
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIonic rectification via electrical double layer modulation at hydrogel interfaces
Yaowen
Ouyang
ab,
Zhong Lin
Wang
acd and
Di
Wei
 *a
*a
aBeijing Institute of Nanoenergy and Nanosystems, Chinese Academy of Sciences, Beijing 101400, P. R. China. E-mail: weidi@binn.cas.cn
bSchool of Nanoscience and Engineering, University of Chinese Academy of Sciences, Beijing 100049, P. R. China
cBeijing Key Laboratory of Micro-Nano Energy and Sensor, Center for High-Entropy Energy and Systems, Beijing Institute of Nanoenergy and Nanosystems, Chinese Academy of Sciences, Beijing 101400, P. R. China
dGeorgia Institute of Technology, Atlanta, GA 30332-0245, USA
First published on 9th June 2025
Abstract
Hydrogel-based iontronics have emerged as key enablers for sustainable energy harvesting and bio-inspired sensing, with applications spanning human–machine interfaces, brain–computer interfaces, and neuromorphic computing. Central to their operation is precise modulation of the electrical double layer (EDL) at hydrogel interfaces, which governs ionic rectification, a critical function for efficient iontronic performance. This review systematically examines EDL modulation strategies for achieving ionic rectification in hydrogel systems, classifying them into four fundamental mechanisms: (1) EDL formation on charged polymer chains in polyelectrolyte hydrogels; (2) nanopore-confined EDL enhanced by hydrogel modification; (3) EDL at hydrogel-based p–n junctions; and (4) asymmetric EDL at hydrogel/electrode interfaces. Representative studies highlighting breakthrough applications of these mechanisms are discussed, alongside an outlook on the future of EDL engineering in hydrogel-based iontronics, emphasizing both opportunities and challenges in optimizing performance.
1. Introduction
Hydrogels have emerged as pivotal materials in iontronics, offering a unique combination of mechanical adaptability and ionic conductivity.1–3 Structurally, they consist of a three-dimensional polymer network encapsulating a large volume of water molecules,4 where polymer entropy elasticity imparts stretchability, while free water molecules facilitate ion transport.5 Beyond their mechanical and conductive properties, hydrogels exhibit excellent biocompatibility and a tunable elastic modulus, enabling seamless integration with biological systems.6 These attributes have positioned hydrogels as a key component in wearable and implantable self-powered sensors,7,8 human–machine interfaces,8–11 brain–computer interfaces,12–14 and neuromorphic computing.15–17 In biological systems, neural signal transmission relies on the selective transport of sodium and potassium ions through ion channels, a process fundamental to nerve excitation.18–20 This ionic selectivity underpins the biomimetic functionality of iontronics, particularly in ionic rectification. The regulation of ion concentration, depletion, flow, and spatiotemporal distribution is crucial for generating, storing, and transmitting ionic signals.21–30 In hydrogel-based iontronics, the distinct treatment of cations and anions is essential, making ionic rectification a fundamental prerequisite for achieving multi-dimensional functionalities.This review explores ionic rectification through the modulation of the electrical double layer (EDL) at hydrogel interfaces. The EDL serves as a crucial medium for interfacial communication, bridging ionic currents in hydrogels with electronic currents in conductors. First introduced by Helmholtz to describe the capacitor-like structure between a charged surface of the conductor electrode and counter ions in a dilute solution,31 the model was later refined by Gouy and Chapman, who incorporated the concept of a diffuse layer to account for surface charge density effects.32,33 Building on these models, the Stern model further delineated the EDL into the Stern and diffuse layers, providing a more accurate representation of surface charge distribution, with the combined width of these layers typically within the Debye length (λD).34 This parameter is critical for characterizing the interaction strength between the conductor surface and the counter ions, which depend on electrolyte concentration and the vacuum dielectric constant. In 2019, Zhong Lin Wang introduced the ‘two-step’ model, highlighting the near-simultaneous occurrence of electron transfer and ion adsorption during contact electrification at dielectric–liquid interfaces.35,36 Recent advances have expanded the EDL concept beyond solid–liquid interfaces to include interactions at liquid–liquid and liquid–air boundaries.37–39 Despite significant progress in ionic rectification via EDL modulation at hydrogel interfaces, a systematic review of this approach remains absent, warranting a comprehensive analysis of its mechanisms and applications.
This review introduces the fundamental concepts and evolution of EDL models. It then explores four key mechanisms of ionic rectification at hydrogel interfaces: (1) EDL on charged polymer chains of hydrogels (Fig. 1a); (2) EDL within nanopores enhanced by hydrogel modification (Fig. 1b); (3) EDL at hydrogel-based junctions, commonly referred to as ionic double layers (IDL) (Fig. 1c); and (4) asymmetric EDL at hydrogel/electrode interfaces (Fig. 1d). Representative recent studies are highlighted, demonstrating the broad applications of EDL-modulated ionic rectification in energy harvesting, self-powered sensors, hydrogel-based diodes and transistors for biological detection, rectifier bridges, ionic logic circuits and neuromorphic devices. Moreover, these mechanisms hold potential for regulating cellular physiological activities. Finally, we discuss the implications of EDL modulation for the advancement of hydrogel-based iontronics and outline key challenges in optimizing ionic rectification for future applications.
 | ||
| Fig. 1 Table of content (ToC), ionic rectification at the hydrogel interface based on EDL modulation. (a) EDL on the charged polymer chain of hydrogels for self-powered intelligent sensors.40 (b) EDL of nanopores enhanced by modifying charged hydrogels for osmotic harvesting.41 (c) Hydrogel p–n junction diode for harvesting the mechanical energy of human motion.42 (d) Ionic rectification induced by modulating the asymmetric EDL at the hydrogel/electrode interface.43 | ||
2. The evolution of EDL models
The EDL typically refers to the spatial arrangement of opposite charges formed at a two-phase interface upon contact. Traditional EDL theories predominantly focus on the physical and chemical properties of interfaces between dilute solutions and conductive electrodes, with applications spanning numerous interdisciplinary fields.44 However, recent advancements have expanded the concept beyond solid–liquid interfaces, with growing attention to EDL phenomena at dielectric solid–liquid, liquid–liquid45–49 and liquid–air50–53 interfaces. Hydrogels, composed of a three-dimensional polymer network encapsulating water molecules, exhibit a solid-like morphology while maintaining a high-water content. The polymer chains on hydrogel surfaces are closely associated with water molecules, making it challenging to classify them as purely solid. Despite this complexity, established EDL concepts, such as the Debye length (λD), remain valuable for understanding and characterizing EDL behavior at hydrogel interfaces.54–562.1 The EDL model at the conductor interface
In 1853, the Helmholtz model was introduced to describe the EDL, proposing that conductive charged surfaces and counterions form a capacitor-like structure with a fixed layer thickness (Fig. 2a). This model established a foundation for studying charge distribution, potential differences, and electrochemical reaction kinetics at electrode surfaces. However, it overlooked the diffusion and adsorption of ions in the solution, as well as complex interactions between the solvent and electrode. In the 1910s, the Gouy–Chapman model was developed, incorporating the concept of a diffuse layer (Fig. 2b). This model assumes that with increased electrode surface charge, the diffuse layer becomes denser. Due to the interplay of electrostatic forces and thermal motion, the ion charge distribution exponentially decreases with distance from the electrode surface. Near the electrode, electrostatic forces dominate, leading to higher charge concentrations, while further from the surface, the influence of these forces weakens, resulting in lower charge densities. In the 1920s, the Stern model refined these theories by dividing the EDL into a compact Stern layer and a diffuse layer (Fig. 2c). This dual-layer model provides a more accurate representation of EDL structure and surface charge distribution, forming the theoretical basis for energy storage, chemical sensing, and environmental monitoring. The Stern layer consists of the inner Helmholtz plane (IHP) and the outer Helmholtz plane (OHP). The IHP, closest to the electrode, is composed of specifically adsorbed ions and solvent molecules, influenced by both electrostatic and chemical interactions. In contrast, the OHP contains solvated ions that interact with the electrode surface through long-range electrostatic forces without specific adsorption. The diffuse layer, consisting of free ions in the solution, extends from the OHP to the bulk solution and is governed by both electrostatic forces and thermal motion. The combined thickness of the Stern and diffuse layers typically falls within the Debye length (λD), a critical parameter representing the interaction strength between ions. The Debye length is influenced by electrolyte concentration and the dielectric constant of the medium.58–60 In diluted electrolyte solutions, λD is relatively thick, while in concentrated solutions, it becomes thinner. Typically, the Debye length ranges from 0.2 to 20 nm (ref. 61 and 62) and can be described using the following equation:63where ε, ε0, F, T, R, nbulk, and Z are the permittivity of solution, the permittivity of vacuum, the Faraday constant, the absolute temperature, the universal gas constant, the concentration of bulk solution, and the charge of the ionic species, respectively.

 | ||
| Fig. 2 The development history of the EDL model.57 (a) The Helmholtz model. (b) The Gouy–Chapman model. (c) The Stern model. | ||
2.2 The ‘two-step’ EDL model at the dielectric solid interface
Previous EDL models were primarily established for conductive solid–liquid interfaces. In 2019, Wang et al. proposed the “two-step” EDL model to describe the EDL at dielectric solid–liquid interfaces, incorporating both electron transfer and ion adsorption processes (Fig. 3).36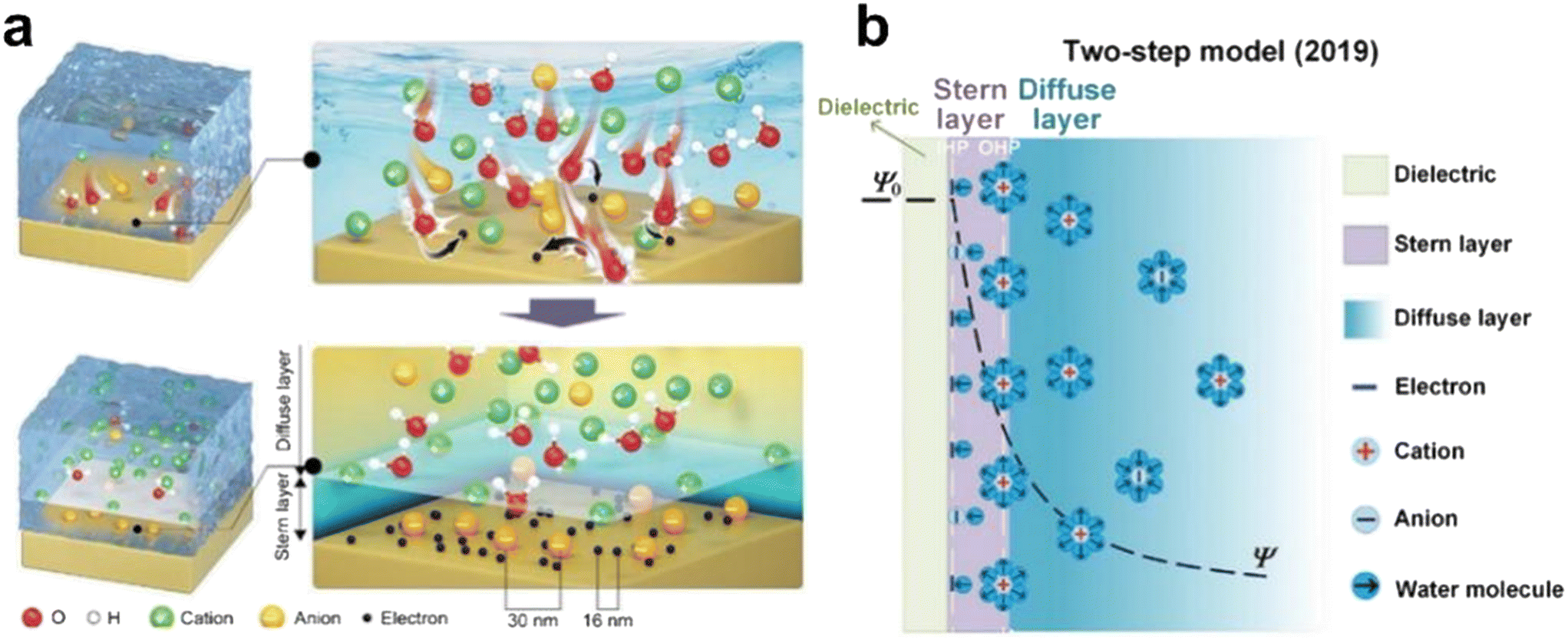 | ||
| Fig. 3 The ‘two-step’ EDL model at the dielectric–liquid interface proposed in 2019. (a) The mechanism of electron transfer and ion adsorption is considered in the ‘two-step’ formation process.64 (b) The ‘two-step’ model of EDL structure and charge composition.65 | ||
This model describes the charge transfer and EDL formation at the dielectric solid–liquid interface. Upon contact, electrons transfer from water molecules to solid atoms due to electronegativity differences, potentially triggering ionization reactions on the solid surface. This results in the concurrent accumulation of electrons and ions, forming the EDL. The “two-step” EDL model integrates electron transfer and ion adsorption processes, offering a framework that elucidates electron–ion interactions. It advances the understanding of solid–liquid interface behavior and drives innovations in interfacial research.
2.3 Dynamic EDL regulation at dielectric interfaces via triboiontronics
The EDL at conductor interfaces can be regulated through electrochemical methods, while adjusting the EDL on dielectric surfaces remains challenging. To address this, Wei et al. utilized a triboelectric field to achieve EDL modulation on dielectric surfaces and proposed the dynamic triboiontronics EDL model in 2023 (Fig. 4).66–71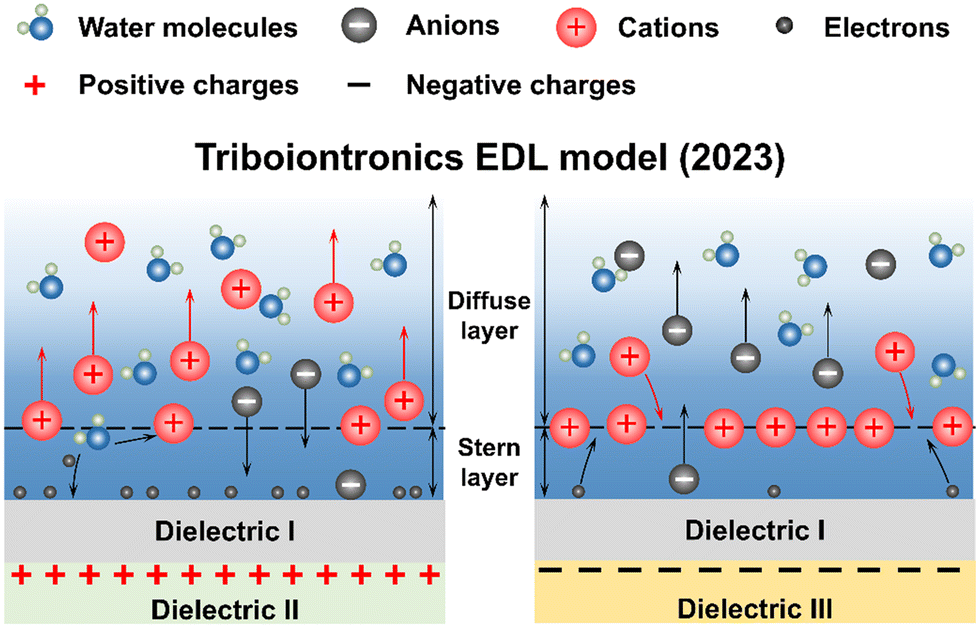 | ||
| Fig. 4 The dynamic triboiontronics EDL model at the dielectric–liquid interface proposed in 2023. The mechanism of dynamic EDL regulation by a triboelectric field. | ||
Considering the mechanism of interfacial charge generation and the characteristics of the EDL, contact electrification is intrinsically linked to EDL properties. Recent advancements have provided deeper scientific insights into EDL behavior, unveiling the potential of ion carrier regulation. This progress not only serves as a novel “probe” for exploring EDL properties but also establishes triboiontronics as a transformative interdisciplinary field with significant research potential. By bridging electron–ion-regulated neural information flow through the EDL's interfacial nature, triboiontronics introduces a new paradigm for interface property control, offering applications in biological sensors, brain–computer interface control and transistor-based logical devices.72–76 Triboiontronic nanogenerators are anticipated to drive advancements in efficient energy harvesting, precise EDL modulation, and the rapid regulation of biomimetic nervous systems.68
3. Ionic rectification mechanisms at hydrogel EDL interfaces
With the rapid advancement of iontronics, ionic–electronic coupling interfaces and controllable ion transport have garnered significant attention. Hydrogels, serving as ion conductors, are extensively applied across various fields, with ionic rectification being a critical prerequisite for their functionality. The EDL is an important medium for modulating ion rectification at the hydrogel interface to achieve a variety of iontronic functions. This review categorizes the mechanisms of ionic rectification through EDL modulation at hydrogel interfaces into four main types: (1) EDL on the charged polymer chain of hydrogels; (2) EDL within nanopores enhanced by hydrogel modification; (3) EDL at hydrogel p–n junctions; and (4) asymmetric EDL at hydrogel/electrode interfaces.3.1 EDL on the charged polymer chains of hydrogels
Polyelectrolyte hydrogels exhibit inherent ionic rectification due to their ion selectivity, which arises from the interfacial interactions between charged polymer chains and counter ions.77 Typically, these polymer chains carry fixed positive or negative charges, attracting counter-ions while repelling co-ions, thus facilitating selective ion transport.78–82 Additionally, the porous network structure of hydrogels provides a size-dependent screening effect on hydrated ions, with smaller pore sizes imposing greater resistance to larger ions.83,84Recently, a heterolayer gel (HLG) comprising an ion-selective hydrogel (ISH) layer and a water-barrier organogel (WBO) layer has been reported (Fig. 5a).85 The unconstrained, flexible three-dimensional ion-selective hydrogel network exhibits a relatively low motion barrier and a dynamically free volume of highly hydrated polyelectrolyte chains, thereby forming a stimulated ion diffusion transport channel. Notably, thermally driven chain fluctuations further induce an ion-hopping concentration effect, reduce the transfer energy barrier, expand the Debye shielding dimension, enhance ion selectivity, and facilitate the internal transport of ion clusters. By mitigating the polarization effect caused by ion concentration blockage, the HLG system improves the efficiency of macro-scale ion-selective transport. In conventional steady-state ion transport theory, the network and charged functional groups impede the stable flow of ions, reducing the transport rate. However, in the HLG, high-density functional groups interact with water molecules via hydrogen bonding, transforming the channel walls into a hydrated and fluctuating state, thereby disrupting the stability of ion traps. The fluctuations of highly hydrated polymer chains dynamically alter the channel size and the spacing of charged groups, further optimizing ion transport (Fig. 5b).88 These changes will affect the absorption and charge distribution of specific ions in the EDL and the aggregation and dispersion of ions. Due to the rearrangement of the EDL, the hopping energy barrier [ΔEhydrated (t), where t represents the duration of fluctuation] will change near the original energy barrier (ΔE0) without considering chain fluctuations accordingly. Importantly, in the thermodynamic system constructed from a soft and unconstrained three-dimensional network, ions will be transported along the path of the minimum energy barrier under the unevenly distributed chemical potential under the ion gradient condition. From a thermodynamic point of view, the total free energy of the system can be minimized. Therefore, the transfer energy barrier experienced by the ions (denoted as ΔEn) will be less than ΔE0. Owing to the cation selectivity of highly space negative-charged hydrogel networks, K+ will diffuse at a faster rate than Cl−, which consequently caused an electrostatic potential along the ion gradient direction. The generated potential and ionic current can be reflected by the interceptions on the voltage and current axes, which were named the open circle voltage (VOC) and short-circle current (ISC), respectively (Fig. 5c). As the concentration gradient increases from 10 to 1000, the corresponding ion selectivity coefficient S (calculated based on the measured VOC and Vredox, where S = 0 represents non-selectivity and S = 1 represents ideal cation selectivity) decreases from 0.98 to 0.92 (Fig. 5d).
 | ||
| Fig. 5 Ion-selective layer on the charged polymer chain of hydrogels. (a) A heterolayer gel (HLG) made up of an ion-selective hydrogel (ISH) layer and a water-barrier organogel (WBO) layer. (b) Ion hopping transport in a three-dimensional hydrated dynamic system constructed from highly hydrated chains. (c) I–V curves of HLG recorded in different KCl concentration gradients. (d) The ultrahigh cation transference number (t+). (a)–(d) Reproduced from ref. 85, Copyright © 2024 American Chemical Society. (e) Schematic of material resources and molecular structures of polysaccharide polyelectrolytes, as well as the gradient structure of resulting hydrogel membranes. (f) I–V curves of the gradient membrane recorded in KCl solution (0.01 M) with the numerical simulation shown in the insert. (g) I–V curves of the gradient CS/SA (chitosan/sodium alginate) membrane under forward and backward saline gradients. (e)–(g) Reproduced from ref. 86, Copyright © 2020 American Chemical Society. (h) PET membrane and heterogeneous membrane that can greatly eliminate concentration polarization. (i) Numerical simulation of the PET membrane and heterogeneous membrane respectively. (j) I–V curve of the 12 μm thick PET membrane before and after 200 μm thick hydrogel covering. (k) Ionic current rectification ratio. (h)–(k) Reproduced from ref. 87, © 2024 Elsevier B.V. All rights reserved. | ||
Bian et al. have prepared a continuous gradient full polysaccharide polyelectrolyte hydrogel membrane, which has been proved to be able to achieve high-performance osmotic power conversion.86 In addition to its inherent high ion conductivity and excellent ion selectivity, the anti-swelling polyelectrolyte gradient membrane also retains the ionic diode effect of an asymmetric membrane to promote unidirectional ion diffusion but avoids adverse interface effects. They can exhibit an ultrahigh power density of 7.87 W m−2. The gradient all-polysaccharide polyelectrolyte hydrogel membranes were fabricated by the reaction–diffusion method.89,90 Once the chitosan (CS) solution came into contact with a polyanion solution (sodium alginate, SA), a thin complex layer formed instantly on the interface of two solutions due to strong electrostatic complexation. Thereafter, the CS gradually diffused across the complex layer and interacted with the polyanion, leading to the growing thickness of membranes. The side close to the low-molecular-weight (LMW) CS solution (referred to as the LD side) can react with more LMW CS, as its electrostatic complexation density is higher than the other side (referred to as the HD side). This shows that for the obtained polyelectrolyte hydrogel film, the LD side has a lower residual negative charge density and more dense network structure than the HD side (Fig. 5e). The molar ratios of the negatively charged and positively charged groups on the LD and HD sides were estimated to be 1.45 and 1.87, respectively, indicating that the negative charges were distributed in a gradient along the thickness of the SA/CS hydrogel film. The increase in potential within the gradient membrane explains the enhancement of transmembrane ionic current under negative bias.91 Through numerical simulations, the ion transport characteristics of gradient membranes have also been studied (Fig. 5f). Cations (e.g. K+) are preferentially attracted by negatively charged channels and transfer in opposite directions under different biases. In the gradient polyelectrolyte hydrogel membrane, there will be an ion depletion region and ion enrichment region. In the electrochemical testing device, artificial seawater (0.5 M KCl) and river water (0.01 M KCl) were used to evaluate the permeability energy conversion performance of the gradient hydrogel membrane. When the HD side is exposed to a 0.5 M KCl solution, the short-circuit current and open circuit voltage are approximately 7.50 mA and 43.52 mV, respectively (Fig. 5g). However, by switching the solution containing 0.5 M KCl to the LD side, the short-circuit current and open circuit voltage were reduced to 4.80 mA and 35.19 mV, respectively, and the calculated internal resistance increased by approximately 26.3%. The huge cation selectivity of the gradient hydrogel membrane is mainly dominated by the HD side. Therefore, cations have a preferential direction from the HD side to the LD side.
In addition, Li et al. reported that a kind of ion selective hydrogel was covered on the porous membrane of polyethylene terephthalate (PET) cylindrical nanochannels to build a heterogeneous membrane.87 This strategy can effectively eliminate the reduction of ion selectivity caused by concentration polarization.92,93 The ionic rectification characteristics of PET/hydrogel heterogeneous films are regulated by their Janus interface wettability and charge density (Fig. 5h). The power density of commercial membranes used for salinity gradient power generation is mostly concentrated in the range of 0.3–1.1 W m−2.94–96 Even if the nanopore density of PET is as high as 5 × 107 cm2, the power density of the PET hydrogel heterogeneous film can still reach 1.92 W m−2 under 50 times salinity gradient. Numerical simulation confirmed that the PET hydrogel heterogeneous film can eliminate concentration polarization. The cation concentration on the low concentration side (orange line) of the PET porous membrane is significantly higher than the volume value (black dashed line), indicating severe concentration polarization, while the ion concentration on the low concentration side (blue line) of the non-homogeneous membrane is almost equal to the volume value (Fig. 5i). When different ion selective hydrogels are covered on both sides of the PET membrane, the movement of anions and cations is inhibited respectively, and the ionic current under positive and negative voltage will decrease (Fig. 5j). To quantify the effect of the hydrogel layer, the ionic rectification ratio (ICR) is defined as the absolute value of the current recorded at +1.5 V to the current recorded at −1.5 V (Fig. 5k). The ICR of the PET film is 1.09 ± 0.18, close to 1. When the single-sided hydrogel is covered, the asymmetry increases, resulting in an ICR of 6.83 ± 0.03 (+, 0) and 9.34 ± 0.43 (−, 0). When the same hydrogel covers both sides, the ICR is close to 1. When the two sides are covered with different hydrogels, because the hydrogels on both sides have opposite charges, the asymmetry of ion transport characteristics will be greatly improved, and the rectification effect is very obvious.
3.2 EDL within nanopores enhanced by hydrogel modification
The ion transport in a charged channel with a size comparable to the λD exhibits different behavior from the bulk phase.97 The preparation of hybrid membranes by combining polyelectrolyte hydrogels with nanopores has proved to be a strategy and method to improve the ionic rectification ratio.98–101 The polyelectrolyte hydrogel can effectively increase the charge density by modification in the nanoconfined space within the nanopore and enhance the ability of the EDL to regulate ions.102–104Chen et al. introduced hydrogels into nanofluids to prepare hydrogel hybrid membranes. The introduction of space charge hydrogel significantly improves ion selectivity (Fig. 6a).105 To construct the hybrid membrane, the precursor solution was injected into the PC membrane, and the precursor solution penetrated the pores of the PC membrane through capillary action (Fig. 6b). After removing the excess solution, the precursor only existed in the pores and fills the space due to the hydrophilicity of the precursor and finally forms a hydrogel network by free radical polymerization. This structure composed of hydrophilic-co-hydrophobic polymer chains can avoid excessive expansion of hydrogel, which will help to maintain the structure and morphology of the hybrid film.107 Theoretical simulation was carried out based on the steady-state Poisson–Nernst–Planck (PNP) equation to study the function of space charge in one-dimensional hydrogel (Fig. 6c).108 A cylindrical model (3D) with surface charge [Fig. 6c(1)] or space charge [Fig. 6c(2)] is designed. The longitudinal section of the surface charge channel shows a higher anion concentration, indicating that its ion selectivity is poor. However, the anion concentration in the space charge channel is low, indicating that the ion selectivity is improved. The conductance of the membrane changes with the decrease of the electrolyte concentration (red curve) (Fig. 6d). The ion conductance of the hydrogel hybrid membrane deviates from the bulk behavior (black curve) obviously when the concentration is below 1 M, showing a charge-governed ion transport. In relatively low concentration, the curve is gentler for the increase of λD.63 The ion selective property of the membrane was assessed with the help of the permeation of fluorescent dye (Fig. 6e). The inset shows the molecular formulas of fluorescent dyes (sulforhodamine and rhodamine 6G), which were selected for their similar structure but opposite charges. As a result, the transmembrane transport speed of Rh(+) is much larger than that of Rh(−), which indicates the excellent cation selectivity of the membrane.
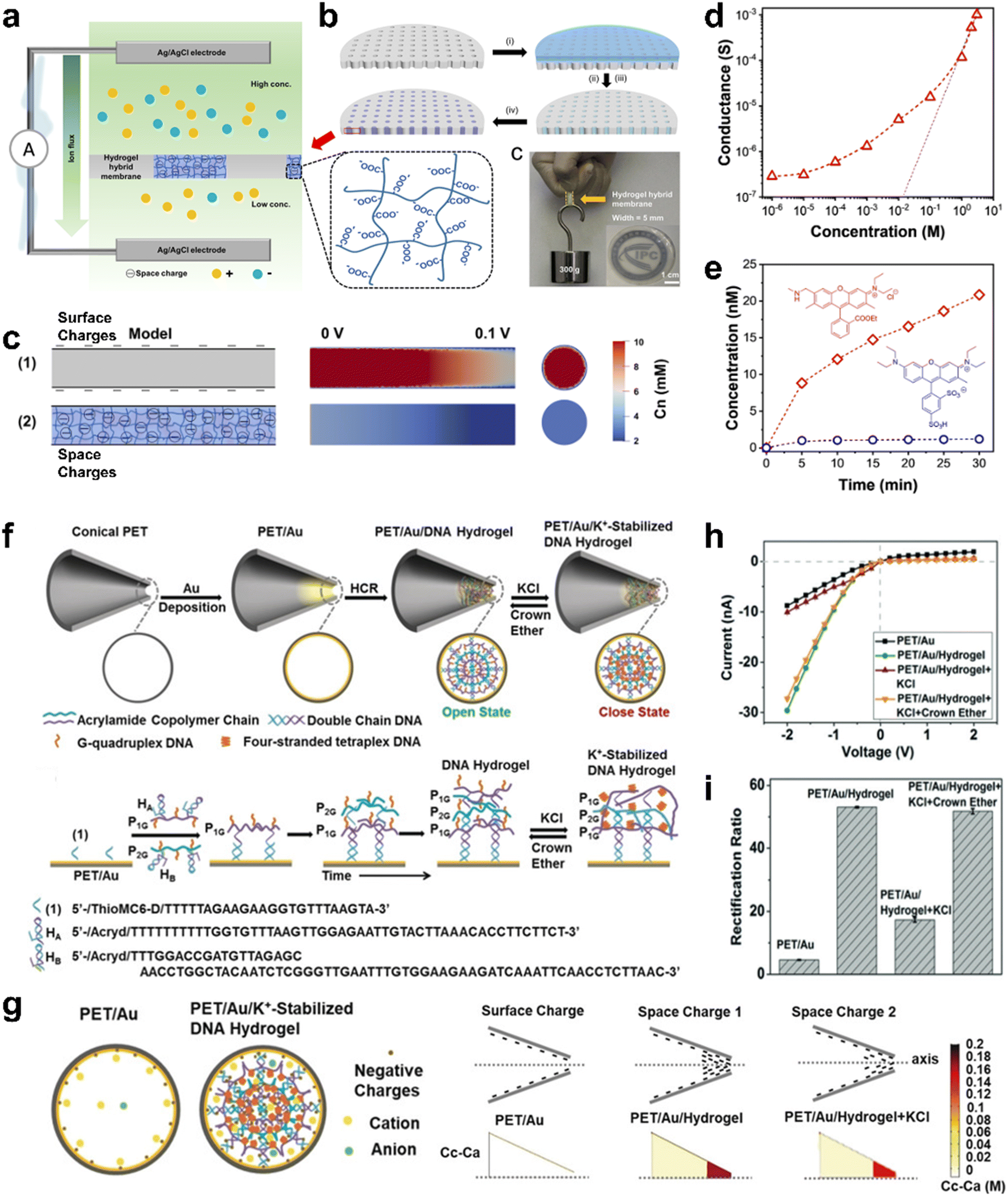 | ||
| Fig. 6 Nanopores modified by polyelectrolyte hydrogel for higher ionic rectification ratios. (a) Schematic depiction of the cation selectivity of the hydrogel hybrid membrane benefiting from space charge. (b) Fabrication processes of the hydrogel hybrid membrane. (c) Simulation of anion concentration profiles of two models reveals the improved ion selectivity of the channel with space charge. (d) Ion conductance of the membrane versus concentration, showing a charge-governed ion transport. (e) Permeability rate curves of rhodamine 6G (Rh(+)) and sulforhodamine ((Rh(−)), which indicate the cation selectivity of the membrane. (a)–(e) Reproduced from ref. 105, Copyright © 2020 American Chemical Society. (f) Illustration of the preparation process of the ion channel. (g) Representation of the cross-sectional charge distributions for the tip side. (h) I–V curves of the conical nanochannel under different conditions. (i) Rectification ratios of the conical nanochannel under different conditions. (f)–(i) Reproduced from ref. 106, © 2018 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
Nanochannels based on smart DNA hydrogels have been demonstrated as stimuli-responsive structures. Unlike other responsive molecules in the nanochannels with monolayer structure, DNA hydrogel is a three-dimensional network with space negative charge, and the ion flux and rectification rate are significantly improved.109,110 The state of the DNA hydrogel can be reversibly switched between non-rigid and rigid networks, providing a nanochannel gating mechanism. Symmetric nanochannels were prepared by surfactant-assisted ion-track etching (Fig. 6f).106 To assemble the DNA hydrogel network onto the gold-plated diopside, acrylamide copolymers P1, P2, P1G and P2G were prepared. The Au-coated diopside is modified by thiolated nucleic acid, forming the composite shown as (1) in Fig. 6f. This composite acts as a promoting chain to initiate the hybridization chain reaction (HCR). Its hybridization with hairpin HA of P1G uncaged HA, leading to the formation of a toehold sequence that is complementary to hairpin HB, which is related to P2G. The subsequent uncaging of HB by the open HA results in a toehold domain that re-opens HA. Thus, DNA hydrogel networks are formed by the promoter-induced cross-opening of hairpins HA and HB onto the tip side. The simulation model and results of ion concentration difference (Cc–Ca) in two-dimensional axisymmetric conical nanochannels were studied (Fig. 6g); Cc and Ca mean concentration of cations and anions respectively. Compared with PET/Au nanochannels, the Cc–Ca and Cc/Ca at the tip of PET/Au/DNA hydrogel nanochannels increased by 54 times and 4.4 times, respectively. For PET/Au/DNA hydrogel + KCl nanochannels, both Cc–Ca and Cc/Ca decreased. The results show that under the action of an external electric field, the ionic current is mainly carried by cations, and the higher the ion concentration, the greater the ionic current. The Au-coated nanochannel reveals a diode like ion transport through the nanochannel (PET/Au), which is ascribed to surface negative chloride anions associated with the channel that assist the transport of Li+. After modification with DNA hydrogels (PET/Au/hydrogel), the ionic current and rectification ratio (±2 V) are greatly enhanced by approximately 3-fold and11-fold over that of the Au-coated nanochannel (Fig. 6h and i).
3.3 EDL at hydrogel p–n junctions
Similar to the p–n junction in silicon-based semiconductors, two polyelectrolyte hydrogels with opposite charges form an ionic EDL (commonly referred to as an ionic double layer, IDL) with fixed ionic charges at the interface. The built-in electric field created at the interface exerts a rectifying effect on the ions. Cayre et al. were among the first to successfully verify this concept by doping agarose gel with sodium poly(styrene sulfonic acid sodium) and poly(diallyldimethylammonium chloride), respectively.111 The mechanism was attributed to the unidirectional current response at the interface between cationic and anionic gels, driven by the anisotropy of movable ionic charges within the gel. As research advanced, this theory has been further refined and developed.A unique hydrogel heterojunction with opposite charges, designed for collecting electrical energy and harvesting fresh water from atmospheric air, has been reported by Lu et al.112 When the polycationic (PC) hydrogel comes into contact with the polyanionic (PA) hydrogel, two hydrogels with opposite charges assemble to form a PA/PC heterojunction (Fig. 7a). The long-range motion of the charged polymer chains is hindered, leading to the diffusion of free counter-ions into the adjacent hydrogel. This results in a depletion region of the flowing counter-ions, thereby forming an IDL at the PA/PC interface (Fig. 7b). Driven by the interfacial electric field, the counter-ions drift in the opposite direction to the ion diffusion, with the electric field at the interface facilitating the separation of counter-ions with opposite charges in each hydrogel. This process ensures the maintenance of a stable ionic gradient and the generation of potential.113,114 Humidity fluctuations promote the hydration and dehydration processes of the hydrogel, leading to changes in the ionic gradient, which are then converted into electrical output. The established interfacial electric field endows the PA/PC heterojunction with ionic current rectification behavior.115 The forward voltage attracts counter-ions to the PA/PC interface, from PA to PC, shrinking the IDL and allowing the current to pass through the junction. When the reverse voltage is applied from PC to PA, the counter-ions are pulled away from the interface, expanding the IDL and hindering the current flow.
 | ||
| Fig. 7 The ionic rectification mechanism of the ionic EDL at the hydrogel p–n junction interface. (a) Schematic representation of the moisture-enabled electricity and water generator made up of hydrogel heterojunctions consisting of two oppositely charged hydrogels: positively charged (PC) and negatively charged (PA) hydrogels. (b) Schematic of the formation of the ionic EDL (also called IDL) and interfacial electric field due to the entropic depletion of mobile ions at the PA/PC junction. (a) and (b) Reproduced from ref. 112, © 2022 Elsevier Ltd. All rights reserved. (c) Schematic illustration of the hydrogel ionic diode. (d) Under the initial state or the diminishing stress and the mechanically released state, illustration showing the electrical behavior and response of the ionic diode including the potential diagram. Quantitative concentration profiles of Na+ and Cl− in the ionic diode, space charge density and potential gradient within the ionic diode simulated in the initial and released state. (c) and (d) Reproduced from ref. 42, © 2021 Wiley-VCH GmbH. | ||
Zhang et al. developed a hydrogel ionic diode to harvest mechanical energy in human motion.42 Two-layer hydrogels were prepared by blending agarose with ionomer sodium polystyrene sulfonate (PSSNa) and polydiallyldimethylammonium chloride (PDACl), respectively. When PSSNa is dissolved in the hydrogel, it exhibits an anionic polystyrene sulfonate skeleton (PSS−) and a movable Na+ cation, which are similar to the receptors and holes in p-type semiconductors, respectively. The main chain of cationic poly (diallyldimethylammonium) (PDA+) and the mobile Cl− anion produced by the dissociation of PDACl are similar to the donor and electron in n-type semiconductors, respectively (Fig. 7c). The working mechanism of the hydrogel ionic diode was explained by the finite element method (FEM) (Fig. 7d). When the hydrogel composites are placed together to form a connection, the mobile Na+ and Cl− ions from the ionomers of PSSNa and PDACl, respectively, diffuse to their corresponding regions through the PTFE membrane driven by entropy. The quantitative concentration distribution calculated by FEM confirms the depletion of Na+ and Cl− ions moving around the junction interface during electrostatic equilibrium. Therefore, the uncompensated charges of PSS− and PDA+ form a space charge region and generate a built-in potential (φbi) on the junction to prevent further diffusion and complete the equilibrium state. The establishment of the barrier was also confirmed by FEM revealing that the contact behavior between the two hydrogel layers was similar to a p–n semiconductor junction.116
3.4 Asymmetric EDL at hydrogel/electrode interfaces
The EDL plays a crucial role in bridging ionic signals and electronic signals. Ionic rectification can be effectively achieved by modulating the differences in the formation of the EDL between hydrogels and various electrode interfaces. Disrupting the formation of the EDL on one side of the electrode surface has been demonstrated as an effective ion rectification strategy. For example, Guo et al. designed a flexible diode composed of hydrogel, Zn, and Ti electrodes, with its working principle attributed to the asymmetric reduction potential of H+ on different electrode surfaces in aqueous solution.117 When a forward bias is applied to the diode, the anions and cations respectively migrate to the surface of the Zn and Ti electrodes and the diode is in an ‘on’ state. Once the reverse bias is applied, the anions and cations respectively migrate to the surface of the Ti and Zn electrodes. However, due to the large overpotential, the reduction reaction of H+ on the surface of the Zn electrode is hindered, and the migration of anions and cations is inhibited, resulting in the failure of the circuit formation, so that the diode is in a ‘off’ state.Additionally, an ionic diode exhibiting light-mediated ionic rectification performance based on light-responsive hydrogels has been demonstrated. The ionic rectification performance can be tuned through ultraviolet (UV) irradiation. By incorporating a photoacid generator (PAG) into polyacrylamide (PAAm) hydrogel, UV irradiation significantly enhances ionic current rectification, with the effect depending on the photoresponse concentration of protons in the hydrogel (Fig. 8a).119 By forming an asymmetric SiO2 nanofilm/PAGs-PAAm hydrogel interface (SNPGI), this ionic diode has a typical rectification ratio (4 × 105). After irradiation, the PAGs-PAAm hydrogel showed obvious color changes, yellow in the merocyanine (MEH) form and colorless in the sulfonato-spiropyran (SP) form (Fig. 8b). The light-mediated H+ release in PAGs-PAAm hydrogel offers an effective approach to control ionic conductivity through reversible trans–cis isomerization. This process enables precise modulation of the hydrogel's ionic properties in response to light, providing a versatile mechanism for tuning ionic conductivity.120 Another ionic diode was developed, consisting of a conductive n-doped Si substrate with a 100 nm SiO2 layer at the top, a photoresponsive PAGs-PAAm hydrogel and a PET-ITO electrode as a counter electrode. The SNPGI was formed by the photo-responsive PAGs-PAAm hydrogel plate encapsulated in silicone rubber between the SiO2 nanofilm and the PET-ITO electrode, allowing selective ion transport [Fig. 8c(i)]. The working principle of the SNPGI diode can be fundamentally attributed to the asymmetric distribution of ions at the nanofilm/hydrogel interface [Fig. 8c(ii)]. When a forward bias is applied to the SNPGI diode, the aqueous cation (H+) in the hydrogel matrix diffuses through the SiO2 layer and is reduced on the Si electrode, resulting in a significant increase in the forward current. At the same time, the aqueous anion (OH−) in the hydrogel matrix migrates to the PET-ITO electrode and completes the circuit under the built-in electric field. As a result, the ionic diode is switched to the ‘on’ state, performing high current under forward bias. On the contrary, when the reverse bias is applied on the SNPGI diode, the water-based cations move to the PET-ITO electrode, but the negatively charged SiO2 layer prevents the migration of the water-based anions. In this case, circuit formation fails, causing the diode to be in a ‘off’ state. Therefore, the selective ion migration of the SNPGI diode at the asymmetric EDL makes it have obvious rectification characteristics.
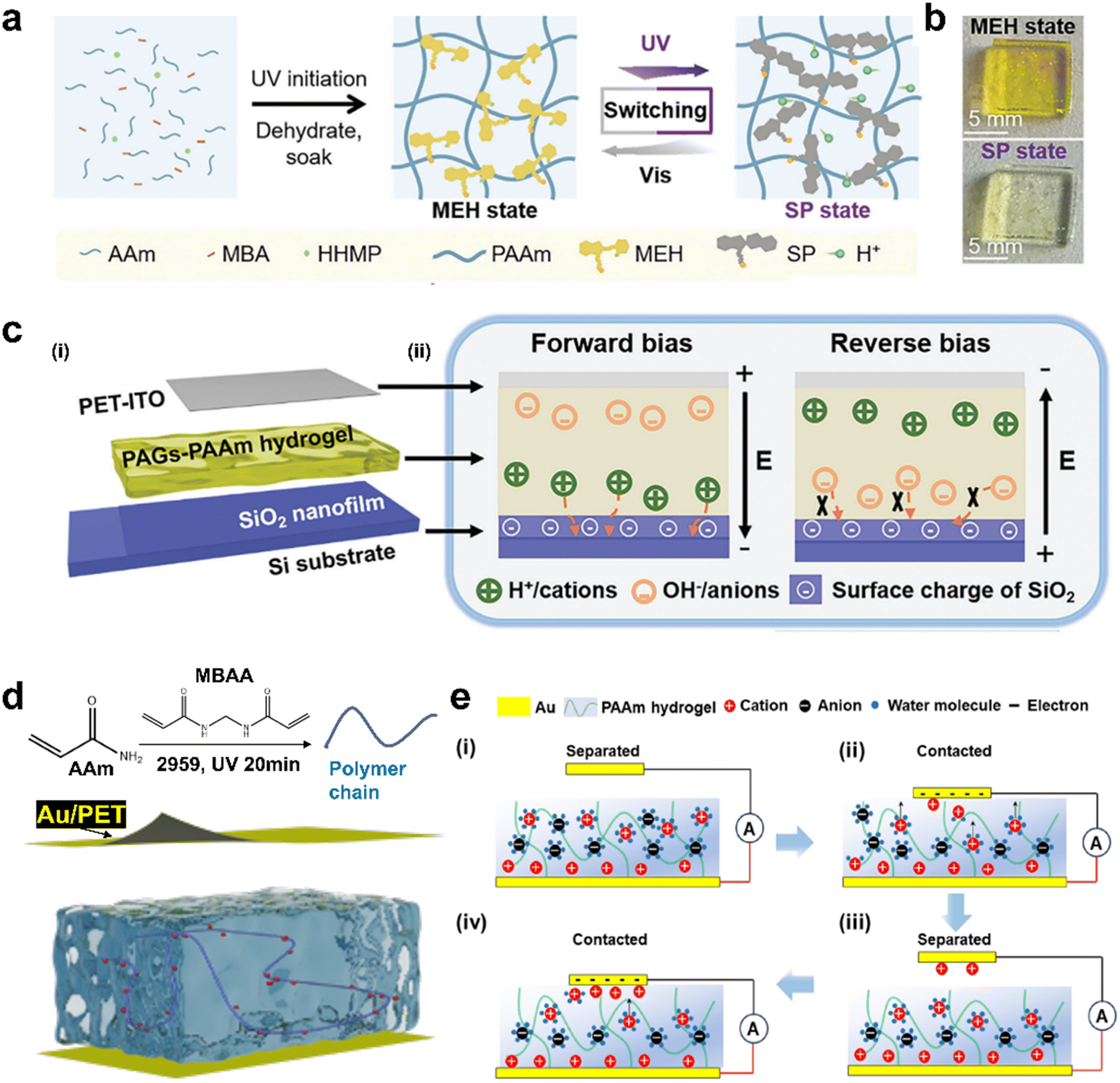 | ||
| Fig. 8 Ionic rectification by modulating asymmetric EDL at the hydrogel/electrode interface. (a) Schematic diagram of the preparation of the photoacid generator-polyacrylamide (PAGs-PAAm) hydrogel, which can be converted between the protonated merocyanine (MEH) form and sulfonato-spiropyran (SP) form. (b) Photographs of the PAGs-PAAm hydrogels in MEH and SP forms. (c) Schematic illustration of the mechanism in this SiO2 nanofilm/PAGs-PAAm gel interface (SNPGI) diode under forward and reverse biases, in which the transport of mobile ions is rectified by the SNPGI asymmetric EDL. (a)–(c) Reproduced from ref. 118, © 2024 Wiley-VCH GmbH. (d) The structure of hydrogel-based iontronics. (e) Schematic diagram of the EDL and ionic current formation mechanism. (d) and (e) Reproduced from ref. 43, Copyright © 2024, American Chemical Society. | ||
Wei's group recently reported a paradigm to achieve ionic rectification based on dynamically modulating the asymmetric EDL between the hydrogel and composite electrode (Au/PET) interface induced by triboelectrification.43 The Au electrode was prepared on a polyethylene terephthalate (PET) substrate by magnetron sputtering. The device adopts a sandwich structure, consisting of two Au/PET electrodes and a piece of polyacrylamide (PAAm) hydrogel (Fig. 8d). The hydrogel is a solidified substance formed by the increase of the volume of a large amount of water in a hydrophilic three-dimensional polymer matrix. Obviously, its surface contains a lot of water molecules. Therefore, the EDL at the interface between the hydrogel and composite electrode can be reasonably approximated as the interaction between water and the Au/PET electrode. The inert metal Au was selected to avoid the redox reaction during the interaction with water. The mechanism of the formation of EDL and the generation of ionic current is shown in Fig. 8e. The electrode fixed on one side of the hydrogel is called the back electrode, and the electrode separated from the surface of the hydrogel by dynamic contact is called the counter electrode. The hydrogel was placed on the back electrode to promote the establishment of a relatively stable EDL. At this time, the counter electrode is not in contact with the hydrogel, preventing the formation of the EDL at the interface [Fig. 8e(i)]. When the counter electrode and the hydrogel come into contact, water molecules promote the transfer of electrons to the counter electrode, and then attract hydrated hydrogen cations, resulting in the EDL [Fig. 8e(ii)]. Therefore, due to the asymmetric EDL between the two electrodes, a unidirectional current appears in the external circuit. Due to the robust stability of EDL adsorption, certain cations persist on the counter electrode surface after separation from the hydrogel [Fig. 8e(iii)], and this retention is accumulated through continuous contact–separation cycles. Considering the limited capacitance of the counter electrode, the establishment of a stable Stern layer means that the contact separation process only affects the ion migration within the diffuse layer, potentially triggering an alternating current signal [Fig. 8e(iv)]. The construction of this asymmetric EDL can continuously realize the unidirectional migration of ions, so as to output ionic direct current in a certain period of time.66
4. Applications of ionic rectification via EDL regulation at hydrogel interfaces
The study of ionic rectification through EDL regulation at the hydrogel interface has a broad range of applications in fields such as energy harvesting,121–123 sensors,124–128 logic circuits,129–131 and neuromorphic devices,132–134 among others. These applications are driving significant advancements in related technologies. In the following sections, we will review some recent representative applications based on ionic rectification.4.1 Energy harvesting
With the rapid development of the Internet of Things, flexible wearable devices, and related fields, the demand for distributed energy in various scenarios has reached new heights. Since most electrical devices require direct current power supply, ionic rectification based on hydrogels has the potential for widespread application across numerous scenarios.Schroeder et al. developed an artificial electrophorus electricus using ion-selective bionic polyelectrolyte hydrogels.135 Four types of hydrogel components were employed as analogues for the main components of electric cells to replicate the anatomical structure of eel. These included membranes with different ion selectivities on the front and rear sides, along with intracellular and extracellular salt chambers. Upon initiating contact, tetrameric repeating units of high-salinity hydrogel, cation-selective gel, low-salinity gel, and anion-selective gel successively formed an ion conduction pathway, establishing an electrolyte ladder with tens to thousands of selective permeation compartments (Fig. 9a). Using the principle of reverse electrodialysis, each of these ‘tetramer gel cells’ generates a voltage of 130–185 mV upon activation, which is equivalent to the potential produced by a single electrolytic cell. As an alternative to the fluid assembly strategy, the artificial device is activated synchronously by initiating mechanical contact between two complementary gel patterns within a large array in a registered and parallel fashion. This design involves printing the precursor solution of the ion-selective membrane onto a lens array on a polyester substrate and similarly printing the precursor solution for the salt conduction pathway (Fig. 9b). The resulting structure generates repeated potential patterns, accumulating to a total voltage of 110 V (Fig. 9c and d).
 | ||
| Fig. 9 The application of ionic rectification in energy harvesting. (a) Electrophorus electricus. (b) Open-circuit voltage and short-circuit current characteristics of printed artificial electric organs as a function of the number of gel cells in a series. (c) Normalized current–voltage relations of various numbers of gel cells added in series or in parallel. (d) Photographs of large complementary arrays of printed hydrogel lenses. Above reproduced from ref. 135, Copyright © 2017, Macmillan Publishers Limited, part of Springer Nature. All rights reserved. (e) Schematic of a hook & loop-like Cu–Zn flexible battery based on hydrogel diodes. (f) The peeling force curves of two as-contacted hydrogel sheets. (g) Power output as a function of time for batteries in different storage forms. (h) The twisted battery composed of six units in series with a voltage of 5.3 V. (e)–(h) Reproduced from ref. 136, © 2018 Elsevier Ltd. All rights reserved. (i) Schematic illustration of the preparation process of the organohydrogel ionic diode. (j) and (k) referred to mechanical energy harvesting measurement with a forward connection for voltage signals and current density signals respectively. (l) Output voltage versus time of the organohydrogel ionic diode under various relative humidity (RH) values. (m) The humidity sensitivity of the self-powered organohydrogel ionic diode. (i)–(m) Reproduced from ref. 137, © 2023 The Authors. SusMat published by Sichuan University and John Wiley & Sons Australia, Ltd.113,114 | ||
Duan et al. reported a hydrogel diode with strong and tunable interfacial adhesion by linking oppositely charged hydrogels together (Fig. 9e).136 The high adhesion energy comes from the strong Coulomb interaction and the effective energy dissipation of the hydrogel matrix.138 By reducing the depletion region in the hydrogel diode, the adhesion force can be easily adjusted. A hook & loop-like Cu–Zn battery was designed using a hydrogel diode as the electrolyte. The battery is rugged, stable and durable, and can power wearable electronics. The battery is split into two half-cells during long-term storage and is easily reconnected together during the working phase, thereby eliminating self-discharge and maintaining capacity. Similar to a semiconductor diode, the hydrogel diode interface features a depletion region due to the spontaneous diffusion of counter-ions driven by the concentration gradient. This depletion region generates an electric field, leading to a strong Coulomb interaction between the opposite net charges within the network. Consequently, the peeling force of the hydrogel diode (∼63 N m−1) is more than an order of magnitude higher than that of two isocharged hydrogels (∼5 N m−1) and is comparable to the tearing force (∼65 N m−1) required to break covalent bonds in a single neutral hydrogel (Fig. 9f). When the battery is stored in an integrated form, due to the spontaneous reaction between the Zn electrode and Cu2+ in the electrolyte,139 the power output decreases rapidly and approaches zero after 8 days. On the contrary, the power output remains constant (0.75 mW) when stored in a separate form. Therefore, the hook & loop-like Cu–Zn battery can eliminate self-discharge by separating into two half-cells during the storage phase and can be easily recombined together during the working phase (Fig. 9g). To demonstrate the wearable application of flexible Cu–Zn batteries, a strip battery with six units in series can still produce a high voltage of 5.3 V under severe distortion (Fig. 9h).
To solve the environmental limitations of single stimulus responsiveness on distributed energy devices, Yin et al. proposed a pressure and humidity dual-responsive ionic diode composed of two oppositely charged organic hydrogels as an energy collector.137 The organic hydrogel ionic diode utilizes the migration of cations and anions to form a depletion region and then enhances the built-in potential along the depletion region under the action of mechanical stress or humidity, converting ultra-low frequency mechanical energy or humidity energy into electrical energy.140 By adding a separator between two composite organic hydrogels containing oppositely charged polymer polyelectrolytes, an organic hydrogel ionic diode with an asymmetric structure was constructed (Fig. 9i). The ionic groups of the two ionic polymers are fixed on the polymer chain, and the counter-ions can be non-localized. MXene nanosheets were introduced into two organic hydrogels to increase conductivity. The hydrophilic polytetrafluoroethylene (PTFE) permeable membrane separator is sandwiched between two organic hydrogel nanocomposites, allowing water and ions to pass through, and also avoiding short circuit due to the merging of organic hydrogel layers containing MXene nanosheets. The electrical output performance is measured by connecting the carbon cloth to both ends of the sandwich structure. An initial potential of about 45.00 mV was observed in the electrical characteristic test, which was attributed to the built-in potential of the junction. When the ionic diode is forwardly connected to the digital multimeter, a voltage of about 26.50 mV is generated under an ultra-low frequency mechanical pressure of about 0.01 Hz (Fig. 9j). In addition, the current output is about 10.10 μA cm−2 (Fig. 9k). The mechanism of water–gas power generation parallels that of pressure-stimulated power generation. Variations in humidity induce the hydration/dehydration process within the organic hydrogel ionic diode, leading to ion gradient fluctuations that are ultimately converted into electrical energy output.141 When exposed to humidity, the voltage of the organic hydrogel ionic diode gradually increased and was stable within about 15 min. In addition, the maximum output voltage of the ionic diode increases with increasing humidity (Fig. 9l), which is attributed to the enhanced dissociation and diffusion of counter-ions at high humidity levels.142 The humidity sensitivity of the self-powered organic hydrogel ion diode is ∼0.93 mV/RH% at low humidity (<90%). When the humidity exceeds 90%, the humidity sensitivity increases to ∼1.35 mV/RH% (Fig. 9m).
4.2 Sensors and flexible circuits
In addition, the ionic rectification at the hydrogel EDL interface has important application scenarios in detection of various substances, physiological signal detection, bridge rectifiers, logic circuits, etc. Some recent representative studies are shown as follows.A monolithic integrated hydrogel ionic diode has been demonstrated for label-free quantitative DNA detection and real-time monitoring of nucleic acid amplification. This is achieved by forming a micron-scale heterojunction of adjacent polycation and polyanion hydrogel segments on a microfluidic chip via a simple photocuring step (Fig. 10a).143 The integrated diode rectified the DNA charge-sensitive ionic current adsorbed on the polycation chain by electrostatic association. This ionic biosensor can perform electrokinetic quantification of DNA in a dynamic range related to typical nucleic acid amplification detection. The polymerase chain reaction (PCR) detection method for a 500 bp DNA fragment of Escherichia coli was evaluated, and the real-time in situ monitoring of an isothermal detection method for amplifying the whole genome of Escherichia coli was performed. The asymmetric ion distribution in the hydrogel junction leads to a certain level of ionic current rectification. Once negatively charged DNA molecules are introduced into the hydrogel, they are adsorbed onto the polycationic chain due to electrostatic interactions. Electrostatic adsorption reduces the net charge of the hydrogel skeleton and affects the ion distribution, resulting in a decrease in ionic current rectification. As a result, the ionic diode instantaneously displays the modulated current–voltage (I–V) characteristics (Fig. 10b). Based on the recorded I–V curves, the intact ionic diode owns an rectification ratio of 4.22 ± 0.53. By continuously monitoring the overall rectification level, the ionic diode can monitor the presence and concentration of nucleic acids in real time.
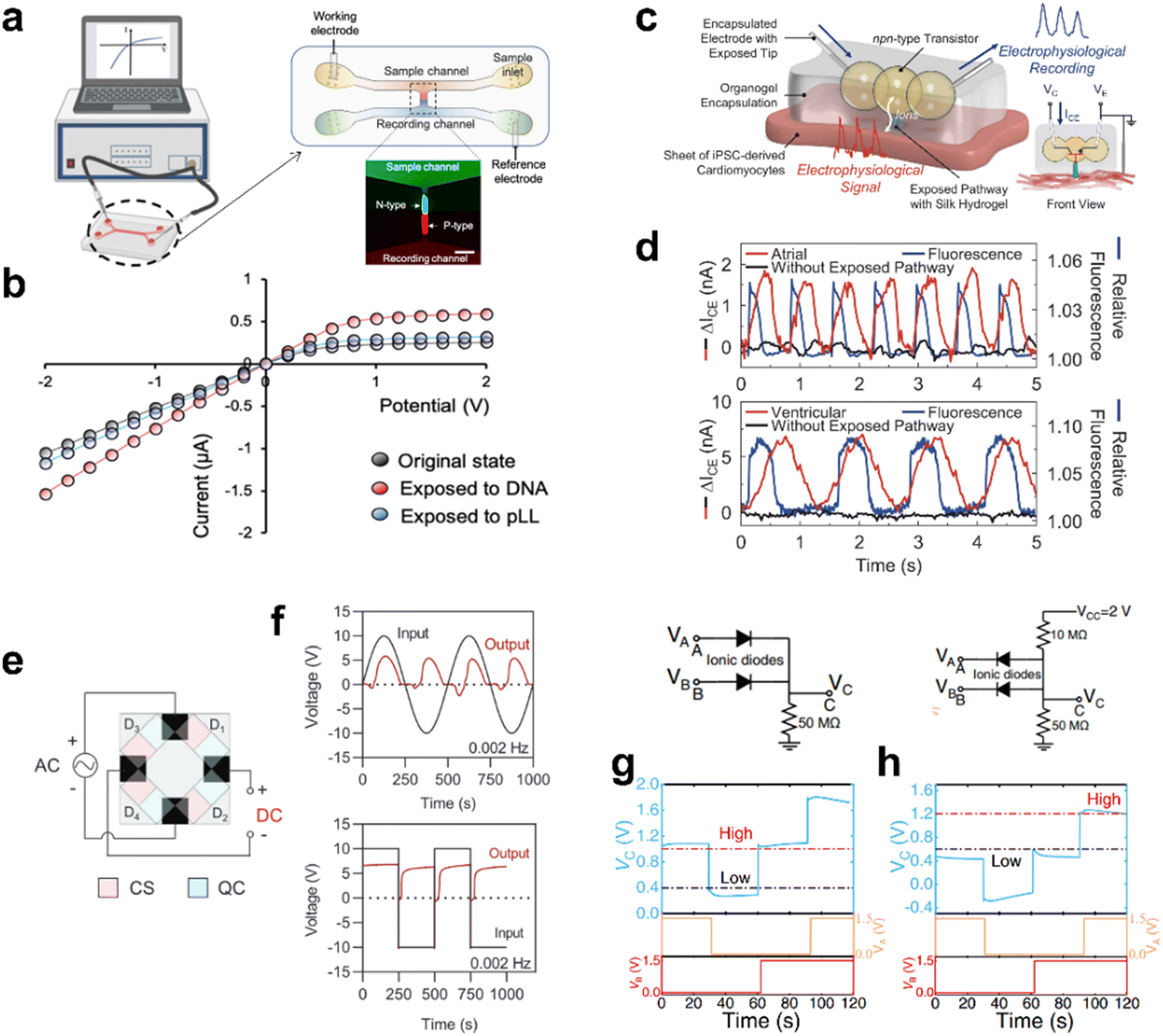 | ||
| Fig. 10 The application of ionic rectification in sensors and flexible circuits. (a) The structure of the monolithic integrated ion diode microchip. (b) The I–V curve when exposed to negative charge. (a) and (b) Reproduced from ref. 143, © 2022 The Authors. published by Elsevier Ltd. (c) Dropletronic transistor for electrophysiological recording. (d) The dropletronic device output and fluorescence imaging of cell membrane potential. (c) and (d) Reproduced from ref. 144, Copyright © 2024 The Authors, some rights reserved; exclusive licensee American Association for the Advancement of Science. (e) Schematic of the 3D-printed ionic full-wave rectifier bridge. (f) Output voltages as response to sinusoidal and square wave input voltages. (e) and (f) Reproduced from ref. 145, © 2023 The Authors. Advanced Functional Materials published by Wiley-VCH GmbH. (g) The circuit diagram and performance of an ionic OR gate. (h) The circuit diagram and performance of an ionic AND gate. (g) and (h) Reproduced from ref. 146, Copyright © 2022, The Author(s). | ||
Zhang et al. reported the use of surfactant-supported self-supporting microscale hydrogel droplet assembly to construct various iontronic modules, circuits, and biological interface.144 The chemical modification of silk fibroin produced a pair of hydrogels with opposite charges. This micro-scale hydrogel droplet ionic transistor can be used as a biocompatible sensor to record electrophysiological signals from human cardiomyocyte sheets. A bipolar junction droplet transistor is encapsulated in a thermoreversible organic gel made of a poly(styrene-b-ethylene-co-butene-b-styrene) triblock copolymer to produce a portable dropletronic (Fig. 10c). The dropletronic was placed on the human induced pluripotent stem cell-derived atrial or ventricular myocardial cell sheet to record the electrophysiological signals of cardiac spontaneous activity. The signal-to-noise ratios of atrial myocytes and ventricular myocytes were 1.3 ± 0.3 and 1.6 ± 0.2 dB, respectively, and remained stable for tens of minutes (Fig. 10d).
Huo et al. developed robust transient ionic junctions using 3D ionic microgel printing.145 This method exhibits excellent printability, enabling the fabrication of diverse ionic junction configurations with high fidelity. By integrating ionic microgels, degradable networks, and highly charged biopolymers, the resulting ionic junctions achieve outstanding electrical performance (current rectification ratio >100). A full-wave rectifier composed of four interconnected ionic junctions has also been demonstrated (Fig. 10e). The printed ionic rectifier successfully proves that this rectification effect gives sine wave and square wave signals (Fig. 10f). The time delay between output and input is attributed to the time required for the input voltage to pass through the threshold voltage. In contrast, the square wave input leads to a shorter time delay due to the instantaneous movement of the junction state, where the input voltage immediately exceeds the threshold during switching.
Jiang et al. assembled polymethyl methacrylate (PMMA) and polyvinylidene fluoride-hexafluoropropylene (PVDF-HFP) gel polymer electrolyte (GPE) into a new type of ionic diode, which realized ionic rectification by simulating the ion diffusion/migration of biological systems.146 This ionic rectification is caused by the different diffusion/migration behaviors of mobile ions in the GPE heterojunction and the rectification ratio is around 23. Basic ‘AND’ and ‘OR’ logic gates were established by this GPE ionic diode. When the applied voltage bias exceeds the 0.7 V threshold, the ionic diode becomes conductive, otherwise. The OR gate circuit based on the GPE ionic diode is shown in Fig. 10g. When the voltage input A and B is higher than the turn-on voltage of the ionic diode, the input signal can be regarded as ‘1’; when there is no voltage input, the input signal will be ‘0’. Similarly, the high voltage measured at the output C is regarded as the output signal ‘1’, while the low output voltage is regarded as the output signal ‘0’. To evaluate the designed ionic diode, the voltage signals with four logical states (0, 0), (0, 1), (1, 0) and (1, 1) are generated by the square waves of A and B. When the input voltage of A or B is 1.5 V (logical ‘1’), the output voltage VC is high or in the logical state ‘1’ (the red dotted line is about 0.9 V). When both VA and VB are 0 V (logic ‘0’), the output VC is low level or logic ‘0’. Similarly, as shown in Fig. 10f, an AND ionic gate based on two GPE ionic diodes is also designed. When the input signal VA or VB is 0 V (logic ‘0’), the two ionic diodes will be connected by VCC (2 V) and conduct the current, resulting in a low output VC of 0.6 V or logic ‘0’ (under the black dotted line). However, when both VA and VB are high voltage 1.5 V, the output VC is high voltage or logic‘1’ (about 1.2 V above the red dotted line).
4.3 Hydrogel-based neuromorphic device
Brain-inspired algorithms, such as artificial neural networks (ANNs), have achieved remarkable success in machine learning.147 However, unlike the brain, conventional computers physically separate memory and processing units, leading to increased latency and high energy consumption. Neuromorphic computing addresses these challenges by developing energy-efficient hardware for information processing, enabling complex task execution with greater efficiency.148 Hydrogel-based artificial synapses serve as critical hardware components in neuromorphic devices.133 This section highlights representative studies on simulating synaptic plasticity through the modulation of the EDL at hydrogel interfaces.Transport behaviors such as selective permeation and hysteretic diffusion of ions resulting from the anion selectivity of hydrogels were mainly responsible for potential relaxation. This relaxation behavior is very similar to the memory behavior of postsynaptic potential (PSPs) in synapses, that is, neural impulses from presynaptic can change the level of PSPs in a short or long time, where the level of PSPs reflects the size of synaptic weight (SWs). Wang et al. described an ionic potential relaxation effect in a hydrogel device prepared by sandwiching a piece of polycationic hydrogel (CH) with anion selectivity between two pieces of neutral hydrogel (NH) without ion selectivity and covering the top and bottom sides with indium tin oxide/poly(ethylene terephthalate) (ITO/PET) flat electrodes to measure ionic signals (Fig. 11a).149 The CH layer was copolymerized from acrylamide (AAm) and methacrylamido propyl trimethylammonium chloride (MAPTAC) by photopolymerization and its anion-selectivity strength adjusted by varying their ratios (Fig. 11b). The classification of handwritten numbers has been successfully realized. First, the features of a 28 × 28 pixel handwritten digital image were extracted through convolution processing, and the extracted features were further linearly transformed into an input pulse current sequence. Then, the encoded pulse sequence was sent to the hydrogel to obtain responsive PSPs. Finally, the obtained PSPs were classified using a fully connected neural network, and the weight matrix in the neural network was trained through linear regression. Fig. 11c shows the coded pulse sequence of the randomly selected number ‘0’ and the corresponding response PSP. The overall classification accuracy reached 95.0% (Fig. 11d and e).
 | ||
| Fig. 11 Neuromorphic devices modulated by the EDL at hydrogel interfaces. (a) Schematic of mimicking PSPs using a trilayer structured hydrogel named P-AxMy. (b) Chemical structure of the polymer skeleton of NH and CH layers, the latter of which possesses anion selectivity due to the positive charge of its skeleton. (c) Encoded pulsed current series and corresponding responsive PSPs for a randomly selected digit “0”. (d) A confusion matrix comparing the counts of predicted outputs and correct outputs. (e) Relationship between classification accuracy and training epochs. (a)–(e) Reproduced from ref. 149, Copyright © 2024, American Chemical Society. (f) Schematic illustration of the artificial synaptic device. (g) Schematic diagram of the optoelectronic neuromorphic circuit and a photovoltaic circuit. (h) Dynamic images encoded in the circuit after various runs of light irradiation, light intensity, illumination time, and illumination frequency. (f)–(h) Reproduced from ref. 150, © The Royal Society of Chemistry 2021. | ||
Han et al. proposed a hydrogel-based electrochemical transistor (HECT). Different synaptic functions can be successfully simulated by coupling the designed transistor with the optical sensor.150 This neuromorphic circuit can output adjustable images according to different trigger lights, achieved by ion rectification through the gate electrode and dynamically regulating the EDL at the interface between hydrogel and PEDOT:PSS. When a positive voltage VG (presynaptic signal) is applied on the gate electrode (presynaptic element), positively charged sodium ions (Na+) in the hydrogel electrolyte (neurotransmitters) are drifted and injected into the active layer (postsynaptic) to compensate for PSS− (acceptor ions). As a result, the conductive state PEDOT+ is reduced to PEDOT0, which is an insulating neutral state. This reaction process can be described by the equation PEDOT+:PSS− + Na+ = PEDOT0 + Na+:PSS− + h+, resulting in a decrease in the IDS of the transistor (inhibitory postsynaptic current). An optoelectronic neuromorphic circuit with similar capability is implemented by coupling a transistor and a photovoltaic divider (Fig. 11g). In this circuit, the photovoltaic divider can convert the light signal into an adjustable voltage and act as the gate voltage to tune the drain current (IDS) of the hydrogel-based transistor. Fig. 11h shows the images encoded based on the IDS of the light-adjustable transistor array, where the optical image input was an ‘L’ pattern. The output images can be gradually enhanced when increasing the light intensity from 1 to 3 mW. Prolonging illumination time was similar to learning behavior, and the output images under different illumination times had been presented (10 s, 20 s, and 30 s). Unlike conventional optical sensors, an intriguing phenomenon was observed wherein the output image of ‘L’ became clearer with increasing illumination time, suggesting that the event duration has an effect equivalent to that of stimulation intensity. Furthermore, a similar trend was noted when the frequency of the input signal increases, indicating that detection accuracy can be enhanced by reducing the pulse interval.
4.4 EDL modulation of cellular physiological activity
Recent studies have shown that polypeptide-based biopolymer hydrogels with critical phase transition temperature also have important applications in the regulation of physiological activities of cells.39 The EDL electric field formed between the dense phase (marked as condensates, formed by polypeptide-based hydrogels) and the dilute phase (bulk solution) can modulate the balance of the H+ concentration gradient between the two phases. This process can generate hydrogen peroxide (H2O2) and affect the process of life activities in the cell, providing a new paradigm for explaining the activity mechanism at the cell level and the application of bioscience.Dai et al. reported that the EDL electric field can be established at the liquid–liquid interface between the condensed state and the bulk solution environment due to the density changes of ions and molecules caused by phase separation. The condensed interface can drive spontaneous redox reactions, thus revealing the basic physical and chemical properties of the condensed interface and the mechanism of interface regulation of biochemical activities.153–157 To achieve the formation of biomolecular condensates, the authors selected a synthetic intrinsically disordered protein (IDP), a resilin-like polypeptide (RLP), which can be used as a single-component driver for condensates through phase separation.158,159 Specifically, RLPWT (Met-Ser-Lys-Gly-Pro-[Gly-Arg-Gly-Asp-Ser-Pro-Tyr-Ser]20-Gly-Tyr) was used, which undergoes an upper critical solution temperature (UCST) phase transition in physiological salt concentration and aqueous buffer in living cells. A phase separation analysis method is designed based on the known phase boundary or the saturation concentration (Csat) of RLPWT, defined as the concentration at which spontaneous phase transition occurs at room temperature and physiological salt concentration. In this method, the protein stock solution in a high-salt buffer is mixed with a diluted (low-salt) buffer to induce condensate formation (Fig. 12a). The reduction-sensitive fluorescent probe resazurin and RLPWT stock solution were mixed with the dilution buffer and the fluorescence signal was evaluated by confocal fluorescence microscopy. The final RLPWT concentration was 10 mM (containing 10 nM resazurin), lower than the Csat of RLPWT and showed no fluorescence signal. In contrast, the sample containing 10 nM resazurin had a RLPWT concentration of 40 mM, which was higher than Csat of RLPWT. The condensate was formed and showed an obvious fluorescence signal, which was initially mainly located at the liquid–liquid interface of the condensate (Fig. 12b). These observations indicate that the liquid–liquid interface of the condensate triggers a reduction reaction, and there may be an interface electric field established by the EDL. Such an electric field can affect the bond dissociation energy and may promote chemical reactions.160 When the surface of biomolecular condensates acquires charge due to its unique molecular structure,161 the excess opposite charge (a counterion) must reside near the surface to maintain charge neutralization, thus forming the EDL (Fig. 12c). This feature also contributes to the size distribution and aggregation tendency of condensates. From the perspective of condensate formation, the biomolecular phase transition leads to the density transition of the system, which can mediate the selective distribution of ions, resulting in charge asymmetry between the two phases. Studies on the ubiquitous electrochemical activity at liquid–gas and liquid–solid interface have shown that the electric field can convert hydroxide ions (OH−) dissociated from water molecules into hydroxyl radicals (OH·) and solvated electrons.162 The recombination reaction between hydroxyl radicals can subsequently generate H2O2 (Fig. 12d). Using the developed RLP-based condensates for programmable intracellular phase separation, RLPWT was expressed in E. coli bacteria from a lac promoter-controlled plasmid. The intracellular H2O2 was tracked by incubating the Peroxy Lucifer 1 (PL-1) probe in the induced growth medium. Confocal microscopy is used to visualize the condensate, and cell regions can be distinguished according to different optical densities. The PL-1 signal was observed at the same position as the condensates, and the signal was higher than that of the cytoplasm (Fig. 12e), confirming that the condensates could produce H2O2 in living cells.
 | ||
| Fig. 12 The physiological activities of cells modulated by EDL interfaces. (a) High concentration of synthetic intrinsically disordered proteins (synIDPs) diluted by a buffer containing resazurin to trigger phase separation. (b) Confocal fluorescence images. (c) Schematic diagram of EDL formation resulting from the surface electrostatic interaction of the condensate. (d) Diagram of H2O2 generation induced by the interface EDL electric field. (e) PL-1 assay for the investigation of spatial distribution of H2O2. (a)–(e) Reproduced from ref. 151, © 2023 Elsevier Inc. (f) The phase transition of biological macromolecules leads to the enrichment or exclusion of specific types of ions, thereby regulating the distribution of ions in cells. (g) Hyperpolarization of cell membranes mediated by condensates. (h) Comparison of the cell growth lag phase and maximum growth rate with/without condensates under different antibiotic treatments. (f)–(h) Reproduced from ref. 152, © 2024 Elsevier Inc. | ||
Recently, another study by this group has shown that the formation of biomolecular aggregates can regulate the electrochemical environment in bacterial cells, thereby affecting global cellular processes. The formation of condensates produces a potential gradient that directly affects the electrochemical properties of cells, including cytoplasmic pH and membrane potential. The regulation of electrochemical balance further controls the interaction between cells and the environment, which directly affects the survival of bacteria under the action of antibiotics. Condensates (the dense phase) and the cytoplasmic environment (the dilute phase) have significantly different pH environments (Fig. 12f). To understand the effect of the condensates on the cytoplasmic ion environment, the same synthetic intrinsically disordered protein (synIDP) was used, consisting of an RLP (amino acid sequence: Ser-LysGly-Pro-[Gly-Arg-Gly-Asp-Ser-Pro-Tyr-Ser]20-Gly-Tyr), and phase separation was performed by the upper critical solution temperature (UCST) phase transition. The synIDP has similar sequence characteristics and the same thermodynamic driving force as many natural IDPs that can drive the formation of condensates. Cells usually control their intracellular ion content through ion channels and regulate membrane potential through ion channels.163,164 Because the membrane potential is sensitive to the intracellular electrochemical environment, a new equilibrium state of ion distribution in the cell is established by the condensed state.165 The formation of the condensed state can regulate the electrochemical balance between the extracellular and intracellular ions, thereby changing the membrane potential.166 The change of membrane potential can have a profound impact on small molecule signals by adjusting the direction and size of small molecule flux. This condensed state-mediated regulatory effect may affect the survival of cells exposed to functional small molecules such as antibiotics, based on their electrostatic charge (Fig. 12g). The growth recovery of reverse-diluted logarithmic phase cells with and without RLP aggregates in M9 medium containing different antibiotics was determined. In contrast, the formation of the condensates magnified the effects of several positively charged antibiotics, gentamicin, chloramphenicol and streptomycin. When acting on cells without condensates (Fig. 12h) at sublethal concentrations, these antibiotics more strongly inhibited the recovery of cells containing aggregates. The lag phase caused by streptomycin in cells containing condensates was three times longer than that in cells without condensates. Gentamicin and chloramphenicol completely inhibited the growth of agglutinate cells. The enhanced sensitivity may be due to the increased uptake of these positively charged antibiotics by condensate-mediated hyperpolarized membrane potential.
5. Conclusion and prospects
In summary, this review systematically examines ionic rectification through the modulation of the EDL at hydrogel interfaces. Notably, the EDL discussed here extends beyond the conventional solid–liquid interface to include liquid–liquid and liquid–solid interfaces. Based on the underlying mechanisms, we classify ionic rectification into four distinct types: (1) EDL on charged polymer chains of hydrogels. The EDL formed along charged polymer molecular chains facilitates rapid transport of counter ions. Thermal fluctuations of these chains further induce ion hopping concentration effects, lower ion transport energy barriers, expand the Debye shielding dimension, enhance ion selectivity, and mitigate Coulomb blockade effects typically observed in rigid nano-confined channels. (2) EDL within nanopores enhanced by hydrogel modification. Introducing polyelectrolyte hydrogels into nanopores or chemically modifying nanopore walls with hydrogels creates composite ion-selective membranes, significantly enhancing ionic rectification. The presence of charged hydrogels increases charge density within confined nanospaces, thickens the EDL, and strengthens ion selectivity. (3) EDL at hydrogel p–n junctions (IDL). When hydrogels bearing oppositely charged polymer chains come into contact, an interfacial depletion layer analogous to a semiconductor p–n junction forms. This IDL establishes a built-in electric field at the interface, permitting ion transport when the applied potential aligns with the field direction while blocking ionic currents when the potential is reversed. (4) Asymmetric EDL at hydrogel/electrode interfaces. By modulating the dynamic EDL between hydrogel–electrode interfaces, selective ion migration within the diffuse layer can be achieved. Ion-selective membranes on electrode surfaces inhibit EDL formation under reverse bias while permitting its formation under forward bias, enabling ionic rectification. Additionally, triboelectrification between composite electrodes (e.g., Au/PET) and hydrogels dynamically modulates the EDL. Differences in EDL capacitance between the two electrode/hydrogel interfaces create a potential difference, driving directional ion migration and further facilitating ionic rectification.Recent representative studies on ionic rectification at hydrogel EDL interfaces have demonstrated their broad applications in various energy devices, including osmotic power sources (such as artificial electric eels), as well as mechanical and moisture energy harvesting. As intelligent sensors, they are widely used for detecting specific substances (e.g., DNA), human electromyographic signals, and motion signals. In flexible circuits, they serve as functional diode components, including bridge rectifiers, logic circuits and neuromorphic integrated circuits. In the biological field, the EDL at the hydrogel interface (between condensates and bulk solution) has been shown to generate H2O2, which regulates cellular physiological activities and enhances the absorption of certain antibiotics by bacteria. Hydrogels serve as excellent carriers for connecting electronics and living organisms due to their outstanding human adaptability and biocompatibility, with the EDL at the hydrogel interface providing a realistic basis for such applications. As a fundamental research direction, ionic rectification through modulating the EDL at the hydrogel interface plays a crucial role in various hydrogel-based iontronics. This not only deepens the understanding of physical and chemical behaviors at the interface but also offers a theoretical foundation and application potential for breakthroughs in multiple technical fields. These include, but are not limited to, bionic interfaces, the development of high-sensitivity biosensors or implantable medical devices, enhancement of interface ion transport and energy storage efficiency in flexible supercapacitors or batteries, advancement of wearable flexible devices, human–machine interaction, and even environmental protection. Despite significant progress in ionic rectification based on the EDL at the hydrogel interface, several challenges remain. In terms of device fabrication, most of the reported devices are manually produced, and scaling up production using industrial equipment presents a significant challenge. Additionally, addressing the performance degradation caused by hydrogel dehydration will require improvements over the long term. Ion contamination may cause crosstalk of multi-mode ion signals. How to improve signal recognition accuracy in complex body environments (such as sweat, blood, temperature gradient, strain, and pH) is also to be solved. In addition, appropriate mechanical properties are conducive to the practical application of hydrogel iontronics, but the increase of polymer crosslinking network density seems to inevitably reduce ionic conductivity. How to solve or balance through appropriate material structure design, processing methods and micro-nano structure design is also a potential problem. The continued development of this field will not only drive the upgrading of existing technologies but may also lead to disruptive innovations, providing new solutions to global challenges such as energy, healthcare, and the environment.
The ICR of the representative work is summarized in Table 1.
| Mechanism type | Electrolyte | Concentration | Bias voltage | ICR | References |
|---|---|---|---|---|---|
| (1) | KCl | 0.1 M | ±1 V | 3–4 | 85 |
| (1) | KCl | 0.01 M | ±2 V | 3 | 86 |
| (1) | KCl | 0.1 M | ±2 V | 100 | 87 |
| (2) | LiCl | 0.1 M | ±2 V | 50 | 106 |
| (3) | — | — | ±5 V | 15 | 111 |
| (3) | — | — | ±2.5 V | 4.5 | 112 |
| (3) | — | — | ±5 V | 13 | 42 |
| (3) | ZnSO4, CuSO4 | 1 M | ±5 V | 50 | 127 |
| (3) | — | — | ±5 V | 10 | 128 |
| (3) | — | — | ±2 V | 4.22 ± 0.53 | 143 |
| (3) | — | — | ±4 V | 215 | 144 |
| (3) | — | — | ±5 V | 43 | 145 |
| (3) | Zn(CF3O3S)2), [EMIM]Cl | — | ±1 V | 23.11 | 146 |
| (4) | LiCl | 1.35 M | ±3 V | 1201 | 117 |
| (4) | — | — | ±10 V | 4 × 105 | 118 |
Data availability
This review does not include any primary research findings, software, or code, nor does it involve the generation or analysis of new data. Data sharing is not applicable to this article.Author contributions
Yaowen Ouyang: writing – review & editing. Zhong Lin Wang: review & editing. Di Wei: writing – review & editing.Conflicts of interest
The authors declare no competing interests.Acknowledgements
This work was supported by the National Natural Science Foundation (grant number 22479016).References
- C. Yang and Z. Suo, Nat. Rev. Mater., 2018, 3, 125–142 CrossRef CAS.
- H. Chun and T. D. Chung, Annu. Rev. Anal. Chem., 2015, 8, 441–462 CrossRef CAS PubMed.
- S. Z. Bisri, S. Shimizu, M. Nakano and Y. Iwasa, Adv. Mater., 2017, 29, 1607054 CrossRef PubMed.
- E. M. Ahmed, J. Adv. Res., 2015, 6, 105–121 CrossRef CAS PubMed.
- C. Keplinger, J.-Y. Sun, C. C. Foo, P. Rothemund, G. M. Whitesides and Z. Suo, Science, 2013, 341, 984–987 CrossRef CAS PubMed.
- J. A. Goding, A. D. Gilmour, U. A. Aregueta-Robles, E. A. Hasan and R. A. Green, Adv. Funct. Mater., 2018, 28, 1702969 CrossRef.
- Z. H. Guo, H. L. Wang, J. Shao, Y. Shao, L. Jia, L. Li, X. Pu and Z. L. Wang, Sci. Adv., 2022, 8, eabo5201 CrossRef CAS PubMed.
- X. Pu, M. Liu, X. Chen, J. Sun, C. Du, Y. Zhang, J. Zhai, W. Hu and Z. L. Wang, Sci. Adv., 2017, 3, e1700015 CrossRef PubMed.
- W. Ding, J. Zhou, J. Cheng, Z. Wang, H. Guo, C. Wu, S. Xu, Z. Wu, X. Xie and Z. L. Wang, Adv. Energy Mater., 2019, 9, 1901320 CrossRef.
- Y. Ouyang, X. Li, Y. Du, Y. Zhang, Z. L. Wang and D. Wei, Small, 2025, e2409998 CrossRef PubMed.
- Y. Du, P. Shen, H. Liu, Y. Zhang, L. Jia, X. Pu, F. Yang, T. Ren, D. Chu, Z. Wang and D. Wei, Sci. Adv., 2024, 10, eadp8681 CrossRef CAS PubMed.
- S. Lin, J. Jiang, K. Huang, L. Li, X. He, P. Du, Y. Wu, J. Liu, X. Li, Z. Huang, Z. Zhou, Y. Yu, J. Gao, M. Lei and H. Wu, ACS Nano, 2023, 17, 24487–24513 CrossRef CAS PubMed.
- M. Hu, J. Ren, Y. Pan, L. Cheng, X. Xu, C. L. Tan, H. Sun, Y. Shi and S. Yan, Adv. Funct. Mater., 2024, 34, 2407926 CrossRef CAS.
- W. U. Khan, Z. Shen, S. M. Mugo, H. Wang and Q. Zhang, Chem. Soc. Rev., 2025, 54, 2832–2880 RSC.
- S. Dai, X. Liu, Y. Liu, Y. Xu, J. Zhang, Y. Wu, P. Cheng, L. Xiong and J. Huang, Adv. Mater., 2023, 35, 2300329 CrossRef CAS PubMed.
- W. Tang, Q. Sun and Z. L. Wang, Chem. Rev., 2023, 123, 12105–12134 CrossRef CAS PubMed.
- S. J. Yoon, J. T. Park and Y. K. Lee, Soft Sci., 2024, 4, 30 Search PubMed.
- P. Peng, F. Yang, X. Li, S. Li, Z. Wang and D. Wei, Cell Rep. Phys. Sci., 2024, 5, 101824 CrossRef CAS.
- H. Qian, D. Wei and Z. Wang, Nano Res., 2023, 16, 11718–11730 CrossRef CAS.
- F. Yang, P. Peng, Z.-Y. Yan, H. Fan, X. Li, S. Li, H. Liu, T.-L. Ren, Y. Zhou and Z. L. Wang, Nat. Energy, 2024, 9, 263–271 CrossRef CAS.
- K. Chen and D. Ho, Aggregate, 2024, 5, e425 CrossRef CAS.
- Y. Dobashi, D. Yao, Y. Petel, T. N. Nguyen, M. S. Sarwar, Y. Thabet, C. L. Ng, E. Scabeni Glitz, G. T. M. Nguyen and C. Plesse, Science, 2022, 376, 502–507 CrossRef CAS PubMed.
- D. Wei, Sci. Rep., 2015, 5, 15173 CrossRef CAS PubMed.
- P. Peng, P. Shen, H. Qian, J. Liu, H. Lu, Y. Jiao, F. Yang, H. Liu, T. Ren, Z. Wang and D. Wei, Device, 2025, 3, 100574 CrossRef.
- H. Qian, P. Peng, H. Fan, Z. Yang, L. Yang, Y. Zhou, D. Tan, F. Yang, M. Willatzen, G. Amaratunga, Z. Wang and D. Wei, Angew. Chem., Int. Ed., 2024, 63, e202414984 CrossRef CAS PubMed.
- P. Peng, F. Yang, Z. Wang and D. Wei, Adv. Energy Mater., 2023, 13, 2302360 CrossRef CAS.
- D. Wei, M. R. Scherer, M. Astley and U. Steiner, Nanotechnology, 2015, 26, 225501 CrossRef PubMed.
- D. Wei, M. R. Astley, N. Harris, R. White, T. Ryhänen and J. Kivioja, Nanoscale, 2014, 6, 9536–9540 RSC.
- D. Wei, P. Hiralal, H. Wang, H. Emrah Unalan, M. Rouvala, I. Alexandrou, P. Andrew, T. Ryhänen and G. A. J. Amaratunga, Nano Energy, 2013, 2, 1054–1062 CrossRef CAS.
- D. Wei, S. Haque, P. Andrew, J. Kivioja, T. Ryhänen, A. Pesquera, A. Centeno, B. Alonso, A. Chuvilin and A. Zurutuza, J. Mater. Chem. A, 2013, 1, 3177–3181 RSC.
- H. v. Helmholtz, Ann. Chim. Phys., 1853, 165, 353–377 CrossRef.
- M. Gouy, J. Phys. Theor. Appl., 1910, 9, 457–468 CrossRef.
- D. L. Chapman, Philos. Mag. A, 1913, 25, 475–481 CrossRef.
- O. Stern, Z. Elektrochem. Angew. Phys. Chem., 1924, 30, 508–516 CrossRef CAS.
- S. Lin, L. Xu, A. Chi Wang and Z. L. Wang, Nat. Commun., 2020, 11, 399 CrossRef CAS PubMed.
- Z. L. Wang and A. C. Wang, Mater. Today, 2019, 30, 34–51 CrossRef CAS.
- J. W. Kim, D. Lee, H. C. Shum and D. A. Weitz, Adv. Mater., 2008, 20, 3239–3243 CrossRef CAS.
- R. Aveyard, B. P. Binks and J. H. Clint, Adv. Colloid Interface Sci., 2003, 100, 503–546 CrossRef.
- Y. Dai, L. You and A. Chilkoti, Nat. Rev. Bioeng., 2023, 1, 466–480 CrossRef CAS PubMed.
- M. N. Biutty, H. Kim, P. L. Handayani, Y. Eom, U. H. Choi, J. H. Kim, M. H. Kim and S. I. Yoo, Chem. Eng. J., 2024, 495, 153565 CrossRef.
- C. Zhu, Y. Teng, G. Xie, P. Li, Y. Qian, B. Niu, P. Liu, W. Chen, X.-Y. Kong and L. Jiang, Chem. Commun., 2020, 56, 10767–10767 RSC.
- Y. Zhang, C. K. Jeong, J. Wang, X. Chen, K. H. Choi, L. Q. Chen, W. Chen, Q. Zhang and Q. Wang, Adv. Mater., 2021, 33, 2103056 CrossRef CAS PubMed.
- Y. W. Ouyang, X. Li, S. X. Li, Z. L. Wang and D. Wei, ACS Appl. Mater. Interfaces, 2024, 16, 18236–18244 CrossRef CAS PubMed.
- H.-J. Butt, K. Graf and M. Kappl, Physics and chemistry of interfaces, John Wiley & Sons, 2023 Search PubMed.
- I. Kosuke, A.-S. Kazuhiro, L. Y. Norifumi, Y. Yuko, S. Tetsuo and N. Naoya, Electrochim. Acta, 2025, 513, 145563 CrossRef.
- W. Yang, Q. Han, W. Li, M. Wu, J. Yao, M. Zhao and X. Lu, Chem. Eng. J., 2022, 448, 137731 CrossRef CAS.
- A. D. Pendergast, S. Gutierrez-Portocarrero, R. Noriega and H. S. White, J. Am. Chem. Soc., 2024, 146, 30464–30473 CrossRef CAS PubMed.
- P. He, Y. Shao, Z. Yu, X. Liang, J. Liu, Y. Bian, Z. Zhu, M. Li, C. M. Pereira and Y. Shao, Anal. Chem., 2022, 94, 9801–9810 CrossRef CAS PubMed.
- Z. Xiuzhong, L. Xiao, Z. Qiwei, F. Lin, Z. Li, C. Xiangyu and W. Zhong Lin, Nano Energy, 2021, 87, 106191 CrossRef.
- S. Zhang, R. Song, H. Zeng, N. Wu, H. Duan and L. Wang, Droplet, 2024, 3, e110 CrossRef.
- D. Jing, D. Li, Y. Pan and B. Bhushan, Langmuir, 2016, 32, 11123–11132 CrossRef CAS PubMed.
- Z. Xu, M. Zhao, Y. Zhang, P. Wang, Y. Wu, L. Li, X. Cui, N. Sun and C. Dai, Chem. Eng. J., 2023, 454, 140393 CrossRef CAS.
- J. Dong, J. Chen, W. Wang, Z. Wei, Z.-Q. Tian and F. R. Fan, J. Am. Chem. Soc., 2024, 146, 2227–2236 CrossRef CAS PubMed.
- J. N. Israelachvili and R. M. Pashley, J. Colloid Interface Sci., 1984, 98, 500–514 CrossRef CAS.
- J. N. Israelachvili, in Hydrocolloids, Elsevier Science, Amsterdam, 2000, pp. 3–21 Search PubMed.
- Y. Min, T. F. Alig, D. W. Lee, J. M. Boggs, J. N. Israelachvili and J. A. Zasadzinski, Biophys. J., 2011, 100, 1490–1498 CrossRef CAS PubMed.
- X. Li, Z. L. Wang and D. Wei, Adv. Funct. Mater., 2024, 34, 2405520 CrossRef CAS.
- A. Hernández, J. Arcos, J. Martínez-Trinidad, O. Bautista, S. Sánchez and F. Méndez, Int. J. Heat Mass Transfer, 2022, 187, 122522 CrossRef.
- I. M. Bhattacharyya and G. Shalev, ACS Sens., 2019, 5, 154–161 CrossRef PubMed.
- P. Gu, S. Yang, X. Liu and G. Yang, Soil Sci. Soc. Am. J., 2020, 84, 494–501 CrossRef CAS.
- S. Das, S. Chakraborty and S. K. Mitra, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2012, 85, 051508 CrossRef PubMed.
- A. Vacic, J. M. Criscione, N. K. Rajan, E. Stern, T. M. Fahmy and M. A. Reed, J. Am. Chem. Soc., 2011, 133, 13886–13889 CrossRef CAS PubMed.
- R. B. Schoch, J. Han and P. Renaud, Rev. Mod. Phys., 2008, 80, 839–883 CrossRef CAS.
- S. Lin, X. Chen and Z. L. Wang, Chem. Rev., 2021, 122, 5209–5232 CrossRef PubMed.
- X. Li, Z. L. Wang and D. Wei, NanoTrends, 2024, 8, 100062 Search PubMed.
- X. Li, R. Li, S. Li, Z. L. Wang and D. Wei, Nat. Commun., 2024, 15, 6182 CrossRef CAS PubMed.
- X. Li, S. Li, X. Guo, J. Shao, Z. L. Wang and D. Wei, Matter, 2023, 6, 3912–3926 CrossRef.
- L. Zhang and D. Wang, Matter, 2023, 6, 3698–3699 CrossRef CAS.
- Y. Wei, X. Li, Z. Yang, J. Shao, Z. L. Wang and D. Wei, Mater. Today, 2024, 74, 2–11 CrossRef CAS.
- R. Li, X. Li, Z. Zhang, M. Willatzen, Z. L. Wang and D. Wei, Adv. Funct. Mater., 2025, 35, 2416457 CrossRef CAS.
- S. Li, Z. Zhang, F. Yang, X. Li, P. Peng, Y. Du, Q. Zeng, M. Willatzen, Z. L. Wang and D. Wei, Device, 2024, 2, 100332 CrossRef.
- W. Tang, Q. Sun and Z. L. Wang, Chem. Rev., 2023, 123, 12105–12134 CrossRef CAS PubMed.
- X. Yang, J. Han, J. Yu, Y. Chen, H. Zhang, M. Ding, C. Jia, J. Sun, Q. Sun and Z. L. Wang, ACS Nano, 2020, 14, 8668–8677 CrossRef CAS PubMed.
- G. Gao, J. Yu, X. Yang, Y. Pang, J. Zhao, C. Pan, Q. Sun and Z. L. Wang, Adv. Mater., 2019, 31, 1806905 CrossRef PubMed.
- M. Ding, D. Ji and W. Hu, ACS Appl. Electron. Mater., 2024, 6, 8655–8670 CrossRef CAS.
- J. Yu, S. Qin, H. Zhang, Y. Wei, X. Zhu, Y. Yang and Q. Sun, Research, Washington, D.C., 2021, vol. 2021, p. 9840918 Search PubMed.
- W. Chen, Q. Wang, J. Chen, Q. Zhang, X. Zhao, Y. Qian, C. Zhu, L. Yang, Y. Zhao and X.-Y. Kong, Nano Lett., 2020, 20, 5705–5713 CrossRef CAS PubMed.
- D. Wei, F. Yang, Z. Jiang and Z. Wang, Nat. Commun., 2022, 13, 4965 CrossRef CAS PubMed.
- L. Yang, L. N. Cao, S. Li, P. Peng, H. Qian, G. Amaratunga, F. Yang, Z. L. Wang and D. Wei, Nano Energy, 2024, 129, 110076 CrossRef CAS.
- Y. Liu, P. Peng, F. Yang, Z. L. Wang and D. Wei, Energy Mater., 2025, 5, 500059 CAS.
- Y. Ouyang, X. Li, S. Li, P. Peng, F. Yang, Z. L. Wang and D. Wei, Nano Energy, 2023, 116, 108796 CrossRef CAS.
- D.-K. Kim, C. Duan, Y.-F. Chen and A. Majumdar, Microfluid. Nanofluid., 2010, 9, 1215–1224 CrossRef CAS.
- S. Wang, T. Yang, D. Zhang, Q. Hua and Y. Zhao, Adv. Mater., 2024, 36, 2405391 CrossRef CAS PubMed.
- F. Li, X. Cai, G. Liu, H. Xu and W. Chen, Adv. Funct. Mater., 2023, 33, 2300701 CrossRef CAS.
- W. Chen, K. Zhou, Z. Wu, L. Yang, Y. Xie, X. Meng, Z. Zhao and L. Wen, J. Am. Chem. Soc., 2024, 146, 13191–13200 CrossRef CAS PubMed.
- G. Bian, N. Pan, Z. Luan, X. Sui, W. Fan, Y. Xia, K. Sui and L. Jiang, Am. Ethnol., 2021, 133, 20456–20462 Search PubMed.
- J. Li, C. Li, H. Dou, X. Zhang, Y. Dai and F. Xia, J. Membr. Sci., 2024, 698, 122644 CrossRef CAS.
- T. A. Manz and D. S. Sholl, J. Chem. Theory Comput., 2010, 6, 2455–2468 CrossRef CAS PubMed.
- H. Cui, N. Pan, W. Fan, C. Liu, Y. Li, Y. Xia and K. Sui, Adv. Funct. Mater., 2019, 29, 1807692 CrossRef.
- N. Pan, M. Lin, H. Cui, W. Fan, C. Liu, F. Chen, C. Fan, Y. Xia and K. Sui, Chem. Mater., 2020, 32, 8442–8449 CrossRef CAS.
- M. Wang, H. Meng, D. Wang, Y. Yin, P. Stroeve, Y. Zhang, Z. Sheng, B. Chen, K. Zhan and X. Hou, Adv. Mater., 2019, 31, 1805130 CrossRef PubMed.
- W. Xin, Z. Zhang, X. Huang, Y. Hu, T. Zhou, C. Zhu, X.-Y. Kong, L. Jiang and L. Wen, Nat. Commun., 2019, 10, 3876 CrossRef PubMed.
- S. Mazinani, A. Al-Shimmery, Y. J. Chew and D. Mattia, J. Membr. Sci., 2022, 644, 120137 CrossRef CAS.
- J. Choi, S. Yang, N.-J. Jeong, H. Kim and W.-S. Kim, Langmuir, 2018, 34, 10837–10846 CrossRef CAS PubMed.
- J. G. Hong and T.-W. Park, J. Electroanal. Chem., 2018, 817, 134–140 CrossRef CAS.
- J. G. Hong, T.-W. Park and Y. Dhadake, J. Electroanal. Chem., 2019, 850, 113437 CrossRef CAS.
- H. Daiguji, P. Yang and A. Majumdar, Nano Lett., 2004, 4, 137–142 CrossRef CAS.
- X. He, K. Zhang, Y. Liu, F. Wu, P. Yu and L. Mao, Angew. Chem., 2018, 130, 4680–4683 CrossRef.
- E. T. Acar, S. F. Buchsbaum, C. Combs, F. Fornasiero and Z. S. Siwy, Sci. Adv., 2019, 5, eaav2568 CrossRef CAS PubMed.
- G. Laucirica, M. E. Toimil-Molares, C. Trautmann, W. Marmisollé and O. Azzaroni, ACS Appl. Mater. Interfaces, 2020, 12, 28148–28157 CrossRef CAS PubMed.
- S. Zhang, L. Yang, D. Ding, P. Gao, F. Xia and M. L. Bruening, Anal. Chem., 2021, 93, 4291–4298 CrossRef CAS PubMed.
- T. Ma, E. Balanzat, J.-M. Janot and S. Balme, ACS Appl. Mater. Interfaces, 2019, 11, 12578–12585 CrossRef CAS PubMed.
- S. Zhang, L. Song, B. Liu, Y.-D. Zhao and W. Chen, Anal. Chim. Acta, 2023, 1251, 341000 CrossRef CAS PubMed.
- C.-Y. Lin, C. Combs, Y.-S. Su, L.-H. Yeh and Z. S. Siwy, J. Am. Chem. Soc., 2019, 141, 3691–3698 CrossRef CAS PubMed.
- W. Chen, Q. Zhang, Y. Qian, W. Xin, D. Hao, X. Zhao, C. Zhu, X.-Y. Kong, B. Lu and L. Jiang, ACS Cent. Sci., 2020, 6, 2097–2104 CrossRef CAS PubMed.
- Y. Wu, D. Wang, I. Willner, Y. Tian and L. Jiang, Angew. Chem., Int. Ed., 2018, 57, 7790–7794 CrossRef CAS PubMed.
- S. Kondo, T. Hiroi, Y. S. Han, T. H. Kim, M. Shibayama, U. i. Chung and T. Sakai, Adv. Mater., 2015, 27, 7407–7411 CrossRef CAS PubMed.
- X. Liu and B. Lu, Phys. Rev. E, 2017, 96, 062416 CrossRef PubMed.
- F. Wang, X. Liu and I. Willner, Angew. Chem., Int. Ed., 2015, 54, 1098–1129 CrossRef CAS PubMed.
- D. Y. Zhang and G. Seelig, Nat. Chem., 2011, 3, 103–113 CrossRef CAS PubMed.
- O. J. Cayre, S. T. Chang and O. D. Velev, J. Am. Chem. Soc., 2007, 129, 10801–10806 CrossRef CAS PubMed.
- W. Lu, T. Ding, X. Wang, C. Zhang, T. Li, K. Zeng and G. W. Ho, Nano Energy, 2022, 104, 107892 CrossRef CAS.
- S.-M. Lim, H. Yoo, M.-A. Oh, S. H. Han, H.-R. Lee, T. D. Chung, Y.-C. Joo and J.-Y. Sun, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 13807–13815 CrossRef CAS PubMed.
- H. J. Kim, B. Chen, Z. Suo and R. C. Hayward, Science, 2020, 367, 773–776 CrossRef CAS PubMed.
- H. R. Lee, J. Woo, S. H. Han, S. M. Lim, S. Lim, Y. W. Kang, W. J. Song, J. M. Park, T. D. Chung and Y. C. Joo, Adv. Funct. Mater., 2019, 29, 1806909 CrossRef.
- Y. Hou, Y. Zhou, L. Yang, Q. Li, Y. Zhang, L. Zhu, M. A. Hickner, Q. Zhang and Q. Wang, Adv. Energy Mater., 2017, 7, 1601983 CrossRef.
- Z. H. Guo, H. L. Wang, Y. Shao, L. Li, L. Jia and X. Pu, Adv. Funct. Mater., 2022, 32, 2112432 CrossRef CAS.
- W. Ren, H. Jing, S. Ding, J. Dan, Z. Xu, T. Guo, H. Wei, Y. Liu and Y. Liu, Small, 2024, 20, 2404874 CrossRef CAS PubMed.
- H.-J. Koo, S.-T. Chang and O. D. Velev, Small, 2010, 6, 1393–1397 CrossRef CAS PubMed.
- Z. Shi, P. Peng, D. Strohecker and Y. Liao, J. Am. Chem. Soc., 2011, 133, 14699–14703 CrossRef CAS PubMed.
- R. Zhu, P. Sun, G. Cui, Y. Yu, S. Ke and J. Zhao, J. Mater. Chem. A, 2024, 12, 14559–14568 RSC.
- Z. Zhang, L. He, C. Zhu, Y. Qian, L. Wen and L. Jiang, Nat. Commun., 2020, 11, 875 CrossRef CAS PubMed.
- Y. Wang, H. Jiang, Y. Zhang, Z. Song, Z. Chen, K. Sui, W. Fan and P. Qi, Chem. Eng. J., 2024, 481, 148512 CrossRef CAS.
- M. Xia, N. Pan, C. Zhang, C. Zhang, W. Fan, Y. Xia, Z. Wang and K. Sui, ACS Nano, 2022, 16, 4714–4725 CrossRef CAS PubMed.
- Y.-L. Liu, S.-Y. Yu, J.-H. Chen, C.-S. Wang, H.-Y. Li, D. Jiang, D. Ye and W.-W. Zhao, ACS Sens., 2022, 7, 3272–3277 CrossRef CAS PubMed.
- B. Ying, Q. Wu, J. Li and X. Liu, Mater. Horiz., 2020, 7, 477–488 RSC.
- M. Du, D. Zhang, W. Fan, K. Zhao, Y. Xia, Z. Nie and K. Sui, Mater. Today Phys., 2022, 26, 100744 CrossRef CAS.
- H. Wang, M. Du, H. Jiang, R. Zhou, Y. Wang, K. Sui and W. Fan, Adv. Funct. Mater., 2024, 34, 2407639 CrossRef CAS.
- S. H. Han, S. I. Kim, H.-R. Lee, S.-M. Lim, S. Y. Yeon, M.-A. Oh, S. Lee, J.-Y. Sun, Y.-C. Joo and T. D. Chung, ACS Appl. Mater. Interfaces, 2021, 13, 6606–6614 CrossRef CAS PubMed.
- S. Woo, H. Kim, J. Kim, H. Ryu and J. Lee, Adv. Electron. Mater., 2024, 10, 2300653 CrossRef CAS.
- Y. Wei, Y. Zhao, Q. Ding, J. Zhao, Y. Yang, Y. Liu, J. Dong, D. Zhang, S. Han and P. Huo, Chem. Eng. J., 2024, 498, 155655 CrossRef CAS.
- S. H. Han, S. I. Kim, M.-A. Oh and T. D. Chung, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2211442120 CrossRef CAS PubMed.
- J. Yan, J. P. K. Armstrong, F. Scarpa and A. W. Perriman, Adv. Mater., 2024, 36, 2403937 CrossRef CAS PubMed.
- Z. Lei and P. Wu, Matter, 2023, 6, 429–444 CrossRef CAS.
- T. B. H. Schroeder, A. Guha, A. Lamoureux, G. VanRenterghem, D. Sept, M. Shtein, J. Yang and M. Mayer, Nature, 2017, 552, 214–218 CrossRef CAS PubMed.
- J. Duan, W. Xie, P. Yang, J. Li, G. Xue, Q. Chen, B. Yu, R. Liu and J. Zhou, Nano Energy, 2018, 48, 569–574 CrossRef CAS.
- J. Yin, N. Liu, P. Jia, Z. Ren, Q. Zhang, W. Lu, Q. Yao, M. Deng and Y. Gao, SusMat, 2023, 3, 859–876 CrossRef CAS.
- C. K. Roy, H. L. Guo, T. L. Sun, A. B. Ihsan, T. Kurokawa, M. Takahata, T. Nonoyama, T. Nakajima and J. P. Gong, Adv. Mater., 2015, 27, 7344–7348 CrossRef CAS PubMed.
- B. W. Petley, Metrologia, 1995, 31, 481 CrossRef.
- Y. Zhou, Y. Hou, Q. Li, L. Yang, Y. Cao, K. H. Choi, Q. Wang and Q. M. Zhang, Adv. Mater. Technol., 2017, 2, 1700118 CrossRef.
- Z. Sun, L. Feng, X. Wen, L. Wang, X. Qin and J. Yu, Mater. Horiz., 2021, 8, 2303–2309 RSC.
- D. Lei, Q. Zhang, N. Liu, T. Su, L. Wang, Z. Ren, Z. Zhang, J. Su and Y. Gao, Adv. Funct. Mater., 2022, 32, 2107330 CrossRef CAS.
- X. Chenwei, L. Jie, L. Luyao, C. Long, Z. Rong, M. Xianqiang and L. Yifan, Mater. Today Bio, 2022, 15, 100281 CrossRef PubMed.
- Y. Zhang, C. M. J. Tan, C. N. Toepfer, X. Lu and H. Bayley, Science, 2024, 386, 1024–1030 CrossRef CAS PubMed.
- R. Huo, G. Bao, Z. He, X. Li, Z. Ma, Z. Yang, R. Moakhar, S. Jiang, C. Chung-Tze-Cheong, A. Nottegar, C. Cao, S. Mahshid and J. Li, Adv. Funct. Mater., 2023, 33, 2213677 CrossRef CAS.
- F. Jiang, W. C. Poh, J. Chen, D. Gao, F. Jiang, X. Guo, J. Chen and P. S. Lee, Nat. Commun., 2022, 13, 6669 CrossRef CAS PubMed.
- D. Hassabis, D. Kumaran, C. Summerfield and M. Botvinick, Neuron, 2017, 95, 245–258 CrossRef CAS PubMed.
- D. Marković, A. Mizrahi, D. Querlioz and J. Grollier, Nat. Rev. Phys., 2020, 2, 499–510 CrossRef.
- L. Wang, S. Wang, G. Xu, Y. Qu, H. Zhang, W. Liu, J. Dai, T. Wang, Z. Liu, Q. Liu and K. Xiao, ACS Nano, 2024, 18, 29704–29714 CrossRef CAS PubMed.
- S. Han, S. Yu, S. Hu, H.-j. Chen, J. Wu and C. Liu, J. Mater. Chem. C, 2021, 9, 11801–11808 RSC.
- Y. Dai, C. F. Chamberlayne, M. S. Messina, C. J. Chang, R. N. Zare, L. You and A. Chilkoti, Chem, 2023, 9, 1594–1609 CAS.
- Y. Dai, Z. Zhou, W. Yu, Y. Ma, K. Kim, N. Rivera, J. Mohammed, E. Lantelme, H. Hsu-Kim, A. Chilkoti and L. You, Cell, 2024, 187, 5951–5966.e5918 CrossRef CAS PubMed.
- S. Lhee, J. K. Lee, J. Kang, S. Kato, S. Kim, R. N. Zare and H. G. Nam, Sci. Adv., 2020, 6, eaba0181 CrossRef CAS PubMed.
- J. K. Lee, D. Samanta, H. G. Nam and R. N. Zare, J. Am. Chem. Soc., 2019, 141, 10585–10589 CrossRef CAS PubMed.
- B. Chen, Y. Xia, R. He, H. Sang, W. Zhang, J. Li, L. Chen, P. Wang, S. Guo, Y. Yin, L. Hu, M. Song, Y. Liang, Y. Wang, G. Jiang and R. N. Zare, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2209056119 CrossRef CAS PubMed.
- L. Zhao, X. Song, C. Gong, D. Zhang, R. Wang, R. N. Zare and X. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2200991119 CrossRef CAS PubMed.
- J. K. Lee, K. L. Walker, H. S. Han, J. Kang, F. B. Prinz, R. M. Waymouth, H. G. Nam and R. N. Zare, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 19294–19298 CrossRef CAS PubMed.
- M. Dzuricky, B. A. Rogers, A. Shahid, P. S. Cremer and A. Chilkoti, Nat. Chem., 2020, 12, 814–825 CrossRef CAS PubMed.
- Y. Dai, M. Farag, D. Lee, X. Zeng, K. Kim, H.-i. Son, X. Guo, J. Su, N. Peterson, J. Mohammed, M. Ney, D. M. Shapiro, R. V. Pappu, A. Chilkoti and L. You, Nat. Chem. Biol., 2023, 19, 518–528 CrossRef CAS PubMed.
- S. Shaik, D. Danovich, J. Joy, Z. Wang and T. Stuyver, J. Am. Chem. Soc., 2020, 142, 12551–12562 CrossRef CAS PubMed.
- M. Farag, S. R. Cohen, W. M. Borcherds, A. Bremer, T. Mittag and R. V. Pappu, Nat. Commun., 2022, 13, 7722 CrossRef CAS PubMed.
- H. Xiong, J. K. Lee, R. N. Zare and W. Min, J. Phys. Chem. Lett., 2020, 11, 7423–7428 CrossRef CAS PubMed.
- T. J. Jentsch, Nat. Rev. Mol. Cell Biol., 2016, 17, 293–307 CrossRef CAS PubMed.
- D. C. Gadsby, Nat. Rev. Mol. Cell Biol., 2009, 10, 344–352 CrossRef CAS PubMed.
- L. Galera-Laporta, C. J. Comerci, J. Garcia-Ojalvo and G. M. Süel, Cell Syst., 2021, 12, 497–508 CrossRef CAS PubMed.
- X. Jin, J.-E. Lee, C. Schaefer, X. Luo, A. J. M. Wollman, A. L. Payne-Dwyer, T. Tian, X. Zhang, X. Chen, Y. Li, T. C. B. McLeish, M. C. Leake and F. Bai, Sci. Adv., 2021, 7, eabh2929 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2025 |