
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProduction of gelatin methacrylate by flow chemistry and investigation of its suitability as a biomaterial†
Pallavi
Sengupta
ab,
Emma
Salisbury
bc,
Anil
Kumar
 a,
Tanja
Junkers
*d and
Neil R.
Cameron
*bef
a,
Tanja
Junkers
*d and
Neil R.
Cameron
*bef
aDepartment of Chemistry, IITB-Monash Research Academy, Indian Institute of Technology Bombay, Mumbai, 400076, India
bDepartment of Materials Science and Engineering, Monash University, 20 Research way, Clayton, Victoria 3800, Australia. E-mail: neil.cameron@monash.edu
cDepartment of Chemistry, University of Warwick, Coventry CV4 7AL, UK
dSchool of Chemistry, Monash University, Clayton, Victoria 3800, Australia. E-mail: tanja.junkers@monash.edu
eSchool of Engineering, University of Warwick, Coventry CV4 7AL, UK
fNanotechnology and Catalysis Research Centre, Universiti Malaya, Kuala Lumpur, Malaysia
First published on 12th June 2025
Abstract
Biopolymers such as gelatin, hyaluronic acid, and chitin are used in the form of hydrogels as scaffolds for tissue engineering and as bioinks for bioprinting. The biopolymers themselves tend to be weak and hence are usually chemically functionalized to improve their stability and tenacity. The chemical functionalization is currently conducted using batch methods, which are time-consuming, difficult to scale up, and have batch-to-batch variation. Flow chemistry, on the other hand, is more efficient, safer, reproducible, easy to scale up, and can give much higher space–time yields compared to batch reactions. In this study, a flow chemistry protocol was developed for the synthesis of the commonly used biomaterial gelatin methacrylate (GelMA), and the resulting GelMA was used in bioprinting and as a hydrogel in cell culture studies to investigate its ability to support cell attachment and expansion. It was found that conversion of gelatin into GelMA proceeded rapidly and optimally at 60 °C, giving reproducible and high degrees of substitution (65–85%) and high yields in up to 20 minutes of reaction. Scale-up of the reaction was also demonstrated. The resulting GelMA was characterized by oscillatory shear rheometry and was found to be capable of extrusion bioprinting, yielding self-supporting and defect-free hydrogel patterns. The GelMA hydrogels were also found to be able to support the proliferation of primary endometrial cells over 6 days of culture. The GelMA produced by flow chemistry, therefore, was shown to be suitable for use as a bioink and as a hydrogel substrate for cell culture, demonstrating the potential of flow chemistry as an efficient method to produce biomaterials for bioprinting and tissue engineering applications.
Introduction
Biopolymers are polymers obtained from biological sources such as plants, animals, and microorganisms. Frequently biocompatible and biodegradable, biopolymers have been widely used as biomaterials for a variety of biological applications, including drug delivery, bioprinting, and tissue engineering. Other desirable properties of a biopolymer for use in tissue engineering are high porosity for efficient facilitation of nutrient diffusion to the cells, an interconnected porous structure for 3D cell propagation, and appropriate mechanical properties for cell growth. However, unmodified biopolymers often suffer from low mechanical strength and rapid degradation.1,2 To make biopolymers stable and sufficiently robust for use as a biomaterial, the biopolymer chain is chemically modified with a crosslinking group. Examples include gelatin methacrylate (GelMA), obtained by modifying gelatin with a methacrylating agent such as methacrylic anhydride;3 hyaluronic acid-tyrosine conjugate (HA-Tyr), which is obtained by coupling hyaluronic acid with tyramine;4 and chemically modified heparan sulphate.5Gelatin is produced from the partial denaturing of the triple helical structure of collagen, which is obtained from animal sources such as porcine skin, bovine skin, fish scales, bone, or connective tissue. It is a linear protein with a range of molecular weights between 15–250 kDa. It has a similar amino acid composition to collagen and contains the repetitive tripeptide sequence Gly-X-Y, where X and Y are commonly proline and hydroxyproline. Gelatin has several beneficial features as a biomaterial: it is cheap, easily available as a commercial product, water-soluble, does not cause any unwanted immune response, non-toxic, biodegradable, similar in composition to the cell extracellular matrix (ECM) and is rich in the linear tripeptide sequence arginine–glycine–aspartic acid (RGD) which can bind to many cell integrin proteins and help in cell attachment, migration, and proliferation. It contains sequences of matrix metalloproteinase (MMP) enzymes that aid in cell remodeling. As gelatin is obtained from collagen, it has several similarities to the structural composition of collagen and hence is an ideal choice of biomaterial for collagenous tissues like skin, tendons, ligaments, cartilage, and bone.6,7
Pure gelatin, however, has its shortcomings as a biomaterial, such as poor mechanical strength, low thermal stability, susceptibility to proteases, and quick degradation. Hence, gelatin has to be modified with an extra functional group or a modifier to increase its durability and stability for use as a biomaterial. There are two main chemically modified forms of gelatin: gelatin methacrylate (GelMA)7,8 and gelatin norbornene (GelNB).9 The chemical modification used in this study is the methacrylation of gelatin to produce GelMA. GelMA can be obtained when the methacrylate groups of methacrylic anhydride attach to the primary amines in the lysine amino acid side-chains in gelatin8 while gelatin norbornene (GelNB) is obtained by treating gelatin with carbic anhydride. The carbonyl group from the dicarboxylic anhydride on carbic anhydride undergoes a nucleophilic attack from the primary amine of gelatin to form an amide linkage.10 GelNB is a newer modification of gelatin which has recently begun to attain importance over the last few years, while GelMA is quite popular and has been in the market for quite a significant amount of time,11 hence, for the present studies on biopolymer synthesis via continuous flow chemistry, the more popular GelMA has been selected. GelMA has also exhibited better extrusion and smoother filaments in bioprinting.29 GelMA, when exposed to UV light and in the presence of a photoinitiator, can photo-crosslink to form a hydrogel which is stable at physiological temperature (37 °C), and which is stronger and more stable compared to gelatin.12 GelMA solution has a low viscosity, is photo-crosslinkable, and is shear-thinning, which allows it to be easily injected into defect sites, after which it can be photo-crosslinked to form a stable scaffold.13
Batch reactions have various advantages, for example, they can easily be optimized, they have a lower requirement for complex infrastructure, and they can be used for different reactions without having to undergo any major changes. However, batch methods also have various disadvantages. The productivity rate of a batch reaction decreases at larger scales, indicating that batch reactions are less effective in scalability. There can be a variability in the amount of product formation with each batch, indicating that the reaction may not be reproducible. The heat generated by exothermic reactions is not dissipated effectively, which can be dangerous for strongly exothermic reactions and/or large-scale processes, as both the products and reactants are “locked” inside the reactor. There can also be a tendency for unwanted side reactions to take place, which can produce by-products, reducing the yield of the desired product.14,15
In GelMA synthesis, the batch-to-batch variation16 and limited scalability are well-documented issues in the literature. GelMA synthesis requires the reaction of gelatin with methacrylic anhydride in an aqueous solvent. Methacrylic anhydride undergoes an exothermic reaction with water to produce methacrylic acid. While this exothermicity is mild in a small-scale reaction (up to 100 ml), its effects in a large-scale reaction in a stagnant batch reactor (a few litres) are unknown. There is a possibility that high exothermicity due to the large scale could result in explosions, as is mentioned in the Safety Data Sheets of methacrylic anhydride. Also, due to the accumulation of methacrylic acid, the overall yield can be reduced, and the final product will require a very thorough purification for many days, which can be time-consuming. Hence, processes like continuous flow can be an alternative approach to overcome some of the difficulties of batch reactions. In continuous flow reactions, both the reactants will be in a continuous motion without being confined inside a closed reactor, which can reduce the exothermicity and hot spot formation to a great extent. The reaction can be allowed to run for as much time as desired, with the flow rates under control, and hence, as many sample as is required can be collected, unlike batch, wherein the reaction is very strictly dependent on the time.17
In a continuous flow reaction, tubes or coils are utilized as reactors, and the reaction takes place in the form of a continuous stream. The reactants are pumped into the flow reactor, and the product is continuously collected, with the waste being removed constantly. Continuous flow reactions have several advantages compared to batch reactions, for example, they are faster, safer, and easier to conduct with better scalability and reproducibility. Tube reactors have a higher surface-to-volume ratio, which allows more efficient collision of reactant molecules, which, in turn, leads to better mass and heat transfer, rapid and more efficient mixing, better reproducibility, and easy scalability (especially for industrial levels of production). Flow reactors typically have a small size ranging between 1 ml to a few tens of ml, hence they are easy to manage and are effective for the safe handling of harmful chemicals.15,16 Flow systems can be pressurized with the help of a back-pressure regulator, which allows reactions in solution phase to take place at a temperature above the boiling point of the solvent. Small-diameter tube reactors are stable at high pressure, which allows reactions to take place at higher temperatures, increasing the rate of the reaction.17,18 This increase in the rate of reaction at a higher temperature, above the boiling point of the solvent and without solvent evaporation, leads to a rise in the productivity along with a reduction in the apparatus footprint, creating a more cost-effective process. The small size of the reactors, as well as the continuous flow of the reactants and products, prevents the formation of unwanted, hazardous byproducts as the reactants and products are not confined inside a closed container for a long time. It also prevents the formation of “hot spots” in the case of exothermic reactions, thus giving rise to much safer reactions.18 Flow systems can also include inline purification and analysis processes, for example, inline dialysis19 and inline NMR spectroscopy,20 as well as other techniques including HPLC, GC-MS, FTIR spectroscopy, or degassing.17 This allows the creation of fully automated and integrated chemical synthesis platforms.
Several hazardous chemical reactions have been performed using flow chemistry over the years such as highly exothermic reactions, for example, the synthesis of a histamine receptor antagonist NBI-75043,21 various catalytic hydrogenation reactions22 such as the synthesis of 2-amino-4′-chlorobiphenyl, an important intermediate in the industrial manufacture of the fungicide Boscalid,23 oxidation reactions of catalyst-free generation of diimide by hydrazine monohydrate oxidation with molecular oxygen,24 and other hazardous reactions such as the reduction of esters using neat borane dimethylsulphide complex.25 Flow chemistry has also enabled a catalyst-free reaction of benzoyl chloride and methanol.26
In GelMA synthesis via flow chemistry, the reactants are continuously pumped under a controlled temperature through the tube. This ensures that every single molecule of both the gelatin and the methacrylic anhydride receives the same temperature, which in turn increases the reproducibility, unlike batch, where there is a constant variation in the environment of the reactants that can result in batch-to-batch variability. Continuous flow techniques allow large-scale GelMA production without the requirement of huge vessels. The tube reactors (as used in this study) are more compact and need a lesser space compared to vessels, and large quantities of GelMA can easily be produced within an optimized space in a shorter time. The flow chemistry process is continuous and can be manually stopped when required, increasing the quantity of product that can be collected. The exothermicity of the reaction between methacrylic anhydride and water can be reduced to a great extent in flow chemistry since they are not in contact for prolonged periods, thus making the reaction safer during large-scale synthesis with more efficient heat dissipation and safer operations.27 A flow synthesis is much more flexible because the flow rates can be easily altered between the reactions to obtain GelMA of different degrees of substitution, unlike batch, where different reactions have to be set up to obtain different degrees of substitution. Considering all the above advantages as well as the results obtained via the small-scale flow synthesis of GelMA, the potential use of flow chemistry to synthesize GelMA has been explored in this paper.
This paper (Fig. 1) focuses on the synthesis of GelMA via a small-scale flow chemistry setup using peristaltic pumps, a PTFE tube reactor in an oil bath, and a micromixer, followed by its purification by dialysis and analysis by NMR spectroscopy. The GelMA obtained through flow chemistry is then studied to determine its suitability for biological applications, including bioprinting29 and cell culture,8 to evaluate its performance in comparison to material produced by batch methods. The properties of the produced GelMA hydrogel material were also studied via oscillatory shear rheometry.28
 | ||
| Fig. 1 Schematic representation of the experimental protocol followed in this study. | ||
Materials and methods
Reagents
Carbonate–bicarbonate buffer, gelatin from bovine skin (Type B), methacrylic anhydride (MA), deuterium oxide (D2O), and lithium phenyl(2,4,6-trimethylbenzoyl phosphinate; LAP) were purchased from Sigma-Aldrich (Truganina, Australia). Stromal cell culture media: DMEM/F12 medium (Gibco Dulbecco's Modified Eagle Medium) containing 10% Foetal Bovine Serum (FBS) (Bovogen), 1% antibiotic–antimycotic (Life Technologies), 2 mM glutamine, Cell Titer 96® AQueous One Solution Cell Proliferation Assay (Promega) containing [3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium] (MTS), and PES (phenazine ethosulphate).GelMA synthesis by the conventional batch method
GelMA was synthesized by the traditional batch method.30 A 10% gelatin solution was prepared by dissolving 1.0 g of gelatin in 10 ml of 0.5 M CB buffer, and the pH was adjusted to 9.0. It was stirred at 50 °C until completely dissolved. 1 ml of methacrylic anhydride was then added to the solution, and the reaction was allowed to proceed for 3 hours. After 3 hours, the reaction was quenched by reducing the pH to 7.0 using 6 M HCl. The reaction mixture was then transferred to a dialysis membrane of 1 kDa molecular weight cut-off, and dialysis was conducted against distilled water for 3 days at 40 °C under stirring to remove any unreacted salts and byproducts. The dialyzed solution was then frozen at −80 °C and freeze-dried for 2 days till a white, porous, foam-like material was obtained. This material was then stored at −20 °C for further use.31–33GelMA synthesis by flow techniques (PTFE coiled tube reactor)
25 ml of the gelatin solution (10% solution diluted to 2% to prevent the gel formation inside the tube) was taken in a 50 ml Falcon tube, into which was placed the end of the inlet tube of one of the SF10 peristaltic pumps. Due to gelatin being a viscous solution, and to limit its chances of gelling during the reaction, the Falcon tube with the gelatin solution was placed inside a hot water bath (50 °C). 25 ml of methacrylic anhydride emulsion solution in water was taken in a 50 ml Falcon tube, and the end of the inlet tube of the other SF10 pump was placed into it. The outlet was connected to the other opening of the micromixer and tightened. The other end of the reactor was inserted into a vial, which acts as the sample collector (Fig. 2). The oil bath was set to a temperature of 50 °C. Once the temperature became stable, the pumps were turned on at a flow rate of 0.1 ml min−1 based on a residence time of 10 min. After a desired amount of product sample was collected, the flow rate was reduced to 0.025 ml min−1 based on an increase in the residence time to 20 min at 50 °C. The temperature was then increased, at first to 60 °C, and then to 70 °C, and the experiment was repeated with the same flow rates based on the residence times of 10 min (0.1 ml min−1) and 20 min (0.025 ml min−1). The reaction was quenched by reducing the pH to 7.0 using 6 M HCl. The reaction mixture was then transferred to a dialysis membrane of 1 kDa molecular weight cut-off, and dialysis was conducted against distilled water for 3 days at 40 °C under stirring to remove any unreacted salts and byproducts. The dialyzed solution was then frozen at −80 °C and freeze-dried for 2 days until a white, porous, foam-like material was obtained. This material was then stored at −20 °C for future use. | ||
| Fig. 2 The continuous flow reactor scheme with the pumps, micromixer, and tube reactors. | ||
Determination of degree of functionalization (DoF) of GelMA by 1H NMR spectroscopy
50 mg of GelMA was dissolved in 1 ml of deuterium oxide, and 1H NMR spectroscopy was conducted using a 400 MHz Bruker NMR spectrometer. The aromatic amino acid peak at 7.0–7.5 ppm was considered the reference. The peak area of the lysine methylene protons at 2.9–3.0 ppm was used to calculate the degree of functionalization or DoF as per the spectra shown in Fig. 3(B). The spectra were analyzed using MestreNova software. The formula below was used for the calculation:7,8,34 | ||
| Fig. 3 (A) Reaction of gelatin and methacrylic anhydride to form gelatin methacrylate (GelMA). (B) 1H NMR spectra of (A) unreacted gelatin and (B) GelMA obtained after the batch reaction. | ||
Degree of functionalization of GelMA:
 | (1) |
Preparation of GelMA-gelatin hydrogels for bioprinting
Hydrogels were prepared by dissolving 1.0 g of the freeze-dried GelMA in 10 ml of CB buffer (10%) at 40 °C. Gelatin was added in varying concentrations of 1%, 2%, 5%, and 7% as the solidifier. To this solution, 0.5% LAP (photoinitiator) was added to initiate UV crosslinking. Once the hydrogel precursor was prepared, it was poured into the printing cartridge and kept at 4 °C for solidifying.35Printability of GelMA-gelatin hydrogels
Different printing patterns were generated using GeSIM Robotics software. GelMA hydrogels with varying concentrations of gelatin were printed, in the form of the patterns generated, on 6-well cell culture plates with the help of a 5 ml syringe containing a nozzle of diameter 0.01 mm at a printing speed of 10 mm s−1 using a GeSiM BioScaffolder 3.2 bioprinter. Printing pressures in the range of 10–65 kPa were used. Once the printing was complete, the scaffolds were exposed to UV light of 365 nm for 1–2 minutes for photo-crosslinking to harden and stabilize the scaffolds. The entire printing process was conducted inside a sterile laminar air flow chamber.35Rheometry
The rheological analysis of the hydrogel was carried out using an MCR 702 Anton-Paar rheometer in parallel plate mode. 0.1 g was placed onto the lower plate, and the upper plate of 10 mm diameter was lowered onto the sample, maintaining a gap of 1 mm. Paraffin oil was placed in a ring around the sample to prevent dehydration, and any extra material protruding from the measuring plate was removed using Kimwipe tissue paper. The linear viscoelastic region (LVER) for each hydrogel was determined by performing an amplitude sweep in the range of 0.1 to 503% shear strain at a constant frequency of 1 rad s−1, followed by a frequency sweep in the range of 0.1 to 100 rad s−1 at a constant shear of 5%. Subsequently, a time sweep was conducted using shear and frequency values from within the LVER (typically 5% and 1 rad s−1, respectively).28,34Cell culture
Primary endometrial stromal cells were isolated from endometrial tissue samples collected from patients undergoing surgery. All human tissues were collected following ethical approval from Monash Health and Monash University Human Research Ethics Committees (HREC). Stromal cells were expanded in DMEM-F12 containing 10% DCC-FBS, 1% L-glutamine, and 1% antibiotic–antimycotic solution. Stromal cells were used experimentally at passage 2.Cell encapsulation in GelMA hydrogels
150 mg of the freeze-dried GelMA was dissolved in 1 ml of DMEM/F12 media. 0.1% of LAP photoinitiator was added. Both LAP and GelMA solutions were sterilised using a 0.2 μm syringe filter in preparation for cell culture. Stromal cells were trypsinized with TrypLE™ (Invitrogen) and counted using a haemocytometer and trypan blue stain. 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cells were centrifuged at 1200 rpm for 5 min. The media was carefully removed, and the cell pellet resuspended in 25 μl GelMA-LAP solution. The suspension was aliquoted in 5 μl droplets in 5 wells of a 96-well cell culture plate. The plate was cured under 365 nm UV light for 3 minutes before the addition of 200 μl culture media to each well. Collagen hydrogels were used as a control for stromal cell culture; the cell pellet was resuspended in ice-cold Collagen and aliquoted in a 96-well plate as above. Plates were incubated at 37 °C for 45 minutes before the addition of media. Culture media was refreshed every 48 h. The entire cell culture setup was incubated in a humidified CO2 chamber at 37 °C.36
000 cells were centrifuged at 1200 rpm for 5 min. The media was carefully removed, and the cell pellet resuspended in 25 μl GelMA-LAP solution. The suspension was aliquoted in 5 μl droplets in 5 wells of a 96-well cell culture plate. The plate was cured under 365 nm UV light for 3 minutes before the addition of 200 μl culture media to each well. Collagen hydrogels were used as a control for stromal cell culture; the cell pellet was resuspended in ice-cold Collagen and aliquoted in a 96-well plate as above. Plates were incubated at 37 °C for 45 minutes before the addition of media. Culture media was refreshed every 48 h. The entire cell culture setup was incubated in a humidified CO2 chamber at 37 °C.36
Cell viability analysis
On days 1 and 6, stromal cell viability was checked using the MTS cell viability assay kit, according to the manufacturer's instructions. The assay detects cellular metabolic activities resulting in a colour change that can be quantified by measuring absorbance. 20 μl of MTS tetrazolium reagent was added to each well and incubated for 2 hours at 37 °C and the soluble formazan product was quantified using a microplate reader (SpectraMax Plus384; Molecular Devices) at 490 nm. The five wells were considered as five technical replicates (repeated measurements of the same sample). On day 1 and day 6, the cells were imaged using brightfield microscopy.37Results and discussion
Synthesis of GelMA using continuous flow
GelMA was initially synthesized by a traditional batch method using a 10% gelatin solution and methacrylic anhydride at 50 °C for 3 hours at a pH of 9.0. Gelatin reacts with methacrylic anhydride to form gelatin methacrylate (GelMA) (Fig. 3A). The anhydride substitution takes place on the amino group in the side chain of the lysine amino acids present in the polypeptide backbone of gelatin, and hence, 1H NMR spectroscopy was used to estimate the degree of functionalization. A 1H NMR spectrum of pure, unreacted gelatin was taken as the control. The aromatic amino acid peak between 7.0 and 7.5 ppm was taken as the reference. It is observed that at points between 5.0 ppm and 6.0 ppm, there are new methacryloyl peaks formed in GelMA, which are absent in the gelatin control. It is also observed that there is a reduction in the size of the lysine methylene peak at 2.9 ppm due to the substitution of amino group of lysine by the methacryloyl groups38,39 (Fig. 3B). Under batch conditions, an optimized degree of substitution (DS) of 80% and a recovery of 50% was reproducibly obtained.Having demonstrated successful synthesis of GelMA under batch conditions, continuous flow synthesis was investigated at residence times of 10 min and 20 min and temperatures of 50, 60, and 70 °C for both. The reaction time in flow was reduced to 10 min and 20 min from the time of 3 hours used in batch. The lowest starting residence time was taken as 10 min, as less than this was considered to be too low, where the reaction might not have sufficient time to take place. When GelMA synthesis was carried out in flow at 50 °C and a residence time of 10 min, there was no significant difference observed in the NMR spectrum of the recovered product with that of gelatin, indicating that the reaction did not take place (Fig. 4A). When the temperature was increased to 60 °C, there was evidence of methacrylate peaks between 5.0 ppm and 6.0 ppm, however, low peak intensity indicates that degree of substitution was low. When the temperature was increased to 70 °C, the formation of methacrylate peaks was observed more clearly, along with new peaks that could be seen between 6.0–6.5 ppm and between 5.5–6.0 ppm, which indicates the possibility of the presence of unwanted byproducts. The new peaks formed on the NMR spectra of the reaction at 70 °C suggest a possible formation of byproducts. The peaks are between 5.9 and 6.4 ppm, which indicates vinylic protons. Due to the higher temperature, some extra vinyl groups might have formed in GelMA on another amino acid, along with the usual lysine. The presence of extra vinylic groups changes the overall structure of GelMA and can lead to increased and excessive crosslinking, making the GelMA hydrogel very stiff. Excessively stiff GelMA hydrogels can affect the cytocompatible properties of GelMA, such as its ability to support cell growth and proliferation.38 These results show that 10 min is too short a residence time for GelMA synthesis in flow; the reactants need more time inside the reactor for the reaction to take place, and hence, in the next step, the residence time was increased to 20 min.
 | ||
| Fig. 4 (A) 1H NMR spectra of (a) unreacted gelatin, (b) GelMA at 50 °C, (c) GelMA at 60 °C, (d) GelMA at 70 °C, at residence time of 10 min. (B) 1H NMR spectra of (a) Unreacted gelatin, (b) GelMA at 50 °C, (c) GelMA at 60 °C, (d) GelMA at 70 °C, at a residence time of 20 min. (C) GelMA synthesis reaction at 60 °C at a residence time of 20 min (highest degree of substitution) using a 6 ml reactor. | ||
When the residence time was increased to 20 min, the reaction was observed to proceed to a greater extent. The NMR spectra (Fig. 4B) show a reduction in the lysine peak at 2.9 ppm, along with the appearance of new methacrylate peaks at 5.5–6.0 ppm, which have significantly higher intensity than for a residence time of 10 min. The best result was observed at 60 °C (Fig. 4Bc) compared to the others. These conditions yielded a degree of substitution (DS) of 84% and a yield of 66%, which was higher than the batch yield of 50%, while the DS remained similar (the DS in batch was 80%). New peaks also appeared in the region of 6.0–6.5 ppm at 70 °C with a 20-minute residence time (Fig. 4Bd). Hence, a residence time of 20 min at 60 °C was determined to be the optimum condition for conducting a PTFE tube flow reaction to synthesize GelMA (Fig. 4Bc and 5).
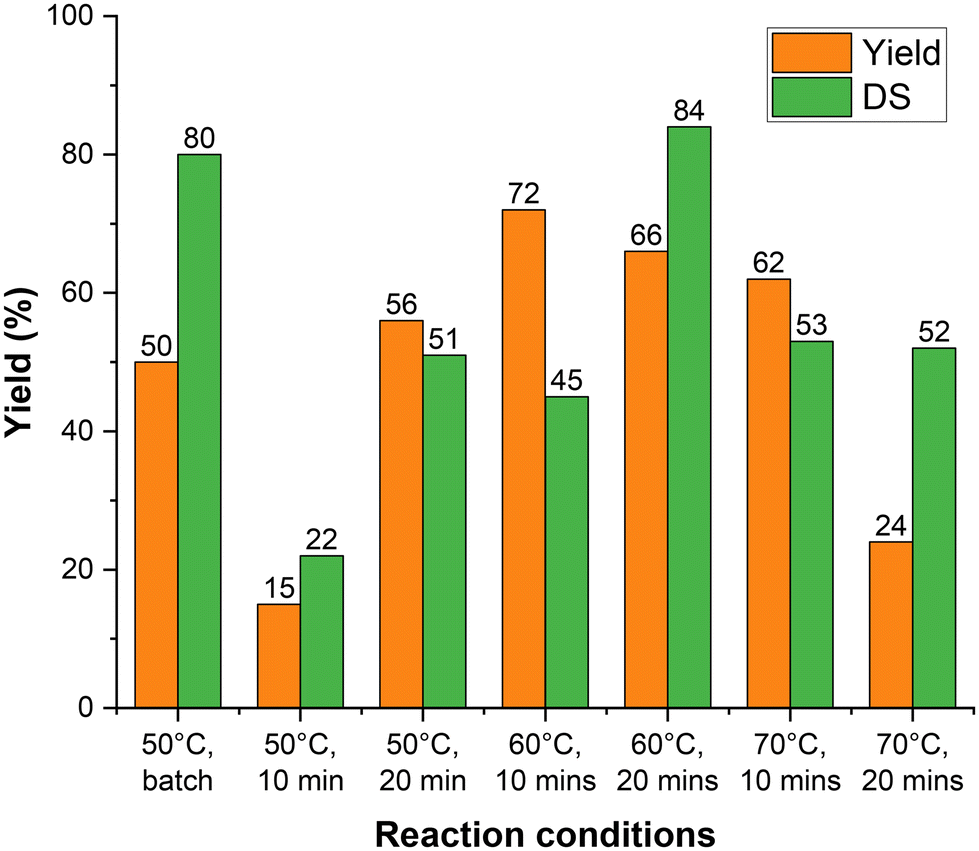 | ||
| Fig. 5 Graph depicting the yield and DS% of GelMA synthesis via flow under different temperatures and residence times. | ||
A GelMA flow reaction was next carried out using a larger reactor volume (6 ml) with the same parameters of 20 min and 60 °C to investigate the possibility of scaling up the synthesis of GelMA. The NMR spectrum of the recovered GelMA is shown in Fig. 4C. Methacrylate peaks can clearly be seen in the range 5.5–6.0 ppm, indicating successful substitution. The degree of substitution and the recovery of the purified GelMA product for each of the continuous flow conditions investigated are shown in Table 1. The space–time yield (STY) of the 6 ml reactor setting was found to be 40 g h−1 L−1 while that of the batch was found to be 6.4 g h−1 L−1, indicating a possible scalability of the reaction.
| Temperature (°C) | Reaction time (min) | Integral value of lysine methylene proton peak in gelatin methacrylatea | Degree of substitution (%) | Yield (%) |
|---|---|---|---|---|
| a The integral value of the lysine methylene proton peak in gelatin was 90.7. | ||||
| 50 | 10 | 70.9 | 22 | 15 |
| 20 | 48.5 | 47 | 56 | |
| 60 | 10 | 30.3 | 67 | 72 |
| 20 | 15.1 | 84 | 66 | |
| 70 | 10 | 42.7 | 53 | 62 |
| 20 | 43.7 | 52 | 24 | |
| 60 (6 ml reactor) | 20 | 29.5 | 68 | 66 |
3D bioprinting of GelMA synthesized by continuous flow processes
GelMA hydrogels were prepared with varying concentrations of gelatin as the modifier (1, 2, 5, and 7%) and 0.5% LAP in 10 ml of CB buffer and were printed using a 5 ml syringe with a 0.01 mm diameter nozzle at a speed of 10 mm s−1. GelMA, in its free form, was found to be of too low viscosity to be printable, and the scaffold patterns were found to be irregular and distorted. Hence, a modifier was added to enhance the viscosity of GelMA. In this study, similar to previous investigations, gelatin has been used as the modifier due to its low antigenicity and high solubility.35 Printing parameters were optimized for the GelMA obtained through flow. The solution concentration of GelMA was kept at 10%. The parameters varied were added gelatin concentration and printing pressure. For 1% and 2% added gelatin, the scaffold was found to be irregular and amorphous, which cannot be considered a good bioink. The fibres produced by the nozzle were very liquid-like and thick, with filaments breaking in between, which were unable to form the desired, well-defined bioink pattern. This shows that low concentrations of gelatin have a low viscosity, and hence, the printed features were not able to retain their desired pattern. When the gelatin concentration was increased to 5% and 7%, very uniform and clear bioink filaments were observed, with the best results obtained at a concentration of 10% GelMA plus 7% gelatin and a pressure of 65 kPa (Fig. 6B). Microscopic images were taken of the 7% gelatin-GelMA printed scaffolds in which it is observed that the 65 kPa and 60 kPa scaffolds are quite smooth, clear, and uniform while the 45 kPa and 40 kPa scaffolds are quite irregular and non-uniform with scaffold filaments breaking in between (Fig. 7). Thus, the GelMA obtained through flow chemistry has the best printability with 7% gelatin at 65 kPa. | ||
| Fig. 6 (A) The desired pattern to be bioprinted with flow GelMA, generated by GeSIM Robotics software; (B) the different scaffolds bioprinted using flow GelMA with different pressures. The blue colour arises from a food dye added to aid visibility. | ||
 | ||
| Fig. 7 Microscopic images of 7% GelMA-gelatin hydrogel under 5× magnification (scale bar – 500 μm). | ||
Rheological investigation of GelMA synthesized by continuous flow processes
The GelMA-7% gelatin hydrogel was subjected to an amplitude sweep oscillatory shear rheometry experiment at a constant frequency of 1 rad s−1 to check the stability of the sample. From the amplitude sweep curve, it can be concluded that the LVER (linear viscoelastic region) of the gelatin-GelMA hydrogel is in the shear strain range of 0.397% to 73%. It is observed that the material remains quite stable over a wide range of shear strain, indicating its tenacity. This is important in a bioink because, during bioprinting, when the bioink is extruded from the nozzle under pressure, there could be an increase in the overall shear stress due to the applied pressure and it is crucial for the bioink to retain its stability and have a smooth flow, even under pressure for proper and efficient printing. G′ and G′′ are the most important parameters in determining the viscoelastic nature of a hydrogel. G′ indicates the solid or elastic nature of the gel, and G′′ determines the liquid or viscous nature of the gel. In this study, G′ is found to be greater than G′′, which indicates the gel nature of the GelMA-gelatin hydrogel (Fig. 8A). A frequency sweep was then conducted while keeping a constant shear strain of 5%, which was within the LVE region from the previous amplitude sweep test. It is seen that, as the oscillation frequency increases, G′ and G′′ remain constant up to 20 rad s−1, after which an increase was observed, indicating shear-induced hardening. Gel-like bioinks (higher solid component) tend to have better retention of filament shapes and a higher material strength compared to viscoelastic inks (higher liquid component). G′ was observed to be greater than G′′ at all frequency values, which shows the gel nature of the material under these conditions (Fig. 8B). An oscillatory time sweep was then performed to investigate the stability of the gelatin-GelMA hydrogel with time. G′ and G′′ were found to be invariant over 10 min, indicating that the gel is quite stable over time (Fig. 8C). Stability with time is an important characteristic trait of bioinks.40,41 The storage modulus (G′) of the gelatin-GelMA hydrogel under these optimized rheological conditions was found to be 351 Pa, which is in the range suitable for tissue engineering hydrogels. | ||
| Fig. 8 Rheological analysis of GelMA-gelatin hydrogel used in bioprinting. (A) Amplitude sweep at constant frequency of 1 rad s−1. (B) Frequency sweep at constant shear strain of 5%. (C) Time sweep for 10 min (shear strain 5%, frequency 1 rad s−1). | ||
Cell viability studies
The hydrogels obtained from GelMA, synthesized using flow chemistry techniques were assessed for tissue engineering and cell culture applications.Primary human endometrial stromal cells encapsulated in the hydrogels were imaged using bright-field microscopy on days 1 and 6 of culture. The cells appear elongated by day 6, indicating that they have successfully spread within the gel droplet (Fig. 9A). In contrast, the control collagen gel had degraded by day 6. This shows the tenacity of the GelMA produced by flow chemistry as well as its ability to support cell growth over a period of time.
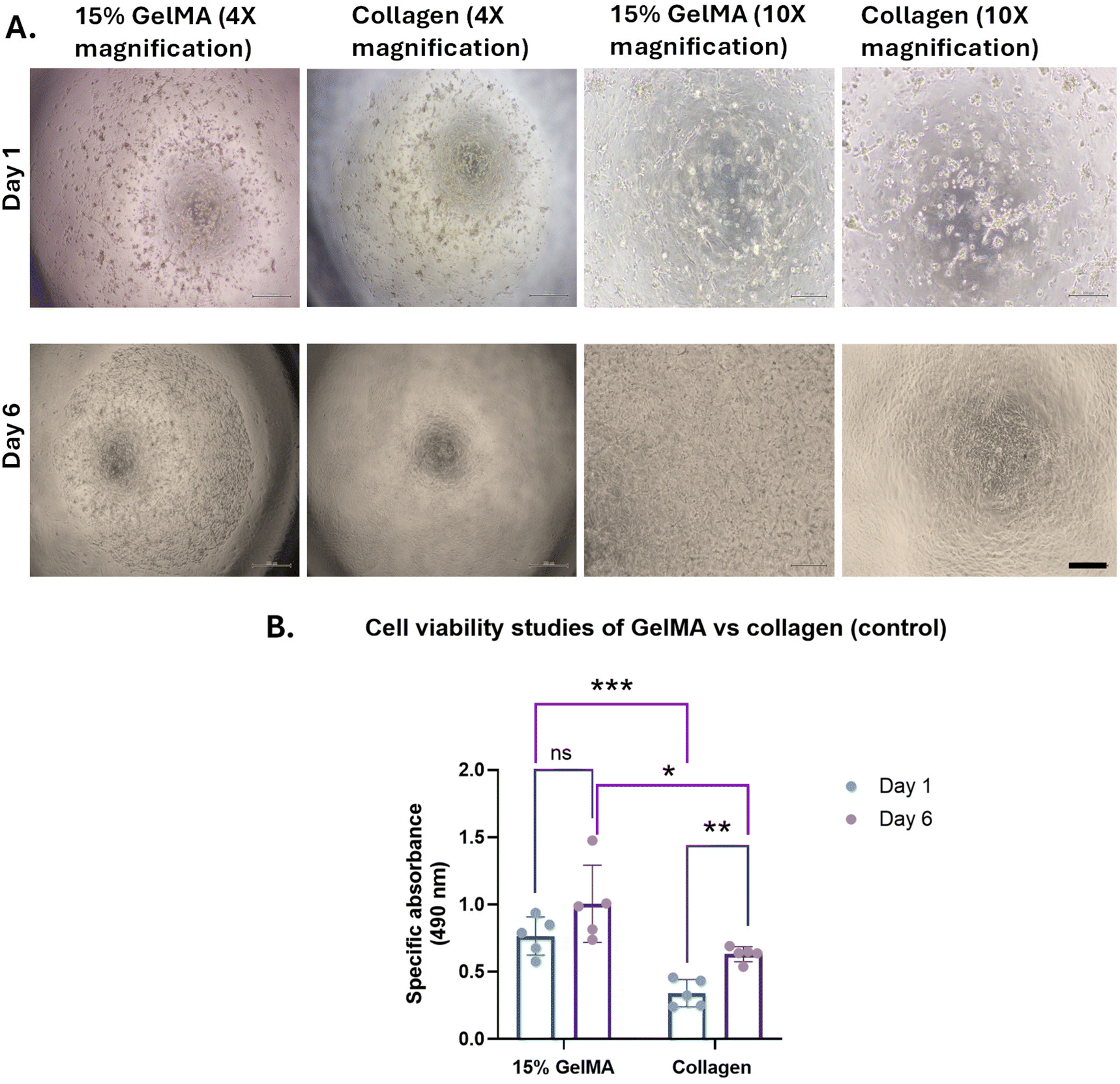 | ||
| Fig. 9 Cell culture studies of GelMA obtained from flow chemistry and collagen. (A) Brightfield cell imaging to check the cell growth on flow GelMA on day 1 and 6 at 4× and 10× magnification (scale bar for 4× – 500 μm, for 10× – 200 μm). (B) Cell viability studies of flow GelMA and collagen. The blue bar denotes cell viability on day 1, and the purple bar denotes cell viability on day 6. Data was taken in the form of five replicates each for GelMA and collagen. Parametric data were analyzed using two-tailed paired Student's T-test for the same material at day 1 and day 6 (GelMA at day 1 and day 6, collagen at day 1 and day 6) and Welch's unpaired T-test for different materials at the same time point (GelMA and collagen at day 1 and day 6). The significance was indicated as ns (not statistically significant), *(p < 0.05), **(p < 0.01), and ***(p < 0.001). | ||
The viability of endometrial stromal cells was assessed using an MTS cell viability assay kit. Data were collected in five replicates for each hydrogel condition, with GelMA and collagen as the control. The specific absorbance directly correlates with the number of viable cells. The cell viability in GelMA at day 1 and day 6 did not show a significant difference while there was a significant difference observed between the cell viability of GelMA and collagen on the same day (day 1 or day 6) (Fig. 9B). The cell viability in the GelMA hydrogels was high (approximately 90%) and was maintained over the 6-day culture period. A slight increase in the absorbance is observed, which may indicate cell proliferation over 6 days (Fig. 9B). The high cell viability indicates the biocompatibility of GelMA produced by flow chemistry and is an indication of its suitability as a biomaterial for tissue engineering, similar to its batch counterpart. The higher cell viability in GelMA produced by flow chemistry, compared to the control collagen, also confirms its superiority as a biomaterial, indicating that there is no change in its properties with a change in the synthesis method from batch to flow. While there have been many studies on the biocompatibility of GelMA produced by batch synthesis, these cell culture experiments demonstrate that the new method of GelMA synthesis by flow chemistry does not adversely affect its biocompatibility.
Conclusions
This work developed an optimized flow chemistry protocol for GelMA synthesis, with a residence time of 20 min and a temperature of 60 °C. These conditions represent an improvement on batch synthesis of GelMA, which proceeds over 3 h. To the best of our knowledge, this is the first attempt at continuous flow synthesis of GelMA. Flow synthesis provided GelMA with a degree of substitution (DS) of 68% and which was capable of scale-up to manufacture usable quantities of material for subsequent investigations. This GelMA was used to produce hydrogels, with varying concentrations of gelatin, which were assessed for extrusion printability for potential applications in bioprinting. 65 kPa was found to be the ideal pressure for the bioprinting of the flow-synthesized GelMA. The rheological characteristics of the GelMA obtained through flow chemistry were studied by amplitude sweep, frequency sweep, and time sweep for confirmation of its properties, which will be beneficial in its use as a potential bioink. The amplitude sweep showed that the LVER (linear viscoelastic region) of the gelatin-GelMA hydrogel is in the shear strain range of 0.397% to 73%, wherein it is seen that the material remains quite stable over a high range of shear strain, indicating its tenacity. The frequency sweep indicates the gel-like nature of the material, while the time sweep shows its stability over time. The GelMA obtained from flow chemistry was found to support the growth of primary human endometrial cells with high cell viability after 6 days. This shows its possible application as a biomaterial in tissue engineering, resulting in hydrogel scaffolds that can support cell growth in the same manner as GelMA produced by batch synthesis methods.Conflicts of interest
The authors declare no conflict of interest.Data availability
NMR spectra, rheology data and cell viability data (absorbances from MTS assays) for this article are available at Monash University Bridges at the following https://doi.org/10.26180/28646165.Acknowledgements
PS would like to thank the IITB-Monash Research Academy for their funding support in the form of a stipend. NRC acknowledges funding from the Australian Research Council for the Industrial Transformation Training Centre for Cell and Tissue Engineering Technologies (CTET), grant number IC190100026. The European Union is gratefully acknowledged for funding through the project SURE-Poly (Marie Skłodowska-Curie grant agreement No. 101085759).References
- R. Jain, S. Shetty and K.S Yadav, J. Drug Delivery Sci. Technol., 2020, 57, 1–15 Search PubMed.
- T. Biswal, Mater. Today: Proc., 2021, 41, 397–402 CAS.
- S. Abdollahi Baghban, M. Ebrahimi, S. Bagheri-Khoulenjani and M. Khorasani, RSC Adv., 2021, 11, 14996–15009 RSC.
- F. Lee, J. E. Chung and M. Kurisawa, Soft Matter, 2008, 4, 880–887 RSC.
- S. Anand, S. Mardhekar, R. Raigawali, N. Mohanta, P. Jain, C. D. Shanthamurthy, B. Gnanaprakasham and R. Kikkeri, Org. Lett., 2020, 22, 3402–3406 CrossRef CAS PubMed.
- A. B. Bello, D. Kim, D. Kim, H. Park and S. H. Lee, Tissue Eng., Part B, 2020, 26, 1–48 Search PubMed.
- K. Elkhoury, M. Morsink, Y. Tahri, C. Kahn, F. Cleymand, S. R. Shin, E. Arab-Tehrany and L. Sanchez-Gonzalez, Int. J. Biol. Macromol., 2021, 183, 918–926 CrossRef CAS PubMed.
- C. Ma, J. B. Choi, Y. S. Jang, S. Y. Kim, T. S. Bae, Y. K. Kim, J. M. Park and M. H. Lee, ACS Omega, 2021, 6, 17433–17441 CrossRef CAS PubMed.
- Z. Mũnoz, H. Shih and C. C. Lin, Biomater. Sci., 2014, 2, 1063–1072 RSC.
- C. Lin, E. Frahm and F. O. Afolabi, Macromol. Biosci., 2024, 24, 1–13 CrossRef PubMed.
- T. Göckler, S. Haase, X. Kempter, R. Pfister, B. R. Maciel, A. Grimm, T. Moliter, N. Willenbacher and U. Schepers, Adv. Healthcare Mater., 2021, 10, 1–13 Search PubMed.
- B. Lee, N. Lum, L. Seow, P. Lim and L. Tan, Materials, 2016, 9, 1–13 Search PubMed.
- Y. Piao, H. You, T. Xu, H. P. Bei, I. Z. Piwko, Y. Y. Kwan and X. Zhao, Eng. Regener., 2021, 2, 47–56 Search PubMed.
- E. Leal, F. Morales-Leal, J. Ancheyta, F. Alonso and P. Torres-Mancera, Ind. Eng. Chem. Res., 2023, 62, 17631–17645 CrossRef.
- B. Charleux and M. Cunningham, Polym. Sci., 2012, 3, 463–499 CAS.
- V. H. S. Rodríguez, Masters' thesis, Tecnològico de Monterey, 2019, pp. 1–76.
- N. Zaquen, M. Rubens, N. Corrigan, J. Xu, P. B. Zetterlund, C. Boyer and T. Junkers, Prog. Polym. Sci., 2020, 107, 1–35 CrossRef.
- B. Gutmann, D. Cantillo and C. O. Kappe, Angew. Chem., Int. Ed., 2015, 54, 6689–6728 CrossRef PubMed.
- P. J. Voorter, G. Dev, A. L. Buckinx, J. Dai, P. Subramanian, A. Kumar, N. R. Cameron and T. Junkers, Chem. Sci., 2023, 14, 8466–8473 RSC.
- G. D. Ammini, J. P. Hooker, J. V. Herck, A. Kumar and T. Junkers, Polym. Chem., 2023, 14, 2708–2716 RSC.
- T. D. Gross, S. Chou, D. Bonneville, R. S. Gross, P. Wang, O. Campopiano, M. A. Ouellette, S. E. Zook, J. P. Reddy, W. J. Moree, F. Jovic and S. Chopade, Org. Process Res. Dev., 2008, 12, 929–939 CrossRef CAS.
- M. Irfan, T. N. Glasnov and C. O. Kappe, ChemSusChem, 2011, 4, 300–316 CrossRef CAS PubMed.
- T. N. Glasnov and C. O. Kappe, Adv. Synth. Catal., 2010, 352, 3089–3097 CrossRef CAS.
- B. Pieber, T. S. Martinez, D. Cantillo and C. O. Kappe, Angew. Chem., Int. Ed., 2013, 52, 10241–10244 CrossRef CAS PubMed.
- S. B. Ötvös and C. O. Kappe, ChemSusChem, 2020, 13, 1800–1807 CrossRef PubMed.
- F. E. A. V. Waes, J. Drabowicz, A. Cukalovic and C. V. Stevens, Green Chem., 2012, 14, 2776–2779 RSC.
- J. Salaklang, V. Maes, M. Conradi, R. Dams and T. Junkers, React. Chem. Eng., 2018, 3, 41–47 RSC.
- I. Pepelanova, K. Kruppa, T. Scheper and A. Lavrentieva, Bioengineering, 2018, 5, 1–15 CrossRef PubMed.
- T. J. Tigner, S. Rajput, A. K. Gaharwar and D. L. Alge, Biomacromolecules, 2020, 21, 454–463 CrossRef CAS PubMed.
- V. D. Bulcke AI, B. Bogdanov, N. De Rooze, E. H. Schacht, M. Cornelissen and H. Berghmans, Biomacromolecules, 2000, 1, 31–38 CrossRef PubMed.
- J. H. Wang, C. W. Tsai, N. Y. Tsai, C. Y. Chiang, R. S. Lin, R. F. Pereira and Y. C. E. Li, Int. J. Biol. Macromol., 2021, 185, 441–450 CrossRef CAS PubMed.
- M. Zhu, Y. Wang, G. Ferracci, J. Zheng, N. J. Cho and B. H. Lee, Sci. Rep., 2019, 9, 1–13 CrossRef CAS PubMed.
- K. Zhang, J. Yang, Y. Sun, Y. Wang, J. Liang, J. Luo, W. Cui, L. Deng, X. Xu, B. Wang and H. Zhang, Friction, 2022, 10, 232–246 CrossRef CAS.
- J. Yin, M. Yan, Y. Wang, J. Fu and H. Suo, ACS Appl. Mater. Interfaces, 2018, 10, 6849–6857 CrossRef CAS PubMed.
- H. Rastin, R. T. Ormsby, G. J. Atkins and D. Losic, ACS Appl. Bio Mater., 2020, 3, 1815–1826 CrossRef CAS PubMed.
- E. Salisbury, T. M. Rawlings, S. Efstathiou, M. Tryfonos, K. Makwana, H. C. Fitzgerald, C. E. Gargett, N. R. Cameron, D. M. Haddleton, J. J. Brosens and A. M. Eissa, ACS Appl. Mater. Interfaces, 2024, 16, 39140–39152 CrossRef CAS PubMed.
- Y. Wang, D. T. Nguyen, G. Yang, J. Anesi, J. Kelly, Z. Chai, F. Ahmady, F. Charchar and J. Golledge, Assay Drug Dev. Technol., 2021, 19, 184–190 CrossRef CAS PubMed.
- R. N. Ghosh, J. Thomas, B. R. Vaidehi, N. Devi, A. Janardanan, P. K. Namboothiri and M. Peter, Mater. Adv., 2023, 4, 5496–5529 RSC.
- H. Shirahama, B. H. Lee, L. P. Tan and N. J. Cho, Sci. Rep., 2016, 6, 1–11 Search PubMed.
- L. Ning, C. Gil, B. Hwang, A. S. Theus, L. Perez, M. L. Tomov, H. Bauser-Heaton and V. Serpooshan, Appl. Phys. Rev., 2020, 7, 1–22 Search PubMed.
- Z. Huang, X. Wang, S. Chi, Z. Hua and C. Bi, Int. J. Agric. Biol. Eng., 2021, 14, 255–260 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5lp00078e |
| This journal is © The Royal Society of Chemistry 2025 |