
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAmplification-free CRISPR/Cas based dual-enzymatic colorimetric nucleic acid biosensing device†
Guodong
Tong
a,
Pabitra
Nath
 b,
Yuki
Hiruta
a and
Daniel
Citterio
*a
b,
Yuki
Hiruta
a and
Daniel
Citterio
*a
aDepartment of Applied Chemistry, Faculty of Science and Technology, Keio University, 3-14-1 Hiyoshi, Kohoku-ku, Yokohama 223-8522, Japan. E-mail: citterio@applc.keio.ac.jp; Tel: +81 45 566 1568
bDepartment of Physics, Tezpur University, Sonitpur, Napaam, Assam 784028, India
First published on 2nd January 2025
Abstract
Nucleic acid testing (NAT) is widely considered the gold standard in analytical fields, with applications spanning environmental monitoring, forensic science and clinical diagnostics, among others. However, its widespread use is often constrained by complicated assay procedures, the need for specialized equipment, and the complexity of reagent handling. In this study, we demonstrate a fully integrated 3D-printed biosensensing device employing a CRISPR/Cas12a-based dual-enzymatic mechanism for highly sensitive and user-friendly nucleic acid detection. A plastic probe stick was designed to host small-sized gold nanoparticles, enhancing enzyme labeling density. Alkaline phosphatase (ALP) was then conjugated via single-stranded DNA, requiring only a single enzyme substrate addition to generate a simple visual signal change. This approach eliminates the need for amplification or centrifugation steps, achieving a limit of detection (LOD) as low as 10 pM – among the highest sensitivities reported for amplification-free colorimetric nucleic acid detection. Furthermore, we developed a device that incorporates this probe stick, integrates all necessary reagents, and features a smartphone-compatible accessory for quantitative analysis. This allows end-users to perform visual or quantitative DNA analysis with simple operations, achieving a visual detection limit of approximately 100 pM, comparable to other CRISPR-based non-amplified nucleic acid detection methods. Additionally, the system successfully distinguished perfectly matched from mismatched nucleic acid sequences, demonstrating its specificity and versatility. Although certain design limitations affected the sensitivity of the integrated device compared to the probe stick alone, the simplicity and portability of this device make it a promising tool for rapid nucleic acid screening in clinical diagnostics, environmental monitoring, and food safety control. This study paves the way for the development of practical biosensors for point-of-care testing (POCT) applications.
Introduction
Nucleic acid testing (NAT) has been widely used in various fields, such as environmental DNA monitoring,1–3 forensic science,4 food safety,5–7 and, most importantly, clinical diagnostics.8–10 To date, the gold standard of NAT involves amplification methods including polymerase chain reaction (PCR), loop-mediated isothermal amplification (LAMP), and recombinase polymerase amplification (RPA), among others. In recent years, the clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated protein (Cas) system has gained significant attention in the field of analytical chemistry.11,12 The unique collateral nuclease activity of some CRISPR/Cas systems, activated by the recognition of target nucleic acids, enables highly efficient collateral cleavage, termed trans-cleavage, of surrounding non-target nucleic acids. Notably, by editing and utilizing CRISPR RNA (crRNA), the CRISPR/Cas system can flexibly recognize any desired complementary sequence.13,14 Based on this mechanism, “SHERLOCK”, one of the first analytical assay methods to utilize the CRISPR/Cas system, was reported in 2017.15 In this approach, the Cas protein is guided to target nucleic acids by a complimentary crRNA sequence, triggering collateral activity to cleave short reporter nucleic acids labeled with a quencher and fluorophore at each end, ultimately generating a strong fluorescence signal.16–19 Some CRISPR/Cas systems have achieved unprecedented high sensitivity in NAT for clinical diagnostics, with limits of detection (LOD) reaching at least femtomolar or attomolar levels, surpassing most conventional methods.20,21 Furthermore, when combined with nucleic acid pre-amplification, the use of CRISPR/Cas also addresses issues such as false-positive signals caused by amplicon cross-contamination or inappropriate primer design. However, the pre-amplification step, which relies on trained operators and highly specialized, often bulky instruments, limits its application in point-of-care testing (POCT).To overcome these drawbacks, many researchers are utilizing the CRISPR/Cas system to develop detection methods for non-amplified nucleic acids.22,23 For example, CRISPR/Cas has been combined with electrochemical sensing,24,25 microchambers,26,27 electrochemiluminescence,28 surface-enhanced Raman spectroscopy,29 and metal-enhanced fluorescence.30 These methods provide high sensitivity with LODs comparable to amplification-based methods. Nonetheless, electrochemical sensing often requires lengthy detection times, and fluorescence signals depend on costly and precise optical instruments, limiting their feasibility for widespread on-site applications.
Colorimetry-based sensing methods consistently exhibit characteristics such as simplicity, low cost, and the ability for direct visual discrimination. Yuan et al. reported a gold nanoparticle (AuNP)-based naked-eye gene detection method, where the target-induced collateral cleavage activity of CRISPR/Cas12a triggers either the aggregation or dispersion of DNA-labeled AuNP probes.31 In addition, many other reports have utilized secondary enzymes that directly trigger significant colour changes upon the addition of substrates.32–35 However, additives such as antibodies, hydrogen peroxide, and pH indicators are typically required, along with complex multi-step procedures including for example centrifugation. These requirements limit their practical application for POCT to some extent. Plastic has been used as a substrate in POCT platforms, not only because of its lightweight nature, ease of molding, and suitability for commercial manufacturing – meeting the ASSURED conditions stipulated by the WHO (affordable, sensitive, specific, user-friendly, rapid and robust, equipment-free, deliverable) – but also for its utility as miniaturized containers for liquid storage and release, as well as its function as a moisture vapor barrier for easy use.36,37 Matsumoto et al. reported the covalent immobilization of nanoparticles and biomolecules on an acrylic resin material,38 which served as inspiration for this work. Building on this, a fully integrated device for a simple colorimetric NAT method has been developed. A prototype device was fabricated using 3D printing technology, which offers great flexibility in creating complex designs. The device integrates the CRISPR/Cas12a target recognition system with alkaline phosphatase (ALP) as a secondary enzyme, significantly enhancing the colorimetric signal in the presence of small amounts of target double-stranded DNA. This characteristic enables users to visually determine assay results by the naked eye. All the reagents necessary for the detection process are pre-integrated into the device, allowing users to perform the assay by simply adding the sample, rotating and pressing the cap. The incubation takes place in a sealed environment, eliminating concerns about solution evaporation. Furthermore, a smartphone-based detection system has been incorporated for quantitative analysis. Users can obtain quantitative data via an app by inserting the device into a specially designed smartphone accessory. Although the sensitivity of this method may not yet match the gold standard of amplification-based nucleic acid tests, its simplicity reduces the reliance on specialized equipment operated by trained experts, enabling preliminary screening performed by laypersons at the point-of-care. This method is potentially applicable for detecting nucleic acids with any sequence, making it an effective tool for disease outbreak control and improving public health responses.
Experimental section
Materials and instruments
Firstly, all nucleic acids utilized in this work, including double-stranded target DNA (tgDNA), single-mismatched dsDNA, double-mismatched dsDNA, crRNA, ssDNA for ALP conjugation and NE buffer, were purchased from Integrated DNA Technologies (Singapore). Their specific sequences are summarized in Table S1.† LbaCas12a was purchased from New England Biolabs, Inc. (Ipswich, MA, USA). Streptavidin-conjugated ALP was obtained from Jackson Immuno Research Laboratories, Inc. (West Grove, PA, USA). BCIP/NBT phosphatase substrate (1-component system) was purchased from Kirkegaard & Perry Laboratories, Inc. (Gaithersburg, MD, USA). Tween-20 was purchased from MP Biomedicals (California, USA), and 2-aminoethyl methacrylate hydrochloride (AEMA-HCl) was purchased from Sigma-Aldrich (St. Louis, MO, USA). Sucrose, bovine serum albumin (BSA), sodium dihydrogenphosphate dihydrate, disodium hydrogenphosphate dodecahydrate, potassium chloride, trisodium citrate dihydrate, sodium hydroxide, sodium chloride, 2,2′-azobis(isobutyronitrile) (AIBN), di-t-butyl dicarbonate (Boc2O), methyl methacrylate (MMA) and hydrochloric acid were purchased from FUJIFILM Wako Pure Chemical Corporation (Osaka, Japan). Ethyl acetate, Tris-EDTA buffer solution (1 M, pH = 8.0), deionized and sterilized water, tris(2-carboxyethyl) phosphine hydrochloride (TCEP), poly(vinyl alcohol) (98% hydrolyzed, degree of polymerization: 2000) and hydrogen tetrachloroaurate(III) tetrahydrate were purchased from Nacalai Tesque (Kyoto, Japan). X-ray photoelectron spectroscopy (XPS-TR) (Japan Electron Optics Laboratory Ltd., Tokyo, Japan) was used to evaluate the AuNPs coating on the probe stick. A BioSpec-nano Life Science Spectrophotometer (Shimadzu, Kyoto, Japan) was used to determine the optical density (OD) when preparing the AuNPs. A CanoScan 9000F Mark II scanner (Canon, Tokyo, Japan) was used to acquire colour images of the probe stick. Quantitative colorimetric data analysis was performed using the Image J software (NIH, Bethesda, MD, USA). A Silhouette CAMEO3 electronic cutting tool from GRAPHTEC (Lindon, UT, USA) was applied to cut laminate film used as a platform for polyvinyl alcohol (PVA) membrane formation. An Objet30 Prime 3D printer with VeroClear resin from Stratasys (Eden Prairie, MN, USA) was used for fabrication of 3D-printed parts. Disposable UV/vis spectrometry cuvettes (for visible light) Semimicro Type (1-2855-02) and vial flat bottom inserts (2-865-15) were purchased from AS ONE (Osaka, Japan). An Oppo A74 smartphone model was used for smartphone-based signal readout.Labeling of AuNPs with ALP-modified ssDNA
Different sizes of AuNPs were synthesized following the general sodium citrate reduction method. All glassware was rinsed with aqua regia before use. A 2.5 mM solution of HAuCl4 (50 mL) was added to a 100 mL round-bottom flask and heated to 100 °C in an oil bath with rapid stirring. Subsequently, 5 mL of 44 mM trisodium citrate solution, also heated to 100 °C, was added. After reacting for 20 minutes, stirring was continued while the solution was allowed to cool to ambient temperature, resulting in uniform AuNPs. For the preparation of 30 nm AuNPs, 50 mL of 2.2 mM trisodium citrate solution was added to a 100 mL round-bottom flask and heated to boiling. To initiate Au seed formation, 333 μL of heated 25 mM HAuCl4 solution was added. After 20 minutes, an additional 333 μL of heated 25 mM HAuCl4 solution and 333 μL of 60 mM trisodium citrate solution were added for two cycles, with a 20 minute interval between each addition. Finally, the size of the AuNPs was observed using a transmission electron microscope (TEM-120) (Tecnai FEI Company, USA) and a ZetasizerNanoS (Malvern, UK). For further experiments, 210 μL of 13 nm AuNPs with OD of 1 was prepared in a PCR-clean microcentrifuge tube. To this, 30 μL of thiol- and biotin-terminated ssDNA (100 bases) at a concentration of 1800 nM was added. The mixture was gently vortexed for 3 hours to allow conjugation. Then, 60 μL of 0.5% (w/v) BSA was added as a surface blocking agent. After vortexing for 10 minutes, the mixture was centrifuged at 9500 rcf for 30 minutes to obtain nanoparticle precipitates. Next, 300 μL of a solution of streptavidin-conjugated ALP at a concentration of 1.0 μg mL−1 was added and vortexed for 2 hours. A 0.01 M Tris buffer (pH = 8.0) was prepared as a washing solution. The washing steps were repeated three times to ensure complete removal of non-specifically adsorbed ALP. The ALP–ssDNA–AuNP precipitate and the washing solution from the 3rd wash were separated. Notably, the washing solution was subjected to further centrifugation at 15000 rcf for 30 minutes to completely remove any tiny particles. Finally, BCIP/NBT solution was added to observe colour changes.Colorimetric signal creation based on CRISPR/Cas cleavage of ALP–ssDNA–AuNPs
The Cas12a enzyme and crRNA were mixed and pre-incubated at ambient temperature for 45 minutes. This was followed by an additional 15 minutes of incubation after adding double-stranded target DNA at final concentrations of 0, 0.01, 0.1, 1, and 10 mM. The goal was to form a sufficiently active CRISPR/Cas12a complex, with final concentrations of Cas12a and crRNA at 50 nM and 100 nM, respectively. The ALP–ssDNA–AuNP conjugate was mixed with the CRISPR/Cas12a complex. After a 30 minute incubation, the mixture was centrifuged at 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rcf for 30 minutes. A 150 μL aliquot of the supernatant was transferred to a microplate and mixed with 40 μL of BCIP/NBT solution. The absorbance was then measured using a microplate reader.
000 rcf for 30 minutes. A 150 μL aliquot of the supernatant was transferred to a microplate and mixed with 40 μL of BCIP/NBT solution. The absorbance was then measured using a microplate reader.
Design of 3D-printed biosensors
The CAD files for the 3D printing of the device components were created using Shapr3D software (Budapest, Hungary). The complete device was printed on an Object30 Prime 3D printer (Stratasys, USA) and consists of the following components: a buffer lid, a Cas disk modified with freeze-dried Cas protein and crRNA, a container, a probe stick, a disposable plastic cuvette (not 3D-printed), a piece of PVA membrane (not 3D-printed), a washer, a cap and the BCIP/NBT substrate (not 3D-printed) (Fig. 1). The scale and detailed specifications of the 3D-printed components are provided in Fig. S2.† | ||
| Fig. 1 Overall design and components of the 3D-printed biosensor; the blue frames show zoomed-in sections. | ||
Fabrication and basic evaluation of the probe stick
Poly(MMA-r-(Boc)AEMA) (PMBA) was synthesized following the procedure reported by Matsumoto et al.38 The probe stick, primarily composed of acrylic resin was prepared using the 3D printer. It should be noted that the exact resin composition is not revealed by the printer manufacturer. The synthesis steps and evaluations are detailed in the ESI.† A 150 μL volume of PMBA, dissolved in ethyl acetate at a concentration of 3% (w/w) was added to a vial insert. The probe stick was immersed in the solution for 15 seconds and then dried overnight in a vacuum desiccator at ambient temperature. Subsequently, 24 probe sticks were placed into a 10 mL vial, followed by the addition of 2 mL of 4 M HCl. The system was incubated at 40 °C, for 4 hours to remove the Boc protecting group and expose the amino groups. After washing three times with deionized water, 1.5 mL of 13 nm AuNPs at ODs of 0.5, 1, 1.5, and 2.0 were added and incubated for 30 min. For comparison, the same amount of 30 nm AuNPs at an OD of 2.0 was used. The AuNPs-coated probe sticks were lightly rinsed with deionized water and dried at 100 °C overnight.Preparation of ALP-labeled probe stick and its optimization
A 150 μL volume thiol- and biotin-terminated ssDNA (30 bases or 100 bases) solution, prepared in 50 mM PBS containing 1 M KCl, was added to vial inserts at concentrations of 2000, 3000, and 4000 nM. The AuNPs-coated probe stick was immersed in that solution for 2 hours with gentle vortexing. After ssDNA conjugation, the probe sticks were washed twice with 500 μL each of 50 mM PBS (containing 1 M KCl) and 0.01 M Tris buffer, respectively. Subsequently, 150 μL streptavidin-conjugated ALP solution at concentrations of 10, 20, and 30 μg mL−1 was added to the vial inserts, following by immersing the ssDNA-conjugated probe sticks for 2 hours. The ALP-labeled probe sticks were prepared for use by washing them three times with 500 μL of 0.01 M Tris buffer containing 0.15 M NaCl and 0.05% (v/v) Tween-20, rinsing them twice with deionized water, and finally three cycles of washing with 150 μL of NE buffer while rinsing with 1000 μL of 0.01 M Tris buffer not containing NaCl and Tween-20 between each NE buffer washing.Fabrication of PVA membrane
As a platform for molding PVA membranes, a 150 μm thick lamination film was used. Circular holes with a diameter of 6.5 mm were cut into a single layer of the lamination film pouch using the electronic cutting tool, followed by lamination with a sheet of transparency film to create individual small compartments (Fig. S3†). These compartments were subsequently used as molds for casting and drying the PVA membranes. PVA powder was prepared as a 5% (w/w) solution by stirring on a hot magnetic stirrer. A volume of 55 μL of the solution was cast into the compartments using a two-stage drying process. First, the membranes were left at ambient temperature overnight to allow initial formation, followed by drying in an oven at 80 °C overnight to achieve better mechanical strength. Finally, the PVA membranes were stored in a desiccator at room temperature (<20% humidity).Colorimetric analysis with the integrated device and smartphone-based quantitative detection
A volume of 4.5 μL of 5 μM Cas protein and 10 μM crRNA solution was mixed with 9 μL 20 wt% sucrose solution as a stabilizer, followed by freeze-drying onto the Cas disk. A volume of 450 μL of target DNA in NE buffer solution was infused into the buffer lid. The pillars on the container were fitted into the corresponding grooves of the lid, forming a first-stage seal. After a 10 minute incubation, the device was inverted 180° to allow the solution to transfer into the container. After incubating for 60 minutes, the lid was rotated 90° to allow the pillars to fully insert into the holes on the lid. By this push action, the liquid broke through the PVA membrane and flowed from the container into the cuvette, followed by a 60 minute ALP enzyme reaction with pre-freeze-dried BCIP/NBT substrate. The designed adaptor for the smartphone-based quantitative detection platform contained a cuvette holder, a light shelter, a convex lens holder, and an LED holder (Fig. S4†). The platform was designed using Shapr 3D software, in analogy to the device components, and printed using the Object30 Prime 3D printer. After modelling, all the parts were sprayed with a black coating to block light. Upon completed reaction, the cuvette was inserted into the cuvette holder and integrated with the other optical components (smartphone, convex lens, pinhole, diffuser and LED light) (Fig. S5†). The smartphone itself, a laptop, or a mobile battery served as the power supply for the LED light. The “Pro mode” settings were used for photographing with the following parameters: ISO = 100; shutter speed = 1/3200; white balance (WB) = 8000. Colour intensities were measured using the RGB Colour Detector app (Fig. S6†).Results and discussions
Design and working principle of the 3D-printed device
To achieve maximum user-friendliness, all necessary reagents have been integrated into the device. The concept of dual-enzymatic reaction eliminates the need for complex amplification procedures, while enabling highly sensitive nucleic acid detection through just a few simple operational steps. The sample solution used for this experiment simulated the composition of samples obtained from commercially available DNA rapid purification kits.39 These kits are designed to purify and concentrate DNA targets from initial samples into a salt-free or low-salt buffer, ensuring compatibility with downstream detection processes.First, the probe stick was surface modified with deprotected PMBA, which exhibits a strong affinity for AuNPs. The ssDNA probe was functionalized with a thiol group at one terminal to bind to AuNPs through thiol–gold interactions. The other terminal featured a biotin moiety, which binds to the streptavidin-modified ALP (Fig. 2A). Here, the ALP enzyme was selected as a secondary enzyme, since it requires only a single substrate addition. This contrasts with HRP, which in addition to a colorimetric substrate also depends on the presence of hydrogen peroxide with poor stability. ALP offers a more straightforward alternative for nucleic acid detection that is well-suited for on-site applications.
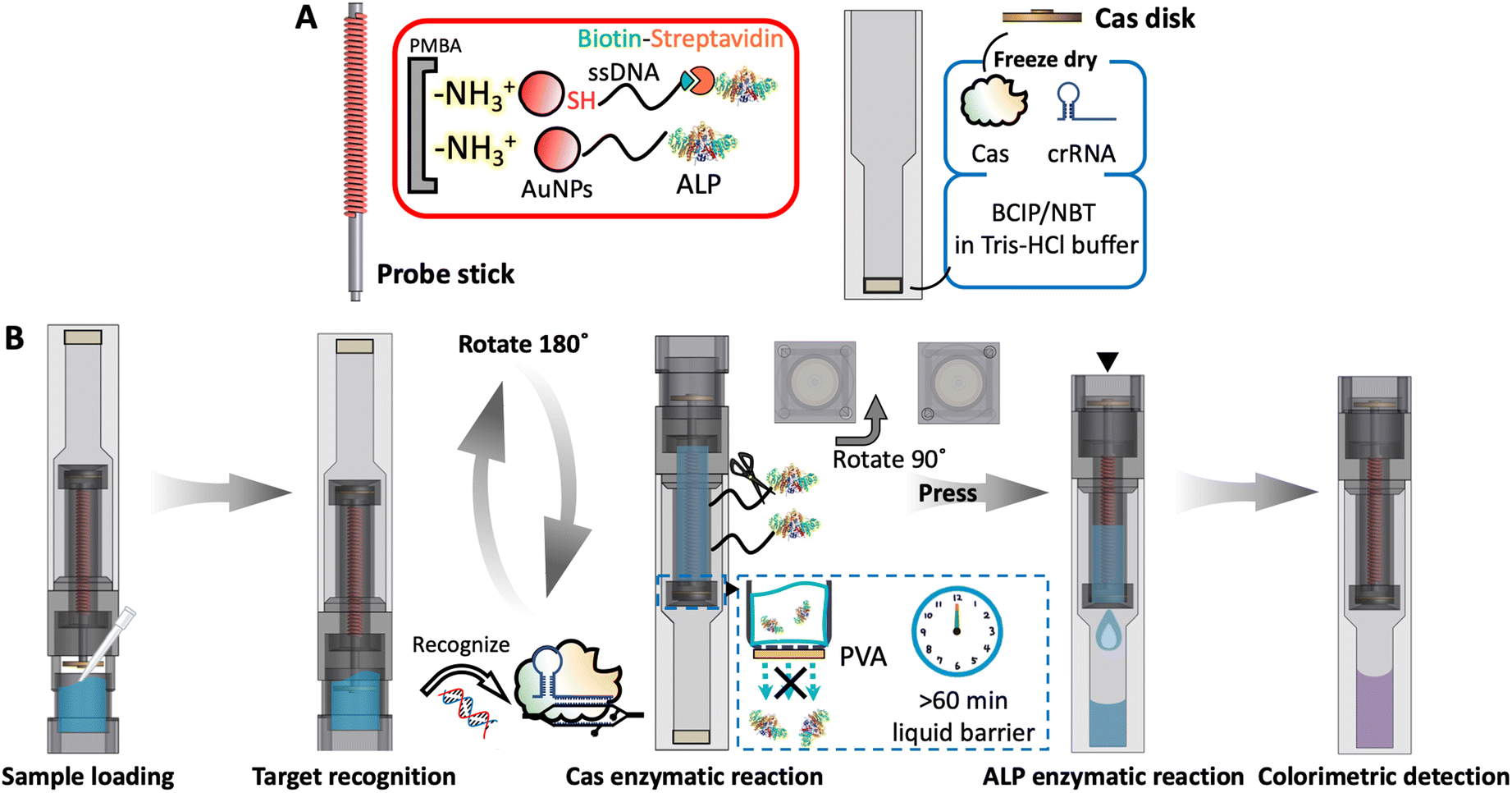 | ||
| Fig. 2 (A) ALP immobilization method on the probe stick and outline of reagent integration into the 3D-printed device; (B) operation and working principle of the 3D-printed nucleic acid biosensing device. | ||
The buffer lid is initially positioned downward, and 450 μL of a sample in NE buffer is introduced (Fig. 2B). The container is then closed to the first stage, embedding the pillars into the corresponding grooves on the lid to create a semi-closed condition, allowing the Cas disc to immerse into the solution. The lyophilized CRISPR/Cas complex becomes rehydrated and initiates the recognition of target DNA. After a 10 minute incubation period, the entire device is inverted 180°, positioning the cuvette downward and allowing the solution to flow from the buffer lid into the main container, where it interacts with the probe stick. In the presence of target DNA, the activated CRISPR/Cas system results in the trans-cleavage of ssDNA on the probe stick, releasing the immobilized ALP enzyme into the solution. The CRISPR/Cas enzymatic reaction is allowed to proceed for 60 minutes, with a PVA membrane acting as a liquid barrier to maintain the solution within the container. After the incubation period, the buffer lid is rotated 90°, aligning and pushing the pillars to fully insert into designated holes on the lid. This action increases air pressure, causing the softened PVA membrane to break and allowing the solution containing ALP in its freed state to flow down to the cuvette at the bottom of the device. The pre-lyophilized BCIP/NBT substrate in the cuvette reacts with the released ALP, resulting in the development of a purple colour. A stronger colour change indicates a larger amount of ALP cleaved and released, corresponding to a higher initial concentration of target DNA. This visible colour change can be assessed qualitatively by the naked eye or quantitatively using the smartphone adapter to measure absorbance accurately. The specific operation steps are shown in ESI† Video S1, using a blue dye (20 μL of 1 mM acid blue) dried on the Cas disk instead of the CRISPR/Cas complex and a red dye (50 μL of 1 mM acid red) pre-dried in the cuvette instead of BCIP/NBT substrate for visualization with 450 μL of NE buffer used as a sample solution.
Properties of AuNPs–ssDNA–ALP conjugates
To establish proof of concept, experiments were carried out using microtubes to evaluate the dual-enzymatic detection approach for target DNA in aqueous solutions. Two sizes of gold nanoparticles (13 nm and 30 nm) were synthesized, and their TEM images as well as hydrodynamic diameter profiles are provided in the ESI† (Fig. S7). In the initial study, 13 nm AuNPs were selected due to their simpler synthesis procedure and ease of preparation. During the conjugation process, it was observed that AuNPs labeled with biomolecules become more challenging to completely separate using mild centrifugation. To minimize residual AuNPs–ssDNA–ALP conjugates in the supernatant, a higher centrifugal force (15000 rcf) was applied, resulting in improved separation and reduced background signal (noise). To confirm ALP immobilization and assess its enzymatic activity, the absorbance of labeled AuNPs was measured. A wavelength shift from 520 nm to 526 nm was observed between the unmodified and modified AuNPs, indicating successful adsorption of biomolecules onto the AuNPs surface (Fig. 3A-I), similar to previously reported work.40 After centrifugation of the AuNPs–ssDNA–ALP conjugates, both the supernatant and precipitate were collected (Fig. 3A-II and III). A distinct colour change was observed upon BCIP/NBT substrate addition to the precipitate (Fig. 3A-III), while no colour change occurred in the supernatant (Fig. 3A-II). This confirmed that ALP was effectively immobilized onto the surface of the gold nanoparticles. To evaluate the Cas-mediated cleavage reaction, supernatants were collected from samples with and without the target DNA (Fig. 3A-IV and V), and the substrate was added. Significant colour development was only observed in the presence of target DNA (Fig. 3A-IV). All corresponding absorption spectra are shown in Fig. 3B. Verification using varying concentrations of target DNA showed consistent trends, with significant absorbance changes correlating with increasing target DNA concentrations (Fig. 3C). Control experiments, in which either crRNA or Cas enzymes were omitted, resulted in no observable colour change, confirming that cleavage could not occur in the absence of these components. In conclusion, these results demonstrate the successful synthesis of AuNP–ssDNA–ALP conjugates and validate their functionality using the CRISPR/Cas system. These findings establish the foundation for effective dual-enzyme-based detection of nucleic acids using this approach.
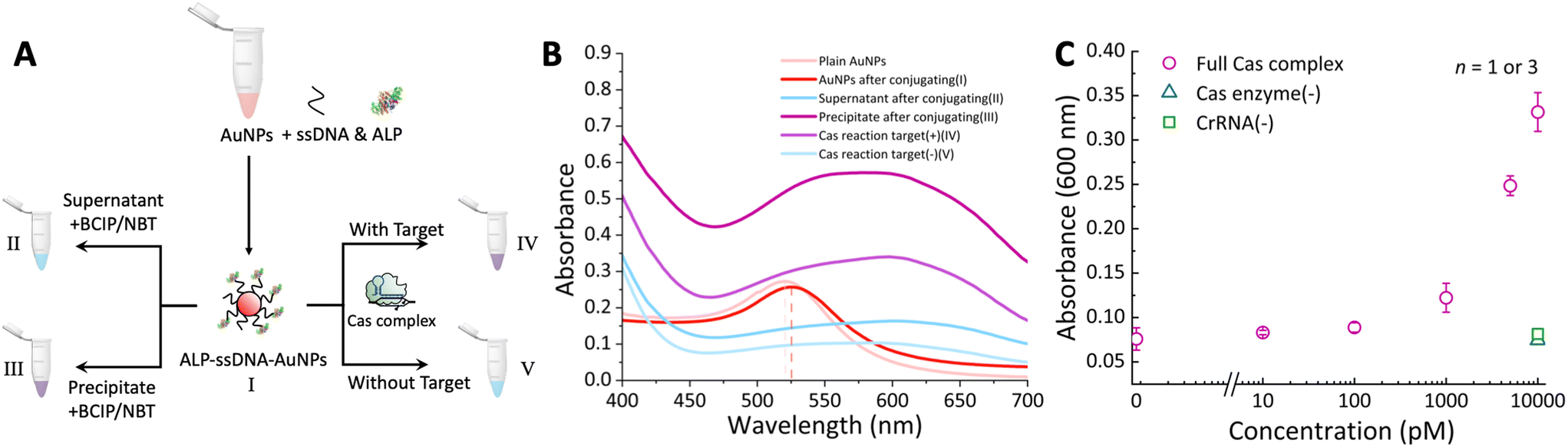 | ||
| Fig. 3 (A) Workflow of evaluating AuNPs–ssDNA–ALP conjugation and target DNA responsiveness; (B) absorption spectra for each solution described in (A); (C) target DNA concentration-dependent response curve after 30 min of Cas cleavage reaction and 20 min of ALP enzymatic reaction in aqueous solutions. | ||
Design and fabrication of probe sticks
The performance of the biosensor developed in this study depends on various factors, with the most important one arising from the structure of the solid support itself.41 A screw-like structure was designed to maximize the surface area, thereby enhancing sensitivity. While there are multiple design options, focus was set on balancing mechanical robustness and surface area. The probe stick used in this study featured a surface area of 300.5 mm2 (Fig. S8†). The conditions for forming AuNPs–ssDNA–ALP conjugates on the surface of the acrylic resin probe stick platform obtained in 3D printing differ substantially from those in solution, particularly regarding parameters such as AuNP size, ssDNA length, and enzyme loading amount. To maximize the functional performance of AuNPs–ssDNA–ALP conjugates immobilized on the probe stick for achieving the highest sensitivity, each process step was optimized. In the first step, the immobilization of the as synthesized AuNPs (13 nm) on the probe stick was examined using X-ray photoelectron spectroscopy (XPS). Three different surface states were compared: unmodified probe sticks, non-deprotected PMBA-coated probe sticks, and deprotected PMBA-coated probe sticks. The results indicated that gold signals were observed on probe sticks coated with PMBA featuring terminal amino groups (Fig. S9†), while the acrylic resin surface of the unmodified 3D-printed material did not retain the AuNPs. Interestingly, weak gold peaks were also detected on the non-deprotected PMBA coating. This could be attributed to hydrophobic interactions between AuNPs and the polymer, which are expected to be less stable at elevated temperatures used in the drying process. These findings highlighted the necessity of PMBA with free amino groups for the effective immobilization of AuNPs and led to the selection of deprotected PMBA for further studies. Subsequently, the immobilization efficiency of 13 nm AuNPs on the probe stick was investigated at varying concentrations (OD = 0.5, 1.0, 1.5, 2.0). After incubation and drying at elevated temperature, the probe sticks were scanned using a scanner, and the images were analyzed using ImageJ to evaluate the grayscale intensity (Fig. S10†). The results demonstrated significant differences in coloration depending on the AuNP concentration, with the darkest colour observed at OD = 2.0. This corresponds to a higher density of immobilized AuNPs. The increased AuNP loading facilitates more effective conjugation of ssDNA and ALP, thereby enhancing the potential for highly sensitive downstream analysis. Notably, efforts to produce higher concentrations of 13 nm AuNPs resulted in increased polydispersity, which can reduce the reproducibility of the experiments. Therefore, it was concluded that AuNPs with an OD of 2.0 provided the optimal condition for this work. Furthermore, the influence of the size of AuNPs and the length of ssDNA used for conjugation was investigated. The probe sticks were first coated with 13 nm AuNPs, followed by immobilization of either 30-base or 100-base ssDNA and ALP enzyme. After performing Cas enzyme-mediated cleavage reactions, the colorimetric signals were analyzed after adding BCIP/NBT substrate to the post-reaction solution (Fig. 4). The results showed that, 100-base ssDNA conjugated to 30 nm AuNPs demonstrated poor cleavage efficiency and low reproducibility (Fig. 4A). Similarly, regardless of the tgDNA concentration (1 or 10 nM), 30-base ssDNA could not be cleaved effectively to release ALP (Fig. 4B). It was hypothesized that the trans cleavage reaction requires sufficient accessibility of the activated Cas enzyme complex to the ssDNA probe, and excessive steric hindrance caused by large AuNPs or short ssDNA significantly inhibits the trans cleavage process. This observation aligns with similar conclusions reported in previous works.42,43 Finally, the concentrations of ssDNA and ALP were optimized (Fig. S11†). For Cas enzymes activated by 1 nM target DNA, increasing ALP concentration enhanced the colorimetric signal without substantially increasing background noise, owing to thorough washing procedures. However, increasing the concentration of ssDNA lead to greater variability in the results. We propose two possible reasons for this: (i) ALP release requires only a single cleavage event, and higher ssDNA concentrations may increase competition between cleavage of residual fragments and cleavage of intact ALP-labeled ssDNA, and (ii) non-specific adsorption of ssDNA strands introduces increased variability in both the Cas cleavage and ALP release steps. Consequently, we selected 2000 nM ssDNA in 150 μL solution per probe stick as the optimal ssDNA concentration, achieving a balance between efficiency and reproducibility. Focusing on that concentration of ssDNA, the use of 30 μg mL−1 of ALP exhibited the smallest error bar, and only a minor signal increase was observed between 20 and 30 μg mL−1 of ALP. Therefore, 150 μL of 30 μg mL−1 ALP solution was selected as the optimal concentration for subsequent experiments. | ||
| Fig. 4 Signal and noise (background absorbance) obtained with probe sticks in dual-enzymatic reactions depending on (A) size of AuNPs used for probe stick modification with 100 base ssDNA, and (B) length of ssDNA used for conjugation of ALP to 13 nm AuNPs. | ||
Influence of reaction time
The relationship between dual-enzymatic reaction time and signal intensity was investigated, as shown in Fig. S12.† It was observed that extending the CRISPR reaction time significantly improved the S/N ratio at low tgDNA concentrations of 1 pM and 100 pM. This trend aligns with the kinetics of the CRISPR reaction, as previously reported.23 Additionally, increasing the ALP reaction time also enhanced the signal intensity at lower target DNA concentrations; however, this improvement was accompanied by an unexpected increase in background signal. Therefore, extending the CRISPR reaction time was identified as a critical strategy for improving detection limits at lower target DNA concentrations while minimizing background interference. To retain the liquid in the main container to secure sufficient CRISPR/Cas trans cleavage reaction time before transfer to the cuvette for subsequent ALP enzyme reaction, a water-soluble membrane fabricated from PVA with a molecular weight of 88000 and a saponification degree of over 98.5% was used. This membrane serves as a temporary liquid barrier effectively blocking water flow for over 60 minutes. At the same time, it absorbs water and softens. By rotating and pressing the lid, the pillar on the container is enabled to fully insert into the designated holes on the lid, allowing the liquid to easily break through the PVA barrier.
Probe stick property evaluation and DNA sensing with integrated device
The performance of the probe sticks with the optimized parameters was evaluated using a 43 bp fragment of human papillomavirus HPV-18 DNA as a model target. As shown in Fig. 5A, the absorbance increased with increasing concentrations of target DNA under the condition of both 60 min reaction time for Cas cleavage and ALP reaction. The LOD was estimated to be approximately 10 pM, based on the lowest measured target DNA concentration providing a signal above the 3σ line shown in Fig. 5A. This represents one of the highest sensitivities compared to other reported amplification-free colorimetric nucleic acid detection methods.34,35 Despite the ssDNA probe being immobilized on a 2D surface, which inherently limits the efficiency of enzyme–substrate interactions, the use of the screw-type probe stick with small gold nanoparticles provided a significantly large surface area, enabling a high density of ssDNA and ALP enzyme labeling. This high labeling capacity is believed to be a key factor contributing to the highly sensitive target detection. | ||
| Fig. 5 Target DNA concentration-dependent response curves: (A) after 60 min of trans cleavage reaction time and 60 min of ALP reaction time with probe stick alone (microplate readout); (B) after 60 min of trans cleavage reaction time and 20 min of ALP reaction time with integrated device (smartphone readout); and (C) red intensity recorded in the absence of tgDNA, or with 1 nM perfectly matched and 1-base or 2-base mismatched DNA sequences (smartphone readout); the dotted lines represent the limit of detection (LOD), calculated as the mean value of the blank + 3 σblank. | ||
Furthermore, the immobilization and use of ALP enzymes with pre-freeze-dried substrate already in place greatly simplify experimental procedures, demonstrating potential for a fully integrated device capable of user-friendly analysis. Considering the diffusion of trace amounts of non-immobilized, semi-permanently adsorbed ALP enzymes into the blank solution, which results in a gradual increase in background signal over time and affects naked-eye judgment, the ALP enzymatic reaction was limited to 20 minutes following a 60 minute Cas cleavage reaction. The quantitative results were analyzed using the smartphone-based adaptor (Fig. 5B). The LOD, estimated from Fig. 5B according to the same method as mentioned above, was below 100 pM. Moreover, a significant visual colour change was observed from concentrations around 100 pM upwards, consistent with previous reports of non-amplified colorimetric nucleic acid detection.32,44 While this method demonstrated the feasibility of measurements using the integrated device, significant variability was observed at certain target DNA concentrations, attributed to two main factors: first, the device design did not ensure a consistent volume of solution being transferred from the main container to the cuvette at the bottom of the device for the second-stage enzymatic reaction. Surface tension effects caused some liquid to remain on the container walls upstream, which affected the final concentration of the BCIP/NBT substrate (resolubilized from the freeze-dried state) in the cuvette and altered the ALP reaction velocity. Second, the use of PVA membranes to retain the liquid in the main container until completed Cas reaction, lead to some of the PVA being dissolved into the reaction solution, potentially influencing the activity of the Cas and/or the ALP enzyme and reducing reproducibility. To investigate the influence of the presence of dissolved PVA residues on the Cas and ALP enzyme reactions, additional experiments have been performed (procedure described in the ESI†). For both the Cas and the ALP catalyzed reactions, there was no significant difference in the signal intensities comparing samples free of or containing PVA (Fig. S13†). However, samples containing PVA exhibited significantly larger error bars compared to the ones without PVA. The error of the Cas reaction is carried into the second ALP enzymatic reaction, and its impact further amplified. These results support the assumption that the matrix effect caused by the partially dissolved PVA membrane influences the reproducibility of the enzymatic reactions.
To compare the performance of the smartphone-based signal readout with that obtained with a microplate reader, 10 μL of 4 M HCl solution was added to the cuvette as a stop solution, followed by transferring an aliquot from the cuvette to a microplate (Fig. S14†). The S/N ratio measured on the microplate reader (absorbance ratio at 600 nm in presence and absence of target DNA), showed a target concentration-dependent signal trend similar to the S/N ratio measured with the smartphone setup (red intensity ratio in presence and absence of target DNA). Although the smartphone setup provided incident light at a single wavelength (600 nm), this proved suitable to serve as a quantitative tool in place of a conventional microplate reader.
Specificity test
Finally, the specificity of the assay performed using the integrated device was evaluated by comparing its response to perfectly complementary nucleic acids, or to those containing 1-base or 2-base mismatches at a concentration of 1 nM. Quantitative comparison was performed using the smartphone-based setup. As shown in Fig. 5C, in the presence of 1 nM of perfectly matched HPV-18 DNA, the red intensity decreased by 3.0-fold and 2.9-fold compared to 1-base mismatched or 2-base mismatched sequences, respectively. These results demonstrate the capability of this nucleic acid biosensor to effectively discriminate between perfectly matched sequences and those with minor mismatches, confirming its suitability for high-specificity POCT for nucleic acids.Conclusions
In this study, a fully integrated 3D-printed biosensor for POCT of nucleic acids has been successfully developed. The biosensor utilizes a probe stick labeled with AuNPs–ssDNA–ALP, enabling a nucleic acid pre-amplification-free colorimetric detection based on a CRISPR/Cas and ALP dual-enzymatic reaction mechanism. The use of small-sized gold nanoparticles combined with a large surface area probe stick enhanced the enzyme labeling density, thereby improving detection sensitivity. Additionally, the immobilized probe conjugation eliminated complex multi-step experimental procedures, such as centrifugation. The probe stick-based assay coupled with microplate readout achieved an LOD as low as 10 pM, one of the highest sensitivities reported for non-amplification colorimetric nucleic acid detection methods. To ensure usability in resource-limited environments without specialized equipment or trained operators, a 3D-printed assembly was used to integrate all necessary reagents into the system. Moreover, a smartphone accessory was adapted for quantitative signal readout. This design enables end-users to perform nucleic acid detection through simple operations with naked-eye visible results or to acquire quantitative data via a smartphone app. Furthermore, the specificity of the integrated system against mismatched nucleic acid sequences was evaluated, demonstrating its ability to reliably differentiate between perfectly matched and mismatched targets. While the device's detection sensitivity did not fully match that of the single probe stick used alone due to design limitations, its simplicity and ease of use highlight its potential for rapid nucleic acid screening. The smartphone-based detection system offers a quantitative analysis solution that aligns with the ASSURED criteria, making it suitable for decentralized testing. Despite these advantages, the device developed in the current study has still limitations. It has been demonstrated to be applicable to DNA samples simulating the composition obtained from commercially available nucleic acid rapid purification kits and does not include preprocessing steps, such as cell or viral lysis, DNA extraction, and purification. Future work will focus on integrating sample preprocessing steps directly into the biosensing device, as well as improving the reproducibility and stability of the device through design optimizations to further enhance its practicality for real-world applications. Overall, this study establishes a highly sensitive, colorimetry-based nucleic acid detection method and represents a significant step toward practical, high-sensitivity point-of-care nucleic acid testing applications.Data availability
Data created and analysed in this study are included in the article and the ESI† file. Raw data supporting the findings of this study are available from the corresponding author upon reasonable request.Author contributions
Guodong Tong: conceptualization, methodology, investigation, data curation, formal analysis, writing – original draft. Pabitra Nath: validation, writing – review & editing, supervision. Yuki Hiruta: validation, writing – review & editing, supervision, funding acquisition. Daniel Citterio: conceptualization, methodology, validation, writing – review & editing, supervision, project administration, funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge financial support by a Grant-in-Aid for Scientific Research (B) (grant no. 22H02109) from the Japan Society for the Promotion of Science (JSPS), and the Nakatani Foundation for Advancement of Measuring Technologies in Biomedical Engineering Grant Program for Research Study to D. C. P. N. acknowledges financial support by JSPS through an Invitational Fellowship. G. T. acknowledges partial funding through a scholarship from the Keio Engineering Foundation and JST Spring (grant no. JPMJSP2123). The authors acknowledge assistance by Ms. Yubuki Nakamura and Mr. Rei Kumada of Keio University for synthesis.Notes and references
- S. F. E. Bell, L. Coffey, J. Debattista, S. G. Badman, A. M. Redmond, D. M. Whiley, J. Lemoire, O. D. Williams, C. Howard, C. F. Gilks and J. A. Dean, Sex. Health, 2020, 17, 359–367 CrossRef PubMed.
- R. Paul, A. C. Saville, J. C. Hansel, Y. Ye, C. Ball, A. Williams, X. Chang, G. Chen, Z. Gu, J. B. Ristaino and Q. Wei, ACS Nano, 2019, 13, 6540–6549 CrossRef CAS PubMed.
- J.-H. Kim and S.-W. Oh, Food Control, 2021, 121, 107575 CrossRef CAS.
- P. A. Barrio, A. Fernández-Rodríguez, P. Martín, C. Fernández, L. Fernández and A. Alonso, Forensic Sci. Int., 2021, 323, 110775 CrossRef CAS PubMed.
- Y. Shang, J. Sun, Y. Ye, J. Zhang, Y. Zhang and X. Sun, Crit. Rev. Food Sci. Nutr., 2020, 60, 201–224 CrossRef CAS PubMed.
- J. L. Mckillip and M. Drake, J. Food Prot., 2004, 67, 823–832 CrossRef CAS PubMed.
- J. Vidic, P. Vizzini, M. Manzano, D. Kavanaugh, N. Ramarao, M. Zivkovic, V. Radonic, N. Knezevic, I. Giouroudi and I. Gadjanski, Sensors, 2019, 19, 1100 CrossRef CAS PubMed.
- B. Oliveira, B. Veigas, A. R. Fernandes, H. Águas, R. Martins, E. Fortunato and P. V. Baptista, Sensors, 2020, 20, 1624 CrossRef CAS PubMed.
- R. Ben-Ami, A. Klochendler, M. Seidel, T. Sido, O. Gurel-Gurevich, M. Yassour, E. Meshorer, G. Benedek, I. Fogel, E. Oiknine-Djian, A. Gertler, Z. Rotstein, B. Lavi, Y. Dor, D. G. Wolf, M. Salton, Y. Drier, A. Klochendler, A. Eden, A. Klar, A. Geldman, A. Arbel, A. Peretz, B. Shalom, B. L. Ochana, D. Avrahami-Tzfati, D. Neiman, D. Steinberg, D. B. Zvi, E. Shpigel, G. Atlan, H. Klein, H. Chekroun, H. Shani, I. Hazan, I. Ansari, I. Magenheim, J. Moss, J. Magenheim, L. Peretz, L. Feigin, M. Saraby, M. Sherman, M. Bentata, M. Avital, M. Kott, M. Peyser, M. Weitz, M. Shacham, M. Grunewald, N. Sasson, N. Wallis, N. Azazmeh, N. Tzarum, O. Fridlich, R. Sher, R. Condiotti, R. Refaeli, R. Ben Ami, R. Zaken-Gallili, R. Helman, S. Ofek, S. Tzaban, S. Piyanzin, S. Anzi, S. Dagan, S. Lilenthal, T. Sido, T. Licht, T. Friehmann, Y. Kaufman, A. Pery, A. Saada, A. Dekel, A. Yeffet, A. Shaag, A. Michael-Gayego, E. Shay, E. Arbib, H. Onallah, K. Ben-Meir, L. Levinzon, L. Cohen-Daniel, L. Natan, M. Hamdan, M. Rivkin, M. Shwieki, O. Vorontsov, R. Barsuk, R. Abramovitch, R. Gutorov, S. Sirhan, S. Abdeen, Y. Yachnin and Y. Daitch, Clin. Microbiol. Infect., 2020, 26, 1248–1253 CrossRef CAS PubMed.
- X. Li, M. Ye, W. Zhang, D. Tan, N. Jaffrezic-Renault, X. Yang and Z. Guo, Biosens. Bioelectron., 2019, 126, 596–607 CrossRef CAS PubMed.
- M. M. Kaminski, O. O. Abudayyeh, J. S. Gootenberg, F. Zhang and J. J. Collins, Nat. Biomed. Eng., 2021, 5, 643–656 CrossRef CAS PubMed.
- J. E. van Dongen, J. T. W. Berendsen, R. D. M. Steenbergen, R. M. F. Wolthuis, J. C. T. Eijkel and L. I. Segerink, Biosens. Bioelectron., 2020, 166, 112445 CrossRef CAS PubMed.
- L. T. Nguyen, B. M. Smith and P. K. Jain, Nat. Commun., 2020, 11, 4906 CrossRef CAS PubMed.
- S.-Y. Li, Q.-X. Cheng, J.-K. Liu, X.-Q. Nie, G.-P. Zhao and J. Wang, Cell Res., 2018, 28, 491–493 CrossRef CAS PubMed.
- J. S. Gootenberg, O. O. Abudayyeh, J. W. Lee, P. Essletzbichler, A. J. Dy, J. Joung, V. Verdine, N. Donghia, N. M. Daringer, C. A. Freije, C. Myhrvold, R. P. Bhattacharyya, J. Livny, A. Regev, E. V. Koonin, D. T. Hung, P. C. Sabeti, J. J. Collins and F. Zhang, Science, 2017, 356, 438–442 CrossRef CAS PubMed.
- L. Li, S. Li, N. Wu, J. Wu, G. Wang, G. Zhao and J. Wang, ACS Synth. Biol., 2019, 8, 2228–2237 CrossRef CAS PubMed.
- M. J. Kellner, J. G. Koob, J. S. Gootenberg, O. O. Abudayyeh and F. Zhang, Nat. Protoc., 2019, 14, 2986–3012 CrossRef CAS PubMed.
- S.-Y. Li, Q.-X. Cheng, J.-M. Wang, X.-Y. Li, Z.-L. Zhang, S. Gao, R.-B. Cao, G.-P. Zhao and J. Wang, Cell Discovery, 2018, 4, 1–4 CAS.
- J. S. Chen, E. Ma, L. B. Harrington, M. Da Costa, X. Tian, J. M. Palefsky and J. A. Doudna, Science, 2018, 360, 436–439 CrossRef CAS PubMed.
- A. S. Avaro and J. G. Santiago, Lab Chip, 2023, 23, 938–963 RSC.
- M. Bao, Q. Chen, Z. Xu, E. C. Jensen, C. Liu, J. T. Waitkus, X. Yuan, Q. He, P. Qin and K. Du, ACS Sens., 2021, 6, 2497–2522 CrossRef CAS PubMed.
- Y. Wang, Z. Wang, Y. Shang, J. Wang, Z. Zhu, L. Xi, J. Xie, Q. Wu, Y. Shen and Y. Ding, Coord. Chem. Rev., 2024, 513, 215895 CrossRef CAS.
- D. A. Huyke, A. Ramachandran, V. I. Bashkirov, E. K. Kotseroglou, T. Kotseroglou and J. G. Santiago, Anal. Chem., 2022, 94, 9826–9834 CrossRef CAS PubMed.
- D. Zhang, Y. Yan, H. Que, T. Yang, X. Cheng, S. Ding, X. Zhang and W. Cheng, ACS Sens., 2020, 5, 557–562 CrossRef CAS PubMed.
- Y. Cui, S. Fan, Z. Yuan, M. Song, J. Hu, D. Qian, D. Zhen, J. Li and B. Zhu, Talanta, 2021, 224, 121878 CrossRef CAS PubMed.
- T. Tian, B. Shu, Y. Jiang, M. Ye, L. Liu, Z. Guo, Z. Han, Z. Wang and X. Zhou, ACS Nano, 2021, 15, 1167–1178 CrossRef CAS PubMed.
- H. Shinoda, Y. Taguchi, R. Nakagawa, A. Makino, S. Okazaki, M. Nakano, Y. Muramoto, C. Takahashi, I. Takahashi, J. Ando, T. Noda, O. Nureki, H. Nishimasu and R. Watanabe, Commun. Biol., 2021, 4, 1–7 CrossRef PubMed.
- P.-F. Liu, K.-R. Zhao, Z.-J. Liu, L. Wang, S.-Y. Ye and G.-X. Liang, Biosens. Bioelectron., 2021, 176, 112954 CrossRef CAS PubMed.
- J.-H. Choi, M. Shin, L. Yang, B. Conley, J. Yoon, S.-N. Lee, K.-B. Lee and J.-W. Choi, ACS Nano, 2021, 15, 13475–13485 CrossRef CAS PubMed.
- J.-H. Choi, J. Lim, M. Shin, S.-H. Paek and J.-W. Choi, Nano Lett., 2021, 21, 693–699 CrossRef CAS PubMed.
- C. Yuan, T. Tian, J. Sun, M. Hu, X. Wang, E. Xiong, M. Cheng, Y. Bao, W. Lin, J. Jiang, C. Yang, Q. Chen, H. Zhang, H. Wang, X. Wang, X. Deng, X. Liao, Y. Liu, Z. Wang, G. Zhang and X. Zhou, Anal. Chem., 2020, 92, 4029–4037 CrossRef CAS PubMed.
- J. Ki, H.-K. Na, S. W. Yoon, V. P. Le, T. G. Lee and E.-K. Lim, ACS Sens., 2022, 7, 3940–3946 CrossRef CAS PubMed.
- S. Gong, X. Wang, P. Zhou, W. Pan, N. Li and B. Tang, Anal. Chem., 2022, 94, 15839–15846 CrossRef CAS PubMed.
- M. Broto, M. M. Kaminski, C. Adrianus, N. Kim, R. Greensmith, S. Dissanayake-Perera, A. J. Schubert, X. Tan, H. Kim, A. S. Dighe, J. J. Collins and M. M. Stevens, Nat. Nanotechnol., 2022, 17, 1120–1126 CrossRef CAS PubMed.
- D. Samanta, S. B. Ebrahimi, N. Ramani and C. A. Mirkin, J. Am. Chem. Soc., 2022, 144, 16310–16315 CrossRef CAS PubMed.
- M. J. Pugia, G. Blankenstein, R.-P. Peters, J. A. Profitt, K. Kadel, T. Willms, R. Sommer, H. H. Kuo and L. S. Schulman, Clin. Chem., 2005, 51, 1923–1932 CrossRef CAS PubMed.
- Y. Yazawa, T. Oonishi, K. Watanabe, A. Shiratori, S. Funaoka and M. Fukushima, J. Biosci. Bioeng., 2014, 118, 344–349 CrossRef CAS PubMed.
- M. Matsumoto, K. Kaneko, M. Hara, M. Matsui, K. Morita and T. Maruyama, RSC Adv., 2021, 11, 23409–23417 RSC.
- Easy-DNA™ gDNA Purification Kit, https://www.thermofisher.com/order/catalog/product/jp/ja/K180001, (accessed October 27, 2024).
- J. J. Storhoff, R. Elghanian, R. C. Mucic, C. A. Mirkin and R. L. Letsinger, J. Am. Chem. Soc., 1998, 120, 1959–1964 CrossRef CAS.
- V. Dugas, A. Elaissari and Y. Chevalier, in Recognition Receptors in Biosensors, ed. M. Zourob, Springer, New York, NY, 2010, pp. 47–134 Search PubMed.
- X. Fu, Y. Shi, F. Peng, M. Zhou, Y. Yin, Y. Tan, M. Chen, X. Yin, G. Ke and X.-B. Zhang, Anal. Chem., 2021, 93, 4967–4974 CrossRef CAS PubMed.
- S. Zhou, J. Ran, S. Man, J. Zhang, R. Yuan and X. Yang, Anal. Chem., 2024, 96, 10654–10661 CrossRef CAS PubMed.
- N. Shao, X. Han, Y. Song, P. Zhang and L. Qin, Anal. Chem., 2019, 91, 12384–12391 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and characterization of PMBA; sequences of nucleic acids used in this work; molecular weight of PMBA; NMR chart for PMBA; sketches and detailed scale for each component of the 3D-printed device; PVA membrane molding platform; sketches and detailed scale for each component of the smartphone accessory; optical path scheme for smartphone-based signal detection; screenshot of the app used during smartphone-based signal detection; TEM images of synthesized AuNPs; sketch and detailed scale for the probe stick; XPS charts for probe sticks after AuNP immobilization; probe sticks coated with different concentrations of AuNPs solution; absorbance after dual enzymatic reaction; absorbance after dual enzymatic reaction for different reaction time; comparison of the S/N ratio between results recorded with a microplate reader and with the smartphone setup. See DOI: https://doi.org/10.1039/d4lc01039f |
| This journal is © The Royal Society of Chemistry 2025 |