CuPc/PVP deposited on the Au/Cr-IDE. (c) Demonstration of the substrate's flexibility. (e) EIS spectra of Cl-CuPc/PVP/Au/Cr-IDE at different %RH. | ||
Electrochemical impedance spectroscopy (EIS) measurements are then used to gain insights into the electrical properties of the sensor under varying humidity conditions, as shown in Fig. 5(e). Initially, at low relative humidity (RH) levels of 11%RH, 33%RH, and 54%RH, the Nyquist plots exhibit large semicircles corresponding to high resistances of 28.7 MΩ, 27.3 MΩ, and 19.1 MΩ, and small capacitances of 0.024 nF, 0.024 nF, and 0.026 nF, respectively, indicating dominant resistive behavior. As humidity increases to 75%RH, 84%RH, and 94%RH, the resistance decreases to 7.9 MΩ, 1.7 MΩ, and 0.47 MΩ, while the capacitance increases to 0.27 nF, 32 nF, and 80 nF, reflecting a shift towards capacitive behavior due to the formation of a more continuous moisture layer, which affects the sensor's double-layer capacitance. This necessitates the use of more complex equivalent circuit models to accurately capture the charge dynamics and moisture interactions at these elevated humidity levels, as shown in the inset of Fig. 5(e). Overall, the decrease in resistance and increase in capacitance with rising humidity indicate enhanced conductivity and moisture interaction, facilitated by the adsorption of water molecules onto the sensor's film and the alteration of its dielectric properties.
Fig. 6(a) demonstrates the current–voltage characteristics of the developed Cl-CuPc/PVP sensor under varying humidity levels, ranging from 11%RH to 94%RH, showing an increase in current and a corresponding decrease in resistance with increasing humidity. The relative humidity response (S), calculated according to eqn (1),31–33 is shown in the inset of Fig. 6(a). The humidity response of the Cl-CuPc/PVP reaches a maximum response of 1.2 × 104%, significantly higher than that of the unmodified CuPc/PVP (4.7 × 103%), refer to Fig. S11 in the ESI.† This improvement is attributed to the enhanced adsorption of water molecules onto the Cl-CuPc/PVP film. The chlorine doping in CuPc increases the positive charge on the copper centers, thereby enhancing their affinity for the oxygen atoms in water molecules. This mechanism is supported by the XPS findings discussed in Fig. 3, which indicate an increase in the positive charge on copper centers after modification. Additionally, PVP enhances the sensor's responsiveness through strong interactions with water molecules due to the mesomeric effect of its peptide groups.45 The detection mechanism of the Cl-CuPc/PVP humidity sensor is summarized schematically in Fig. S12 (ESI†).
 | ||
| Fig. 6 (a) Current–voltage characteristics of the Cl-CuPc/PVP sensor across varying relative humidity (%RH) levels. The inset shows the plot of the humidity response (S) as a function of %RH. (b) Reproducibility of the Cl-CuPc/PVP sensor over three cycles at 11%RH and 94%RH. (c) Response and recovery time under fast change of %RH values between 11%RH and 94% RH. (d) Stability of the sensor over 10 days of storage at room temperature. | ||
Fig. 6(b) presents the sensor's reproducibility across three consecutive cycles of exposure to alternating humidity levels (11%RH and 94%RH). The current responses at both 11%RH and 94%RH exhibit good reproducibility during continuous measurements, with relative standard deviations of 4.3% and 4.6%, respectively. Fig. 6(c) provides a detailed examination of a single cycle, where the response and recovery times are determined to be 35 seconds and 243 seconds, respectively. Note that the response time is defined as the duration required for the sensor to achieve a stable reading when the humidity is decreased from 94%RH to 11%RH, while the recovery time is the time needed to stabilize when the humidity is increased from 11%RH back to 94%RH.
The observed difference in response and recovery times can be attributed to the underlying mechanisms. During the response phase, the sensor releases (desorbs) water molecules as the surrounding humidity decreases. This process is relatively rapid because some of the water molecules on the sensor's surface are loosely bound and can easily diffuse out into the drier environment. In contrast, during the recovery phase, the sensor adsorbs water molecules from a more humid atmosphere, which often requires a longer time to rebuild the network of interactions, such as hydrogen bonds, between the water molecules and the sensor surface. This results in a longer recovery time compared to the response time.46
The long-term stability of the sensor stored at room temperature was tested over a 10-day period, as shown in Fig. 6(d), demonstrating excellent stability with relative standard deviations (RSD) of 6.0%, 5.8%, and 5.4% at 11%RH, 54%RH, and 94%RH, respectively. Prolonged storage for 20 and 30 days resulted in increased RSD values of 7.3% and 8.7% at 94%RH, and 6.8% and 8.1% at 54%RH, indicating a gradual decline in performance. Notably, at low humidity (11%RH), the sensor's performance deteriorated significantly, with a reduction in activity exceeding 10% after 10 days. These findings demonstrate that the sensor maintains operational stability for at least 10 days, particularly at moderate to high relative humidity levels, but its functional lifetime is limited under low-humidity environments.
The performance of the developed Cl-CuPc/PVP humidity sensor is compared with that of other humidity sensors in Table 2.
| Electrode | Dynamic range (%RH) | S (%) | Response/recovery time (s) | Ref. |
|---|---|---|---|---|
| AuNP: gold nanoparticles, CNT: carbon nanotubes, MWCNT: multi-walled carbon nanotubes, NFC: cellulose nanofiber, PEG: polyethylene glycol, PI: polyimide, PVP: polyvinylpyrrolidone, rGO: reduced graphene oxide. | ||||
| MoS2/PVP | 11–94 | 80 | 5/2 | 33 |
| MoS2 | 0–35 | 104 | 10/60 | 47 |
| MoS2 | 0–80 | 85 | 0.6/0.3 | 48 |
| MoS2 | 10–95 | 5.3 × 106 | 8/22 | 49 |
| MWCNT/PI | 20–90 | 12.8 | 5/600 | 50 |
| NFC/MWCNT | 11–95 | 69.9 | 330/377 | 32 |
| PEG/AuNP | 1.8–95 | 104 | 1.2/3 | 51 |
| SnO2/rGO | 11–97 | 45.02 | 90/100 | 52 |
| Cl-CuPc/PVP | 11–94 | 1.2 × 104 | 35/243 | This work |
4 Conclusions
This work presents the first utilization and comprehensive elucidation of the roles of halides in the electrochemical modification of layered materials such as CuPc. By employing a halide-mediated process, we successfully optimize the physicochemical and surface properties of CuPc, achieving thinner layers and facilitating halogenation while preserving its structural integrity. These modifications have rendered the functionalized material highly effective as a humidity sensor. We further integrate the enhanced CuPc with polyvinylpyrrolidone on flexible interdigitated electrodes mounted on polyimide substrates. The resulting sensors demonstrate robustness, high sensitivity, excellent reproducibility, and a broad dynamic range, making them well-suited for wearable and unconventional applications. Importantly, the scalable and environmentally friendly nature of this process significantly expands the potential applications of metal phthalocyanines in material science and sensor technology.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This work is financially supported by Suranaree University of Technology Research and Development Fund [grant number: IRD1-102-67-12-28].References
- E. Jungyoon, S. Kim, E. Lim, K. Lee, D. Cha and B. Friedman, Effects of substrate temperature on copper(II) phthalocyanine thin films, Appl. Surf. Sci., 2003, 205(1–4), 274–279 Search PubMed.
- R. Prabakaran, R. Kesavamoorthy, G. Reddy and F. P. Xavier, Structural investigation of copper phthalocyanine thin films using X-ray diffraction, Raman scattering and optical absorption measurements, Phys. Status Solidi B, 2002, 229(3), 1175–1186 CrossRef CAS.
- G. Kumar, V. Nampoori, C. Vallabhan, G. Jose and N. Unnikrishnan, Emission spectral studies of phthalocyanines in borate glass matrix, J. Mater. Sci. Lett., 2000, 19, 1669–1672 CrossRef CAS.
- M. Anghelone, D. Jembrih-Simbürger and M. Schreiner, Identification of copper phthalocyanine blue polymorphs in unaged and aged paint systems by means of micro-Raman spectroscopy and Random Forest, Spectrochim. Acta, Part A, 2015, 149, 419–425 CrossRef CAS PubMed.
- R. F. Salzman, J. Xue, B. P. Rand, A. Alexander, M. E. Thompson and S. R. Forrest, The effects of copper phthalocyanine purity on organic solar cell performance, Org. Electron., 2005, 6(5–6), 242–246 CrossRef CAS.
- T. Schwieger, H. Peisert, M. Golden, M. Knupfer and J. Fink, Electronic structure of the organic semiconductor copper phthalocyanine and K-CuPc studied using photoemission spectroscopy, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 66(15), 155207 CrossRef.
- S. Schumann, R. A. Hatton and T. S. Jones, Organic photovoltaic devices based on water-soluble copper phthalocyanine, J. Phys. Chem. C, 2011, 115(11), 4916–4921 CrossRef CAS.
- X. Li, Y. Jiang, G. Xie, H. Tai, P. Sun and B. Zhang, Copper phthalocyanine thin film transistors for hydrogen sulfide detection, Sens. Actuators, B, 2013, 176, 1191–1196 CrossRef CAS.
- K. Ngamchuea, C. Moonla, A. Watwiangkham, S. Wannapaiboon and S. Suthirakun, Electrochemical and structural investigation of copper phthalocyanine: Application in the analysis of kidney disease biomarker, Electrochim. Acta, 2022, 428, 140951 CrossRef CAS.
- P. Sunon, B. Ngokpho, K. Kaewket, S. Wannapaiboon and K. Ngamchuea, Copper(II) phthalocyanine as an electrocatalytic electrode for cathodic detection of urinary tryptophan, Analyst, 2024, 149(10), 3041–3051 RSC.
- C. Hu, D. Jiang, Y. Zhang, H. Gao, Y. Zeng, N. Khaorapapong, Z. Liu, Y. Yamauchi and M. Pan, Porphyrins-based multidimensional nanomaterials: Structural design, modification and applications, Coord. Chem. Rev., 2025, 523, 216264 CrossRef CAS.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D.-E. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Electric field effect in atomically thin carbon films, Science, 2004, 306(5696), 666–669 CrossRef CAS PubMed.
- W. Liu, C. Wang, L. Zhang, H. Pan, W. Liu, J. Chen, D. Yang, Y. Xiang, K. Wang and J. Jiang, Exfoliation of amorphous phthalocyanine conjugated polymers into ultrathin nanosheets for highly efficient oxygen reduction, J. Mater. Chem. A, 2019, 7(7), 3112–3119 RSC.
- S. Liu, L. Wang, L. Li, Q. Luo and J. Chen, Chemically exfoliated few-layer phthalocyanine-based covalent organic frameworks used as improved energy storage electrode for lithium-ion batteries, J. Energy Storage, 2024, 98, 113124 CrossRef.
- V. Nicolosi, M. Chhowalla, M. G. Kanatzidis, M. S. Strano and J. N. Coleman, Liquid exfoliation of layered materials, Science, 2013, 340(6139), 1226419 CrossRef.
- Y. Tanamura, T. Uchida, N. Teramae, M. Kikuchi, K. Kusaba and Y. Onodera, Ship-in-a-bottle synthesis of copper phthalocyanine molecules within mesoporous channels of MCM-41 by a chemical vapor deposition method, Nano Lett., 2001, 1(7), 387–390 CrossRef CAS.
- Y. Ogawa, T. Niu, S. L. Wong, M. Tsuji, A. T. S. Wee, W. Chen and H. Ago, Self-assembly of polar phthalocyanine molecules on graphene grown by chemical vapor deposition, J. Phys. Chem. C, 2013, 117(42), 21849–21855 CrossRef CAS.
- A. Ambrosi and M. Pumera, Exfoliation of layered materials using electrochemistry, Chem. Soc. Rev., 2018, 47(19), 7213–7224 RSC.
- Y. Yang, H. Hou, G. Zou, W. Shi, H. Shuai, J. Li and X. Ji, Electrochemical exfoliation of graphene-like two-dimensional nanomaterials, Nanoscale, 2019, 11(1), 16–33 RSC.
- N. Liu, P. Kim, J. H. Kim, J. H. Ye, S. Kim and C. J. Lee, Large-area atomically thin MoS2 nanosheets prepared using electrochemical exfoliation, ACS Nano, 2014, 8(7), 6902–6910 CrossRef CAS PubMed.
- S. E. Fosdick, K. N. Knust, K. Scida and R. M. Crooks, Bipolar electrochemistry, Angew. Chem., Int. Ed., 2013, 52(40), 10438–10456 CrossRef CAS PubMed.
- Y. Wang, C. C. Mayorga-Martinez, X. Chia, Z. Sofer and M. Pumera, Nonconductive layered hexagonal boron nitride exfoliation by bipolar electrochemistry, Nanoscale, 2018, 10(15), 7298–7303 RSC.
- S. M. Tan, C. C. Mayorga-Martinez, Z. Sofer and M. Pumera, Bipolar electrochemistry exfoliation of layered metal chalcogenides Sb2S3 and Bi2S3 and their hydrogen evolution applications, Chem. – Eur. J., 2020, 26(29), 6479–6483 CrossRef CAS PubMed.
- I. Khakpour, A. R. Baboukani, A. Allagui, A. A. Hachicha and C. Wang, On the mechanistic pathways of exfoliation-and-deposition of graphene by bipolar electrochemistry, Nanotechnol., 2021, 32(34), 345603 CrossRef CAS PubMed.
- S. M. Beladi-Mousavi, G. Salinas, N. Antonatos, V. Mazanek, P. Garrigue, Z. Sofer and A. Kuhn, Fine-tuning the functionality of reduced graphene oxide via bipolar electrochemistry in freestanding 2D reaction layers, Carbon, 2022, 191, 439–447 CrossRef CAS.
- M. Zhao, C. Casiraghi and K. Parvez, Electrochemical exfoliation of 2D materials beyond graphene, Chem. Soc. Rev., 2024, 53, 3036–3064 RSC.
- M. Sajid, Z. Khattak, K. Rahman, G. Hassan and K. Choi, Progress and future of relative humidity sensors: a review from materials perspective, Bull. Mater. Sci., 2022, 45(4), 238 CrossRef CAS.
- Z. Li, B. Lin, S. Zhang, C. Ding, S. Sun and M. Pan, A cellulose nanocrystal-based dual response of photonic colors and fluorescence for sensitive benzene gas detection, Int. J. Biol. Macromol., 2024, 132706 CrossRef CAS PubMed.
- A. Farzaneh, M. D. Esrafili and S. Okur, Experimental and density functional theory study on humidity sensing properties of copper phthalocyanine (CuPc), Mater. Res. Express, 2019, 6(10), 105901 CrossRef CAS.
- B. Ravel and M. Newville, ATHENA, ARTEMIS, HEPHAESTUS: data analysis for X-ray absorption spectroscopy using IFEFFIT, J. Synchrotron Radiat., 2005, 12(4), 537–541 CrossRef CAS PubMed.
- H. Zhao, T. Zhang, R. Qi, J. Dai, S. Liu and T. Fei, Drawn on paper: a reproducible humidity sensitive device by handwriting, ACS Appl. Mater. Interfaces, 2017, 9(33), 28002–28009 CrossRef CAS PubMed.
- P. Zhu, Y. Liu, Z. Fang, Y. Kuang, Y. Zhang, C. Peng and G. Chen, Flexible and highly sensitive humidity sensor based on cellulose nanofibers and carbon nanotube composite film, Langmuir, 2019, 35(14), 4834–4842 CrossRef CAS PubMed.
- X.-F. Jin, L. Chen, Y. Zhang, X.-J. Zhang, Y.-M. Chen and J.-J. Chen, Inkjet-printed MoS2/PVP hybrid nanocomposite for enhanced humidity sensing, Sens. Actuators, A, 2020, 316, 112388 CrossRef CAS.
- A. C. Cruickshank, C. J. Dotzler, S. Din, S. Heutz, M. F. Toney and M. P. Ryan, The crystalline structure of copper phthalocyanine films on ZnO (1100), J. Am. Chem. Soc., 2012, 134(35), 14302–14305 CrossRef CAS PubMed.
- S. S. Mali, D. Dalavi, P. Bhosale, C. Betty, A. Chauhan and P. Patil, Electro-optical properties of copper phthalocyanines (CuPc) vacuum deposited thin films, RSC Adv., 2012, 2(5), 2100–2104 RSC.
- A. Farag, Optical absorption studies of copper phthalocyanine thin films, Opt. Laser Technol., 2007, 39(4), 728–732 CrossRef CAS.
- K. A. Castro, F. Figueira, F. A. A. Paz, J. P. Tomé, R. S. da Silva, S. Nakagaki, M. G. P. Neves, J. A. Cavaleiro and M. M. Simões, Copper-phthalocyanine coordination polymer as a reusable catechol oxidase biomimetic catalyst, Dalton Trans., 2019, 48(23), 8144–8152 RSC.
- P. Tavlarakis, J. J. Urban and N. Snow, Determination of total polyvinylpyrrolidone (PVP) in ophthalmic solutions by size exclusion chromatography with ultraviolet-visible detection, J. Chromatogr. Sci., 2011, 49(6), 457–462 CAS.
- P. Liewchirakorn and K. Ngamchuea, Benign electrolytic modifications of starch: effects on functional groups and physical properties, RSC Adv., 2023, 13(43), 30040–30051 RSC.
- F. Evangelista, V. Carravetta, G. Stefani, B. Jansik, M. Alagia, S. Stranges and A. Ruocco, Electronic structure of copper phthalocyanine: An experimental and theoretical study of occupied and unoccupied levels, J. Chem. Phys., 2007, 126(12), 124709 CrossRef PubMed.
- C. Lamberti, S. Bordiga, F. Bonino, C. Prestipino, G. Berlier, L. Capello, F. D’Acapito, F. L. i Xamena and A. Zecchina, Determination of the oxidation and coordination state of copper on different Cu-based catalysts by XANES spectroscopy in situ or in operando conditions, Phys. Chem. Chem. Phys., 2003, 5(20), 4502–4509 RSC.
- L. S. Chia, Y. H. Du, S. Palale and P. S. Lee, Interaction of copper phthalocyanine with nitrogen dioxide and ammonia investigation using X-ray absorption spectroscopy and chemiresistive gas measurements, ACS Omega, 2019, 4(6), 10388–10395 CrossRef CAS PubMed.
- D. Zheng, Z. Gao, X. He, F. Zhang and L. Liu, Surface and interface analysis for copper phthalocyanine (CuPc) and indium-tin-oxide (ITO) using X-ray photoelectron spectroscopy (XPS), Appl. Surf. Sci., 2003, 211(1–4), 24–30 CrossRef CAS.
- S. Rattanopas, K. Chansaenpak, K. Siwawannapong, K. Ngamchuea, S. Wet-osot, J. Treekoon, T. Pewklang, T. Jinaphon, K. Sagarik and R. Y. Lai, Synthesis and characterization of push-pull Aza-BODIPY dyes towards application in NIR-II photothermal therapy, ChemPhotoChem, 2020, 4(11), 5304–5311 CrossRef CAS.
- A. A. De Queiroz, D. A. Soares, P. Trzesniak and G. A. Abraham, Resistive-type humidity sensors based on PVP–Co and PVP–I2 complexes, J. Polym. Sci., Part B: Polym. Phys., 2001, 39(4), 459–469 CrossRef CAS.
- A. M. Laera, G. Cassano, E. Burresi, M. L. Protopapa and M. Penza, Flexible humidity sensor based on chemically reduced graphene oxide, Chemosensors, 2024, 12(12), 245 CrossRef.
- J. Zhao, N. Li, H. Yu, Z. Wei, M. Liao, P. Chen, S. Wang, D. Shi, Q. Sun and G. Zhang, Retracted: Highly sensitive MoS2 humidity sensors array for noncontact sensation, Adv. Mater., 2017, 29(34), 1702076 CrossRef PubMed.
- H. Jeong, Y. Noh and D. Lee, Highly stable and sensitive resistive flexible humidity sensors by means of roll-to-roll printed electrodes and flower-like TiO2 nanostructures, Ceram. Int., 2019, 45(1), 985–992 CrossRef CAS.
- N. M. Pereira, N. P. Rezende, T. H. Cunha, A. P. Barboza, G. G. Silva, D. Lippross, B. R. Neves, H. Chacham, A. S. Ferlauto and R. G. Lacerda, Aerosol-printed MoS2 ink as a high sensitivity humidity sensor, ACS Omega, 2022, 7(11), 9388–9396 CrossRef CAS PubMed.
- Q.-Y. Tang, Y. Chan and K. Zhang, Fast response resistive humidity sensitivity of polyimide/multiwall carbon nanotube composite films, Sens. Actuators, B, 2011, 152(1), 99–106 CrossRef CAS.
- C.-H. Su, H.-L. Chiu, Y.-C. Chen, M. Yesilmen, F. Schulz, B. Ketelsen, T. Vossmeyer and Y.-C. Liao, Highly responsive PEG/gold nanoparticle thin-film humidity sensor via inkjet printing technology, Langmuir, 2019, 35(9), 3256–3264 CrossRef CAS PubMed.
- D. Zhang, H. Chang and R. Liu, Humidity-sensing properties of one-step hydrothermally synthesized tin dioxide-decorated graphene nanocomposite on polyimide substrate, J. Electron. Mater., 2016, 45, 4275–4281 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ma01013b |
| This journal is © The Royal Society of Chemistry 2025 |