
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Heavy-atom-free BODIPY-based photodynamic therapy agents activated at long wavelengths†
Jennifer
Soler-Beatty‡
ab,
Edurne
Avellanal-Zaballa‡
c,
Gonzalo
Durán-Sampedro‡
d,
Alba
García-Fernández
 abe,
Antonia R.
Agarrabeitia
df,
Jorge
Bañuelos
*c,
Ramón Martínez
Mañez
*abeg and
María J.
Ortiz
*d
abe,
Antonia R.
Agarrabeitia
df,
Jorge
Bañuelos
*c,
Ramón Martínez
Mañez
*abeg and
María J.
Ortiz
*d
aInstituto Interuniversitario de Investigación de Reconocimiento Molecular y Desarrollo Tecnológico (IDM), Universitat Politècnica de València, Universitat de València, 46022, Valencia, Spain. E-mail: rmaez@quim.upv.es
bUnidad Mixta UPV-CIPF de Investigación en Mecanismos de Enfermedades y Nanomedicina, Centro de Investigación Príncipe Felipe, Universitat Politècnica de València, 46012, Valencia, Spain
cDpto. Química Física, Facultad de Ciencia y Tecnología, Universidad del País Vasco (UPV/EHU), 48080, Bilbao, Spain. E-mail: jorge.banuelos@ehu.es
dDpto. Química Orgánica, Facultad de CC. Químicas, Universidad Complutense de Madrid, 28040, Madrid, Spain. E-mail: mjortiz@quim.ucm.es
eCIBER de Bioingeniería, Biomateriales y Nanomedicina (CIBER-BBN), Instituto de Salud Carlos III, Spain
fSección Departamental de Química Orgánica, Facultad de Óptica y Optometría, Universidad Complutense de Madrid, 28040, Madrid, Spain
gUnidad Mixta de Investigación en Nanomedicina y Sensores. Universitat Politècnica de València, IIS La Fe, 46026, Valencia, Spain
First published on 16th December 2024
Abstract
Photodynamic therapy (PDT) is an emerging clinical tool that uses light as an agent unleashing cytotoxic activity for treating cancer and other diseases, including those showing drug resistance. One of the main areas of research to fully implement PDT as a real alternative to chemo or radiotherapy is the development of improved photosensitizers (PSs). This work aims to contribute to the design of novel PSs able to generate reactive oxygen species (ROS) upon activation with light within the biological window (deep red or near-infrared region) while being non-toxic under dark conditions (heavy-atom-free). For this, we have chosen BODIPY-based covalent dimers directly linked through their 3-position as model structures. This molecular scaffold has been previously tested as a fluorescent probe to stain cells, but not as a PS for ROS generation under red illumination. Using readily available synthetic protocols, we have changed the steric hindrance around the linkage and added functional groups suited to enhance targetable biorecognition and solubility in physiological media. The spectroscopic characterization confirms that these dimers are photoactivated in a spectral window approaching 700 nm and display noticeable fluorescence signals beyond this wavelength, together with a notable generation of singlet oxygen. Encouraged by these photophysical signatures, we conducted in vitro trials in cancer cells. These assays ratify the ability of most of the herein reported dimers to sensitize ROS and induce cell death upon long-wavelength illumination.
Introduction
Photodynamic therapy (PDT)1–4 is a minimally invasive alternative to the currently applied clinical protocols in oncology for the treatment of cancer and in biomedicine to kill bacteria, especially in antimicrobial resistant diseases,5–7 one of the main health challenges facing society, according to the World Health Organization (WHO). PDT relies on the synergy of three interplaying agents: light, photosensitizer (PS) and the presence of oxygen in tissues (Fig. 1). While these components are individually harmless, their combination triggers a cytotoxic response able to kill cells. The use of light for therapeutic purposes has been known since ancient times (phototherapy). Nevertheless, the concept of PDT emerged in the past century when light was combined with PS reagents.8–10 Initially, PDT was tested to treat some skin, breast and lung carcinomas, using mainly porphyrin-based derivatives as PSs.11 In recent years, PDT has gained renewed interest as a real alternative to the existing harsh and non-selective oncological treatments without surgery, based on chemotherapy (delivery of drug combinations) and radiotherapy. These traditional approaches often have severe side effects on humans, whereas PDT enables a more targeted treatment in specific cellular areas owing to light-driven selective sensitization in target tissues. A crucial discovery for the resurgence of PDT was the recognition of the role of oxygen in the therapeutic activity.12–14 Upon light exposure, the PS generates reactive oxygen species (ROS), like singlet oxygen (1O2)15 or other oxygen-based radicals and ions, from tissue oxygen (Fig. 1). These photoinduced cytotoxic ROS are ultimately responsible for causing cell death via apoptosis or necrosis.16–18 Moreover, the formation of ROS from the PS is also unravelled and two alternative mechanisms are proposed (Fig. 1).19 On the one hand, the Type I20 mechanism involves electron transfer between a substrate and the excited PS, leading to the formation of radicals and ions capable of reacting with oxygen and generating ROS (like superoxide radical ions or hydroxide radicals). On the other hand, the Type II21 mechanism involves 1O2 generation after energy transfer from the excited triplet state of the PS to surrounding oxygen (3O2). | ||
| Fig. 1 Photochemical mechanisms involved in PDT for light-driven ROS generation. | ||
Despite its promise, several hurdles remain unsolved before PDT can be fully integrated into clinical practice. A major challenge lies in designing improved PSs. Currently, clinically approved PSs hold some drawbacks related to their biophotonic performance, tedious and complex synthetic access, and limited options for post-functionalization.22,23 An ideal PS should meet several key signatures. First, it should selectively accumulate in target cells or tissues and be activated by long-wavelength light to ensure deep penetration of light into tissues, selective excitation and minimal damage to healthy cells. In this regard, it should absorb efficiently the incoming light to use low irradiation doses and avoid skin photodamage. Besides, it should be photostable (resistant to light-induced degradation) to endure long lasting and repetitive irradiation doses, biocompatible (not harmful to living tissues) to avoid non-desired accumulations, and exhibit low dark toxicity (not toxic in the absence of light). Ideally, the PS should only generate ROS upon illumination and in a region limited to the vicinity of the target cells. Finally, it should be easily eliminated from the body after finishing the PDT treatment.
Many of the hitherto reported PSs are organic owing to their light weight, small size, biocompatibility and ease of processing. Related to this property, BODIPY dyes have aroused as an attractive molecular scaffold to develop PSs susceptible to fulfil the above mentioned requirements (Fig. 2).24–26 Indeed, the BODIPY molecular core can be readily tailored by workable and accessible synthetic routes to add the required functionalities, and thereby modulate at will their photophysical and biochemical properties.27 In essence, pristine BODIPYs are highly fluorescent and have a low triplet state population, but thanks to the rich chemistry of these dyes, they can be endowed with high enough intersystem crossing (ISC),28 for instance anchoring heavy atoms.29 However, those PSs based in heavy-atom-free BODIPYs are receiving more attention for PDT owing to their lower cost, reduced dark toxicity, better photostability, and longer triplet excited state lifetime than the halogenated ones.30,31 In heavy-atom-free PSs, the promotion of intramolecular charge transfer (ICT) mediates in the population of the triplet state.32,33 This photoinduced process is called spin–orbit charge transfer intersystem crossing (SOCT-ISC).34–36 The ICT operating under a symmetry-breaking mechanism (SBCT) is an effective approach to endow such spin–orbit coupling characteristic to the ICT and reach the triplet manifold, rather than the typical electrostatic ICT in push–pull chromophores bearing electron donors and acceptors. The SBCT is not driven by electrostatic forces and can be induced by directly linking two chromophores to promote an orthogonal arrangement between them.37–39 This short distance and geometrical configuration in a weak coupling regime enable charge separation upon excitation, and the ensuing population of the triplet manifold after charge recombination.40
 | ||
| Fig. 2 Representative model molecular structures of heavy-atom-free BODIPY-based dimers with different linkage positions reported in the bibliography. | ||
Directly and covalently linked BODIPY dimers are model molecular architectures undergoing this kind of CT enabling ISC (Fig. 2).41 In fact, BODIPY dimerization is nowadays in the spotlight as a successful strategy to render heavy-atom-free PSs.30,31,42 Most of them involve linkage through at least one meso position of the BODIPY core (the most tested junctions are 3–8′ and 2–8′), owing to the high sensitivity of this position to the substituent stereoelectronic properties (Fig. 2).43,44 However, although these PSs effectively generate ROS, they cannot be photoactivated at long wavelengths. The sterical hindrance-induced orthogonal disposition of the BODIPYs enhances the required SBCT for ISC promotion, but at the same time, it prevents electronic coupling between the BODIPYs. Thus, the light absorption of the dimers matches that of the corresponding single BODIPY building block. Trying to shift the absorption of the heavy-atom-free BODIPY dimers to the red edge, other linkage positions were tested in BODIPY dimers.45 In this regard, the 3–3′ junction46,47 has been highlighted as an ideal linkage to develop long wavelength-emitting dimers as fluorescent probes able to stain lysosomes and track them by bioimaging.48 Nonetheless, the workability of this kind of long wavelength dimers applied for PDT has not been tested yet. The only precedent of the ability of directly linked 3–3′ dimers to generate 1O2 involves sterically hindered derivatives (peralkylated BODIPY chromophores). However, this generation occurs under short wavelength irradiation (standard green light excitation of the BODIPY), owing to the orthogonal disposition of the BODIPY enabling triplet state population but hampering electronic coupling.49
Motivated by these pioneer findings, we became interested in ascertaining whether this kind of 3,3′-BODIPY dimers could be suitable as heavy-atom-free PSs photoactivated under red illumination. To this aim, we have synthesized a series of these dimers (1,482–7, in Fig. 3). On the one hand, the dimers were labelled with functional groups, such as ester moieties, which resemble fatty acid chains found in lipids,50 aiming to improve the specific recognition of cellular targets or sulfonate moieties to improve solubility.51 On the other hand, we varied the steric hindrance around the linkage positions by introducing bulky groups (methyls or bromines) at nearby positions (2 and 6). This allowed us to study the impact of the geometrical arrangement of the chromophoric subunits in the spectral shift and the feasibility of ISC population mediated by ICT. Both features are key photophysical properties in PSs because they trigger the irradiation wavelength for photoactivation and ROS generation, respectively. Moreover, we conducted computationally aided photophysical characterization that provides valuable information about the relationship between the molecular structure and photophysical properties. We have especially focused on the evaluation of competitive processes like fluorescence and singlet oxygen generation. Finally, we carried out in vitro assays to assess the viability of these PSs for specific staining of cellular targets (like lysosomes and lipid droplets) by bioimaging and their ability to generate ROS under selective long wavelength irradiation in cancer cells.
 | ||
| Fig. 3 Molecular structures of compounds 1–7. | ||
Results and discussion
Synthesis
Scheme 1 outlines the chemical steps for the synthesis of the targeted 3–3′ BODIPY dimers. For the preparation of the bis-methylated analogue 2, the previously reported BODIPY dimer 148 was treated with excess dimethyl zinc in the presence of PdCl2(PPh3)2 to afford 2 in high yield. A similar Negishi coupling of 1 with 2 or 6 equivalents of 6-ethoxy-6-oxohexyl zinc bromide rendered the corresponding derivatives 3 and 4, respectively, both in good yields. Subsequently, the selective bromination of the disubstituted derivative 4 at the 2 and 6 positions with N-bromosuccinimide (NBS) in dichloromethane (DCM) led to the tetrabromide derivative 5 in a moderate yield. Afterwards, the polyalkylated derivative 6 was obtained through the straightforward Negishi coupling of 5 with dimethylzinc and PdCl2(PPh3)2/X-Phos as the catalytic system. Finally, the substitution of chlorine of 1 with sodium 2-mercaptoethanesulfonate (MESNA) afforded the target compound 7, with improved solubility, in a 66% yield (Scheme 1). See the ESI† for more details about the synthetic procedure. | ||
Scheme 1 Synthesis of dimers 2–7. Conditions: (i) Me2Zn (molar ratio 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5), PdCl2(PPh3)2, toluene anh., rt, 1 h; (ii) BrZn(CH2)5CO2Et (molar ratio 1:2), PdCl2(PPh3)2, toluene anh., rt, 1 h; (iii) BrZn(CH2)5CO2Et (molar ratio 1:6), PdCl2(PPh3)2, toluene anh., rt, 1 h; (iv) NBS (molar ratio 1:6), DCM anh., rt, 24 h; (v) Me2Zn, toluene anh., PdCl2(PPh3)2/X-Phos, rt, 24 h; (vi) MESNA (molar ratio 1:10), NaHCO3, THF/H2O, rt, 4 h. :5), PdCl2(PPh3)2, toluene anh., rt, 1 h; (ii) BrZn(CH2)5CO2Et (molar ratio 1:2), PdCl2(PPh3)2, toluene anh., rt, 1 h; (iii) BrZn(CH2)5CO2Et (molar ratio 1:6), PdCl2(PPh3)2, toluene anh., rt, 1 h; (iv) NBS (molar ratio 1:6), DCM anh., rt, 24 h; (v) Me2Zn, toluene anh., PdCl2(PPh3)2/X-Phos, rt, 24 h; (vi) MESNA (molar ratio 1:10), NaHCO3, THF/H2O, rt, 4 h. | ||
All the new compounds were fully characterized by 1H-NMR, 13C-NMR, IR and high-resolution mass spectrometry (HRMS) (see the ESI†). Dimer 2 has similar NMR spectra to that of the previously reported dimer 148 (Fig. S1 and S2 in the ESI†). The signals at δ = 2.65 in 1H-NMR spectra and δ = 15.6 in 13C-NMR spectra are due to the two α-pyrrolic methyl groups. The ester moieties in dimers 3, 4, 5 and 6 lead to a quartet signal at δ ∼ 4.1, corresponding to the CH2 group bound to oxygen, and five signals of methylene groups of the alkyl chain in 1H-NMR spectra (Fig. S3, S5, S7 and S9 in the ESI†). The CH3 of ester moieties appears as a triplet at δ ∼ 1.2. The 13C-NMR spectra show a signal at δ ∼ 173.9 (Fig. S4, S6, S8 and S10 in the ESI†), characteristic of the alkoxycarbonyl group. In dimer 5, the 2,6-bromines induce a slight separation of the protons (δ = 6.97 and 6.96) and methyls (δ = 2.21 and 2.15) of the mesityl ring in the 1H-NMR spectrum (Fig. S7 in the ESI†). The same effect is observed in 13C-NMR spectra (δ = 128.5 and 128.4 for the protons and δ = 20.5 and 20.3 for the CH3). Additionally, two C–Br signals appear at δ = 111.7 and 109.5 (Fig. S8 in the ESI†). In the 1H-NMR spectrum of dimer 6 (Fig. S9 in ESI†), the signals of the mesityl group are also split (protons δ = 6.97 and 6.96, and methyls δ = 2.22 and 2.15). The 13C-NMR spectrum shows these signals at δ = 128.1, 128.0, 20.6 and 20.3 ppm (Fig. S10 in the ESI†). In addition, there are two signals at δ = 1.95 and 1.88 in the 1H-NMR spectrum; δ = 11.42 and 11.38 in the 13C-NMR spectrum, corresponding to the four 2,6-methyls. Finally, the presence of the α-MESNA group (7) provides a 1H-NMR spectrum analogous to dimer 1,48 but with two additional signals (at δ = 3.50 and 3.20 as multiplets in the 1H-NMR spectrum and δ = 52.3 and 28.3 in the 13C-NMR spectrum, Fig. S11 and S12 in the ESI†) from the methylenes adjacent to the sulfur atoms.
Photophysical properties
The less constrained (unsubstituted at neighbour positions 2 and 6) 3–3′ dimers 1,482–4 and 7 share similar spectroscopic signatures. Regardless of the attached specific chemical group (chlorine and/or polymethylene chains with end ester groups) at the opposite position (5) of the linkage for dimerization, dimers 1–4 display broad absorption bands but sharp and structured fluorescence bands at the red edge of the visible spectrum (at around 670–685 nm and 720–730 nm, respectively, Fig. 4). These spectral bands are strongly red-shifted relative to the 8-mesityl BODIPY subunit and show enhanced Stokes shifts (up to two times larger, Table S1 in the ESI†). More pronounced bathochromic shifts are achieved upon attachment of the sulphur group, bearing an aliphatic chain containing end anionic sulfonate, at the said chromophoric 5 position (7). Indeed, both absorption and emission reach the near-infrared region (NIR, in particular at 720 nm and 770 nm, respectively, Fig. 4) owing to the resonant effect of sulphur with the dipyrrine. The location of the spectral bands in the red-NIR region suggests a strong electronic coupling between the two BODIPY subunits through the 3–3′ junction. Indeed, the computationally optimized geometries feature a torsion dihedral angle between the dipyrrine planes of 40°. This twist enables a resonant interaction between the chromophoric electronic clouds. In fact, the frontier contour maps involved in the electronic transitions are spread across both BODIPY subunits (Fig. 4). | ||
| Fig. 4 Normalized absorption (thin line) and fluorescence (thick line) spectra of representative 3–3′ BODIPY dimers 2 and 7 (dye concentration 2 μM) in acetonitrile to highlight the photoactive spectral region covered with these dimers. The corresponding HOMO and LUMO contour maps of representative dimer 2 are also included. The rest of spectra in more solvents and for the other dimers are collected in Fig. S19–S21 in the ESI.† | ||
These dimers 1–4 yield high fluorescence efficiencies in the far red-NIR region (around 50–55%), with low sensibility to the solvent polarity, and monoexponential decay curves with lifetimes around 3 ns (Table 1 and Table S1 in the ESI†). The fluorescence efficiency of dimer 7 is lower (not higher than 20%, Table 1) likely due to the higher internal conversion at such long wavelengths, according to the energy gap law. In agreement with this enhancement of non-radiative deactivation (Table S1 in the ESI†), the fluorescence lifetime also decreases to 1.5 ns (Table 1). The low influence of the solvent polarity on the fluorescence response suggests that the non-orthogonal arrangement of the BODIPY is not suitable to induce ICT, and hence the ISC should be also hindered. Despite this, all these 3–3′ dimers are able to generate a gentle amount of singlet oxygen (around 10–18%), reaching a remarkable 28% in the methylated dimer 2 (Table 1). Red-emitting dimer 7 is not soluble in chloroform and dissolves only in polar/protic media where the singlet oxygen lifetime decays faster, hampering the spectroscopic detection of its phosphorescence emission at 1275 nm.52 This is why its 1O2 generation efficiency (18% in Table 1) was recorded in deuterated methanol to extend 1O2 lifetime and allow the detection of its emission for quantification.53 Therefore, the 1O2 generation efficiency of these 3–3′ dimers should be enough to promote PDT action.
| λ ab (nm) | ε max (104 M−1 cm−1) | λ fl (nm) | Φ fl | τ fl (ns) | Φ Δ | |
|---|---|---|---|---|---|---|
| a Data obtained under our experimental conditions. b Photophysical data in ethanol and singlet oxygen generation value in deuterated methanol (not soluble in chloroform).Absorption (λab) and fluorescence (λfl) wavelength, molar absorption at the maximum wavelength (εmax), fluorescence quantum yield (Φfl) and lifetime (τfl), and singlet oxygen generation quantum yield (ΦΔ). | ||||||
| 1 48 , | 674.5 | 5.4 | 720.5 | 0.57 | 3.23 | 0.11 |
| 2 | 686.0 | 5.4 | 731.5 | 0.51 | 2.91 | 0.28 |
| 3 | 685.5 | 3.8 | 726.0 | 0.49 | 3.05 | 0.14 |
| 4 | 681.0 | 5.2 | 724.5 | 0.55 | 3.33 | 0.10 |
| 5 | 575.0 | 8.0 | 676.5 | 0.03 | 0.18 (91%)–0.35 (9%) | 0.97 |
| 501.5 | 10.7 | |||||
| 6 | 575.0 | 3.5 | 676.0 | 0.61 | 3.66 | 0.29 |
| 497.5 | 3.0 | |||||
| 7 | 723.0 | 6.3 | 773.0 | 0.14 | 1.33 | 0.18 |
These results were somehow unexpected. In previously reported BODIPY dimers involving the meso position, the perpendicular arrangement of the chromophore was crucial to enable SBCT and populate the SOC-ISC to reach the triplet manifold and generate 1O2. However, in our 3–3′ dimers, the disposition of the BODIPYs is no longer perpendicular and the torsional angle allows an electron coupling between them. This interaction shifts the spectra deep into the biological window, showing notable fluorescence efficiency together with reasonable 1O2 generation efficiency. These findings pave the way for the implementation of these 3–3′ dimers as PSs activated upon red-NIR illumination without inducing ICT processes, which usually are detrimental for the fluorescence response. Likely, the electronic redistribution promoted by the resonant interaction between the 3–3′ linked BODIPYs cores enhances the ISC and generates 1O2, while still allowing for a remarkable fluorescence response.
These astonishing findings prompted us to ascertain the impact of the twist angle between the chromophores on ICT probability and evaluate its influence on fluorescence and singlet oxygen generation. To this aim, and taking as reference dimer 4, we inserted bulky bromines (5) and methyls (6) in the positions 2 and 6 of both BODIPY subunits. As a result of the induced steric hindrance around the linkage position, both chromophores are forced to adopt a perpendicular disposition (dihedral angle almost 90°, Fig. 5). This geometrical arrangement has a clear impact on the photophysics of the dimers (Table 1). The absorption spectral profile of these constrained dimers 5 and 6 features two clearly distinguishable and sizable peaks (Fig. 5): long wavelength absorption (at around 575 nm), hypsochromically shifted to the parent dimer 4, and short wavelength absorption (at around 500 nm), matching that expected for isolated 8-mesityl BODIPY. The perpendicular disposition of the BODIPYs hampers the resonant interaction between them in dimers 5 and 6 (Fig. 5). Thus, each BODIPY retains its own electronic identity (short wavelength absorption), and just a partial electronic coupling is feasible, leading to long wavelength absorption. However, this peak is not as red-shifted as in the unconstrained dimer 4, where the resonant interaction is not hindered. Note that the chromophoric 3 position is very sensitive to the substituent effect, and hence aromatic moieties grafted at such positions are able to cause pronounced spectral shifts.54 On the other hand, the fluorescence profile of constrained dimers 5 and 6 displays a single broad band (at around 675 nm), without the vibrational resolution of the less constrained dimer 4. This band is also hypsochromically shifted relative to the unconstrained dimer 4 (Fig. 5). The absence of dual emission (in contrast to the registered dual absorption) can be explained by intramolecular excitation energy transfer from the BODIPY alone to the entity formed by the partially electronic interaction of the BODIPYs. Indeed, the same emission spectra are recorded regardless of the excited absorption band.
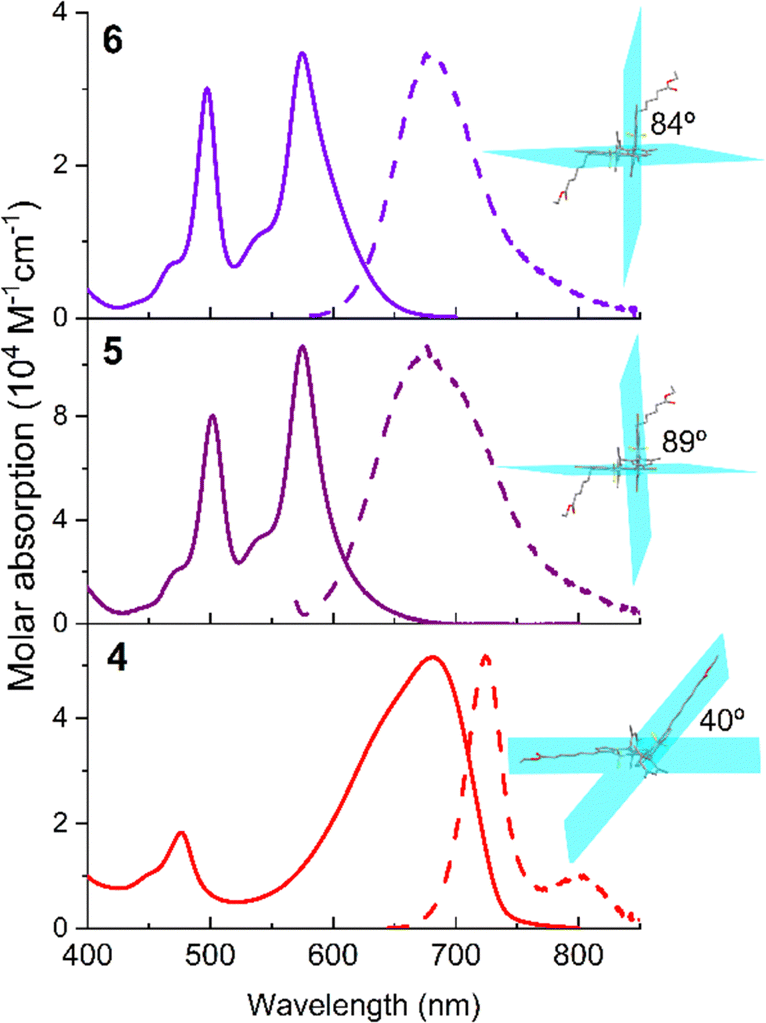 | ||
| Fig. 5 Absorption and normalized fluorescence (dashed) spectra of 3–3′ BODIPY dimer 4 and its corresponding constrained brominated (5) and methylated (6) derivatives in chloroform (dye concentration 2 μM). The corresponding ground optimized geometries are also appended, highlighting the torsional angles between the chromophoric planes. The corresponding spectra in more solvents are recorded as shown in Fig. S20 and S21 of the ESI.† | ||
The fluorescence response of the methylated dimer 6 is affected by the solvent polarity, dropping from around 60% in non-polar solvents to 23% in polar solvents (Table S1 in the ESI†). This is accompanied by faster fluorescence lifetimes in polar solvents (from around 3.5 ns down to 1.7 ns, Table S1 in the ESI†). This trend suggests that the perpendicular arrangement of the chromophores allows SBCT to occur more readily, especially in polar media. Indeed, the fluorescence band shows positive solvatochromism, in contrast to the negative one recorded for the rest of the dimers (typical behaviour of BODIPYs, Fig. S19–S21 in the ESI†). Additionally, the Stokes shift enlarges to 3000 cm−1 in polar media (Table S1 in the ESI†). As a consequence, the ISC is more feasible and the 1O2 generation efficiency increases almost three times with regard to the parent dimer 4 (from 10% to 29%, Table 1). In line with this finding, the brominated dimer 5 is the most efficient 1O2 PS (97% in Table 1) among the herein tested dimers. This value is not only a consequence of the orthogonality-induced higher SBCT probability but also a consequence of the heavy-atom effect of bromine. The synergy of both factors enhances the ISC pathway, as evidenced by the almost complete loss of fluorescent signal (just 3% for dimer 5, Table 1).
Therefore, the unconstrained 3–3′ dimers stand out as promising PSs for ROS generation upon long wavelength (red-NIR) irradiation in preclinical trials directed to PDT. Note that in prior reported studies, this possibility was not considered, and the in vitro assays were focused just on their performance as fluorescent probes for the tracking of lysosomes within cells, neglecting their potential for PDT.48
In vitro PDT experiments
The phototoxic capacity of dimers 1–7 was evaluated in human melanoma SK-Mel-103 cells and cell proliferation was measured with the WST-1 reagent (Fig. 6). SK-Mel-103 cells were treated for 24 h with increasing concentrations of each of the dimers, ranging from the minimum concentration of 0.25 μM to the maximum concentration of 50 μM. Then, cells were irradiated for 30 min with a 36 W LED source at 10 cm using a red filter that only allows the passage of wavelengths λ > 500 nm (16 mW cm−2 light dose, see the ESI† for more details). After irradiation, cells were incubated under dark conditions for 24 h and cell viability was measured. | ||
| Fig. 6 Photocytotoxic assay in SK-Mel-103 cancer cells of 1 (A), 2 (B), 3 (C), 4 (D), 5 (E), 6 (F) and 7 (G). Cells were treated with increasing concentrations of each of the compounds within the range of 0.05 μM to 50 μM for 24 h before irradiation with visible light (λ > 475 nm) for 30 min. DMSO refers to the control cells exclusively treated with the maximum concentration of DMSO (1%). Cell viability was determined 24 h after the irradiation step with the WST-1 reagent. Values are expressed as mean ± SEM, and statistical significance was assessed by one-way ANOVA (n ≥ 3). *p-value < 0.05; **p-value < 0.01; ***p-value < 0.001 and ****p-value < 0.0001. | ||
Cell viability assays show that dimers 4 and 6, despite having the ability to generate 1O2 (Table 1), are not capable of inducing cell death upon irradiation in vitro. In contrast, dimers 1–3, 5 and 7 induced a concentration-dependent reduction in cell viability upon cell irradiation. This reduction in the percentage of cell viability agrees with the results obtained in the singlet oxygen generation analysis because all the compounds have a ΦΔ > 0 (Table 1). All of them exhibit higher efficiency in killing irradiated SK-Mel-103 cells compared to the non-irradiated control (dark control). The formation of ROS in cells under both light and dark conditions will be analysed later on (vide infra).
To quantify the effectiveness of dimers 1–3, 5 and 7 in killing cells, the EC50 values (see the ESI† for details) were calculated from the sigmoidal fitting of the concentration-cell viability curves (Fig. S22 in the ESI†). Dimers 2, 5 and 7 exhibited the strongest phototoxic ability with EC50 values of 0.75 ± 0.18 μM, 0.86 ± 0.22 μM and 2.39 ± 0.14 μM, respectively. In contrast, dimers 1 and 3 were less effective with EC50 values of 7.94 ± 1.38 μM and 5.16 ± 0.81 μM, respectively. It must be mentioned that there is not a direct correlation between the highest values of singlet oxygen formation with a greater phototoxic capacity of the dimers. This finding suggests that other factors besides singlet oxygen generation, such as generation of another type of ROS in the cells, or the PSs’ ability to be internalized in cells, must also play a role in their capability to kill cells by photoactivation. To monitor the cellular uptake (vide infra), we have selected the smallest dimer (2) and the functionalized dimers for enhanced biorecognition (4) and solubility (7) because all of them retain a reasonably high fluorescence efficiency to be tracked by bioimaging (Table 1).
Organelle distribution of 2, 4 and 7
In this section, we studied the internalization of 2, 4 and 7 photoactivatable dimers and their intracellular distribution because the intracellular localization of the photosensitizer also affects the efficiency of apoptosis induction by PDT. The dimers studied show a punctate cytoplasmic distribution in SK-Mel-103 cells after 24 h of incubation (Fig. 7), indicating that these compounds are efficiently internalized by melanoma cells. Afterwards, we studied in more detail whether the lack of cytotoxic activity of compound 4 is due to a low cellular uptake and accumulation, and therefore, its anti-tumor efficiency. As can be seen in the confocal images in Fig. S23 in the ESI,† PS agents 4 and 7 are able to cross the cell membrane and accumulate in the intracellular compartment in the same way. Therefore, the inability of agent 4 to induce cell death must be due to some other reason rather than a poor diffusion and accumulation inside the cells. It should also be noted that the type of PS, the type of cancer cell and the intracellular localization of the PS are crucial to effectively induce apoptosis by PDT. This is because ROS have extremely short diffusion distance within cells (<50 nm),55 so that PSs generally need to be localized in close proximity to the target structure to increase the cytotoxic efficiency. | ||
| Fig. 7 Subcellular co-localization of 2 (A), 7 (B) and 4 (C) in SK-Mel-103 following 24 h of incubation with 5 μm, 10 μM and 10 μM, respectively, and stained with Hoechst 33342 (blue) and green trackers: PM546 (lipid droplets, up) and LysoTracker (lysosomes, down). Images were acquired using the confocal microscope Leica TCS SP8 HyVolution II. Dimers 2, 4 and 7 were excited at 562 nm and the emission was collected at 640–780 nm. Green trackers were excited at 488 and the emission was collected at 490–530 nm. Scale bar 20 μm, R: Pearson's correlation coefficient. | ||
To this end, a co-localization analysis of each of the dimers was carried out using the green subcellular markers LysoTracker (for lysosomes, LDs) and BODIPY dye PM546 (for LDs) and the Pearson's correlation coefficient (R) was calculated based on a pixel-by-pixel based analysis. Fig. 7 shows the subcellular co-localization of 2 (A) and 7 (B) in SK-Mel-103 stained with nuclei (blue) and organelle markers (green). The red emission of 2 and 7 does not co-localize either in lysosomes or in LDs with a R lower than 0.7. Therefore, we can conclude that there is no preferential accumulation of these dimers in these intracellular structures. In contrast, 4 shows a greater co-localization of the fluorescent signal with the lysosome marker and to a lesser extent with the LDs, with R values of 0.72 and 0.68, respectively (Fig. 7C). Therefore, dimer 4, despite not being a theragnostic agent, shows a greater preference than the other compounds tested in this study for accumulating in cellular structures. This is likely due to the grafted ester moieties, which makes it an interesting candidate as a fluorescence bioimaging probe.
For that reason, we can conclude that the cytotoxic capacity of compounds 2 and 7 compared to non-cytotoxic compound 4 may be due to other factors not associated with its intracellular location and cellular uptake. Therefore, these differences could be due to the type of cells treated and the biochemical characteristics of the PS, which affect its pharmacokinetic properties, amphiphilicity and the capacity to trigger pro- or anti-death signalling pathways upon light exposure.
ROS generation in cells
We evaluated the generation of ROS upon irradiation (λ > 500 nm) in cells treated with compound 7, bearing anionic sulfonates for enhanced stability in the physiological media. It was able to induce cell death at low concentrations (EC50 2.39 ± 0.14 μM). To check this behaviour unambiguously, cells were incubated with the ROS detection reagent CM-H2DCFDA green, which becomes fluorescent when it reacts with ROS.56Fig. 8 shows that the fluorescence intensity of the CM-H2DCFDA marker increases in the cells treated with 10 μM of dimer 7 and irradiated for 30 min, compared with the dark controls (without irradiation) and cells untreated (light alone). This result supports that dimer 7 is capable of efficiently producing ROS (such as 1O2, see Table 1) in cells and thus inducing their death when activated by light. | ||
| Fig. 8 Determination of ROS generation in SK-Mel-103 cells after PDT treatment with 7. (A) and (B) Confocal images of SK-Mel-103 cells incubated with 10 μM of 7 during 24 h and irradiated with visible light (λ > 500 nm) (B) for 30 min, compared to control cells maintained in the dark (A). Cells were incubated with 2 mg mL−1 Hoechst 33342 (blue) and 500 nM CM-H2DCFDA probe (green). Scale bar 20 mm. (C) Quantification of mean fluorescence intensity from confocal images of SK-Mel-103 cells incubated with 10 μM of 7 during 24 h and irradiated with visible light (λ > 500 nm) for 30 min, compared to control cells maintained in the dark. Values are expressed as mean ± SEM, and statistical significance was assessed using the two-tailed Student's t-test (n = 3). ****p-value < 0.0001. | ||
Conclusions
The herein conducted study brings BODIPY-based 3–3′ dimers to the spotlight as a molecular scaffold with multifunctional potential for biophotonics. These dimers are activated within the biological window for deep tissue penetration owing to their absorption bands placed in the far red spectral region (close to 700 nm). Besides, they display bright near-infrared fluorescence signals (around 730–770 nm), enabling easier detection within cells by bioimaging. One of the main novel outcomes is that some of them can generate ROS and induce cell death, without the need for heavy atoms or steric hindrance to promote charge transfer processes, as seen in the rest of the related BODIPY-based dimers. Therefore, this molecular design fulfils many of the requirements for an ideal photosensitizer in photodynamic therapy. The in vitro assays in cancer cells validate the photophysical properties because most of these dimers decrease cell viability owing to the induction of ROS-mediated cell death selectively under long wavelength irradiation. Moreover, upon proper functionalization for biorecognition, selected dimers accumulate preferably in lysosomes and to a lesser extent in lipid droplets, as evidenced by bioimaging. Therefore, this remarkable synthetic accessibility and easy adaptability through workable chemistry, together with the inherent chemical robustness and photochemical stability of the BODIPY core, envisage that this kind of BODIPY-based PS should have a superior behaviour compared to porphyrin-based photosensitizers (such as chlorine e6, absorbing and emitting in a similar spectral window) in practical PDT applications.As a future prospect and considering that there are free chromophoric positions amenable to further functionalization in these dimers, we aim to decorate them with the required moieties enabling a more specific targetable staining of cells. We believe that in this way it might be possible to design a fluorescent photosensitizer for bioimaging-guided PDT, selectively activated with deep red light. By combining these functionalities in a single molecule, we envision meeting the requested criteria for an ideal PS for clinical use.
Author contributions
Conceptualization (A. R. A. and M. J. O.); data curation (A. G. F. and J. B.); funding acquisition (J. B., R. M. M. and M. J. O.); investigation (J. S. B., E. A. Z. and G. D. S.); supervision (A. G. F., A. R. A. and J. B.); writing – original draft (A. G. F. and J. B.); writing – review & editing (J. B., R. M. M. and M. J. O.). All authors have given approval to the final version of the manuscript.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge grants PID2020-114755GB-C32 and -C33, and PID2021-128141OB-C22 funded by the Spanish MCIN/AEI/10.13039/501100011033, and grant IT1639-22 funded by the Basque Government.References
- D. E. J. G. J. Dolmans, D. Fukumura and R. K. Jain, Nat. Rev. Cancer, 2003, 3, 380–387 CrossRef CAS PubMed.
- S. Yano, S. Hirohara, M. Obata, Y. Hagiya, S.-I. Ogura, A. Ikeda, H. Kataoka, M. Tanaka and T. Joh, J. Photochem. Photobiol., C, 2011, 12, 46–67 CrossRef CAS.
- J. H. Correia, J. A. Rodriguez, S. Pimenta, T. Dong and Z. Yang, Pharmaceutics, 2021, 13, 1332 CrossRef CAS PubMed.
- I. R. Calori, H. Bi and A. C. Tedesco, ACS Appl. Bio Mater., 2021, 4, 195–228 CrossRef CAS PubMed.
- Y. Feng, C. C. Tonon, S. Ashraf and T. Hasan, Adv. Drug Delivery Rev., 2021, 177, 113941 CrossRef CAS PubMed.
- J. Jiang, X. Lv, H. Cheng, D. Yang, W. Xu, Y. Hu, Y. Song and G. Zeng, Acta Biomater., 2024, 177, 1–19 CrossRef CAS PubMed.
- M. Piksa, C. Lian, I. C. Samuel, K. J. Pawlik, I. D. W. Samuel and K. Matczyszyn, Chem. Soc. Rev., 2023, 52, 1697–1722 RSC.
- J. Moan and Q. Peng, Anticancer Res., 2003, 23, 3591–3600 Search PubMed.
- D. Kessel, J. Clin. Med., 2019, 8, 1851 CrossRef PubMed.
- M. R. Hamblin, Photochem. Photobiol., 2020, 93, 506–516 CrossRef PubMed.
- J. Kou, D. Dou and L. Yang, Oncotarget, 2017, 8, 81591–81603 CrossRef PubMed.
- Z. Zhou, J. Song, L. Nie and X. Chen, Chem. Soc. Rev., 2016, 45, 6597–6626 RSC.
- D. L. Sai, J. Lee, D. L. Nguyen and Y.-P. Kim, Exp. Mol. Med., 2021, 53, 495–504 CrossRef CAS PubMed.
- F. Wei, T. W. Rees, X. Liao, L. Ji and H. Chao, Coord. Chem. Rev., 2021, 432, 213714 CrossRef CAS.
- G. Li, M. Wu, Y. Xu, Q. Wang, J. Liu, Z. Zhou, J. Ji, Q. Tang, X. Gu, S. Liu, Y. Qin, L. Wu and Q. Zao, Coord. Chem. Rev., 2023, 478, 214979 CrossRef CAS.
- J. Soriano, I. Mora-Espí, M. E. Alea-Reyes, L. Pérez-García, L. Barrios, E. Ibañez and C. Nogués, Sci. Rep., 2017, 7, 41340 CrossRef CAS PubMed.
- T. Mishchenko, I. Balalaeva, A. Gorokhova, M. Vedunova and D. V. Krysko, Cell Death Dis., 2022, 13, 455 CrossRef PubMed.
- P. S. Maharjan and H. K. Bhattarai, J. Oncol., 2022, 72111485 Search PubMed.
- S. Kwiatkowski, B. Knap, D. Przystupski, J. Saczko, E. Kedsierska, K. Knap-Czop, J. Kotlinska, O. Michel, K. Kotowski and J. Kulbacka, Biomed. Pharmacother., 2018, 1098–1107 CrossRef PubMed.
- B. Lu, L. Wang, H. Tang and D. Cao, J. Mater. Chem. B, 2023, 11, 4600–4618 RSC.
- L. Yu, Z. Liu, K. Jin, J. Liu, X. Zhu, Y. Zhang and Y. Wu, Acta Pharm. Sin. B, 2024, 14, 1111–1131 CrossRef CAS PubMed.
- X. Zhao, J. Lu, J. Fan, H. Chao and X. Peng, Chem. Soc. Rev., 2021, 50, 4185–4219 RSC.
- T. C. Pharm, V.-N. Nguyen, Y. Choi, S. Lee and J. Yoon, Chem. Rev., 2021, 21, 13454–13619 Search PubMed.
- M. L. Agazzi, M. B. Ballatore, A. M. Durantini, A. N. Durantini and A. C. Tomé, J. Photochem. Photobiol., C, 2019, 40, 21–48 CrossRef CAS.
- D. Wang, X. Wang, S. Zhou, P. Gu, X. Zhu, C. Wang and Q. Zhang, Coord. Chem. Rev., 2023, 482, 215074 CrossRef CAS.
- J. Wang, Q. Gong, L. Jiao and E. Hao, Coord. Chem. Rev., 2023, 496, 215367 CrossRef CAS.
- N. Boens, B. Verbelen, M. J. Ortiz, L. Jiao and W. Dehaen, Coord. Chem. Rev., 2019, 399, 213024 CrossRef CAS.
- K. Chen, Y. Dong, X. Zhao, M. Imran, G. Tang, J. Zhao and Q. Liu, Front. Chem., 2019, 7, 821 CrossRef CAS PubMed.
- W. Hu, R. Zhang, X.-F. Zhang, J. Liu and L. Luo, Spectrochim. Acta, Part A, 2022, 272, 120965 CrossRef CAS PubMed.
- M. A. Filatov, Org. Biomol. Chem., 2020, 18, 10–27 RSC.
- V.-N. Nguyen, Y. Yan, J. Zhao and J. Yoon, Acc. Chem. Res., 2021, 54, 207–220 CrossRef CAS PubMed.
- J. Zhao, K. Chen, Y. Hou, Y. Che, L. Liu and D. Jia, Org. Biomol. Chem., 2018, 16, 3692–3701 RSC.
- M. Lv, X. Wang, D. Wang, X. Li, Y. Liu, H. Pan, S. Zhang, J. Xu and J. Chen, Phys. Chem. Chem. Phys., 2021, 23, 25455–25466 RSC.
- Y. Dong, A. A. Sukhanov, J. Zhao, A. Elmali, X. Li, B. Dick, A. Karatay and V. K. Voronkova, J. Phys. Chem. C, 2019, 123, 22793–22811 CrossRef CAS.
- V.-N. Nguyen, J. Ha, C. W. Koh, B. Ryu, G. Kim., J. H. Park, C.-Y. Kim, S. Park and J. Yoon, Chem. Mater., 2021, 33, 7889–7896 CrossRef CAS.
- Z. Wang, L. Ma, H. Zhao, Y. Wan, X.-F. Zhang, Y. Li, Z. Kuang and A. Xia, Phys. Chem. Chem. Phys., 2023, 25, 24386–24394 RSC.
- L. Estergreen, A. R. Mencke, D. E. Cotton, N. V. Korovina, J. Michl, S. T. Roberts, M. E. Thompson and S. E. Bradforth, Acc. Chem. Res., 2022, 55, 1561–1572 CrossRef CAS PubMed.
- J. H. Golden, L. Estergreen, T. Porter, A. C. Tadle, M. R. Daniel Sylvinson, J. W. Facendola, C. P. Kubiak, S. E. Bradforth and M. E. Thompson, ACS Appl. Energy Mat, 2018, 1, 1083–1095 CrossRef CAS.
- M. Kellogg, A. Akil, D. S. M. Ravinson, L. Estergreen, S. E. Bradforth and M. E. Thompson, Faraday Discuss., 2019, 216, 379–394 RSC.
- Y. Liu, J. Zhao, A. Iagatti, L. Bussotti, P. Foggi, E. Castellucci, M. Di Donato and K.-l Han, J. Phys. Chem. C, 2018, 122, 2502–2511 CrossRef CAS.
- N. A. Bumagina, E. V. Antina, A. A. Ksenofontov, L. A. Antina, A. A. Kalyagin and M. B. Berezin, Coord. Chem. Rev., 2022, 469, 214684 CrossRef CAS.
- L. Wang, Q. Wu, Z. Kang, X. Guo, W. Miao, Z. Li, H. Zuo, H. Wang, H. Si, L. Jiao and E. Hao, Org. Lett., 2023, 25, 5055–5060 CrossRef CAS PubMed.
- Y. Cakmak, S. Kolemen, S. Duman, Y. Dede, Y. Dolen, B. Kilic, Z. Kostereli, L. T. Yildirim, A. L. Dogan, D. Guc and A. U. Akkaya, Angew. Chem., Int. Ed., 2011, 50, 11937–11941 CrossRef CAS PubMed.
- J. Cao, T. Zhang and W. Sun, Dyes Pigm., 2023, 208, 110797 CrossRef.
- K.-X. Teng, W.-K. Chen, L.-Y. Niu, W.-H. Fang, G. Cui and Q.-Z. Yang, Angew. Chem., Int. Ed., 2021, 60, 19912–19920 CrossRef CAS PubMed.
- D. Wang, Q. Wu, X. Zhang, W. Wang, E. Hao and L. Jiao, Org. Lett., 2020, 22, 7694–7698 CrossRef CAS PubMed.
- Q. Gong, X. Zhang, W. Li, X. Guo, Q. Wu, C. Yu, L. Jiao, Y. Xiao and E. Hao, J. Am. Chem. Soc., 2022, 144, 21992–21999 CrossRef CAS PubMed.
- Z. Kang, Q. Wu, X. Guo, L. Wang, Y. Ye, C. Yu, H. Wang, E. Hao and L. Jiao, Chem. Commun., 2021, 57, 9886–9889 RSC.
- B. Ventura, G. Mancorni, M. Bröring, R. Krüger and L. Flamigni, New J. Chem., 2009, 33, 428–438 RSC.
- A. Tabero, F. García-Garrido, A. Prieto-Castañeda, E. Palao, A. R. Agarrabeitia, I. García-Moreno, A. Villanueva, S. de la Moya and M. J. Ortiz, Chem. Commun., 2020, 56, 940–943 RSC.
- D. Kand, P. Liu, M. X. Navarro, L. J. Fischer, L. Rousso-Noori, D. Friedmann-Morvinski, A. H. Winter, E. W. Miller and R. Weinstain, J. Am. Chem. Soc., 2020, 142, 4970–4974 CrossRef CAS PubMed.
- M. Bregnhoj, M. Westberg, F. Jensen and P. R. Ogilby, Phys. Chem. Chem. Phys., 2016, 18, 22946–22961 RSC.
- F. Thorning, P. Henke and P. R. Ogilby, J. Am. Chem. Soc., 2022, 144, 10902–10911 CrossRef CAS PubMed.
- E. Avellanal-Zaballa, L. Gartzia-Rivero, T. Arbeloa and J. Bañuelos, Int. Rev. Phys. Chem., 2022, 41, 177–203 Search PubMed.
- W. Jakubowski and G. Bartosz, Cell Biol. Int., 2000, 24, 757–760 CrossRef CAS PubMed.
- W. M. Sharman, C. M. Allen and J. E. van Lier, Methods Enzymol., 2000, 319, 376–400 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic procedure and characterization of compounds (NMR spectra), details of the experimental techniques and computational calculations, photophysical data, absorption and fluorescence spectra, and cell viability. See DOI: https://doi.org/10.1039/d4ma00951g |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |