
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTarget analyte assisted sensitive electrochemical detection of cocaine on screen printed electrodes†
Ana Gomez
Cardoso
,
Hoda
Mozaffari
,
Syed Rahin
Ahmed
 ,
Herlys
Viltres
,
Greter A.
Ortega
,
Seshasai
Srinivasan
* and
Amin Reza
Rajabzadeh
*
,
Herlys
Viltres
,
Greter A.
Ortega
,
Seshasai
Srinivasan
* and
Amin Reza
Rajabzadeh
*
W Booth School of Engineering Practice and Technology, McMaster University, 1280 Main Street West Hamilton, Ontario L8S 4L7, Canada. E-mail: ssriniv@mcmaster.ca; rajaba@mcmaster.ca
First published on 24th June 2025
Abstract
The use of cocaine leads to several severe health conditions, and overconsumption often leads to death. Currently, cocaine detection devices require incubation periods, highly trained personnel, and expensive practices not suitable for roadside applications. Herein, a novel electrochemical biomolecule-free sensor for cocaine detection in complex matrices is presented using the electroactive characteristics of the cocaine molecule that does not require any biomolecules or chemicals for detection. This study implements the cocaine-modified carbon working electrodes to detect cocaine using cyclic voltammetry in a buffer solution and human saliva. At optimized conditions, the proposed electrochemical sensor enabled the detection of cocaine with a limit of detection of 1.73 ng mL−1 in PBS buffer (pH ∼7.4). Additionally, to facilitate detection in saliva, a machine learning strategy was introduced to analyze sensor analytical responses to overcome saliva-related complications in electrochemical sensing and challenges emanating from saliva-to-saliva variation. The data processing results allowed us to distinguish between cocaine concentrations ranging from 0 to over 50 ng mL−1 in saliva with an accuracy of 85%. Further, the successful detection of cocaine in the presence of various interferences was achieved, revealing that the m-Z-COC sensor is highly specific and a promising sensor for the development of a roadside oral fluid cocaine detection kit.
1. Introduction
Cocaine (COC) is an alkaloid drug obtained from the leaves of the coca plant (Erythroxylum coca). It stimulates the central nervous system, creating a feeling of euphoria.1 The continuous use of cocaine leads to dependence and has severe adverse effects, such as increased blood pressure, depression, and cardiovascular diseases. The psychotropic effects caused by drug abuse have impacted society in the form of accidents, violence, sexual assaults, and drug-related crimes.2 Besides, after administration (oral, nasal, injected, etc.), cocaine is metabolized in the liver into various metabolites.10 The primary metabolites of cocaine are benzoylecgonine (BZ) and ecgonine methyl ester (EME). The metabolites are later excreted in urine and represent 90% of the original cocaine dose; 10% of the initial cocaine dose passed as the intact compound.10 Cocaine is also detectable in saliva up to two days after administration and can be collected less invasively than in blood or urine.3 The maximum concentrations of cocaine in saliva can range from 400 to 1900 ng mL−1 immediately following administration. The half-life of cocaine is six hours (on average, as it varies between individuals), and it is eliminated from the body more rapidly than other illicit substances.3,4 The Substance Abuse and Mental Health Services (SAMSHA) currently has an acceptable threshold of 20 ng mL−1 of cocaine in saliva.2 Furthermore, as per the guidelines from Driving Under the Influence of Drugs, Alcohol, and Medicines (DRUID) and the Roadside Testing Assessment (ROSITA) project, cocaine detection devices should not only have a success rate greater than 80% but should also ideally take less than five minutes for the overall analysis.5,6In cocaine detection, electrochemical sensor approaches exhibit several advantages over other methods (including colorimetry and fluorescence). For instance, we can obtain highly accurate and quantitative results rapidly. Some recent reports on the detection of cocaine include colorimetry sensor with a limit of detection (LoD) of 440 pM (0.15 ng mL−1) in PBS buffer and the recovery in spiked human serum samples was 94.71–98.63%,7 square wave voltammetry (SWV)-based sensor with a LoD of 21 nM (7.13 ng mL−1) in PBS buffer,8 and differential pulse voltammetry (DPV)-based sensor with a LoD of 28.62 nM (9.72 ng mL−1) in water media and sensor was successfully applied for the cocaine analysis in synthetic urine samples.9 Despite ongoing developments, detecting low levels of cocaine in oral fluid within five minutes and without an incubation period has yet to be established. Currently, both Narcocheck and Medical Disposable-Saliva Screen drug tests are used in the market; however, both devices only report qualitative results that take between 10 to 15 minutes.10 Besides, quantitative onsite systems include Securetec Drugwipe (cut off: 30 ng mL−1),11 Draeger DrugTest 5000 Analyzer (cut off: 20 ng mL−1),12 Alere DDS2 (cut off: 30 ng mL−1),13 and RapiScan (cut off: 30 ng mL−1),14 but most of these sensors have detection times of approximately 10 minutes and reported inconveniences that require further adjustments.
On the other hand, one of the major restraints currently in commercializing sensors is issues with accuracy and reliability. Experiments performed in a laboratory setting under near-perfect conditions are difficult to replicate in the field. Machine learning (ML) is a discipline that is becoming increasingly popular in a broad range of applications. Similarly, chemometrics is an area that implements mathematical and statistical methods to analyze chemical data.26 Chemometrics has been widely accepted in analytical chemistry and is one of the most useful approaches to overcome the challenges of electrochemical sensors.15,16 ML algorithms can be generated, so saliva interference is not the most critical factor in analyte detection. Similarly, noise is a considerable challenge found in electrochemical signals, and by training ML models, the analyte signal can be easily distinguished from the signal noise.
This paper reports on the development of a novel biomolecule-free electrochemical sensor to detect cocaine in real saliva and address the various limitations of existing devices. At first, the working electrode (WE) of the screen-printed electrode (SPE) was modified with the same analyte being detected, cocaine. The affinity of the modified SPE surface towards the addition of different concentrated cocaine samples allows the detection of very low-level cocaine in saliva in less than one minute. This work also introduces ML to analyze the data sets obtained from the electrochemical cocaine sensors to overcome the person-to-person variation setbacks and the cross-interference problems in complex saliva samples. The integration of chemometrics to analyze the electrochemical signals resulted in excellent results, with an accuracy of 85% for the detection of cocaine in human saliva.
2. Experimental details
2.1. Materials and equipment
Cocaine hydrochloride, (−)-trans-Δ9-tetrahydrocannabinol (THC), chloroauric acid (HAuCl4·3H2O), sodium borohydride (NaBH4), cysteamine hydrochloride, ethanol (85%), PBS buffer, levamisole hydrochloride, and caffeine in methanol (MetOH) were all purchased from Sigma-Aldrich, Oakville, Ontario, Canada. ELISA Cocaine Oral Fluid Kit was purchased from Neogen Corporation (Lansing, Michigan, USA). Fresh stock solutions were prepared for each experiment to achieve the most accurate results.The electrochemical experiments were performed using a PalmSens 4 Potentiostat (Basi, Bioanalytical Systems Inc., USA) connected to a computer using the PalmSens PSTrace Software. SPE with carbon-based working (3 mm/0.071 cm2), counter electrodes, and silver reference were purchased from Zensor R&D, Taiwan. ELISA measurements were recorded using a BioTek Synergy H1 microplate reader. The X-ray photoelectron spectroscopy (XPS) analyses were carried out with a Kratos AXIS Supra X-ray photoelectron spectrometer using a monochromatic Al K(alpha) source (15 mA, 15 kV). The instrument work function was calibrated to give a binding energy (BE) of 83.96 eV for the Au 4f7/2 line for metallic gold, and the spectrometer dispersion was adjusted to give a BE of 932.62 eV for the Cu 2p3/2 line of metallic copper. The Kratos charge neutralizer system was used on all specimens. Survey scan analyses were carried out with an analysis area of 300 × 700 microns and a pass energy of 160 eV. High-resolution analyses were carried out with an analysis area of 300 × 700 microns and a pass energy of 20 eV. Spectra have been corrected to the C–C and C–H lines of the carbon 1 s spectrum (aliphatic carbon) set to 285.00 eV. Spectra were analyzed using CasaXPS software (version 2.3.14).
2.2. Modification of screen-printed electrodes using cocaine
At first, the electrodes were thoroughly rinsed with Milli-Q water and airdried. Then, 100 μL of PBS was dispensed on the electrode and performed SWV (parameters: equilibration time of 3 s, voltammetric potential scan from 0 to 1.5 V with a frequency of 15 Hz, an amplitude of 25 mV, and a step potential of 5 mV). This pre-treatment step was repeated three times per electrode. After that, a deposition solution (referred to as the COCi solution) composed of cocaine hydrochloride at different concentrations (Table S1.1†) was prepared and drop-casted on the working electrode (WE). The electrodes were air-dried for approximately six minutes and kept in a zipper bag containing oxygen adsorbent until further use (Scheme S1†).2.3. Cocaine attachment to working electrode
A modified version of an ELISA method was used to confirm the attachment of COC on the working electrode (Fig. S1†). Initially, several electrodes were modified using the COCi solution, while a control group was left un-modified. Then, gold nanoparticles (AuNPs) conjugated with a cocaine-binding antibody solution were pipetted onto the working electrode after the COCi solution was absorbed and kept for one hour at room temperature. Following another washing step with PBS, 3,3′,5,5′-tetramethylbenzidine (TMB), and H2O2 were pipetted on the electrode, and hydrogen peroxide (10%) was deposited on the electrode to stop the reactions. The solution on the electrodes was pipetted onto a 96-well plate and analyzed with a BioTek Synergy H1 microplate reader at a wavelength of 450 nm.2.4. Saliva collection
The fresh saliva samples were collected from healthy lab members in microtubes and used to prepare a series of cocaine dilutions (0, 10, 25, 50, 100, 250, 500, and 1000 ng mL−1). Then, an absorbent pad was placed inside the microtube to collect the different concentrated cocaine samples. This pad was then introduced into a syringe (3 mL) containing a glass wool filter. The absorbed pad was squeezed with a plunger, and filtered samples were collected into a new microtube. The recovered cocaine after filtration was analyzed using an ELISA cocaine oral fluid kit.2.5. Cocaine detection in PBS and human saliva
The COC-based electrodes were used for cocaine detection in PBS and saliva. A serial dilution method was used to prepare samples with different concentrations of cocaine hydrochloride in PBS ranging from 0 to 1000 ng mL−1 (Table S1.2†). Then, 100 μL of the samples in PBS were individually pipetted on the electrode to cover the entire area of the sensor. The parameters used for CV were as follows: equilibration time of 5 s, voltammetric potential scan from 0 to 1.5 V, E step of 0.01 V s−1, and a scan rate of 0.1 V s−1. It is expected that the intensity of the current is proportional to the amount of cocaine measured on the electrode. In the specific case of cocaine detection in saliva, once the samples were ready and the electrodes were modified, 65 μL of the sample was dispensed on the electrode and interrogated using the same cyclic voltammetry (CV) parameters stated previously, except for the potential range (0.4–1.9 V).2.6. Interference studies
Interference testing was carried out with popular drugs and common cutting agents found in cocaine use on the street, i.e., THC, ethanol, caffeine, and levamisole. Initial CV analysis was performed with each interferent at a high concentration of 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 ng mL−1 of cutting agents on bare/pristine/unmodified electrodes (P-Z) to identify their oxidation/reduction potential. The parameters of CV were the same as those used for the detection of cocaine. Once the signal of each potential interferent was identified, a lower concentration (50 ng mL−1) of each interferent was analyzed on the pristine electrode (P-Z) to examine analytes interaction at low concentrations on a P-Z. A cocaine concentration of 50 ng mL−1 in PBS, mixed with each adulterant, and in saliva was detected using the modified electrode m-Z-COC.
000 ng mL−1 of cutting agents on bare/pristine/unmodified electrodes (P-Z) to identify their oxidation/reduction potential. The parameters of CV were the same as those used for the detection of cocaine. Once the signal of each potential interferent was identified, a lower concentration (50 ng mL−1) of each interferent was analyzed on the pristine electrode (P-Z) to examine analytes interaction at low concentrations on a P-Z. A cocaine concentration of 50 ng mL−1 in PBS, mixed with each adulterant, and in saliva was detected using the modified electrode m-Z-COC.
3. Results and discussion
3.1. Bare SPE performance for cocaine detection
According to several sources, the oxidation of COC on graphite SPE occurs in the potential range of 1 to 1.1 V.17–19 Depending on the type of electrode used, the potential oxidation range of cocaine can vary. Therefore, the first step towards the detection of cocaine on graphite SPE electrodes was to identify the cocaine oxidation peak. Thus, high concentrations of cocaine dissolved in PBS were interrogated using SWV (5 s for equilibration time, 0.05 V and 0 s as precondition potential, 0.9–1.2 V range for the voltammetric potential scan with a frequency of 15 Hz, 25 mV of amplitude, and 5 mV for the step potential). Concentrations from 0 to 50000 ng mL−1 (50 μg mL−1) of cocaine in PBS were prepared via serial dilution. High concentrations of cocaine in PBS showed sharp and well-defined peaks between 1 and 1.1 V (Fig. 1a). According to the literature, the proposed mechanism focuses on the electrochemical oxidation of the tertiary amine group found in cocaine molecules. Additionally, the oxidation process includes the detachment of an electron from the nitrogen in the amino functional group, followed by a proton loss to form a neutral radical. The neutral radical loses an electron and is then hydrolyzed, producing secondary amine, norcocaine, and an aldehyde.15
 | ||
| Fig. 1 a) SWV signals of different amounts of cocaine and b) calibration curve for cocaine detection in PBS using P-Z. | ||
Besides, as the concentration decreased, the current intensity became visibly weaker on bare electrodes. Also, any concentration lower than 2500 ng mL−1 (2.5 μg mL−1) of cocaine in PBS did not develop a visible signal, and as the concentration decreased further, the signal was no longer visible. On a P-Z electrode, the lowest concentration that can be detected is LODbare electrode = 1269 ng mL−1 (Fig. 1b), which does not comply with any of the cut-offs set by drug safety enforcement organizations such as DRUID or the ROSITA project.5,6 The completion of this analysis evidenced that the modification of the electrodes was necessary to enhance cocaine detection.
3.2. Modified sensor performance
The electrode modification was confirmed by the ELISA method (Fig. S1†). At first, Au NPs of ∼40 nm in size and with an absorbance peak at 532 nm have been synthesized (Fig. S1B†). The nanozymatic activity of as-synthesized Au NPs was utilized to confirm the attachment of cocaine on the SPE surface (Fig. S1C†). A higher absorbance value of the modified electrode compared to the unmodified oen revealed the successful surface modification of SPE by cocaine (Fig. S1C†). Moreover, the SEM image also clearly shows a morphological differences of pristine SPE and cocaine modified SPE (Fig. S1D and E†). The primary purpose of modifying electrodes is to increase the selectivity and sensitivity toward the desired analyte. Previous reports by Ortega et al. and Renaud-Young et al.20,21 demonstrated the use of electrodes being modified by using the same analyte during manufacturing as linker points to facilitate the interaction between the analytes dispersed on the samples and the electrode surface.The improved affinity to the desired analytes could be related to the enhanced physical interactions and/or chemical coupling. After such interaction occurs, a potential rule is applied to an electrochemical reaction that produces a signal characteristic of the analyte and its concentration. Therefore, the design of the novel biomolecule-free sensor for cocaine detection includes the use of the same analyte being detected to modify the electrode. In this case, the electrodes were modified by the deposition of the cocaine solution (COCi) on the working electrode, which is absorbed on the surface of the working electrode. This methodology is accompanied by electrochemical interrogation to immobilize the analyte on the working electrode.8,9 However, during experimental research, it was established that once cocaine oxidizes on the electrode, there are no adduct molecules remaining. The adduct molecules that remain in the case of Renaud-Young et al. and Ortega et al.20,21 facilitate the further oxidation of other molecules found in the samples. Since all the cocaine molecules oxidize under CV interrogation, the improvement of cocaine detection could be related to physical interactions and chemical coupling. The physical adsorption preserves the activity and the stability of the cocaine analyte.22 To the best of the authors' knowledge, this approach has never been implemented previously or manufactured for the electrochemical detection of cocaine. However, similar work has been attempted to detect other drugs.20,21
Two different approaches were evaluated to implement the cocaine deposition on the Zensor working electrode. The first approach included the preparation of a cocaine solution in a mix of H2O:MetOH (9:1) before deposition (m-Z-COC*) (Fig. 2e, I). In the second case, cocaine hydrochloride was dispersed in a mix of PBS:MetOH (9:1) (Fig. 2e, II). XPS and CV techniques were used to elucidate the interaction of cocaine with the WE surface in both conditions (I and II). Two peaks at 399.0 and 401.9 eV, attributed to amine/amide and protonated amine, respectively, were employed to fit the N 1s high-resolution signal after COC/H2O:MetOH deposition on the working electrode (m-Z-COC*) (Fig. 2a, Table S2.4†). However, for this specific case, m-Z-COC*, the atomic percent of the second contribution corresponding to the protonated amine (43.1%) was higher than the one obtained for the m-Z-COC when COC/PBS:MetOH is employed for deposition (Fig. 2b, Table S2.4†). The presence of a higher number of protonated amine (condition I) on the working electrode does not allow the oxidation of the tertiary amine groups on the cocaine structure, avoiding further detection of the cocaine analyte (Fig. 2c and e). Meanwhile, in the deposition based on PBS:MetOH, the buffer fixes a local pH on the working electrode, allowing it to deprotonate the COCi amine, favoring the tertiary amine oxidation (Fig. 2d and e). On the same note, the pH of deionized water ranges from 6 to 6.4, whereas the pH of the PBS solution used was 7.4, which is closer to the pKa of cocaine at 8.6. Therefore, condition II, PBS:MetOH (9:1), will be used to prepare the cocaine solution for further experiments.
 | ||
| Fig. 2 N 1s high-resolution spectra of a) m-Z-COC* and b) m-Z-COC. Cocaine detection using c) m-Z-COC* and d) m-Z-COC electrodes employing CV. e) Schematic representation of the electrochemical oxidation mechanism for cocaine. | ||
The working electrodes of the Zensor SPE were modified with the cocaine solution (COCi) prepared using specifications defined in condition II (Fig. 3a) for further studies and optimization. One of the first parameters to be optimized was the amount of COCi that would be used to modify the working electrode. Fig. 3b demonstrates that when the electrode is not modified with the COCi solution, there is no electrochemical signal; however, the m-Z-COC electrode provides a distinguishable signal between 0 and 50 ng mL−1. In order to optimize the signal and study electrode saturation, various amounts of COCi were used to modify the electrode (100, 150, 200, and 500 ng of cocaine). Other optimized parameters include drying the electrodes after modification, varying scan rates during CV interrogation, and latency periods after depositing the cocaine sample on the m-Z-COC sensor (Fig. S2†). In an attempt to follow the traditional electrodeposition method, immediately after dispensing COCi deposition on the working electrode, the electrodes were interrogated with PBS under SWV parameters to immobilize the cocaine. However, in this case, the cocaine was fully oxidized on the first scan (Fig. 3b). After an initial scan, no cocaine was evidenced, resulting in a loss of amperometric amplitude. Therefore, the electrodeposition step was removed from the protocol, and an absorption deposition was employed, which increased signal strength and helped overcome this issue.
 | ||
| Fig. 3 a) Schematic of electrode modification. b) SWV detection of cocaine (50 ng mL−1) using P-Z and m-Z-COC in PBS. c) Cocaine detection using different amounts of COCi to modify the electrode, and d) calibration curve for cocaine detection in PBS using m-Z-COC (COCi 150 ng). | ||
Before starting the optimization of the amount of cocaine (COCi) to be deposited, the scan rate parameters of CV were optimized since it also affects the intensity of the peak alongside the optimization of how to dry the cocaine solution on the working electrode (Fig. S2B†). Subsequently, different amounts of COCi were used to modify the electrode surface, which drastically impacted the overall results. Fig. 3c shows that using a low amount, such as 100 ng COCi, does not provide accurate results. After testing the 75 ng mL−1 of cocaine in the PBS sample, the intensity decreases for 100 ng mL−1 when, in theory, it should increase since there is more cocaine present. Nonetheless, 150 ng COCi provides excellent results that distinguish between 0 and 50 ng mL−1 easily. Also, the intensity is higher when detecting 100 ng mL−1 of cocaine in PBS (Fig. 3c). Although 200 ng COCi has the greatest intensity with the proper pattern, the difference between the concentrations is not as noticeable as with 150 ng COCi. Additionally, a calibration curve of different cocaine concentrations vs. current was plotted using 150 ng COCi-modified electrodes in PBS (Fig. 3d). The results revealed that cocaine concentration was linearly correlated up to 50 ng mL−1, and the calculated LoD of cocaine in PBS using this novel cocaine-based sensor was 1.73 ng mL−1, which is well below the official cut-off.
On the other hand, once the m-Z-COC was evaluated in the detection of cocaine between 0 to 100 ng mL−1, it was essential to assess if higher concentrations followed the same pattern of current intensity correlating to the analyte concentration. As seen in Fig. 3c, using 500 ng COCi, the hook effect at 100 ng mL−1 was evidenced because the current intensity of the concentration begins to decrease and fall below 50 ng mL−1. Experiments were performed with concentrations up to 1000 ng mL−1 (Fig. S3†). The primary goal of this novel biomolecule-free sensor, m-Z-COC, was to detect from 0 to 50 ng mL−1. Therefore, higher concentrations should not fall below the intensity of 50 ng mL−1. In addition to finding that concentrations up to 1000 ng mL−1 were not below the strict cut-off limit with PBS-spiked samples, data were subsequently collected using saliva as the study medium.
3.3. Electrode characterization
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C aromatic, C–C/C–H, C–OH/C–O–C/C–Cl, and O–CO, respectively (Fig. 4b, Table S2.2†). After COC modification, four signals were also employed to fit the C 1s signals for the rest of the samples (m-Z-COC, m-Z-COC1, and m-Z-COC*). However, in the specific case of m-Z-COC, a decrease in the first contribution (aromatic CC) is observed, which could be related to fewer CC functional groups in the cocaine structure than in the graphite-based ink of the WE.
C aromatic, C–C/C–H, C–OH/C–O–C/C–Cl, and O–CO, respectively (Fig. 4b, Table S2.2†). After COC modification, four signals were also employed to fit the C 1s signals for the rest of the samples (m-Z-COC, m-Z-COC1, and m-Z-COC*). However, in the specific case of m-Z-COC, a decrease in the first contribution (aromatic CC) is observed, which could be related to fewer CC functional groups in the cocaine structure than in the graphite-based ink of the WE.
 | ||
| Fig. 4 a) Element quantification from survey spectra; b) and c), C 1s and d)–f) N 1s high-resolution spectra of P-Z and m-Z-COC. | ||
The high-resolution signal of N 1s was deconvoluted into two contributions for all the samples (P-Z, P-Z-PBS, m-Z-COC, m-Z-COC1, and m-Z-COC*) (Table S2.4†). In the P-Z electrode after PBS cleaning using CV (m-Z-PBS), the first contribution at 399.9 eV was related to amine/amide from the ink of the WE.23 The second contribution at higher binding energies (401.9 eV) is attributed to protonated amines.24 After cocaine electrode modification, slight changes are observed in both contributions. The peak related to amine/amide increased (1.6%) due to the presence of the tertiary amine group in the cocaine structure incorporated on the WE surface (Fig. 4d, Table S2.4†). However, when the COC-based electrode is interrogated using CV in PBS, significant changes are observed in both peaks of N 1s high-resolution signal (Fig. 4f, Table S2.4†). The atomic percent of the first contribution related to amine functional groups increased from 68.4 to 84.9; meanwhile, the percent of the second contribution (protonated amines) decreased from 31.4 to 15.1 (Table S2.4†). This result confirms the oxidation of the tertiary amine group present in the cocaine structure. In the oxidation of this amine group, the abstraction of an electron from the nitrogen is carried out, followed by proton loss, which forms a neutral radical. The neutral radical loses an electron and is hydrolyzed to the products. The oxidation reaction leads to a secondary amine, norcocaine, and an aldehyde17 (Fig. 4e). Thus, XPS corroborated that the amine group plays the main role in the electrochemical oxidation of cocaine molecules on the surface of the WE.
| Ip = (2.69 × 105)Con3/2γ1/2D1/2A, | (1) |
 | ||
| Fig. 5 (a) Configuration of a Zensor electrode. (b) CV response of a P-Z with different scan rates. (c) CV response of m-Z-COC with different scan rates. (d) Magnified cocaine peak in m-Z-COC scans. | ||
From this electrochemical analysis, it was also established that the oxidation of cocaine is an irreversible reaction. As seen in Fig. 5b, the peaks of the voltammogram are close together, indicating the reversibility of the K4[FeCN6] (FeCN) 0.1 mM in KCl 0.1 M reaction on a P-Z. However, Fig. 5c and d exhibit the voltammograms of electrodes that have been modified with COCi, and the distance between each peak is greater than 60 mV, which indicates an irreversible reaction.25
Fig. 5b shows the scans of P-Z, while Fig. 5c represents cocaine peaks at different scan rates when m-Z-COC is employed. In the magnified image (Fig. 5d), the peaks are sharper and more defined for higher scan rates, 0.175 V s−1 and above, whereas lower scan rates develop broad and weak peaks. This information is prevalent and provides a better understanding of how cocaine molecules act in an electrochemical setting. To further analyze the kinetics of the cocaine oxidation of a carbon electrode, CV was used with various scan rates from 0.025 to 0.2 mV s−1 using 50 ng mL−1 of cocaine in PBS. Given that the current increased linearly with the square root of the scan rate, it can be concluded that the cocaine oxidation follows a diffusion-controlled process, which is supported by Mirceski et al.26 The electrodes were further characterized by analyzing the cocaine attachment to the carbon SPE using a modified ELISA (sections S1 and S2 of the ESI†).
3.4. Cocaine detection in saliva
After optimizing the detection of cocaine in PBS and understanding the electrochemical performance of the m-Z-COC, the next step was to assess the designed sensor in human saliva. One of the major steps was to analyze the recovery of cocaine in saliva after filtration using an ELISA test to evaluate samples of cocaine in saliva before and after filtration (Fig. S4†). Results from this experiment demonstrated that the recovery percentage of cocaine after filtering the oral fluid was 95% for concentrations of the drug ranging from 10 to 50 ng mL−1. As the drug concentration in saliva increased (250 to 1000 ng mL−1), the recovery rate decreased to 74%. The overall outcome was the filtering device did not overly impact the detection (S6†).Regarding the detection of cocaine in saliva, after CV interrogation, at first glance, there was no visible peak of cocaine oxidation in the expected potential region of 1–1.1 V (Fig. 6a). The only visible peak was a large and sharp peak at around 1.4 V, which could be attributed to one or a few of the electroactive components found in saliva. In order to obtain a visible signal, a subtraction of the curve's method was implemented to overcome the imminent challenge. While m-Z-COC were scanned with specific concentrations of cocaine in saliva, a P-Z was also scanned with the same spiked samples. This arrangement is like those found in electronic tongues (ET), where sensors work together to identify a specific analyte.25 It has been established that a P-Z cannot detect low concentrations of cocaine either in PBS or in saliva. Hence, the signal obtained from the P-Z scanned with a spiked sample was representative of the electroactive interferents found in saliva. By subtracting the data obtained for the P-Z from the signal received from a m-Z-COC, the signal demonstrates the correlation between current intensity and analyte concentration.
 | ||
| Fig. 6 (a) and (b) Comparison between original signal and signal subtracted from a spiked P-Z (c) SD analysis of various concentrations of cocaine in saliva. | ||
As shown in Fig. 6a, there is no signal for the oxidation of cocaine present. Once the signal from the m-Z-COC is subtracted from the signal of the P-Z, a peak appears precisely where the oxidation peak of cocaine is expected (Fig. 6b). The peak intensity can be visualized after manually evaluating the peak intensity of 0, 25, 50, and 100 ng mL−1 cocaine solutions (Fig. 6c). However, there is no clear separation between 10 ng mL−1 and 25 ng mL−1, but the error bar on 100 ng mL−1 overlaps with the error bar on 50 ng mL−1. The variation that is evidenced in Fig. 6c can be attributed to several factors, including the deposition of the COCi solution and the dispersed concentrations of cocaine in saliva. Overall, the subtraction method was neither efficient nor effective for every saliva sample. While some samples followed a pattern of current intensity correlating with concentration, the data had too much variation. The challenge of saliva-to-saliva variation was pertinent in this type of analysis because saliva can vary according to individual's diets, gender, and age. In analyzing several saliva samples, it was clear that saliva-to-saliva variation was a significant issue (Fig. S5†). To further explore the potential of m-Z-COC, machine learning (ML) was introduced to analyze sensor analytical responses to overcome saliva complications in electrochemical sensing and saliva-to-saliva variation.
3.5. Sensor selectivity
Substances such as caffeine and levamisole are often found in cocaine as adulterants since they can stimulate the effects of cocaine.27 THC and ethanol were also explored since they are substances frequently consumed by cocaine users.16 Initially, each drug was individually scanned on various P-Z at a high concentration (50000 ng mL−1) to identify the direct oxidation signal, which would later be beneficial in analyzing the interference with the identifiable peak of cocaine at 1–1.1 V (Fig. S6†). The peak for THC was the farthest from the oxidation peak of cocaine at around 0.5 V. Similarly, the oxidation peaks for ethanol and caffeine were around 1.4 V, which were far enough from the cocaine peak not interfiering during the analysis. Levamisole, one of the most used cutting agents (found in over 45% of cocaine street samples),27 had a peak very close to cocaine at 1.16 V with a high current intensity of 30.3 μA. After identifying the oxidation peak for each interferent, a smaller concentration of 50 ng mL−1 of each substance in PBS was scanned on a P-Z. As expected, such low concentrations of any substance were not visible on P-Z. Still working with PBS, several m-Z-COC were prepared to examine each substance on a m-Z-COC. Since the electrodes are modified with cocaine, it is expected that they will have a greater affinity for the cocaine samples and treat the samples without cocaine as zeros. However, given that all the substances used are electroactive in nature, it is not rare that their electroactivity would impact the intensity of the current (Fig. S5†).
To further analyze the affinity of the m-Z-COC to cocaine, the oxidation potential range of cocaine (1–1.1 V) was analyzed as the area of interest. The interferents in saliva were interrogated on m-Z-COC, and the difference in current between the interferents and cocaine can be seen in Fig. 7(a and b). Although there is zero cocaine in the samples interrogated when scanning the different interferents, these samples are inherently electroactive, meaning that their current values were not the same as the current value when interrogating just saliva or just PBS. However, the different oxidation potential of these interferents as well as their different current intensities, are a proper indicator that the analyte being detected is not cocaine. ML was also implemented to test the selectivity of the m-Z-COC. It was determined that using binary analysis, the model was able to differentiate between the samples that had cocaine and samples that did not have cocaine (besides the cocaine in the working electrode). For example, one sample of levamisole-spiked saliva interrogated on a modified electrode was classified as not having cocaine by the model. Fig. 7C displays the voltammograms of benzocaine, procaine, lidocaine, ascorbic acid, and uric acid, showing distinct electrochemical signatures compared to cocaine.
 | ||
| Fig. 7 (a) Current intensity difference between adulterant and cocaine. (b) Peak intensities of the different interferents in the sample (zero is a blank with no drug present). (c) Voltammograms of different interferents and cocaine. | ||
3.6. Study of stability and reproducibility of the sensor
The modified SPE electrodes were used for the accelerated stability, reproducibility, and repeatability studies through CV technique (Fig. S7A–C†). The stability of the electrodes was assessed up to 60 days, and the results are presented in Fig. S7A.† No significant drop in electrode current was observed up to 60 days in this study. To check the repeatability of the present study, five separate modified SPEs are utilized. Simultaneously, five separate experiments with the same electrode were executed for the repeatability study. Fig. S7B and C† demonstrate minimal variation in the current responses of individual electrodes, highlighting the excellent repeatability and reproducibility of the modified electrodes.3.7. Machine learning algorithms
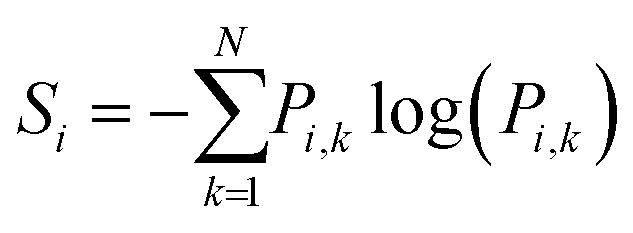 | (2) |
 | (3) |
SVM models are used to separate classes to obtain the most significant margin possible using hyperplanes. Hyperplanes are decision boundaries that aid in the classification of the data points. Support vectors are the nearest points to the hyperplane's margin. The outlier points determine the position and orientation of the hyperplane. Also, the Kernel functions facilitate the transformation of the data required from its original dimension into a higher-dimension space. The functions base the transformations on the similarity and distances found between two data points in their original dimension. The independent features used, as well as the dimension of the databases, drastically impact the performance of SVM methods. This research used the Gaussian radial basis function (RBF) as a kernel function.
One of the splitting processes used was the Gini impurity approach to analyze the data of various concentrations of cocaine in saliva. This approach is used to decide the most effective split from a root node and other subsequent splits. Although there were eight classes (0, 10, 25, 50, 100, 250, 500, 1000), only ternary and quaternary classification was performed (Tables S3.1–S3.4†). Due to the nature of the data, having eight individual classes would introduce too much variability. The distribution for the ternary classification was 30 (0 ng mL−1), 30 (10 ng mL−1), 180 (≥25 ng mL−1), and for quaternary, 58 (0 ng mL−1), 40 (10 ng mL−1), 40 (25 ng mL−1) and 40 (50 ng mL−1). Depending on the classification used (ternary or quaternary), the accuracy of the results changed from 78–85% in the testing set and 99–100% in the training set. The highest accuracy with ternary classification came from distinguishing between 0, 10, and 25 ng mL−1 with 85% accuracy. Using quaternary classification increased the range of concentrations used, and the set with the highest accuracy included 0, 10, 25, and 50 ng mL−1, obtaining 80% accuracy.
4. Conclusions
In this study, a novel biomolecule-free cocaine-based sensor for the detection of cocaine is presented with high sensitivity and selectivity. The electrochemical sensor uses the physical interactions between the carbon working electrode and the cocaine that helped to absorb cocaine on the WE surface. Then, the affinity towards cocaine samples allowed the detection of cocaine at low concentrations in PBS and saliva media. While the detection of cocaine in PBS was straightforward, several issues arose with saliva as it is a recognized complex medium. Unwanted particles in the oral fluid (such as food) were removed from the spiked saliva by filtration with glass wool. Then, ML methods were used to analyze the data due to the noisy nature of sensor signals and saliva interference. The highest accuracy achieved for the detection of cocaine in saliva was 85% using ML methods. Additionally, m-Z-COC can distinguish between adulterants and cocaine with 100% accuracy, which shows the real-life applicability of the proposed novel sensor.Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to acknowledge the support of Collaborative Research and Development Grants and the Discovery grant from the Natural Sciences and Engineering Research Council of Canada, Mitacs, and eye3Concepts Inc. for funding this research.References
- L. Gao, H. Wang, Z. Deng, W. Xiang, H. Shi and B. Xie, et al., Highly sensitive detection for cocaine using an aptamer–cocaine–aptamer method, New J. Chem., 2020, 44, 2571–2577, 10.1039/C9NJ05147C.
- F. P. Busardò, S. Pichini, M. Pellegrini, A. Montana and A. F. Faro, et al., Correlation between Blood and Oral Fluid Psychoactive Drug Concentrations and Cognitive Impairment in Driving under the Influence of Drugs, Curr. Neuropharmacol., 2018, 16, 84, DOI:10.2174/1570159X15666170828162057.
- T. R. Fiorentin, J. N. Scherer, M. C. A. Marcelo, T. R. V. Sousa, F. Pechansky and M. F. Ferrão, et al., Comparison of Cocaine/Crack Biomarkers Concentrations in Oral Fluid, Urine and Plasma Simultaneously Collected From Drug Users, J. Anal. Toxicol., 2018, 42, 69–76, DOI:10.1093/JAT/BKX085.
- K. N. Ellefsen, M. Concheiro, S. Pirard, D. A. Gorelick and M. A. Huestis, Oral fluid cocaine and benzoylecgonine concentrations following controlled intravenous cocaine administration, Forensic Sci. Int., 2016, 260, 95–101, DOI:10.1016/J.FORSCIINT.2016.01.013.
- Rosita n.d. http://www.rosita.org/ (accessed March 1, 2022).
- Driving Under the Influence of Drugs, Alcohol and Medicines in Europe — findings from the DRUID project | https://www.emcdda.europa n.d. https://www.emcdda.europa.eu/publications/thematic-papers/druid_en (accessed March 1, 2022).
- K. Abnous, N. M. Danesh, M. Ramezani, S. M. Taghdisi and A. S. Emrani, A novel colorimetric aptasensor for ultrasensitive detection of cocaine based on the formation of three-way junction pockets on the surfaces of gold nanoparticles, Anal. Chim. Acta, 2018, 1020, 110–115, DOI:10.1016/J.ACA.2018.02.066.
- N. Tavakkoli, N. Soltani and F. Mohammadi, A nanoporous gold-based electrochemical aptasensor for sensitive detection of cocaine, RSC Adv., 2019, 9, 14296–14301, 10.1039/C9RA01292C.
- T. Yilmaz Sengel, E. Guler, M. Arslan, Z. P. Gumus, S. Sanli and E. Aldemir, et al., “Biomimetic-electrochemical-sensory-platform” for biomolecule free cocaine testing, Mater. Sci. Eng., C, 2018, 90, 211–218, DOI:10.1016/J.MSEC.2018.04.043.
- Saliva screening test for 5 drugs - NarcoCheck n.d. https://www.narcocheck.com/en/saliva-drug-tests/multi-drugs-saliva-test-5in1.html (accessed March 1, 2022).
- M. Asbridge, R. Ogilvie and R. Associate, A Feasibility Study of Roadside Oral Fluid Drug Testing, 2015 Search PubMed.
- D. A. Labianca, Non-foolproof nature of slope detection technology in the Dräger Alcotest 9510, Forensic Toxicol., 2017, 36(1), 222–224, DOI:10.1007/S11419-017-0373-X.
- A. J. Krotulski, A. L. A. Mohr, M. Friscia and B. K. Logan, Field Detection of Drugs of Abuse in Oral Fluid Using the Alere™ DDS®2 Mobile Test System with Confirmation by Liquid Chromatography Tandem Mass Spectrometry (LC–MS/MS), J. Anal. Toxicol., 2018, 42, 170–176, DOI:10.1093/JAT/BKX105.
- L. T. Sniegoski, J. Waddell, M. J. Welch, A. A. Fatah, M. Gackstetter and R. Q. Thompson, Evaluation of Oral Fluid Testing Devices, n.d.
- F. Cui, Y. Yue, Y. Zhang, Z. Zhang and H. S. Zhou, Advancing Biosensors with Machine Learning, ACS Sens., 2020, 5, 3346–3364, DOI:10.1021/ACSSENSORS.0C01424.
- D. Ortiz-Aguayo, X. Cetó, K. de Wael and M. del Valle, Resolution of opiate illicit drugs signals in the presence of some cutting agents with use of a voltammetric sensor array and machine learning strategies, Sens. Actuators, B, 2022, 357, 131345, DOI:10.1016/J.SNB.2021.131345.
- J. M. P. J. Garrido, F. Borges, C. M. A. Brett and E. M. P. J. Garrido, Carbon nanotube β-cyclodextrin-modified electrode for quantification of cocaine in seized street samples, Ionics, 2016, 22, 2511–2518, DOI:10.1007/S11581-016-1765-3.
- A. B. D. da Silva, A. S. Castro and M. F. Oliveira, Development of Carbon Paste Electrode Chemically Modified with Schiff Base Complexes for Forensic Analysis of Cocaine, Braz. J. Anal. Chem., 2022, 106–117, DOI:10.30744/brjac.2179-3425.AR-17-2021.
- N. A. Abdelshafi, J. Bell, K. Rurack and R. J. Schneider, Microfluidic electrochemical immunosensor for the trace analysis of cocaine in water and body fluids, Drug Test. Anal., 2019, 11, 492–500, DOI:10.1002/DTA.2515.
- G. A. Ortega, S. R. Ahmed, S. K. Tuteja, S. Srinivasan and A. R. Rajabzadeh, A biomolecule-free electrochemical sensing approach based on a novel electrode modification technique: Detection of ultra-low concentration of Δ9-tetrahydrocannabinol in saliva by turning a sample analyte into a sensor analyte, Talanta, 2022, 236, 122863, DOI:10.1016/J.TALANTA.2021.122863.
- M. Renaud-Young, R. M. Mayall, V. Salehi, M. Goledzinowski, F. J. E. Comeau and J. L. MacCallum, et al., Development of an ultra-sensitive electrochemical sensor for Δ9-tetrahydrocannabinol (THC) and its metabolites using carbon paper electrodes, Electrochim. Acta, 2019, 307, 351–359, DOI:10.1016/J.ELECTACTA.2019.02.117.
- R. J. Gillams, C. D. Lorenz and S. E. Mclain, On the hydration and conformation of cocaine in solution, Chem. Phys. Lett., 2017, 676, 58–64, DOI:10.1016/j.cplett.2017.03.040.
- H. Hantsche, High resolution XPS of organic polymers, the scienta ESCA300 database, ed. G. Beamson and D. Briggs, Wiley, Chichester, 1992, p. 295, DOI:10.1002/adma.19930051035.
- X-ray Photoelectron Spectroscopy (XPS) Reference Pages: Spin Orbit Splitting n.d. http://www.xpsfitting.com/2012/08/spin-orbit-splitting.html (accessed April 25, 2022).
- P. Krishnaveni and V. Ganesh, Electron transfer studies of a conventional redox probe in human sweat and saliva bio-mimicking conditions, Sci. Rep., 2021, 11, 7663, DOI:10.1038/s41598-021-86866-z.
- V. Mirceski, S. Komorsky-Lovric and M. Lovric, Square-Wave Voltammetry: Theory and Application, https://books.google.ca/books?hl=en&lr=&id=EuNbBfNsXn4C&oi=fnd&pg=PA1&dq=info:Id0e2k8fkJEJ:scholar.google.com&ots=9dt34SDdCt&sig=HvvoZDNsoq1iQtO-WenPWKLDO1g&redir_esc=y#v=onepage&q&f=false, (accessed March 1, 2022) Search PubMed.
- M. de Jong, A. Florea, A. M. de Vries, A. L. N. van Nuijs, A. Covaci and F. van Durme, et al., Levamisole: A Common Adulterant in Cocaine Street Samples Hindering Electrochemical Detection of Cocaine, Anal. Chem., 2018, 90, 5290–5297, DOI:10.1021/ACS.ANALCHEM.8B00204/SUPPL_FILE/AC8B00204_SI_001.PDF.
- Y. Vlasov, A. Legin, A. Rudnitskaya, C. di Natale and A. D'Amico, Nonspecific sensor arrays (“electronic tongue”) for chemical analysis of liquids: (IUPAC technical report), Pure Appl. Chem., 2005, 77, 1965–1983, DOI:10.1351/PAC200577111965/MACHINEREADABLECITATION/RIS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5lf00006h |
| This journal is © The Royal Society of Chemistry 2025 |