
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceScalable solution for high dispersibility and low heterogeneity of nuclear-shielding high inorganic filler/polymer composites: a vitrimer via reactive extrusion & mechanochemical intercalation†
Shuangxin
Lai
a,
Qianyue
Tan
a,
Hongli
Xie
a,
Jiliang
Gong
a,
Liang
Xue
b,
Haiping
Liu
b,
Ruiqian
Zhang
d,
Yijun
Li
 a and
Shibing
Bai
*ac
a and
Shibing
Bai
*ac
aNational Key Laboratory of Advanced Polymer Materials, Polymer Research Institute of Sichuan University, Chengdu 610065, China. E-mail: baishibing@scu.edu.cn
bXi'an Yuanchuang Aviation Technology Co., Ltd, Xian 710065, China
cSchool of Chemical Engineering and Technology, Xinjiang University, China
dScience and Technology on Reactor Fuel and Materials Laboratory, Nuclear Power Institute of China, Chengdu 610200, China
First published on 18th June 2025
Abstract
Many are pursuing high-performance polymer composites with ultra-high inorganic fillers, focusing on improving inorganic particle dispersion. However, few studies have explored how to address the heterogeneity introduced by inorganic particles after meeting dispersion requirements. This work proposed that resolving the heterogeneity in polymer composites with high inorganic particle content relies on utilizing the movement of polymer chains for self-adaptation. We employed ultra-high-filled tungsten powder (W)/high-density polyethylene (HDPE) composites as the basic model. A vitrimer was introduced to enhance the intensity of mutual diffusion of HDPE chains during static hot pressing. The vitrimer-modified W/HDPE composites (W/HDPE-v) not only ensured the high dispersion of W but also facilitated intense mutual diffusion of the chains through the bond exchange of the vitrimer under thermal action. This process led to the ordered stacking of the C–C main chains and increased the crystallinity of the composites. Through the chains’ mutual diffusion, fluctuations in the modulus of the HDPE matrix were reduced, and the interfacial layer between the HDPE and W underwent continuous dynamic reorganization. This dynamic reorganization achieved heterogeneity reduction. The introduction of the vitrimer also generated regions within the polymer chains that exhibited different steric hindrances, which were significantly influenced by factors such as the crosslinking agent content and external forces. This resulted in a directional bond exchange of the vitrimer and traction on the polymer chains, promoting the self-aggregation of polymer chains and the rejection of inorganic particles. The final composites exhibited good mechanical properties and gamma-ray shielding effects.
Introduction
Polymers, as typical platform-based materials, possess unique characteristics, such as low-temperature processability, the ability to be compounded with inorganic functional particles, and the ease of designing interface structures.1 These attributes make them easy to compound with various inorganic particles and be applied in various functional fields. Inorganic filler/polymer composites could be classified based on the proportion of inorganic fillers (low or high). Composites with a low proportion of inorganic fillers and designed interface structures are popular in the scientific research community for applications such as thermal interface management materials. In contrast, when the objective is to achieve high functional performance in polymer-based inorganic particle composites, industries frequently opt for such composites, including lead–boron polyethylene,2 precision thermal and electrically conductive adhesives,3 and potting adhesives, where the proportions of inorganic fillers are needed to exceed 60 wt%. To prepare polymer composites with ultra-high proportions of inorganic fillers, achieving high dispersion of inorganic particles is crucial. Because of the strong density difference between the inorganic particles and the polymer matrix, there are non-uniform responses of each phase to external forces. This makes it difficult to mix them, thus presenting the problem of poor dispersion. Key solving strategies include the following: (1) at the equipment level: employing external forces through twin-screw extruders, reciprocating single-screw extruders, or planetary screw extruders for forced dispersion; (2) using compatibility agents like silane coupling agents, surfactants, and maleic anhydride grafts; and (3) spheroidization of inorganic particles and surface heterogeneous modification to enhance flow and dispersion behavior. The above schemes have adequately solved the problem of efficient dispersion of inorganic fillers in the polymer matrix. Therefore, this kind of polymer composite filled with ultra-high inorganic particles has not received much attention in polymer processing research. However, it is often overlooked that no matter how high the dispersion of inorganic particles is achieved, inorganic particles and polymers still belong to a heterogeneous system. This heterogeneity stems from the differences in the intrinsic physical properties between the inorganic particles and the polymer matrix and their inevitable interfacial disparities. This heterogeneity inevitably leads to severe stress concentration and internal stress within the polymer matrix. Therefore, our team posits that the core focus of the next stage of research on polymer composites with ultra-high inorganic filler proportions should be on eliminating or counteracting these heterogeneities.The key to addressing the heterogeneity in polymer matrix composites lies in the self-adaptation of the polymer matrix. This involves the movement of polymer molecular chains in response to external stimuli, adapting to the surrounding inorganic particles. Although polymer chains inherently possess a relaxation effect, the process is often too prolonged and occurs gradually during product use. Therefore, in applications involving polymer composites with ultra-high proportions of inorganic particles, cross-linking and high crystallization are frequently employed to inhibit relaxation and ensure structural stability. Consequently, the polymer matrix must self-adapt to the inorganic particles during processing. Typically, the self-movement of polymer chains is observed in the regular arrangement of molecular chains during isothermal crystallization, with most other movements occurring under applied shear fields. Zhao,4 for the first time, harnessed the self-movement ability of polymer molecular chains during isothermal crystallization to regulate the arrangement of silica particles within the polymer matrix. This was achieved through the exclusion behavior observed during the regular arrangement and crystallization of molecular chains. However, Zhao's study utilized poly(ethylene oxide) (PEO) as the matrix, with an isothermal crystallization period of 7 days. For general-purpose plastics, the isothermal crystallization time is typically less than one hour, making this approach unsuitable for addressing heterogeneity in highly filled composites based on such plastics. Nonetheless, Zhao's work inspired us to consider using the self-movement ability of polymer chains to resolve heterogeneity. Dynamic covalent polymer networks naturally come to mind in this context. The bonding form can be divided into dissociative (Diels–Alder reactions) and associative (vitrimer) types.5 The key point of the dynamic covalent cross-linked network is that it undergoes continuous reorganization under the action of heat (the traditional covalent cross-linked network can cause the molecular chains of the thermoplastic matrix to lose their mobility), which can induce the mutual diffusion movement of the molecular chains in the entire polymer matrix.6 Cao's pioneering work demonstrated the preparation of polymer composites with high inorganic filler content using dynamic covalent bonds from Diels–Alder (D–A) reactions.7,8 This included alumina (60 wt%)/polycaprolactone and NdFeB (85 wt%)/polybutadiene rubber composites, which exhibited excellent mechanical properties and high filler dispersion. The high dispersion was unequivocally achieved as a result of particle surface modification and their direct involvement in the polymerization process. However, these studies did not explore whether the self-movement process of polymer chains under long-term thermal stimulation could reduce heterogeneity. Furthermore, the D–A reaction presents challenges for direct application in the current methods of preparing polymer composites with high inorganic particle content, stemming from the need to design complex monomers for the D–A reaction.
In this study, we utilized polymer-based nuclear shielding composites as a fundamental model to investigate the mitigation of heterogeneity effects induced by ultra-high inorganic fillers in thermoplastic matrices through the engineering of molecular chain dynamics. Considering that the most common commercial polymer nuclear shielding material is lead–boron polyethylene, in this paper, we chose HDPE as the matrix and used environmentally friendly tungsten powder (70 wt%) as the functional filler for nuclear shielding. We developed gamma-ray shielding components that exhibit high dispersion and low heterogeneity by integrating dynamic covalent bonds with robust mechanochemical intercalation capabilities. Considering the ease of constructing dynamic covalent cross-linked structures in a polymer matrix, we built a vitrimer structure in the HDPE matrix. The vitrimer dynamic structure in the HDPE matrix was constructed using the method reported in Kar's work.9 This approach only required the maleation of HDPE or the direct use of commercially available maleic anhydride-grafted HDPE pellets. Adding a 1 wt% catalyst and 0.5–3 wt% cross-linking agent allowed construction via twin-screw extrusion, making the process convenient and well-suited for direct industrial scale-up. For tungsten powder, we selected commercially available powder modified with KH560. We utilized solid-state shear milling (S3M) to intercalate tungsten powder into the vitrimer-modified HDPE effectively and to homogenize the vitrimer cross-linked structure formed during reactive extrusion. Furthermore, the combination of reactive extrusion and mechanochemical intercalation was conducive to achieving a high degree of dispersed compounding of KH560-modified tungsten powder and the vitrimer-modified HDPE substrate, weakening the phase agglomeration caused by viscosity during subsequent melt extrusion. A twin-screw extruder was then used to facilitate powder plasticization, and prolonged hot pressing was utilized to induce efficient self-movement of the HDPE's carbon–carbon main chains. This process successfully constructed low-heterogeneity polymer composites with highly dispersed ultra-high inorganic fillers. This entire method aligns fully with the production processes typical of any polymer modification factory, streamlining the path to rapid industrialization and production. Consequently, this approach achieves a breakthrough from the theoretical exploration of dynamic covalent bonds to practical industrial application.
Experiments
Materials
The high-density polyethylene grafted with maleic anhydride (HDPE-g-MA), with a grafting rate of 1%, was purchased from Dow Co., Ltd. First, we need to emphasize that the choice of HDPE or HDPE-g-MA as the matrix has little impact on this work. If HDPE is chosen as the matrix, HDPE-g-MA can be easily prepared by simply blending HDPE, maleic anhydride, and dicumyl peroxide (DCP) using a twin-screw extruder. In this study, we purchased commercially available HDPE-g-MA for experimental convenience. Kh560-modified Tungsten nanoparticles, with a particle size of 500 nm, were purchased from Shanghai Yao Tan Nano Material Co., Ltd. Bisphenol A diglycidyl ether (DGEBA) and zinc acetylacetonate hydrate (Zn(acac)2) were procured from Sigma Aldrich.Preparation of composite materials
The experimental process is shown in Fig. 1. First, the modification of MA-g-HDPE has been relatively mature. For the consideration of simplifying the experimental process, we directly utilized commercially available HDPE-g-MA to construct high-density polyethylene vitrimer (HDPE-v) with different DGEBA contents of 0.5 wt%, 1 wt%, and 3 wt% in a twin-screw extruder (SHJ-20, Nanjing Giant Machinery Co., Ltd, China), which was named HDPE-v-X% (X = 0.5, 1, 3). The temperature of the twin-screw extruder was set at 180 °C, 185 °C,190 °C, 190 °C, 190 °C, and 185 °C, and the screw speed was maintained at 30 rpm. Then, the high tungsten powder (70 wt%) – filled HDPE composite powder was prepared by S3M using HDPE-v as the matrix. The internal temperature of the S3M equipment was set at 15 °C; the pressure between the milling discs was set at 3, 4, 5 MPa, and the rotating speed of the moving milling disc was controlled at 50, 100, and 150 rpm, respectively, which was named W/HDPE-v-X%–Y–Z (Y = 50, 100, 150 rpm; Z = 3, 4, 5, 6 MPa). The material enters the equipment from the feed port and completes the milling after discharging. W/HDPE-v composites can be obtained as a powder after 10 milling cycles. Pellets were then prepared in a twin-screw extruder. The temperature of the twin-screw extruder was set at 180 °C, 185 °C, 190 °C, 190 °C, 190 °C, and 185 °C, and the screw speed was maintained at 30, 80, 130, or 200 rpm respectively, which was named W/HDPE-v-X%–Y–Z–T (T = 30, 80, 130, or 200 rpm). Finally, the samples were made by hot pressing using a plate vulcanizer at 200 °C and 10 MPa for different times (20 min, 1 h, 2 h, 6 h), which was named W/HDPE-v-X%–Y–Z–T–H (H = 20 min, 1 h, 2 h, 6 h). For the comparison samples, MA-g-HDPE and KH560 modified tungsten powder were extruded and granulated using a twin-screw extruder (50 rpm, 200 °C), and then samples were prepared by hot pressing (200 °C) for 20 minutes, which was named HDPE-g-MA + W. The optimal process parameters are detailed in Table S2.†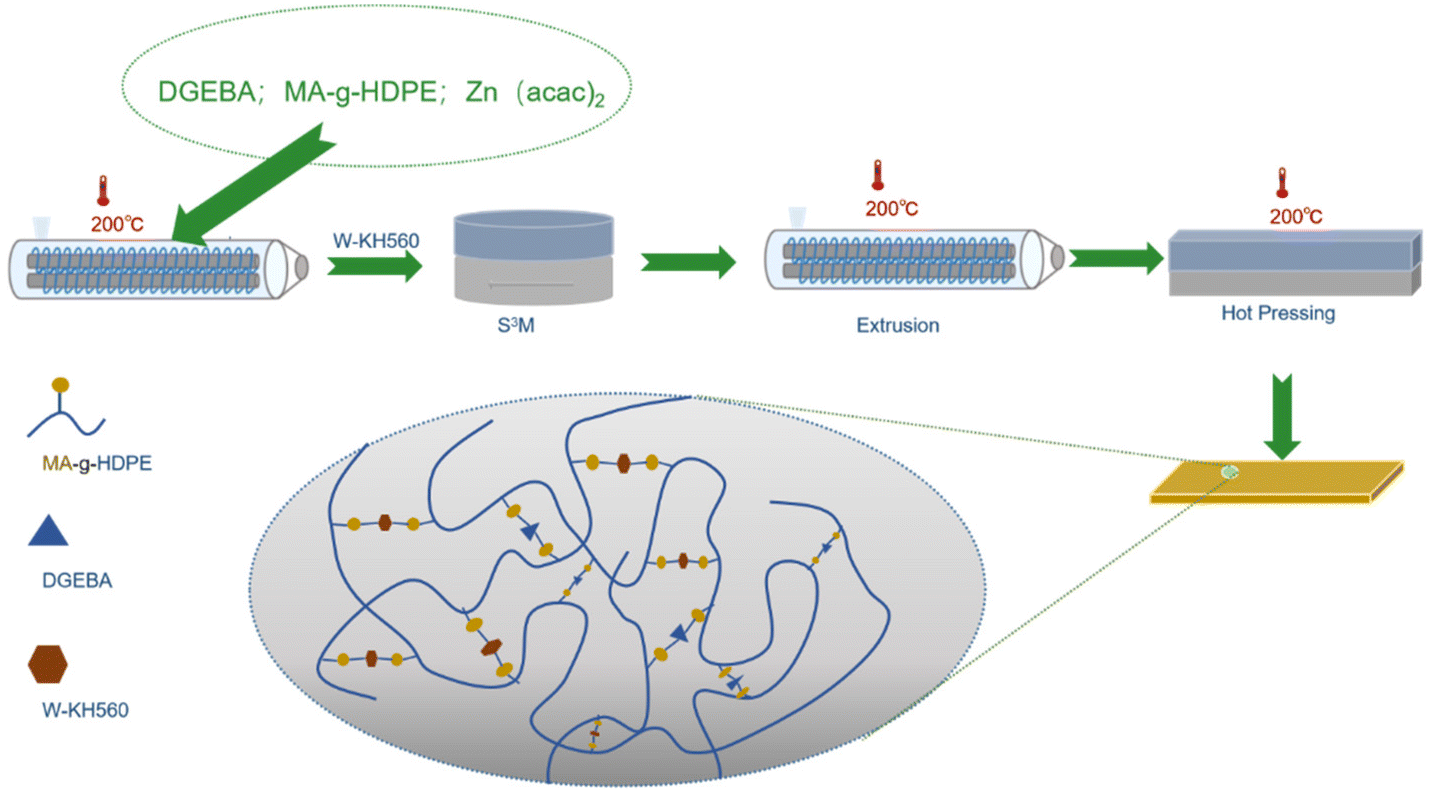 | ||
| Fig. 1 The schematic of the preparation process for W/HDPE-v composites. | ||
Characterization
Scanning electron microscopy (SEM) was performed on a scanning electron microscope (FEI Instrument Co. Ltd) to observe the morphology. Before SEM evaluation, the samples were sputter-coated with gold to prevent charging during the test. Transmission electron microscopy (TEM) was performed on a transmission electron microscope (JEOL JEM-100CX). Composites were cut into 80–100 nm thin sections at a low temperature using a LEICA EM FC6 frozen ultramicrotome, and the samples were then placed on copper grids. Rheological properties were analyzed on an AR 2000ex stress-controlled rheometer (TA Instruments) with 25 mm parallel plate fixtures. A dynamic temperature sweep from 140 to 250 °C was employed, and samples were maintained at 1 Hz. The behaviors of crystallization of the prepared composites were measured by a differential scanning calorimeter (DSC-204-Netzsch Instrument, Germany) under a nitrogen atmosphere. The samples, weighing 7–9 mg, were heated from 40 °C to 250 °C at a speed of 10 °C min−1 and then cooled down to 40 °C at a speed of 10 C min−1. Similar temperature profiles were followed for all the samples. In the first cycle of the temperature scan the sample was heated to 250 °C and kept isothermally for 5 min to erase the thermal history. The crystallization temperature was determined from the exothermic peak observed while cooling the sample to 30 °C. A second heating cycle was performed to determine the melting temperature from the endothermic peak. An atomic force microscope or AFM (MFP-3D, Asylum Research, Santa Barbara, CA, USA) was used for performing indentations on the prepared samples to characterize the heterogeneity of the composites. The DMA 850 was employed for the measurement of thermo-mechanical properties. A temperature ramp was conducted in tensile mode on rectangular specimens with dimensions of 15 mm–5 mm–0.9 mm at 2 °C min−1 and frequency of 1 Hz with 0.01% strain in the temperature ranges of −120–120 °C for W/HDPE-v. Prior to the temperature ramp, the DMA was equilibrated at 40 °C for 5 min. The tensile strength of various samples was tested using a universal testing machine (Instron 5567) at room temperature with a cross-head speed of 50 mm min−1 according to ASTM D638 standards. The γ-ray shielding test was performed using a Cs-137 source and the energy of the γ-ray was 661.6 keV.Results and discussion
Introduction of vitrimer and verification of dynamics
Dynamic covalent bonds have rapidly advanced and made significant impacts in numerous emerging fields, such as intelligent materials,10 controllable synthesis,11 and the recycling of thermosetting resins.12 However, few studies have explored the integration of dynamic covalent crosslinking networks with common polymer materials like polyethylene (PE) and polypropylene (PP). Notable examples include the Diels–Alder (D–A) reaction with PP,13 borate ester bonds with PE,14 and transesterification vitrimer structures with both polypropylene and polyethylene.9 While the incorporation of dynamic crosslinking networks has endowed common plastics like polyethylene with enhanced mechanical properties, improved regeneration performance of heterogeneous mixed plastics, and superior performance in highly filled resin-based composites, general-purpose thermoplastics such as polyethylene can readily achieve these functionalities without such modifications. For instance, introducing a trace amount of dicumyl peroxide (DCP) as a crosslinking agent can maintain the processability of crosslinked modified polyethylene while enhancing its mechanical performance. Mixed plastics can be recycled by producing high-thickness products through pressure extrusion. Additionally, thermoplastics like polyethylene can easily facilitate the preparation of composites with high inorganic filler content without complex modification processes, as exemplified by lead–boron polyethylene. Therefore, it is imperative to clarify the significance of introducing dynamic covalent crosslinking networks into thermoplastic matrices such as polyethylene. This clarification forms the foundation of our subsequent work.From the perspective of polymer processing, the mobility of polymer molecular chains exists at two extremes. At one extreme is screw extrusion, where molecular chains rapidly disentangle and move swiftly. At the other extreme are methods like hot pressing and selective laser sintering (SLS), characterized by a static state of molecular chains with only localized interfacial thermal motion. The introduction of dynamic covalent bonds, such as those in vitrimers, imparts molecular chains with the ability to diffuse and move under static processing conditions (Fig. 2). This supplements a new forming paradigm that utilizes the mobility of the mid-sections of polymer chains.
 | ||
| Fig. 2 The significance of introducing dynamic covalent crosslinks into thermoplastic polymer matrices. | ||
In this study, we adopted the scheme reported by Kar for constructing vitrimer structures within a polypropylene (PP) matrix.9 By adjusting the material feed rate, rotational speed, and temperature of a traditional twin-screw extruder—and with a material residence time of just one minute—we successfully prepared high-density polyethylene (HDPE) with a vitrimer micro-crosslinked structure. The HDPE vitrimer (HDPE-v) particles obtained through twin-screw reactive extrusion appeared pale yellow, and their surfaces displayed pronounced uneven flow morphology due to crosslinking (Fig. 3a′). The FTIR-ATR spectroscopy (Fig. 3a) confirmed that the intensity of the maleic anhydride groups (MA) in samples prepared by reactive extrusion with varying amounts of the DGEBA crosslinking agent with maleic anhydride-grafted HDPE (HDPE-g-MA) gradually decreased. This observation verified the formation of vitrimer micro-crosslinked structures within the HDPE matrix.
 | ||
| Fig. 3 Verification of the vitrimer structure and dynamics of the W/HDPE-v composite material: the reactive extrusion preparation of HDPE-v-X (X = 0.5, 1, 3) % with a vitrimer micro-crosslinked structure: (a) the peak intensity changes after the crosslinking reaction of maleic anhydride groups in FTIR-ATR; (a′) optical photographs of the particle morphology of HDPE-v with different crosslinking densities prepared by reactive extrusion. Mechanochemical intercalation of different W contents with HDPE-v-0.5% via S3M: (b–d) SEM images of the composite powder surface morphology and the magnified counterpart (b′–d′). Viscosity vs. temperature curve of 70%W/HDPE-v-1% (e). | ||
Although numerous studies15 have reported that vitrimers can undergo multiple thermal processing under the influence of heat, it is important to recognize that vitrimers are fundamentally crosslinked materials. Their presence inevitably leads to a significant decrease in the fluidity of the matrix. Our previous work16 demonstrated that, with the aid of the powerful pulverization and compounding effects of S3M, we could achieve room-temperature embedding and dispersion of inorganic particles with the polymer and disrupt the crosslinking network of the vitrimer to improve subsequent melt processing flow behavior. We investigated the powder morphology resulting from the co-milling of HDPE-v particles with varying contents of KH560-modified tungsten powder (Fig. 3b–d and b′–d′). We found that the HDPE-v particles were severely deformed, transforming into rough, flake-shaped powders. Moreover, in the composite powder containing 50% tungsten, the tungsten was dispersed and embedded within the HDPE-v matrix. When the tungsten powder content increased to 70%, large areas of tungsten powder adhered to the surface of the HDPE-v powder, and no sporadic tungsten particles were observed between the voids of the HDPE-v powders. This indicated that the enforced embedding effect of S3M achieved an efficient combination of tungsten powder and HDPE-v. The obtained composite powder was plasticized using a twin-screw extruder and formed by hot pressing, and we investigated the change in viscosity of the product with temperature. It was found that the viscosity of the HDPE-g-MA + W composite without vitrimer decreased significantly with increasing temperature. However, the viscosity of the 70% W/HDPE-v composite product with a vitrimer micro-crosslinked structure only began to decrease above 220 °C, and the overall viscosity remained at the high-viscosity state characteristic of crosslinked materials. This successfully verifies the dynamics of the vitrimer micro-crosslinked structure in the composite containing only 30% HDPE-v matrix.7 Moreover, samples prepared under different hot-pressing times showed little difference in viscosity, indicating that the crosslinking density in the W/HDPE-v composite material did not change significantly. This also confirms the characteristic of the vitrimer structure's unchanged crosslinking density.17
Effect of vitrimer on inorganic particle dispersion and heterogeneity
Blending tungsten powder, polyethylene (PE), KH560 (a silane coupling agent), and POE-g-MA in an internal mixer, followed by compression molding, resulted in composites with a severe macroscopic agglomeration of tungsten powder, uneven microscopic dispersion, and numerous defects in the prepared parts. Although this method is a conventional solution for preparing masterbatches in the industry, it primarily relies on modifiers like coupling agents to act as bridging agents, considering the polarity difference between inorganic particles and the plastic matrix. However, there is no strong bonding between the inorganic particles and the plastic matrix. Consequently, industrial practices often employ equipment such as reciprocating single-screw, triple-screw, or planetary screw extruders to apply strong external forces that promote the dispersion of inorganic particles. Unfortunately, these devices are typically not available under laboratory conditions. In our work, we addressed this limitation by constructing a vitrimer structure to induce a strong bonding effect between tungsten powder and the high-density polyethylene (HDPE) matrix, thereby enhancing the dispersion of tungsten powder within the matrix. As shown in Fig. 4(c–f and c′–f′), in the W/HDPE-v composite material, tungsten powder exhibits a well-dispersed distribution at both macroscopic and microscopic scales, similar to the previous works.7,8 Many tungsten particles are dispersed as individual entities within the HDPE-v matrix (Fig. S1†), outperforming the HDPE-g-MA + W and the standard blend of tungsten powder with polyethylene. Moreover, even after prolonged hot-pressing treatment, the tungsten powder maintains a high degree of single-particle dispersion within the HDPE-v matrix. | ||
| Fig. 4 Dispersion of KH560-modified tungsten powder in an HDPE-v matrix: SEM graphs of cross-sections of samples (a and a′) 70%W/PE composites with the addition of KH560 and POE-MA-modified agents; (b and b′) HDPE-g-MA + W; (c and c′) 70%W/HDPE-v-0.5%–100 rpm–6 MPa–200 rpm–20 min; (d and d′) 70%W/HDPE-v-0.5%–100 rpm–6 MPa–200 rpm–1 h; (e and e′) 70%W/HDPE-v-0.5%–100 rpm–6 MPa–200 rpm–2 h; (f and f′) 70%W/HDPE-v-0.5%–100 rpm–6 MPa–200 rpm–6 h. | ||
DSC was used to investigate the movement behavior of the entire molecular chain to fully elucidate the influence of the vitrimer structure's dynamics on the W/HDPE-v composites. When comparing the two matrices—HDPE and HDPE-g-MA (Fig. 5c and f)—the most significant difference observed was a small melting peak at 165 °C in the melting curve of HDPE-g-MA. However, the cooling curve did not exhibit a corresponding crystallization peak near this temperature, indicating that this peak is not associated with the traditional melting and crystallization of the crystalline regions composed of C–C main chains. Considering that the melting point and boiling point of maleic anhydride are 52 °C and 202 °C, respectively, with a decomposition temperature as high as 300 °C, this small peak cannot be attributed to the decomposition of residual maleic anhydride in HDPE-g-MA or to the degradation of HDPE-g-MA itself. We reasonably infer that this small peak (referred to as the “MA peak”) represents the manifestation of strong cohesive energy between molecular chains induced by the polar groups of maleic anhydride. This cohesive interaction only diminishes at temperatures significantly above the melting point of the matrix, at which point the molecular chains containing strong polar groups begin to move freely.
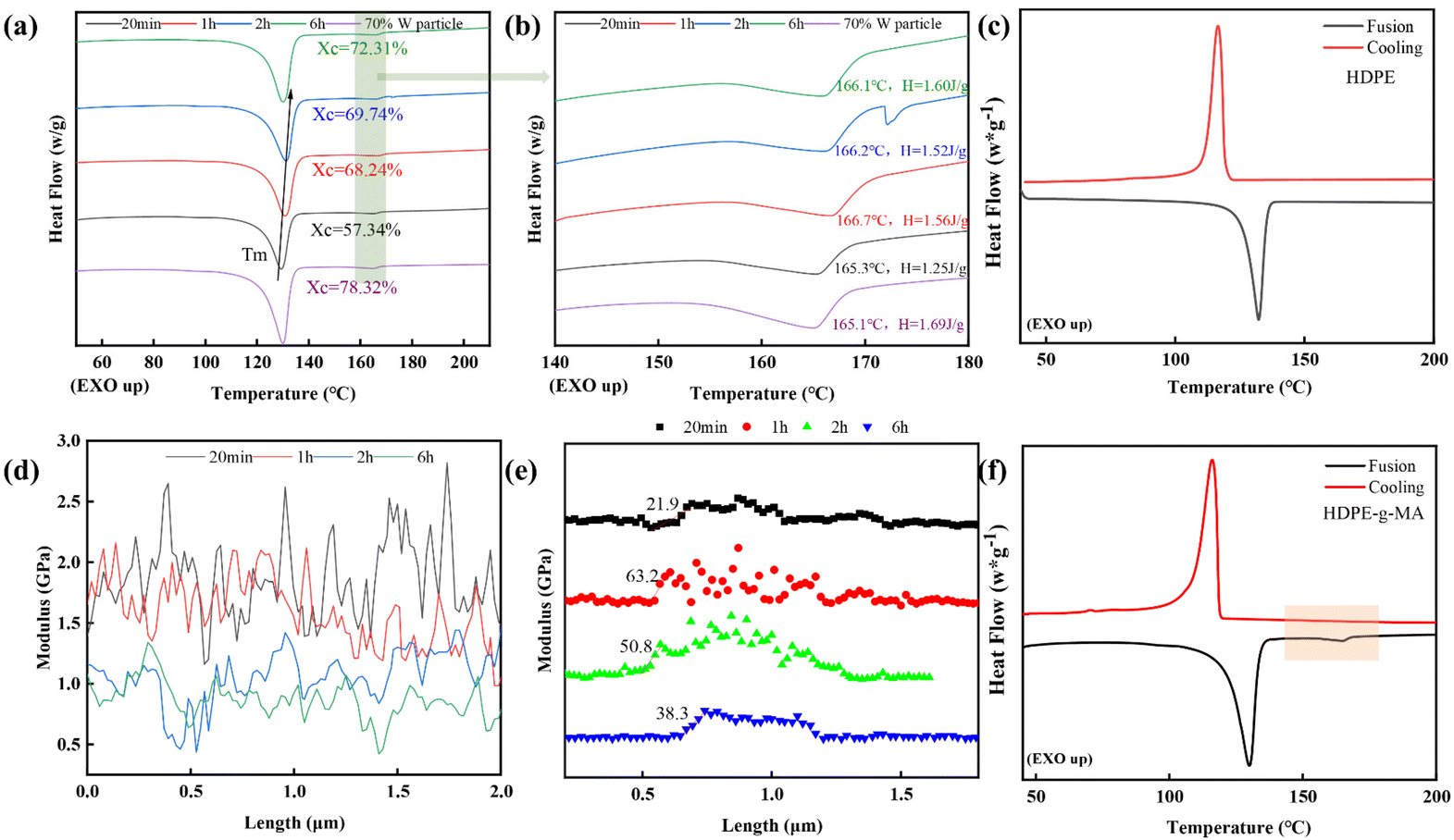 | ||
| Fig. 5 Role of the vitrimer structure in W/HDPE-v composites: (a) DSC curves of 70%W/HDPE-v-0.5%–100 rpm–6 MPa–200 rpm–(20 min–6 h) and twin-screw plasticized particles (70%W/HDPE-v-0.5%–100 rpm–6 MPa–200 rpm); (b) the magnified graphs of DSC curves of the MA peak; (c) DSC curves of the HDPE matrix; AFM results from Fig. S2:† (d) changes in the HDPE-v matrix modulus with hot-pressing time; (e) modulus transition at the interface between the HDPE-v matrix and tungsten particles; (f) DSC curves of the HDPE-g-MA matrix. | ||
To fully elucidate the influence of the vitrimer structure's dynamics on the W/HDPE-v composites, we investigated the DSC melting curves of the W/HDPE-v-0.5%–100 rpm–6 MPa–200 rpm twin-screw plasticized particles and the W/HDPE-v-0.5% products subjected to different hot-pressing times. It was observed that the melting point of the blends shifted to higher temperatures with prolonged hot pressing, and the change in melting enthalpy was notably significant. The crystallinity of the twin-screw plasticized particles was 78.3%, substantially higher than the initial crystallinity of HDPE-g-MA (55.1%), indicating that the 70% tungsten powder in the HDPE-v matrix exhibited significant heterogeneous nucleation ability (Fig. 5a). After an initial hot pressing for 20 min, the crystallinity of the product decreased markedly, suggesting that a crosslinking reaction occurred in W/HDPE-v-0.5%, which disrupted the crystallization of the matrix. However, with extended hot-pressing times, the crystallinity of the composite material began to recover. As shown in Fig. 3e, the vitrimer crosslinking structure existed in the samples at all hot-pressing times and did not disappear with prolonged pressing. Therefore, the recovery of crystallinity in the W/HDPE-v-0.5% composite must result from the continuous bond exchange of the vitrimer under sustained thermal conditions.
This bond exchange could occur within the HDPE-v matrix and at the interface between the HDPE-v and the KH560-modified tungsten particles. The question arises: which of these two types of bond exchanges leads to the recovery of crystallinity? First, considering the exchange at the interface between the HDPE-v matrix and the KH560-modified tungsten particles, if the crosslinking bonds in the HDPE-v matrix are completely transferred to the surface of the tungsten particles through the bond exchange, the crystallinity of the matrix would indeed recover due to the disappearance of cross-linking networks. According to Fig. 5e, this type of bond exchange has a minimal impact on crystallinity. The transition of modulus between the matrix and tungsten particles in the W/HDPE-v-0.5%–20 min product was the smoothest, indicating that the interfacial bonding strength of the 20-minute sample was higher. When the hot-pressing time was extended to 1 hour, the interface transition became relatively sharp, suggesting that the strong interface bonding achieved during the 20-minute hot pressing has weakened—that is, the direction of bond exchange between the W particles and the matrix shifted from the particles back to the matrix. As the hot-pressing time extends, the interface transition gradually smoothed out again. This indicated that the bond exchange between the HDPE-v matrix and the KH560-modified tungsten powder was a continuous two-way process, which could help eliminate the interface differences between the plastic matrix and the inorganic particles. Moreover, this two-way bond exchange process suggested that the crosslinking network of the composite material was largely maintained. Therefore, it was not the primary factor contributing to the recovery of the crystallinity. The continuous bond exchange within the HDPE-v matrix was likely the main reason for the observed recovery in sample crystallinity. In polymers, an increase in crystallinity often results from the stacking of molecular chains, fundamentally stemming from the regular arrangement facilitated by polymer chain movement. The ongoing bond exchange of the vitrimer under prolonged hot pressing inevitably caused the deformation of the C–C main chains of the matrix. Considering both the recovery of crystallinity and the deformation of the C–C main chains, it can be reasonably inferred that the extent of this deformation is much greater than that reported in previous simulations.18–20 The C–C main chains can undergo significant mutual diffusion and adjustment, ultimately leading to a regular arrangement of polymer chains. Additionally, the strong polar groups in the HDPE-v matrix play a guiding role during the mutual adjustment of the molecular chains. As shown in Fig. 5b, the change in the melting enthalpy associated with the MA peak of the samples aligned with the change in crystallinity. In conclusion, we have verified that the vitrimer structure imparts the polymer with the ability for mid-chain movement under static processing conditions. This provided a strategy to achieve a high degree of dispersion of inorganic particles within the plastic matrix while reducing heterogeneity.
Impact of kinetic factors on W/HDPE-v vitrimer composite fabrication
Several questions must be addressed regarding the W/HDPE-v composites containing vitrimer structures: 1. What changes occur to the vitrimer structure during the twin-screw plasticization process and the hot-pressing stage? 2. Where are the main reaction sites for bond exchange within the HDPE-v matrix and at the interface between the HDPE-v matrix and KH560-modified tungsten powder? 3. What are the effects of introducing different contents of DGEBA crosslinking agent into the composite material? 4. What is the specific role of S3M in preparing the composite material? First, we studied the influence of varying contents of the DGEBA crosslinking agent on the W/HDPE-v composites while significantly reducing the processing intensity of S3M and the shear strength during twin-screw plasticization extrusion. As shown in Fig. S3c–f,† the cross-sections of particles obtained under conditions of 50 rpm and 3 MPa for S3M, and a twin-screw rotational speed of 30 rpm, exhibited polymer agglomeration phases composed entirely of HDPE-v without any tungsten particles. This phenomenon was also observed in the cross-sections of subsequent hot-pressed products (Fig. 6a–c). Notably, the higher the content of the DGEBA crosslinking agent, the more pronounced the polymer agglomeration regions became. Polymer agglomeration regions could be attributed to viscosity differences in the HDPE-v matrix caused by the vitrimer crosslinking network, leading to uneven flow during melt extrusion. This phenomenon also indicated that the vitrimer micro-crosslinking network prepared via twin-screw reactive extrusion was not uniform. Therefore, when the DGEBA content was too high (3%), it was more likely to result in pronounced cross-sectional defects (Fig. 6c) and reduced mechanical properties (Fig. S3b†). Among the samples, the product with a 1% crosslinking agent displayed the most significant performance fluctuations. We suspected this might be due to insufficient hot-pressing time and inadequate bond exchange within the composite material. Therefore, we selected the 1% crosslinking agent product as the variable model for subsequent experiments. Upon prolonging the hot-pressing time, the polymer agglomeration regions in the cross-section of the 1% product did not disappear and even showed a slight increase (Fig. 6d–f). Interestingly, the presence of these polymer agglomeration regions and the extended hot-pressing treatment did not affect the thermal stability of the W/HDPE-v product (Fig. 6g and h). | ||
| Fig. 6 At a low twin-screw rotational speed (30 rpm) and a low degree of S3M treatment, the influence of different contents of the DGEBA crosslinking agent on W/HDPE-v: the cross-section morphology of W/HDPE-v-(0.5% (a), 1% (b); (3%) (c))–50 rpm–3 MPa–30 rpm–20 min; the cross-section morphology of W/HDPE-v-1%–50 rpm–3 MPa–(1 h (d), 2 h (e), 6 h (f)); (g and h) the influence of different hot-pressing times on the thermal stability of W/HDPE-v-1%–50 rpm–3 MPa–30 rpm–(20 min–6 h), W/HDPE-v-1%–50 rpm–3 MPa–30 rpm twin-screw extruded particles, and the HDPE-g-MA matrix. | ||
Increasing the shear strength of the twin-screw extruder is typically the first approach to address the issue of polymer agglomeration due to viscosity differences. However, as shown in Fig. 7, both the cross-sections of the particles obtained at high twin-screw rotational speeds and the cross-sections of the hot-pressed products exhibited regions of polymer agglomeration. Compared to Fig. S4(b and e–g†), the area of polymer agglomeration at high twin-screw rotational speeds has increased, indicating a heightened inhomogeneity in the vitrimer micro-crosslinking network within the HDPE-v matrix. Possible reasons for this were the short residence time of the HDPE-v matrix in the thermal field. Thus, there was a low degree of vitrimer bond exchange in the HDPE-v matrix. These factors resulted in the vitrimer structure of the HDPE-v matrix exhibiting more characteristics of a traditional covalent crosslinking network, thereby restricting the exchange of molecular chains in the W/HDPE-v composites within the thermal field. Although increasing twin-screw shear strength did not eliminate polymer agglomeration, it was still advantageous for enhancing the dispersion of tungsten particles in the HDPE-v matrix (Fig. 8).
 | ||
| Fig. 7 The cross-section morphology of W/HDPE-v-1%–50 rpm-3 MPa–80 and 130 rpm twin-screw particles: (a) 130 rpm, (b) 80 rpm and hot-pressed samples: (c) 130 rpm-20min, (c′) 130 rpm-1h, (c′′) 130 rpm-2h, (c′′′) 130 rpm-6h; (d) 80 rpm-20 min, (d′) 80 rpm-1h, (d′′) 80 rpm-2h, (d′′′) 80 rpm-6h. | ||
 | ||
| Fig. 8 The dispersion of tungsten powder in W/HDPE-v-1%–50 rpm–3 MPa–6 h composites from SEM graphs of cross-section: (a) 30 rpm; (b) 80 rpm; (c) 130 rpm. | ||
To ensure the efficient dispersion of tungsten particles and eliminate polymer agglomeration regions, it is essential to fully disrupt the vitrimer micro-crosslinking network in the HDPE-v matrix before twin-screw plasticization. Then, we could utilize the dynamics of the vitrimer structure to allow its self-reorganization during the hot-pressing stage. Our previous works demonstrated that S3M effectively could disrupt the crosslinking network and promote polymer chain exchange in rubber-plastic products.6 Accordingly, we intensified the co-milling treatment while blending tungsten powder with HDPE-v-0.5% particles and further achieved rapid plasticization in a twin-screw extruder at a rotational speed of 130 rpm. As a result, there were no polymer agglomeration regions in the cross-section of the extruded particles (Fig. 9a), and the tungsten powder was well dispersed throughout the particles (Fig. 9a′). This outcome underscores the importance of employing S3M for plastic matrices characterized by uneven vitrimer crosslinking networks at room temperature, particularly when combined with high-density inorganic particles. Following this, the plasticized extruded particles underwent hot-pressing treatment. After hot-pressing for both 20 minutes and 1 hour, no polymer agglomeration regions appeared in the cross-sections of the products, and the tungsten powder particles remained highly dispersed. However, when the hot-pressing time was extended to 2 hours, 6 hours, and 12 hours, a small number of polymer agglomeration regions reemerged (as indicated by the red circles (Fig. 9d–f)). As the hot-pressing time increased—especially at 12 hours—the number of polymer agglomeration regions also increased, leading to enhanced agglomeration of tungsten powder particles. This phenomenon suggested that the continuous C–C main chain exchange movement in the W/HDPE-v composite material, containing vitrimer, may lead to the rejection of inorganic fillers during long-term hot pressing. This behavior resembled the rejection seen during isothermal crystallization, as noted by Zhao;4 however, a more detailed study of how polymer agglomeration zones are generated is necessary.
 | ||
| Fig. 9 The cross-section morphology of W/HDPE-v-0.5%–150 rpm-5 MPa–130 rpm twin-screw particles (a–a′) and hot-pressed samples with different thermal processing time: (b–b′) 20 min, (c–c′) 1h, (d–d′) 2h, (e–e′) 6h, (f–f′) 12 h. | ||
The DSC data (Table S1†) for each sample prepared by regulating the content of the DGEBA crosslinking agent, adjusting the processing intensity of S3M, and varying the shear strength of the twin-screw indicated that the changes in crystallinity with increased hot-pressing time and the corresponding variations in the melting enthalpy of the MA peak were consistent with previous findings (Fig. 5a and b). These results confirmed the existence of the vitrimer structure and its associated functionality in each sample.
The dynamic behaviors of W/HDPE-v samples under different kinetic variables were characterized by dynamic mechanical analysis (DMA). As shown in Fig. 10(a–c), the α-phase transition21 temperatures of samples with varying DGEBA crosslinking agent contents, twin-screw shear strengths, and S3M treatment strengths overall exhibited a trend of low-temperature shifts as the hot-pressing time increased. This further proved that the dynamic vitrimer structure was capable of strong mutual diffusion with the C–C main chain, forming the crystalline region under long-term thermal stimulation. It was also beneficial for enhancing the activity of the C–C main chain. Comparing the average temperatures of the α-transition peaks of the 1% and 0.5% products revealed that the higher crosslinking agent sample had a lower temperature and demonstrated stronger molecular chain activity. Along with the cross-sectional differences observed in Fig. S4(a–c†) with varying DGEBA contents, this strongly suggested that altering the amount of DGEBA added during the reactive extrusion process not only introduced vitrimer covalent crosslinking networks with different crosslinking densities but also facilitated the incorporation of additional dynamic units. After modifying the twin-screw and S3M treatment intensities, significant changes in the standard deviation of the peak temperature were observed. The sample (1%–130 rpm) exhibited the smallest standard deviation, followed by the samples (0.5%–150 rpm–5 MPa–130 rpm). In contrast, the sample (30 rpm) showed the highest standard deviation. This finding indicated that high twin-screw shear strength enhanced the mutual diffusion of polymer chains, resulting in a lower barrier for mutual exchange between molecular chains during subsequent hot-pressing, leading to more stable changes reflected in the smaller fluctuations of the storage and loss moduli of each hot-pressed sample (Fig. 10b and c). Conversely, the sample with a twin-screw rotational speed of 30 rpm demonstrated low twin-screw shear strength and insufficient molecular chain mutual diffusion. This deficiency led to a more intense degree of vitrimer bond exchange and mutual diffusion among C–C main chains during subsequent hot-pressing, continuing until a stable molecular chain mixed state was achieved under long-term thermal stimulation. This was manifested as a continuous decrease in the storage modulus in the early stages, followed by recovery to the same modulus state as the 1%–130 rpm and 0.5%–130 rpm products after 6 hours of hot-pressing (the same observed for changes in loss modulus) (Fig. 10d and e).
 | ||
| Fig. 10 DMA graphs: the loss modulus of W/HDPE-v-1%–50 rpm–3 MPa–30 rpm–(20 min–6 h) (a); W/HDPE-v-1%–50 rpm–3 MPa–130 rpm–(20 min–12 h) (b); and W/HDPE-v-0.5%–150 rpm–5 MPa–130 rpm–(20 min–12 h) (c). Comparison of storage modulus among W/HDPE-v-1%–50 rpm–3 MPa–30 rpm, W/HDPE-v-1%–50 rpm–3 MPa–130 rpm, and W/HDPE-v-0.5%–150 rpm–5 MPa–130 rpm (d); comparison of loss modulus among W/HDPE-v-1%–50 rpm–3 MPa–30 rpm, W/HDPE-v-1%–50 rpm–3 MPa–130 rpm, and W/HDPE-v-0.5%–150 rpm–5 MPa–130 rpm (e). | ||
However, a very interesting phenomenon was observed. According to DMA analysis, a high twin-screw shear strength led to better uniformity in polymer chain distribution. However, the SEM (Fig. 7a–d) revealed that high twin-screw rotational speeds caused more polymer agglomeration regions in the sample cross-section. Notably, the 1%–30 rpm samples and the products subjected to high S3M treatment exhibited fewer polymer agglomeration regions. However, they demonstrated a higher standard deviation of the α-transition temperature. This indicated that as long as there was a certain degree of bonding between the polymer agglomeration regions and the ordinary W/HDPE-v phase regions, an increase in the barrier for the mutual diffusion movement of molecular chains during long-term hot-pressing would occur, leading to greater volatility. This could be understood by comparing the positions of the equal-length black arrows in Fig. 10b and c. In the 1%–130 rpm product, the black arrow just reached the lowest curve peak from the highest curve peak. In contrast, in the 0.5%–150 rpm–5 MPa–130 rpm product, this equal-length black arrow started from the highest curve peak but failed to reach the lowest curve peak, further confirming that the 0.5%–130 rpm sample exhibited relatively higher volatility. Furthermore, this finding also suggested that steric hindrance to molecular chain movement in the polymer agglomeration region was higher. Of course, when molecular chains with high and low steric hindrances became entangled, the mutual diffusion movement driven by the dynamics of the vitrimer was difficult.
At high twin-screw rotational speeds (130 rpm), the residence time of the materials was limited, and the mutual diffusion movement of molecular chains was more inclined to occur with molecular chains that had similar steric hindrances. Consequently, the degree of molecular chain diffusion between the high-steric-hindrance vitrimer crosslinking region and the low-steric-hindrance W/HDPE-v region was low, causing the vitrimer crosslinking region to exist as an independent phase within the sample cross-section. The polymer agglomeration region exhibited a high vitrimer crosslinking density. Thus, the internal bond exchange under thermal action could be significant, causing a weak degree of polymer chain exchange with the surrounding material. As a result, with the extension of the hot-pressing time, the size of the polymer agglomeration region changed little (Fig. 7c–c′′′, d–d′′′).
In the case of the 0.5%–150 rpm–5 MPa–130 rpm sample, the high-crosslinking region was disrupted by S3M and fully combined with the low-steric-hindrance phase region. The polymer chains containing more dynamic vitrimer units also tended to mix. The low-steric-hindrance molecular chains instead acted as obstacles. This resulted in the 0.5%–150 rpm–5 MPa–130 rpm sample only being able to generate small-sized polymer agglomeration regions under long-term thermal stimulation. For the 1%–30 rpm sample, the low twin-screw rotational speed allowed the vitrimer crosslinking region and the low-steric-hindrance polymer chain region more time for a certain degree of diffusion. This resulted in only a few polymer agglomeration regions in the product (Fig. S3d and 4b†). However, as the hot-pressing time extended, these zones transformed into larger-sized polymer agglomeration regions, which means that the bond exchange of the vitrimer could also pull the entangled molecular chains with different movement steric hindrances to form polymer agglomeration regions. In conclusion, the bond exchange between the vitrimer structures and the resulting polymer chain mixing movement was the main reason for the formation and expansion of polymer agglomeration regions.
The complex molecular chain changes in the composites can be understood with the help of the following schematic diagram (Fig. 11). When the tungsten powder particles are well dispersed, the molecular chain exchange caused by the vitrimer structure under the action of heat could promote the regular and uniform arrangement of HDPE molecular chains, reducing the impact of tungsten powder particles on the heterogeneity of the polymer matrix. However, when there are some regions in the matrix where the tungsten powder particles are relatively agglomerated, long-term hot-pressing will cause the HDPE molecular chains in the tungsten powder agglomeration regions to approach each other and transform into polymer agglomeration regions.
 | ||
| Fig. 11 The schematic diagram of interfacial bonding, chain movement, and phase realignment in W/HDPE composites: (a and a′) the situation of good W particle dispersion in the HDPE matrix; (b and b′) the situation of worse W particle dispersion in the HDPE matrix. | ||
Increasing the treatment intensities of the twin-screw and S3M, as well as prolonging the hot-pressing time, helped reduce the strength of the composite material and improve the elongation (Fig. 12a and b). The elongation of the 0.5%–150 rpm–5 MPa–130 rpm sample was the best. Although there was a strong fluctuation, it was better than that of the HDPE-g-MA + W control sample. At the same time, the gamma-ray shielding effect of the vitrimer-modified W/HDPE-v composite material was better (Fig. 12c), which was mainly due to the better dispersion effect of tungsten powder in the composites.
 | ||
| Fig. 12 The mechanical properties of each sample: (a) yield stress, (b) strain; gamma-ray shielding effects: HDPE-g-MA + W, W/HDPE-v-0.5%–150 rpm–5 MPa–130 rpm–20 min, and W/HDPE-v-0.5%–150 rpm–5 MPa–130 rpm–6 h. (c) Comparison of gamma-ray shielding effects of each sample. | ||
Conclusion
This work successfully constructed a vitrimer structure in the HDPE matrix using twin-screw reactive extrusion and utilized S3M to prepare a composite powder with an ultra-high proportion of tungsten powder, as well as to achieve the homogenization of the uneven vitrimer crosslinking network. Subsequently, hot-pressing was performed to prepare samples using the twin-screw plasticized particles as raw materials. The influence of the dynamic vitrimer structure on the structure and properties of the W/HDPE-v composites was thoroughly investigated, resulting in the successful preparation of composites with high mechanical properties and enhanced gamma-ray shielding effects. The vitrimer-modified W/HDPE-v composites not only achieved a high dispersion effect with a significant proportion of filled tungsten powder but also facilitated the continuous mutual diffusion movement of polymer chains under static thermal processing. This process promoted the regular stacking of molecular chains above the melting point, increasing the crystallinity of the composites. Furthermore, this continuous mutual diffusion movement of molecular chains could mitigate the impact of high inorganic fillers on the heterogeneity and internal stress concentration within the resin matrix, thereby reducing fluctuations in the modulus of the resin matrix. This enhancement in molecular chain mobility during static processing compensates for the lack of molding methods that permit mid-stage molecular chain movement in traditional polymer processing. In this work, by adjusting the content of the introduced crosslinking agent, the twin-screw plasticization shear strength, and the S3M treatment intensity, it was found that a high content of the crosslinking agent led to the formation of polymer agglomeration regions. These regions were primarily composed of polymer chains with high movement steric hindrance, resulting from a high vitrimer crosslinking density. Simply increasing the twin-screw shear strength improved the uniformity of the matrix but did not eliminate the polymer agglomeration regions. Instead, due to differences in the steric hindrance of the polymer chains, phase separation occurred, leading to the generation of more agglomeration phases. Within the polymer agglomeration regions, numerous dynamic vitrimer units were present. Under prolonged thermal action, these units tended to undergo bond exchange themselves, resulting in weak mutual diffusion with surrounding molecular chains that had low movement steric hindrance. Consequently, after long-term hot-pressing, the size of the polymer agglomeration regions remained unchanged. When the polymer agglomeration regions with high movement steric hindrance became entangled with polymer chains of low movement steric hindrance in the W/HDPE-v matrix through external force, the directionality of vitrimer bond exchange facilitated the reformation and expansion of the polymer agglomeration regions. After thoroughly studying the influence of the dynamic vitrimer structure on the molecular chains of the composite material, this work successfully prepared a composite material that exhibited superior mechanical properties and enhanced gamma-ray shielding effects.Considering the capabilities of industrial-grade twin-screw extruders and solid-state shear milling, we believed that this composite material could be relatively easily applied to large-scale production. Industrial twin-screw extruders feature optimized length-to-diameter ratios (≥40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) and extended residence time (30–1200 s), enabling efficient catalyst–monomer–HDPE interactions. Their forced-conveying mechanism eliminates localized heat/monomer accumulation, while specialized screw elements (e.g., kneading blocks and reverse flights) ensure homogeneous tungsten dispersion and process safety without energy-intensive treatment. The experimental-scale solid-state shear milling (S3M) can process 20–50 kilograms of tungsten powder–HDPE composite powder per hour, consuming 20 kilowatt-hours. Industrial-scale solid-state shear milling devices can process 1000–2000 kilograms per hour, consuming 150 kilowatt-hours. Therefore, from the perspective of equipment, there is no problem with industrial-scale upscaling.
:1) and extended residence time (30–1200 s), enabling efficient catalyst–monomer–HDPE interactions. Their forced-conveying mechanism eliminates localized heat/monomer accumulation, while specialized screw elements (e.g., kneading blocks and reverse flights) ensure homogeneous tungsten dispersion and process safety without energy-intensive treatment. The experimental-scale solid-state shear milling (S3M) can process 20–50 kilograms of tungsten powder–HDPE composite powder per hour, consuming 20 kilowatt-hours. Industrial-scale solid-state shear milling devices can process 1000–2000 kilograms per hour, consuming 150 kilowatt-hours. Therefore, from the perspective of equipment, there is no problem with industrial-scale upscaling.
Author contributions
Prof. Shibing Bai conceived the idea and supervised the project. Dr Shuangxin Lai and Qianyue Tan designed the experiments, analyzed the data, and wrote the paper. Dr Haiping Liu, Liang Xue, Yijun Li, and Ruiqian Zhang contributed to the discussion of the experimental results. Hongli Xie and Jiliang Gong conducted SEM and DSC analyses.Data availability
Data are available on request from the authors.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This work is financed by the National Natural Science Foundation of China (52173041) and the Institutional Joint Innovation Fund from Sichuan University and the Nuclear Power Institute of China (SCU&NPIC-LHCX-15).References
- R. B. J. Chandra, B. Shivamurthy, S. D. Kulkarni and M. S. Kumar, Mater. Res. Express, 2019, 6, 082008 CrossRef CAS.
- L. Zhang, M. Jia, J. Gong and W. Xia, Radiat. Eff. Defects Solids, 2017, 172, 643–649 CrossRef CAS.
- X.-S. Li, X.-Z. Xiang, L. Wang and X.-J. Bai, Rare Met., 2018, 37, 191–195 CrossRef CAS.
- D. Zhao, V. Gimenez-Pinto, A. M. Jimenez, L. Zhao, J. Jestin, S. K. Kumar, B. Kuei, E. D. Gomez, A. S. Prasad, L. S. Schadler, M. M. Khani and B. C. Benicewicz, ACS Cent. Sci., 2017, 3, 751–758 CrossRef CAS PubMed.
- W. Zou, J. Dong, Y. Luo, Q. Zhao and T. Xie, Adv. Mater., 2017, 29, 1606100 CrossRef PubMed.
- S. Lai, C. Cheng, X. Su, Y. Liao, Q. Tan and S. Bai, J. Cleaner Prod., 2023, 412, 137324 CrossRef CAS.
- Y. Cao, M. Z. Rong and M. Q. Zhang, Compos. Part A, 2021, 151, 106647 CrossRef CAS.
- Y. Cao, M. Zhu, M. Z. Rong and M. Q. Zhang, Adv. Compos. Hybrid Mater., 2023, 6, 38 CrossRef CAS.
- G. P. Kar, M. O. Saed and E. M. Terentjev, J. Mater. Chem. A, 2020, 8, 24137–24147 RSC.
- Y. Amamoto, H. Otsuka, A. Takahara and K. Matyjaszewski, Adv. Mater., 2012, 24, 3975–3980 CrossRef CAS PubMed.
- Y. Zhang, Q. Tang, Z. Li and K. Zhang, Angew. Chem., Int. Ed., 2023, 135, e202302527 CrossRef.
- B. Qin, S. Liu, Z. Huang, L. Zeng, J.-F. Xu and X. Zhang, Nat. Commun., 2022, 13, 7595 CrossRef CAS PubMed.
- H. Muljana, S. Arends, K. Remerie, G. Boven, F. Picchioni and R. K. Bose, Polymers, 2022, 14, 1176 CrossRef CAS PubMed.
- L.-M. Peng, Z. Xu, W.-Y. Wang, X. Zhao, R.-Y. Bao, L. Bai, K. Ke, Z.-Y. Liu, M.-B. Yang and W. Yang, ACS Appl. Energy Mater., 2021, 4, 11173–11182 CrossRef CAS.
- Z. Chen, Y.-C. Sun, J. Wang, H. J. Qi, T. Wang and H. E. Naguib, ACS Appl. Mater. Interfaces, 2020, 12, 8740–8750 CrossRef CAS PubMed.
- S. Lai, C. Cheng, B. Yuan, Y. Liao, X. Su and S. Bai, Waste Manage., 2023, 158, 153–163 CrossRef CAS PubMed.
- D. Montarnal, M. Capelot, F. Tournilhac and L. Leibler, Science, 2011, 334, 965–968 CrossRef CAS PubMed.
- Z. Li, H. Zhao, P. Duan, L. Zhang and J. Liu, Langmuir, 2024, 40, 7769–7780 CrossRef CAS PubMed.
- H. Zhao, X. Wei, Y. Fang, K. Gao, T. Yue, L. Zhang, V. Ganesan, F. Meng and J. Liu, Macromolecules, 2022, 55, 1091–1103 CrossRef CAS.
- H. Zhao, P. Duan, Z. Li, Q. Chen, T. Yue, L. Zhang, V. Ganesan and J. Liu, Macromolecules, 2023, 56, 9336–9349 CrossRef CAS.
- H. A. Khonakdar, U. Wagenknecht, S. H. Jafari, R. Hässler and H. Eslami, Adv. Polym. Technol., 2004, 23, 307–315 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5lp00097a |
| This journal is © The Royal Society of Chemistry 2025 |