
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Systematic investigation of the role of the epoxides as substrates for CO2 capture in the cycloaddition reaction catalysed by ascorbic acid†
Thalía
Ortiz-García
a,
Sergio
Posada-Pérez
*ab,
Layla
El-Khchin
a,
David
Dalmau
 c,
Juan V.
Alegre-Requena
*c,
Miquel
Solà
a,
Valerio
D'Elia
*d and
Albert
Poater
*a
c,
Juan V.
Alegre-Requena
*c,
Miquel
Solà
a,
Valerio
D'Elia
*d and
Albert
Poater
*a
aInstitut de Química Computacional i Catàlisi and Departament de Química, Universitat de Girona, c/Maria Aurèlia Capmany 69, 17003, Girona, Catalonia, Spain. E-mail: albert.poater@udg.edu
bDepartment of General Chemistry: Algemene Chemie (ALGC), Vrije Universiteit Brussel, Pleinlaan 2, 1050 Brussel, Belgium
cDepartment of Inorganic Chemistry, Instituto de Síntesis Química y Catálisis Homogénea (ISQCH), CSIC—Universidad de Zaragoza, Pedro Cerbuna 12, 50009 Zaragoza, Spain
dDepartment of Materials Science and Engineering, VISTEC Advanced Laboratory for Environment-Related Inorganic and Organic Syntheses, Vidyasirimedhi Institute of Science and Technology (VISTEC), 21210, Payupnai, Wang Chan, Thailand
First published on 30th April 2025
Abstract
This work establishes a comprehensive theoretical framework for synthesizing cyclic organic carbonates, crucial for the polymer industry, through the organocatalytic cycloaddition of carbon dioxide (CO2) to epoxides under mild pressure and temperature conditions. Using advanced computational techniques, the study examines the thermodynamic and kinetic aspects of the reaction, with a particular focus on epoxide substrates featuring diverse substituents. Detailed analysis reveals activation energy barriers and identifies the rate-determining step (rds), offering crucial insights into the molecular processes governing the reaction. An automated data-driven workflow revealed that the buried volume of the epoxide O atoms was among the most influential molecular features affecting reaction barriers. Overall, the findings align with experimental data, offering insights into substrate design for optimized CO2 utilization. This work calls for a systematic exploration of ascorbic acid-based catalyst modifications to optimize energy barriers and improve overall reaction performance, paving the way for rational catalyst design and predictive catalysis in CO2 valorization. The computational study is not limited to basic research or ascorbic acid but is applicable to most catalysts capable of carrying out this reaction in the polymer industry.
Keywords: Epoxide; CO2 activation; Sustainable catalysis; Data-driven workflows; DFT calculations; Predictive catalysis; Cycloaddition.
1 Introduction
The valorisation of CO2 as a renewable source of carbon is fundamental according to the raised atmospheric CO2 levels. While carbon capture and storage offer mitigation potential, their deployment is hindered by high energy demands,1 economic constraints, and reliance on energy-intensive reagents.2,3Nevertheless, CO2 capture and utilization remains a privileged approach for further development.4–8 In CO2 capture, various techniques, including chemical and physical absorption9,10 by metal organic frameworks11,12 and membrane separation13 are being explored. Ionic liquids (ILs) show promise as green solvents for CO2 absorption,14–17 yet current materials have limited efficiency and pose environmental concerns like high regeneration costs and not enough stability.9,10,18 Different approaches have been utilized to enhance the characteristics of ionic liquids for carbon capture,19 utilization, and storage (CCUS).20 These include the development of ionic liquids with favourable properties like amino acid ILs,21–23 amine-functionalized ILs,24–26 phosphonium-based ILs,22,27 and protic ILs.28–30 Certain types of ionic liquids are reusable, offering a potential solution to mitigate environmental concerns linked to their usage.31–35 On the other hand, CO2 capture alone is limited by the need for suitable storage.36 Therefore, researchers are actively seeking methodologies and materials with improved properties to address the challenge of CO2 capture and conversion.37–41 In this context, the main nonreductive approach to CO2 conversion is represented by the cycloaddition of CO2 to highly available epoxide substrates that can be produced synthetically or even have biobased origin.42–44 The functionalization of CO2 by epoxides to afford cyclic carbonates leads to a wide selection of applications, as aprotic polar solvents45 that can be found in electrolytes in lithium ion batteries,46 polymer precursors47,48 and fuel additives. Furthermore, they are vital organic intermediates or precursors in the production of pharmaceutical, agricultural, and fine chemical products.49 Moreover, the cycloaddition of CO2 to epoxides can be carried out under mild conditions by using extremely simple biobased compounds50 mainly as hydrogen bond donors such as amino acids,51 polysaccharides,52 lignin,53,54 and even ascorbic acid38 when used in the presence of a nucleophilic compound (see Scheme 1 for mechanistic pathway using ascorbic acid and tetrabutylammonium iodide, TBAI, as the catalytic system).55 Ascorbic acid, in particular, serves for the cycloaddition of CO2 to epoxides under ambient conditions and it has been recently developed into a single component system for application as a recoverable ionic catalyst in biphasic media.56 Recent works have recognized ascorbic acid as a rare example of viable biobased catalysts for CO2–epoxide cycloaddition57 and it is often used as the hydrogen bond donor of choice for the preparation of challenging multicarbonate monomers as precursors of sustainable polyhydroxyurethanes.58,59
 | ||
| Scheme 1 Cycloaddition of epoxides (R = substituent; cat = ascorbic acid; Nu = nucleophile, being F, Cl, Br, I, or TBAI) and CO2. | ||
In parallel to the development of new catalytic systems and concepts, researchers have been looking into gaining a deeper understanding in the mechanistic features of the cycloaddition reaction through mechanistic60,61 and/or computational studies.62,63 Generally, in such computational studies, just a few epoxide substrates are used, most typically, ethylene and propylene oxide for the sake of simplicity, i.e., some of the most reactive epoxides, together with epichlorohydrin and glycidyl ethers, a priori, due to limited steric hindrance. However, such an approach does not render the whole picture of the extremely different reactivity of the diverse range of available epoxides that ultimately depends on their substitution pattern and which, in turn, leads to very different conversion values from substrate to substrate.64 Studies involving a systematic investigation of the reactivity of multiple (six) substrates are extremely rare,65 and yet limited to the case of metal-based catalytic systems, without including other crucially important epoxides for various applications such as those bearing halogen substituents66 and glycidyl ethers.67 Here, by density functional theory (DFT) calculations, we carry out a systematic study involving a large library of epoxides to investigate the effect of the substrate structure and electronic features on the efficiency of the catalytic process by using an organocatalytic systems (ascorbic acid/TBAI). This work explores as well the role of the counter-cation of the nucleophilic halide, which is generally overlooked in literature-reported calculations. Additionally, we have used a data-driven workflow including automated featurization and machine learning (ML) modelling to analyse the most important molecular parameters influencing reaction barriers. Through this comprehensive approach, the project aims to provide an efficient, sustainable methodology with practical applications in cyclic carbonate synthesis, and the results are extrapolable to any of the broad range of studies that lead to those compounds from CO2, from ionic liquids68,69 to the complexity and encapsulation of metallic organic frameworks (MOFs).70
2 Results and discussion
As commonly reported in the literature,71 our investigation uses ascorbic acid as the hydrogen bond donor and primarily an iodide ion as the nucleophile, initially neglecting the role of the counter-cation. Specifically, a quaternary ammonium salt was omitted from the reaction mechanism due to its negligible coordinating ability. This study focuses on various epoxide substrates included in Table 1 interacting with CO2.| System | R1 | R2 | R3 |
|---|---|---|---|
| EPO-H | –H | –H | –H |
| EPO-CH 3 | –H | –CH3 | –H |
| EPO-(CH 3 ) 2 | –CH3 | –CH3 | –H |
| EPO-cis(CH 3 ) 2 | –H | –CH3 | –CH3 |
| EPO-trans(CH 3 ) 2 | –CH3 | –H | –CH3 |
| EPO-tBu | –H | –C(CH3)3 | –H |
| EPO-CH 2 -nBu | –H | –(CH2)4–CH3 | –H |
| EPO-OH | –H | –OH | –H |
| EPO-CH 2 -OH | –H | –CH2–OH | –H |
| EPO-Glyc | –H | –CH2–O–C(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)–(CH2)CH3 O)–(CH2)CH3 |
–H |
| EPO-Ph | –H | –Ph | –H |
| EPO-(Ph) 2 | –Ph | –Ph | –H |
| EPO-cis(Ph) 2 | –H | –Ph | –Ph |
| EPO-trans(Ph) 2 | –Ph | –H | –Ph |
| EPO-CH 2 Ph | –H | –CH2–Ph | –H |
| EPO-CF 3 | –H | –CF3 | –H |
| EPO-C(CF 3 ) 3 | –H | –C(CF3)3 | –H |
| EPO-CH 2 F | –H | –CH2F | –H |
| EPO-CH 2 Cl | –H | –CH2Cl | –H |
| EPO-CH 2 Br | –H | –CH2Br | –H |
| EPO-Cyc | –H | –(CH2)4– | |
Table 2 provides a comprehensive breakdown of the relative energies associated with the pathway of the cycloaddition reaction, as illustrated in Scheme 1. Table 2 provides a comprehensive overview of the relative energies associated with the different steps of the reaction of all the epoxide substrates (see Table 1), offering insights into the thermodynamics and kinetics of the molecular processes involved, referenced from the catalyst, i.e., ascorbic acid (all the xyz coordinates can be found in the ESI†).
| Substrate | A | TS-AB | B | TS-BC | C | TS-CD | D | E |
|---|---|---|---|---|---|---|---|---|
| EPO-H | 1.7 | 17.9 | 8.0 | — | 5.7 | 21.2 | −1.5 | −0.1 |
| EPO-CH 3 | 1.3 | 18.3 | 10.7 | 19.8 | 8.6 | 23.1 | −0.8 | 0.0 |
| EPO-(CH 3 ) 2 | 1.2 | 18.9 | 13.2 | 23.3 | 14.2 | 27.5 | −0.2 | −0.2 |
| EPO-cis(CH 3 ) 2 | 0.9 | 20.8 | 14.5 | 23.4 | 12.6 | 27.8 | 1.0 | −1.3 |
| EPO-trans(CH 3 ) 2 | 1.3 | 21.9 | 16.3 | 21.5 | 13.5 | 27.3 | 0.6 | −0.9 |
| EPO-tBu | 0.2 | 19.8 | 14.0 | — | 11.9 | 25.7 | −0.1 | −2.0 |
| EPO-CH 2 -nBu | 1.7 | 19.5 | 8.6 | 22.4 | 9.4 | 25.5 | 0.9 | −6.1 |
| EPO-OH | 1.5 | 17.9 | 7.8 | — | 7.5 | 23.0 | −0.1 | 5.9 |
| EPO-CH 2 -OH | 0.9 | 18.0 | 11.6 | — | 10.0 | 24.3 | 5.5 | −0.7 |
| EPO-Glyc | −0.5 | — | 9.9 | 11.5 | 21.4 | 0.0 | −0.9 | |
| EPO-Ph | 2.3 | 20.8 | 12.7 | — | 9.4 | 24.6 | 2.3 | 1.3 |
| EPO-(Ph) 2 | 3.8 | 20.6 | 16.6 | — | 22.6 | 31.9 | 4.9 | 3.5 |
| EPO-cis(Ph) 2 | 5.0 | 26.0 | 17.0 | 19.2 | 18.2 | 29.3 | 5.6 | 5.4 |
| EPO-trans(Ph) 2 | 3.3 | 21.2 | 16.8 | — | 17.2 | 24.2 | 6.7 | 3.7 |
| EPO-CH 2 Ph | 1.8 | 18.7 | 14.2 | 14.6 | 11.3 | 26.6 | 3.5 | 1.4 |
| EPO-CF 3 | 3.1 | 17.2 | 8.0 | — | 8.6 | 25.4 | 2.3 | 3.2 |
| EPO-C(CF 3 ) 3 | 2.2 | 21.8 | 12.2 | — | 15.6 | 32.7 | 4.2 | 3.2 |
| EPO-CH 2 F | 2.1 | 18.2 | 10.5 | — | 9.5 | 22.7 | −0.5 | 1.0 |
| EPO-CH 2 Cl | 2.1 | 18.1 | 11.7 | — | 8.3 | 23.4 | 0.2 | 1.4 |
| EPO-CH 2 Br | 2.0 | 18.2 | 12.1 | — | 10.7 | 24.0 | 0.4 | 1.9 |
| EPO-Cyc | 0.9 | 23.0 | 20.0 | — | 10.6 | 29.1 | 1.2 | −2.4 |
For the EPO-H (unsubstituted epoxide; ethylene oxide) the binding of ascorbic acid to the epoxide (A), despite the presence of two hydrogen bonds between the acid and the epoxide oxygen, has a cost of 1.7 kcal mol−1, attributed to the entropic component. Subsequently, the nucleophilic agent, represented exclusively by the iodide ion in this case, initiates the reaction by attacking the less substituted position of the epoxide, resulting in its ring-opening. The corresponding transition state corresponds to an energy barrier of 16.2 kcal mol−1, but speaking of the kinetic cost, it should be referenced to the lowest energy intermediate, which in this case would correspond to the substrate and catalyst separately, i.e., the point of 0.0 kcal mol−1. Thus, following the scheme described by Kozuch and Shaik,72,73 the kinetic cost of TS-AB is 17.9 kcal mol−1. This transition state leads to an intermediate B, relatively unstable, specifically 6.3 kcal mol−1 relative to intermediate A. The following step in the reaction mechanism involves the insertion of CO2 from intermediate B. It is noteworthy that for the EPO-H, the corresponding transition state could not be identified, although the subsequent intermediate C was successfully located. This insertion step is exergonic, with an energy release of 2.3 kcal mol−1. The inability to locate the CO2 insertion transition state is not uncommon, as it typically features very low negative frequencies that are often challenging to identify and are therefore frequently omitted in reaction mechanism descriptions. From there, the TS-CD for the closing of the cyclic carbonate, with the expansion of the initial epoxide ring from 3 to 5 members, guides us in an exergonic step of 7.2 kcal mol−1, that is still also exergonic relative to the starting materials in agreement with the thermodynamically favourable nature of the cycloaddition process.74 It involves a kinetic cost of 21.2 kcal mol−1, and therefore, it is the rate-determining state (rds) of the reaction, although relative to intermediate C, it would only have an energy barrier of 15.5 kcal mol−1, therefore, lower than the corresponding barrier for TS-AB. Finally, additionally, intermediate E is simply obtained from D eliminating the iodide nucleophilic ion that is no longer bonded to any carbon.
As for the methylated epoxide (EPO-CH3), the binding of the co-catalyst (TS-AB) presents a similar high barrier, but slightly higher, with a value of 18.3 kcal mol−1. This small increase of 0.4 kcal mol−1 may already suggest that steric hindrance could be the reason. Then for this substrate, TS-BC was located, thus ruling out the insertion of CO2 as a rate-limiting step since it is 3.3 kcal mol−1 lower than TS-CD, which is 1.9 kcal mol−1 higher than for EPO-H. Since also TS-BC was located for EPO-CH3, the three corresponding transition states for this substrate are displayed in Fig. 1 to investigate their nature (and in Fig. S1† including the intermediates, as well). It should be highlighted that ascorbic acid primarily stabilizes the epoxide through two hydrogen bonds via its enediol hydroxyls to the epoxide oxygen. However, one aspect to note is that once the iodide attack cleaves the C–O bond of the epoxide in TS-AB, it entails the transfer of a hydrogen from ascorbic acid, thus protonating the oxygen of the epoxide momentarily in intermediate B. It is interesting to note that the possible protonation of the alkoxide intermediate by hydrogen bond donors, which is further validated by the difference in pKa of the species, was considered in previous studies. The protonation of the alkoxide was suggested as a possible factor affecting the catalytic activity of strongly acidic hydrogen bond donors such as carboxylic acids.75 It is interesting to note that the possible protonation of the alkoxide intermediate by the hydrogen bond donor, despite being seldom discussed in the literature, is expected due to the difference in pKa of the species.63 On the other hand, it should be noted that already in the next transition state, TS-BC, the proton automatically returns to ascorbic acid. In fact, Fig. S1† also includes an adduct of intermediate B with a CO2 molecule, called B + CO2, where ascorbic acid regains the proton. This species also serves to show that the CO2 entry barrier is mostly minimal, and in fact for EPO-CH3, it is only 0.2 kcal mol−1 in gas-phase optimization, i.e., only by geometry, but once the solvent effect is incorporated, it becomes negative by 1.1 kcal mol−1. This also explains why the transition state TS-BC was only observed for a few epoxide substrates. As previously commented, the imaginary frequency for this transition state is very small just 7.2i cm−1. In intermediate C, it is notable that ascorbic acid continues to form two hydrogen bonds through two of its hydroxyl groups. However, these interactions now involve both oxygen atoms of the bonded CO2 molecule, rather than being limited to a single oxygen atom. In the transition state corresponding to the cyclization of the cyclic carbonate (TS-CD), ascorbic acid once again establishes two hydrogen bonds, but this time exclusively with a single oxygen atom of the forming carbonate (see Fig. 1 and S2†).
 | ||
| Fig. 1 Transition states of the cycloaddition of carbon dioxide (CO2) to epoxide EPO-CH3 (selected distances in Å and imaginary frequencies included within brackets in cm−1). | ||
The explored substituents on the epoxide, which increase the steric hindrance, include –(CH3)2, –tBu and –Ph2. Specifically, for EPO-(CH3)2, the reaction presents larger energy barriers. The nucleophilic attack of the iodide anion (TS-AB) presents a kinetic cost of 18.9 kcal mol−1, the insertion of CO2 (TS-BC) of 23.3 kcal mol−1, but the most significant increase is found in TS-CD, with a value of 27.5 kcal mol−1. The rigidity of the epoxy backbone is considered to be somewhat significant, as the rds barrier with EPO-Cyc increases to 29.1 kcal mol−1, at least 1.3 kcal mol−1 higher than the analogous system, EPO-cis(CH3)2, for comparison.
Regarding EPO-tBu, this TS-CD is slightly more disfavoured compared to EPO-(CH3)2, up to 2 kcal mol−1. Therefore, the effect of disubstitution at the same carbon atom seems less detrimental than increasing the bulkiness of a single substituent. However, this finding cannot be generalized, as the EPO-Ph and EPO-(Ph)2 systems exhibit energy barriers of 24.6 and 31.9 kcal mol−1, respectively. Therefore, disubstitution emerges as a critical factor in some cases.
EPO-CH 2 Cl shows an energy barrier comparable to EPO-CH3, while EPO-CH2Br has a slightly larger barrier than EPO-CH2Cl — less than 1 kcal mol−1. EPO-CH2F with a relatively smaller and highly electronegative fluorine atom has a even lower upper reaction barrier than EPO-CH3 confirming the high reactivity of this substrate.56 This effect of the halide substituent becomes even clearer when comparing the values of TS-CD of 22.7, 23.4, and 24.0 kcal mol−1 for EPO-CH2F, EPO-CH2Cl, and EPO-CH2Br, respectively. In fact, comparing the experimental reactivity of the last two substrates, both clearly emerge at a disadvantage compared to other more structurally simple substrates, such as EPO-H or EPO-CH3. Therefore, the DFT computed energy barriers confirm the past experimental results.76,77
In Fig. 2, the TS-CD structure for some of the other studied epoxides is displayed (all included in Fig. S2†). For the formation distance of the C⋯O bond and the breaking C⋯I bond, it can be observed how bulky groups tend to modify the corresponding energy, but no substantial difference is observed. Therefore, in the following sections, an additional effort must be made in the structural study, as well as to provide insights into the electronic factors influencing the rds.
 | ||
| Fig. 2 Transition state TS-CD of the cycloaddition of CO2 for EPO-tBu, EPO-CH2F and EPO-CF3 (selected distances in Å and imaginary frequencies included within brackets in cm−1). | ||
Since the transition state for the closure of the cyclic carbonate ring (TS-CD) is found as the most energy-demanding step of the reaction we plotted in Fig. 3 how the kinetic cost ranges between the different epoxide candidates.
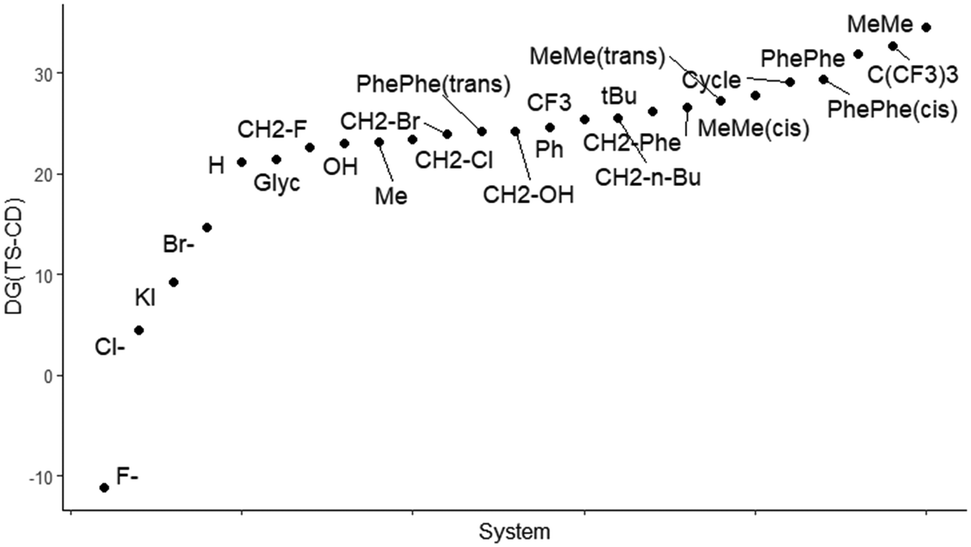 | ||
| Fig. 3 Plot of all the studied epoxides with respect to the relative energy of TS-CD with respect to reactant complex A (Gibbs energies in kcal mol−1). | ||
One key factor in explaining the high energy barriers observed in Fig. 3. for some of the substrates at this transition state is the bond distance between the carbon of the epoxide bearing the iodine and the oxygen of the inserted CO2. Based on the data in Table 3 and the Hammond postulate,78 it is expected that the smaller the C–I distance is, the more reactant-like the TS-CD and the more favoured the ring closure would be, and consequently, the energy barrier at this step of the reaction would be smaller. At the same time, the C–C distance of the former epoxide should be considered. For the TS-CD, the distances of the forming C–O bond and the breaking C–I bond are inconclusive and do not generate a clear trend, although it is perceived that the shorter both distances are, the more feasible the closure of the cyclic carbonate is. Using the SambVca2.1 program,79,80 steric maps have been generated for the different substrates. Table 3 also includes the values of the overall %VBur (by quadrants in Tables S1 and S2†) of the free epoxide and TS-CD. It should be noted that quantitatively the hindrance increases in the transition state, but qualitatively by the steric maps collected in Fig. 4 the analysis leads to similar conclusions. Thus, globally, the %VBur increases noticeably with the presence of bulky groups on the carbon that facilitates the C–O bond formation. This effect is even more pronounced on the alpha carbon, especially when this position is doubly substituted.
| Substrate | Distances | %VBur | ||||
|---|---|---|---|---|---|---|
| d(C–O) epoxide | d(C–C) epoxide | d(C⋯O) TS-CD | d(C⋯X) TS-CD | Epoxide | TS-CD | |
| EPO-H | 1.430 | 1.478 | 2.060 | 2.721 | 32.1 | 40.6 |
| EPO-CH 3 | 1.434 | 1.480 | 2.065 | 2.735 | 36.9 | 46.0 |
| EPO-(CH 3 ) 2 | 1.446 | 1.483 | 2.090 | 2.749 | 41.9 | 50.9 |
| EPO-cis(CH 3 ) 2 | 1.440 | 1.485 | 2.098 | 3.002 | 46.5 | 55.1 |
| EPO-trans(CH 3 ) 2 | 1.440 | 1.481 | 2.121 | 2.886 | 46.8 | 55.3 |
| EPO-tBu | 1.437 | 1.478 | 2.076 | 2.767 | 42.5 | 51.1 |
| EPO-CH 2 -nBu | 1.434 | 1.479 | 1.996 | 2.832 | 38.7 | 49.1 |
| EPO-OH | 1.445 | 1.469 | 2.033 | 2.765 | 35.8 | 44.8 |
| EPO-CH 2 -OH | 1.437 | 1.476 | 2.089 | 2.719 | 38.3 | 47.5 |
| EPO-Glyc | 1.441 | 1.477 | 1.951 | 2.839 | 41.7 | 46.5 |
| EPO-Ph | 1.430 | 1.488 | 2.039 | 2.750 | 39.9 | 49.2 |
| EPO-(Ph) 2 | 1.447 | 1.490 | 2.042 | 2.807 | 48.1 | 57.2 |
| EPO-cis(Ph) 2 | 1.433 | 1.502 | 2.133 | 2.956 | 58.1 | 66.3 |
| EPO-trans(Ph) 2 | 1.435 | 1.502 | 2.046 | 3.042 | 59.0 | 67.7 |
| EPO-CH 2 Ph | 1.436 | 1.477 | 2.010 | 2.828 | 40.7 | 50.8 |
| EPO-CF 3 | 1.436 | 1.476 | 2.006 | 2.798 | 40.5 | 49.4 |
| EPO-C(CF 3 ) 3 | 1.439 | 1.477 | 2.028 | 2.815 | 49.2 | 56.6 |
| EPO-CH 2 F | 1.436 | 1.477 | 2.040 | 2.750 | 38.1 | 46.1 |
| EPO-CH 2 Cl | 1.438 | 1.476 | 2.038 | 2.752 | 39.2 | 46.2 |
| EPO-CH 2 Br | 1.437 | 1.476 | 2.035 | 2.753 | 39.6 | 46.3 |
| EPO-Cyc | 1.436 | 1.486 | 2.133 | 2.934 | 50.9 | 59.4 |
 | ||
| Fig. 4 %VBur and topographic steric maps in Å (XY plane) for TS-CD, using the carbon forming the C–O bond as the center, with a radius of 3.5 Å and with the Z axis defined by the C–O bond, the other carbon of the original epoxide in the XZ plane. | ||
Additionally, the asymmetry of the steric maps should be highlighted, as this can help determine whether the steric hindrance is confined to a specific quadrant. Fig. 4, with the steric maps, clarifies that although there may be a heavily hindered quadrant, the rest is accessible,81,82 especially in cases of monosubstitution rather than disubstitution of the carbon atom, as seen in EPO-(Me)2 or EPO-(Ph)2.
Moreover, the nature of the nucleophilic ion has also been studied. As shown in Table 4, the rds remains the same for Br, Cl, and F, and the kinetics also detect TS-CD as the rds, with barriers changing for EPO-CH3 from 23.1 kcal mol−1 for I− to 16.2, 20.4, and 29.5 kcal mol−1 with Br−, Cl−, and F−, respectively (see Tables S3–S5† for further structural and electronic details). Thus, it is revealed that Cl− and even more so Br− would perform as better nucleophiles than I−. However such an observation is at variance with what is generally observed experimentally in the cycloaddition reaction catalysed by hydrogen bond donors and halide salts where the iodide anion is the most efficient nucleophile,63,75,83 and calls for further clarification, probably in terms of solvation in the reaction mixture. Thus, there must be this or some other factor/s. Actually, smaller halides are supposed to be better nucleophiles in polar aprotic solvent, but epoxides do not seem to follow such a trend experimentally, and the inclusion of the countercation TBA+ might be crucial.
| Nucleophile | A | A + X − | TS-AB | B | TS-BC | C | TS-CD | D |
|---|---|---|---|---|---|---|---|---|
| I− | 0.0 | 1.3 | 18.3 | 10.7 | 19.8 | 8.6 | 23.1 | −0.8 |
| Br− | 0.0 | 8.6 | 14.0 | −0.1 | — | −1.5 | 14.7 | −2.0 |
| Cl− | 0.0 | −15.9 | −22.7 | −12.8 | — | −14.9 | 4.5 | −8.7 |
| F− | 0.0 | −40.6 | −8.3 | −41.3 | — | −39.7 | −11.1 | −12.7 |
| KI | 0.0 | −11.7 | 9.0 | −6.5 | — | −5.7 | 9.2 | −20.8 |
| TBAI | 0.0 | 8.2 | — | 5.9 | — | 9.7 | — | −0.2 |
On the other hand, it should be mentioned that what changes the most in our calculations when varying the halide is the exergonicity of the process, and thus, the adduct of ascorbic acid and the epoxide, when the halide is at a non-coordination distance, opens a stability difference window of over 40 kcal mol−1. It is important to clarify at this point that the origin of the kinetic barrier is not the same for the computational set. Due to the endothermicity revealed by the CO2 cycloaddition process using I− as nucleophile for all the substrates included in our setup, the kinetic cost of the process was calculated as the difference between the energy of the transition state TS-CD with respect to the reference (A). The barrier from the intermediate C to the TS-CD is slightly lower but rigorously speaking the larger energy difference must be taken as the kinetic cost of the process. Nevertheless, the use of F−, Cl−, and Br− as nucleophiles implies changes in the thermodynamics. Anions of more electronegative halides stabilize intermediates and transition states along the reaction mechanism and as reported in Table 4 the energy difference between A and TS-CD becomes much lower, even negative. In this scenario, the kinetic cost of the reaction must be accounted from intermediate C, since the rds of the process is to overcome the TS-CD from C intermediate.
The large difference in the reaction thermodynamics as function of the selected nucleophile prompted us to try to reduce the anionic character weight of the halide by studying what happens when instead of using the I− ion, we use a salt such as KI. To note, in the discussion above, the countercation of the nucleophilic halide, that is TBA+, was omitted. This omission has two reasons, firstly, the high computational cost of including the tetra-n-butylammonium cation, but also to avoid distorting the results since this cation lacks coordinating nature and hence could be located at almost any position. It is worth noting that such unpredictability of the cation position greatly complicates its inclusion in DFT static calculations, and dynamic calculations would likely be necessary. Nevertheless, the disparity in its position throughout the reaction mechanism could also hinder the proper comparison between different epoxide substituents. However, we have simplistically attempted to include the whole TBAI molecule in the calculations in Table 4, as well. In detail, in the presence of TBAI, the intermediates were located, but attempts to locate the connecting transition states failed. Therefore, the much simpler KI was used, for which it is indeed demonstrated that the TS-CD is maintained as the rds with almost the same kinetic cost (see Fig. 5), decreasing only from 23.1 to 20.9 kcal mol−1, while the opening of the epoxide requires 20.9 kcal mol−1 compared with 18.3 kcal mol−1 when using the I− anion alone (see Tables S6–S9† for further energetic, structural, and electronic details).
 | ||
| Fig. 5 Transition states TS-AB (left) and TS-CD (right) of the cycloaddition of CO2 for EPO-CH3 (selected distances in Å). | ||
The role of the potassium countercation is not insignificant, as Fig. 5 demonstrates how the structure of ascorbic acid folds towards the epoxide, while being strongly bound to the iodide anion. It should be noted that TBAI would diminish this countercation strength, given its less binding nature compared to a K cation. In addition, Fig. 5 also shows the evidence of the role of the side chain of hydroxyl groups.
Next, we analysed reactivity trends using electronic and steric descriptors derived from the starting epoxide substrates. These parameters were generated from SMILES strings using an automated featurization workflow implemented within the AQME program.84 In this protocol, a wide array of 34 electronic properties was generated using the PTB and GFN methods of xTB,85 combined with 4 steric descriptors from MORFEUS,86 and the octanol/water partition coefficient (MolLogP) obtained with RDKit (Fig. 6a).87
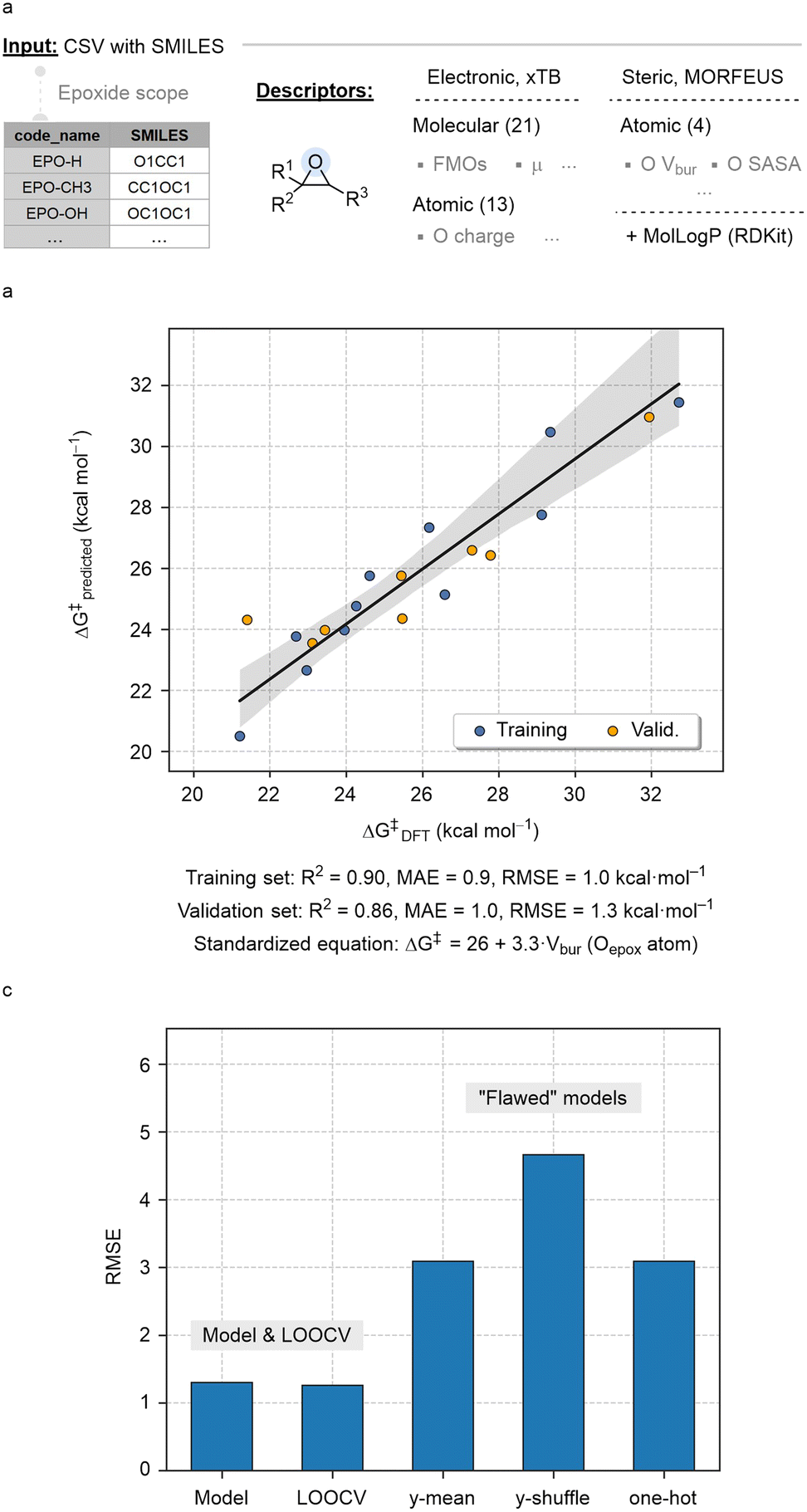 | ||
| Fig. 6 (a) Summary of the molecular featurization workflow; (b) calculated vs. predicted ΔG‡ (kcal mol−1) obtained with the optimal linear model; (c) RMSE of model, LOOCV and “flawed” models. | ||
The collection of descriptors in Fig. 6a integrates a diverse range of molecular properties along with specific features of the oxygen atom in the epoxide, which is central to the rds and thus expected to influence reaction kinetics. The descriptor database was employed to build machine learning models using the ROBERT software,88 including random forest, neural network, gradient boosting and linear regression. After excluding two preliminary outliers (EPO-(CH3)2 and EPO-trans(Ph)2), we obtained a linear model with 19 data points, utilizing %VBur as the sole parameter to estimate activation barriers. As shown in Fig. 6b, the model shows high coefficients of determination between predicted and measured values (R2 of 0.90 and 0.86 in the training and validation sets) and low root mean squared errors (RMSE of 1.0 and 1.3 in the training and validation sets).
No significant overfitting was observed in the leave-one-out cross-validation test (LOOCV; Fig. 6c, left). Additionally, to ensure the model captures meaningful trends in the data, three “flawed” models were evaluated: (i) y-mean, which assesses accuracy when predicted y values are fixed to the mean y value, (ii) y-shuffle, which evaluates accuracy using a model trained on randomly shuffled y values, and (iii) one-hot, which tests accuracy when all descriptors are replaced with binary values (0 s and 1 s).89 In all cases, the resulting RMSE was significantly higher than those of the original model and the LOOCV (Fig. 6c, right).
The predictive and interpretative performance of the model was further assessed using the ROBERT score (ROBERT_report PDF file of the ESI†).88 This score, rated out of ten, evaluates models based on multiple criteria, including correlation between predictions and measurements, human interpretability, error distribution, sensitivity to features, and resilience against overfitting and underfitting.90 The score of 7 achieved in this case is a satisfactory result for this low-data scenario, indicating that %VBur is likely among the most influential descriptors affecting reactivity. This automated SMILES-based workflow also underscores the potential of combining cost-effective descriptors with automation to analyse reactivity trends even in datasets with limited datapoints.91
3 Conclusions
This study reports a detailed and enhanced investigation of the reaction pathway of CO2 cycloaddition to epoxides, focusing on the dual catalytic role of ascorbic acid and the iodide ion as the primary nucleophile. The investigation focuses on the interaction of various epoxides substrates with CO2 with a particular emphasis on the thermodynamics and kinetics of the molecular processes. Through comprehensive computational analysis, the findings highlight the interplay of thermodynamic and kinetic factors governing the reaction pathway, with particular emphasis on the transition states and intermediate species. For unsubstituted ethylene oxide (EPO-H), the cyclization of the carbonate is confirmed as the rds, with a substantial energy barrier that aligns with experimental observations. Comparatively, methyl-substituted (EPO-CH3) and bulkier epoxides show an increase in steric hindrance, leading to a higher kinetic cost, particularly at the rds. In detail, steric maps reveal that the steric hindrance increases from the initial free epoxides to transition states (TS-CD). The %VBur values rise with bulkier groups on the carbon facilitating the C–O bond formation, especially when the alpha carbon is doubly substituted. Notably, steric maps exhibit asymmetry, indicating that steric hindrance may be confined to specific quadrants. In monosubstituted cases, certain quadrants remain accessible, unlike in disubstituted scenarios such as EPO-(Me)2 or EPO-(Ph)2.The study demonstrates the successful integration of electronic and steric descriptors into a machine learning framework to predict reactivity trends in epoxide substrates. Utilizing the AQME program for molecular featurization, a broad set of descriptors was compiled, emphasizing features of the O atom critical to the rds. This dataset facilitated the construction of a linear model via the ROBERT software,88 achieving a strong predictive performance and no significant overfitting. The use of %VBur as the sole parameter underscores its significance in governing activation barriers. These results illustrate the potential of combining automated, cost-effective molecular descriptors with machine learning to analyze reactivity trends, even with limited datasets.
4 Computational details
4.1 Density functional theory (DFT) calculations
Geometry optimizations using DFT were conducted employing the BP86 level of the generalized gradient approximation,92,93 implemented in the Gaussian16 software.94 To account for dispersion corrections, the D3 version of Grimme was incorporated.95 The electronic structure of the systems was modelled using the valence double-ζ with polarization (SVP) basis set for main group atoms,96 while for iodine the quasi-relativistic SDD effective core potential of Stuttgart/Dresden was utilized.97 Gibbs energies were determined from single-point energy calculations on the BP86-D3/SVP∼sdd geometries using the B3LYP functional,98 Grimme's dispersion, and the cc-pVTZ basis set.99 Solvent effects, with propylene oxide (PO) as the solvent, were included using the polarization continuum model (PCM).100 Electronic energies from the latter single point energy calculations in solvent were then converted to Gibbs energy, incorporating zero-point energy and thermal corrections from gas-phase frequency calculations at the BP86/SVP∼sdd level.4.2 Molecular featurization
The AQME program (v1.7.1) was used to generate Boltzmann-averaged descriptors from a CSV file containing SMILES strings of the epoxide substrates.84 This automated workflow initially performs conformational searches with RDKit, geometry optimization with xTB,85 and descriptor generation with xTB (GFN2 and PTB), MORFEUS,86 and RDKit.87 Both molecular and atomic descriptors were considered, and all the data was stored in a CSV database. The command line used was:“python -m aqme --qdescp --input “epox.csv” --qdescp_atoms “[O1CC1]””
In this command, epox.csv refers to the initial CSV file of SMILES strings and substrate names (SMILES and code_name columns, respectively). The option “--qdescp_atoms “[O1CC1]”” specifies the creation of atomic descriptors for the three elements forming the epoxide rings. The program generated three CSV files with varying number of descriptors (“denovo”, “interpret”, and “full”), from which the “interpret” level was selected as our descriptor database. The atomic descriptors for the O atom were kept, while the descriptors for the two C atoms were removed (see discussion below).
4.3 ML workflow
The ROBERT program (v1.3.0)88 used the “interpret” descriptor database created with AQME as an input, including also the target values (activation barriers, g_barr). ROBERT performed data curation, ML model screening, testing model reliability, and feature importance analysis, among other protocols. The command line used to execute ROBERT was:“python -m robert --csv_name “AQME-ROBERT_interpret.csv” --ignore “[SMILES]” --names “code_name” --y “g_barr””.
ROBERT generated a PDF report containing comprehensive information about the models, enhancing transparency and providing instructions for reproducibility. Both this PDF file and the input descriptor database are available as part of the ESI.†
Data availability
The data supporting this article have been included in the main manuscript and as part of the ESI.†Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
We thank the Spanish Ministerio de Ciencia, Innovación y Universidades (MCIN/AEI/https://doi.org/10.13039/501100011033/FEDER, UE) for projects PID2021-127423NB-I00 (to A. P.), PID2022-140159NA-I00 (to J. V. A.-R.), PID2023-147424NB-I00 (to M. S.), and RED2022-134939-T (to M. S.), and the Generalitat de Catalunya for project 2021SGR623, predoctoral FISDUR 2023 grant to T. O.-G. and ICREA Academia Prizes to A. P. (2019) and M. S. (2024). A. P. is a Serra Húnter Fellow. S. P.-P. acknowledges co-funding from the European Union's Horizon 2020 research and innovation Maria Skłodowska-Curie Actions, under grant agreement number 945380. D. D. and J. V. A.-R. acknowledge Gobierno de Aragón-Fondo Social Europeo (Research Group E07_23R). We acknowledge Angie Sacoto for helpful initial testing calculations. V. D. E. thanks the National Research Council of Thailand, grant no. N42A650196, for research support. European Union's Recovery and Resilience Facility-Next Generation (MMT24-ISQCH-01) in the framework of the General Invitation of the Spanish Government's public business entity Red.es to participate in talent attraction and retention programmes within Investment 4 of Component 19 of the Recovery, Transformation and Resilience Plan (MOMENTUM).References
- C. Seidel, A. Jörke, B. Vollbrecht, A. Seidel-Morgenstern and A. Kienle, Kinetic modeling of methanol synthesis from renewable resources, Chem. Eng. Sci., 2018, 175, 130–138 CrossRef CAS.
- A. W. Kleij, M. Northa and A. Urakawa, CO2 catalysis, ChemSusChem, 2017, 10, 1036–1038 CrossRef CAS PubMed.
- N. von der Assen and A. Bardow, Life cycle assessment of polyols for polyurethane production using CO2 as feedstock: Insights from an industrial case study, Green Chem., 2014, 16, 3272–3280 RSC.
- G. A. Bhat and D. J. Darensbourg, Progress in the catalytic reactions of CO2 and epoxides to selectively provide cyclic or polymeric carbonates, Green Chem., 2022, 24, 5007–5034 RSC.
- W. Natongchai, J. A. Luque-Urrutia, C. Phungpanya, M. Solà, V. D'Elia, A. Poater and H. Zipse, Cycloaddition of CO2 to epoxides by highly nucleophilic 4-aminopyridines: establishing a relationship between carbon basicity and catalytic performance by experimental and DFT investigations, Org. Chem. Front., 2021, 8, 613–627 RSC.
- Q.-W. Song, R. Ma, P. Liu, K. Zhang and L.-N. He, Recent progress in CO2 conversion into organic chemicals by molecular catalysis, Green Chem., 2023, 25, 6538–6560 RSC.
- Z. Lv, S. Chen, X. Huang and C. Qin, Recent progress and perspective on integrated CO2 capture and utilization, Curr. Opin. Green Sustainable Chem., 2023, 40, 100771 CrossRef CAS.
- C.-H. Yu, C.-H. Huang and C.-S. Tan, A review of CO2 capture by absorption and adsorption, Aerosol Air Qual. Res., 2012, 12, 745–769 CrossRef CAS.
- S. Hussain, H. Dong, S. Zeng, M. U. Ahmad, F. K. Shehzad, H. Wu and Y. Zhang, Investigation uncovered the impact of anions on CO2 absorption by low viscous ether functionalized pyridinium ionic liquids, J. Mol. Liq., 2021, 336, 116362 CrossRef CAS.
- S. C. Balchandani, A. Dey, B. Mandal, A. Kumar and S. Dharaskar, Elucidating the important thermophysical characterization properties of amine activated hybrid novel solvents for designing post-combustion CO2 capture unit, J. Mol. Liq., 2022, 355, 118919 CrossRef CAS.
- E. Coronado, M. Giménez-Marqués, G. Mínguez Espallargas, F. Rey and I. J. Vitórica-Yrezábal, Spin-crossover modification through selective CO2 sorption, J. Am. Chem. Soc., 2013, 135, 15986–15989 CrossRef CAS PubMed.
- J. Poater, M. Gimferrer and A. Poater, Covalent and ionic capacity of MOFs to sorb small gas molecules, Inorg. Chem., 2018, 57, 6981–6990 CrossRef CAS PubMed.
- L.-Y. Wang, Y.-L. Xu, Z.-D. Li, Y.-N. Wei and J.-P. Wei, CO2/CH4 and H2S/CO2 selectivity by ionic liquids in natural gas sweetening, Energy Fuel, 2018, 32, 1023 Search PubMed.
- M. Aghaie, N. Rezaei and S. Zendehboudi, A systematic review on CO2 capture with ionic liquids: Current status and future prospects, Renewable Sustainable Energy Rev., 2018, 96, 502–525 CrossRef CAS.
- J. F. Brennecke and E. J. Maginn, Ionic liquids: Innovative fluids for chemical processing, AIChE J., 2001, 47, 2384–2389 CrossRef CAS.
- M. J. Earle and K. R. Seddon, Ionic liquids. Green solvents for the future, Pure Appl. Chem., 2000, 72, 1391–1398 CrossRef CAS.
- L. A. Blanchard, D. Hancu, E. J. Beckman and J. F. Brennecke, Green processing using ionic liquids and CO2, Nature, 1999, 399, 28–29 CrossRef.
- R. W. Baker and K. Lokhandwala, Natural gas processing with membranes: An overview, Ind. Eng. Chem. Res., 2008, 47, 2109–2121 CrossRef CAS.
- A.-H. Liu, R. Ma, C. Song, Z.-Z. Yang, A. Yu, Y. Cai, L.-N. He, Y.-N. Zhao, B. Yu and Q.-W. Song, Equimolar CO2 capture by N-substituted amino acid salts and subsequent conversion, Angew. Chem., Int. Ed., 2012, 51, 11306–11310 CrossRef CAS PubMed.
- Q. Zhu, Y. Zeng and Y. Zheng, Overview of CO2 capture and electrolysis technology in molten salts: Operational parameters and their effects, Ind. Chem. Mater., 2023, 1, 595–617 RSC.
- A. R. Shaikh, H. Karkhanechi, E. Kamio, T. Yoshioka and H. Matsuyama, Quantum mechanical and molecular dynamics simulations of dual-amino-acid ionic liquids for CO2 capture, J. Phys. Chem. C, 2016, 120, 27734–27745 CrossRef CAS.
- J. Zhang, S. Zhang, K. Dong, Y. Zhang, Y. Shen and X. Lv, Supported absorption of CO2 by tetrabutylphosphonium amino acid ionic liquids, Chem. – Eur. J., 2006, 12, 4021–4026 CrossRef CAS PubMed.
- S. Onofri, H. Adenusi, A. Le Donne and E. Bodo, CO2 capture in ionic liquids based on amino acid anions with protic side chains: A computational assessment of kinetically efficient reaction mechanisms, ChemistryOpen, 2020, 9, 1153–1160 CrossRef CAS PubMed.
- J. Chau, G. Obuskovic, X. M. Jie, T. Mulukutla and K. K. Sirkar, Solubilities of CO2 and helium in an ionic liquid containing poly(amidoamine) dendrimer gen 0, Ind. Eng. Chem. Res., 2013, 52, 10484–10494 CrossRef CAS.
- B. Cao, J. Du, S. Liu, X. Zhu, X. Sun, H. Sun and H. Fu, Carbon dioxide capture by amino-functionalized ionic liquids: DFT based theoretical analysis substantiated by FT-IR investigation, RSC Adv., 2016, 6, 10462–10470 RSC.
- B. E. Gurkan, J. C. de la Fuente, E. M. Mindrup, L. E. Ficke, B. F. Goodrich, E. A. Price, W. F. Schneider and J. F. Brennecke, Equimolar CO2 absorption by anion-functionalized ionic liquids, J. Am. Chem. Soc., 2010, 132, 2116–2117 CrossRef CAS PubMed.
- N. D. Harper, K. D. Nizio, A. D. Hendsbee, J. D. Masuda, K. N. Robertson, L. J. Murphy, M. B. Johnson, C. C. Pye and J. A. C. Clyburne, Survey of carbon dioxide capture in phosphonium-based ionic liquids and end-capped polyethylene glycol using DETA (DETA = Diethylenetriamine) as a model absorbent, Ind. Eng. Chem. Res., 2011, 50, 2822–2830 CrossRef CAS.
- T. L. Greaves and C. J. Drummond, Protic ionic liquids: Properties and applications, Chem. Rev., 2008, 108, 206–237 CrossRef CAS PubMed.
- K. Huang, X. Zhang, Y. Xu, Y. Wu, X. Hu and Y. Xu, Protic ionic liquids for the selective absorption of H2S from CO2: Thermodynamic analysis, AIChE J., 2014, 4232–4240 CrossRef CAS.
- X. Zhang, W. Xiong, L. Peng, Y. Wua and X. Hu, Highly selective absorption separation of H2S and CO2 from CH4 by novel azole-based protic ionic liquids, AIChE J., 2020, 66, e16936 CrossRef CAS.
- M. A. Hussain, Y. Soujanya and G. N. Sastry, Evaluating the efficacy of amino acids as CO2 capturing agents: A first principles investigation, Environ. Sci. Technol., 2011, 45, 8582–8588 CrossRef CAS PubMed.
- D. S. Firaha and B. Kirchner, Tuning the carbon dioxide absorption in amino acid ionic liquids, ChemSusChem, 2016, 9, 1591–1599 CrossRef CAS PubMed.
- A. R. Shaikh, E. Kamio, H. Takaba and H. Matsuyama, Effects of water concentration on the free volume of amino acid ionic liquids investigated by molecular dynamics simulations, J. Phys. Chem. B, 2015, 119, 263–273 CrossRef CAS PubMed.
- A. R. Shaikh, S. Posada-Pérez, A. Brotons-Rufes, J. J. Pajski, G. Vajiha, A. Kumar, A. Mateen, M. Poater, M. Chawla Solà and L. Cavallo, Selective absorption of H2S and CO2 by azole based protic ionic liquids: A combined density functional theory and molecular dynamics study, J. Mol. Liq., 2022, 367, 120558 CrossRef CAS.
- A. R. Shaikh, M. Ashraf, T. AlMayef, M. Chawla, A. Poater and L. Cavallo, Amino acid ionic liquids as potential candidates for CO2 capture: Combined density functional theory and molecular dynamics simulations, Chem. Phys. Lett., 2020, 745, 137239 CrossRef CAS.
- R. M. Cuéllar-Franca and A. Azapagic, Carbon capture, storage and utilisation technologies: A critical analysis and comparison of their life cycle environmental impacts, J. CO2 Util., 2015, 9, 82–102 CrossRef.
- T. Kessaratikoon, T. Theerathanagorn, D. Crespy and V. D'Elia, Organocatalytic polymers from affordable and readily available building blocks for the cycloaddition of CO2 to epoxides, J. Org. Chem., 2023, 88, 4894–4924 CrossRef CAS PubMed.
- S. Arayachukiat, P. Yingcharoen, S. V. C. Vummaleti, L. Cavallo, A. Poater and V. D'Elia, Cycloaddition of CO2 to challenging N-tosyl aziridines using a halogen-free niobium complex: Catalytic activity and mechanistic insights, Mol. Catal., 2017, 443, 280–285 CrossRef CAS.
- A. A. Marciniak, K. J. Lamb, L. P. Ozorio, C. J. A. Mota and M. North, Heterogeneous catalysts for cyclic carbonate synthesis from carbon dioxide and epoxides, Curr. Opin. Green Sustainable Chem., 2020, 26, 100365 CrossRef.
- B. Shao, Y. Zhang, Z. Sun, J. Li, Z. Gao, Z. Xie, J. Hu and H. Liu, CO2 capture and in-situ conversion: Recent progresses and perspectives, Green Chem. Eng., 2022, 3, 189–198 CrossRef.
- I. S. Metcalfe, M. North, R. Pasquale and A. Thursfield, An integrated approach to energy and chemicals production, Energy Environ. Sci., 2010, 3, 212–215 RSC.
- A. J. Kamphuis, F. Picchioni and P. P. Pescarmona, CO2-fixation into cyclic and polymeric carbonates: Principles and applications, Green Chem., 2019, 21, 406–448 RSC.
- F. Della Monica and A. W. Kleij, Mechanistic guidelines in nonreductive conversion of CO2: The case of cyclic carbonates, Catal. Sci. Technol., 2020, 10, 3483–3501 RSC.
- V. D'Elia and A. W. Kleij, Surface science approach to the heterogeneous cycloaddition of CO2 to epoxides catalyzed by site-isolated metal complexes and single atoms: A review, Green Chem. Eng., 2022, 4, 210–227 CrossRef.
- M. Sathish, K. Jan Sreeram, J. Raghava Rao and B. Unni Nair, Cyclic carbonate: A recyclable medium for zero discharge tanning, ACS Sustainable Chem. Eng., 2016, 4, 1032–1040 CrossRef CAS.
- J.-H. Yang, Q. Liu, K. Z. Pupek, T. L. Dzwiniel, N. L. Dietz Rago, J. Cao, N. Dandu, L. Curtiss, K. Liu, C. Liao and Z.-C. Zhang, Molecular engineering to enable high-voltage lithium-ion battery: From propylene carbonate to trifluoropropylene carbonate, ACS Energy Lett., 2021, 6, 371–378 CrossRef CAS.
- C. Carré, Y. Ecochard, S. Caillol and L. Avérous, From the synthesis of biobased cyclic carbonate to polyhydroxyurethanes: A promising route towards renewable non-isocyanate polyurethanes, ChemSusChem, 2019, 12, 3410–3430 CrossRef PubMed.
- T. Theerathanagorn, T. Kessaratikoon, H. U. Rehman, V. D'Elia and D. Crespy, Polyhydroxyurethanes from biobased monomers and CO2: A bridge between sustainable chemistry and CO2 utilization, Chin. J. Chem., 2023, 42, 652–685 CrossRef.
- W. Guo, J. E. Gómez, À. Cristòfol, J. Xie and A. W. Kleij, Catalytic transformations of functionalized cyclic organic carbonates, Angew. Chem., Int. Ed., 2018, 57, 13735–13747 CrossRef CAS PubMed.
- C. Claver, M. B. Yeamin, M. Reguero and A. M. Masdeu-Bultó, Recent advances in the use of catalysts based on natural products for the conversion of CO2 into cyclic carbonates, Green Chem., 2020, 22, 7665–7706 RSC.
- J. Tharun, K. R. Roshan, A. C. Kathalikkattil, D.-H. Kang, H.-M. Ryu and D.-W. Park, Natural amino acids/H2O as a metal- and halide-free catalyst system for the synthesis of propylene carbonate from propylene oxide and CO2 under moderate conditions, RSC Adv., 2014, 4, 41266–41270 RSC.
- W. Chen, L.-X. Zhong, X.-W. Peng, R.-C. Sun and F.-C. Lu, Chemical fixation of carbon dioxide using a green and efficient catalytic system based on sugarcane bagasse—An agricultural waste, ACS Sustainable Chem. Eng., 2015, 3, 147–152 CrossRef CAS.
- L. Guo, R. Dou, Y. Wu, R. Zhang, L. Wang, Y. Wang, Z. Gong, J. Chen and X. Wu, From lignin waste to efficient catalyst: Illuminating the impact of lignin structure on catalytic activity of cycloaddition reaction, ACS Sustainable Chem. Eng., 2019, 7, 16585–16594 CrossRef CAS.
- W. Jaroonwatana, T. Theerathanagorn, M. Theerasilp, S. Del Gobbo, D. Yiamsawas, V. D’Elia and D. Crespy, Nanoparticles of aromatic biopolymers catalyze CO2 cycloaddition to epoxides under atmospheric conditions, Sustainable Energy Fuels, 2021, 5, 5431–5444 RSC.
- W. Natongchai, D. Crespy and V. D'Elia, CO2 fixation: Cycloaddition of CO2 to epoxides using practical metal-free recyclable catalysts, Chem. Commun., 2025, 61, 419–440 RSC.
- T. Theerathanagorn, A. Vidal-López, A. Comas-Vives, A. Poater and V. D'Elia, Cycloaddition of CO2 to epoxides “around water”: A strategy to apply and recycle efficient water-soluble bio-based organocatalysts in biphasic media, Green Chem., 2023, 25, 4336–4349 RSC.
- A. Brzęczek-Szafran, A. Siewniak and A. Chrobok, Assessment of green chemistry metrics for carbon dioxide fixation into cyclic carbonates using eutectic mixtures as catalyst: Comprehensive evaluation on the example of a tannic acid-derived system, ACS Sustainable Chem. Eng., 2023, 11, 11415–11423 CrossRef.
- X. Yang, S. Wang, X. Liu, Z. Huang, X. Huang, X. Xu, H. Liu, D. Wang and S. Shang, Preparation of non-isocyanate polyurethanes from epoxy soybean oil: Dual dynamic networks to realize self-healing and reprocessing under mild conditions, Green Chem., 2021, 23, 6349–6355 RSC.
- X. Yang, C. Ren, X. Liu, P. Sun, X. Xu, H. Liu, M. Shen, S. Shang and Z. Song, Recyclable non-isocyanate polyurethanes containing a dynamic covalent network derived from epoxy soybean oil and CO2, Mater. Chem. Front., 2021, 5, 6160–6170 RSC.
- M. North and R. Pasquale, Mechanism of cyclic carbonate synthesis from epoxides and CO2, Angew. Chem., Int. Ed., 2009, 48, 2946–2948 CrossRef CAS PubMed.
- V. D'Elia, A. A. Ghani, A. Monassier, J. Sofack-Kreutzer, J. D. A. Pelletier, M. Drees, S. V. C. Vummaleti, A. Poater, L. Cavallo, M. Cokoja, J.-M. Basset and F. E. Kühn, Dynamics of the NbCl5-catalyzed cycloaddition of propylene oxide and CO2: Assessing the dual role of the nucleophilic Co-catalysts, Chem. – Eur. J., 2014, 20, 11870–11882 CrossRef PubMed.
- T.-T. Wang, Y. Xie and W.-Q. Den, Reaction mechanism of epoxide cycloaddition to CO2 catalyzed by Salen-M (M = Co, Al, Zn), J. Phys. Chem. A, 2014, 118, 9239–9243 CrossRef CAS PubMed.
- C. J. Whiteoak, A. Nova, F. Maseras and A. W. Kleij, Merging sustainability with organocatalysis in the formation of organic carbonates by using CO2 as a feedstock, ChemSusChem, 2012, 5, 2032–2038 CrossRef CAS PubMed.
- Z. Guo, Q. Jiang, Y. Shi, J. Li, X. Yang, W. Hou, Y. Zhou and J. Wang, Tethering dual hydroxyls into mesoporous poly(ionic liquid)s for chemical fixation of CO2 at ambient conditions: A combined experimental and theoretical study, ACS Catal., 2017, 7, 6770–6780 CrossRef CAS.
- F. Castro-Gómez, G. Salassa, A. W. Kleij and C. Bo, A DFT study on the mechanism of the cycloaddition reaction of CO2 to epoxides catalyzed by Zn(Salphen) complexes, Chem. – Eur. J., 2013, 19, 6289–6298 CrossRef PubMed.
- K. Norseeda, P. Yingcharoen, P. Nimnual, S. Puchum, S. Arayachukiat, T. Piromchart, M. Wagner, H. Zipse and V. D'Elia, Discovery of a phosphonium ionic liquid phase from the reaction of trialkylphosphines and epichlorohydrin carbonate and application as a CO2-based triphasic demulsifier of crude oil, J. Mol. Struct., 2023, 1292, 136122 CrossRef CAS.
- R. Morales-Cerrada, B. Boutevin and S. Caillol, Glycerol carbonate methacrylate: A cross-linking agent for hydroxyurethane-acrylate coatings, Prog. Org. Coat., 2021, 151, 106078 CrossRef CAS.
- X.-L. Meng, Z.-J. Ju, S.-J. Zhang, X.-D. Liang, N. von Solms, X.-C. Zhang and X.-P. Zhang, Efficient transformation of CO2 to cyclic carbonates using bifunctional protic ionic liquids under mild conditions, Green Chem., 2019, 21, 3456–3463 RSC.
- T. Ying, X. Tan, Q. Su, W.-G. Cheng, L. Dong and S.-J. Zhang, Polymeric ionic liquids tailored by different chain groups for the efficient conversion of CO2 into cyclic carbonates, Green Chem., 2019, 21, 2352–2361 RSC.
- P. Rani, R. Das and C. M. Nagaraja, A review on framework (MOF/COF/POP)-based materials for efficient conversion of CO2 to bio-active oxazolidinones, Inorg. Chem. Front., 2025, 12, 430–478 RSC.
- S. Arayachukiat, C. Kongtes, A. Barthel, S. V. C. Vummaleti, A. Poater, S. Wannakao, L. Cavallo and V. D'Elia, Ascorbic acid as a bifunctional hydrogen bond donor for the synthesis of cyclic carbonates from CO2 under ambient conditions, ACS Sustainable Chem. Eng., 2017, 5, 6392–6397 CrossRef CAS.
- S. Kozuch and S. Shaik, How to conceptualize catalytic cycles? The energetic span model, Acc. Chem. Res., 2011, 44, 101–110 CrossRef CAS PubMed.
- S. Kozuch and J. M. L. Martin, The rate-determining step is dead. Long live the rate-determining state!, ChemPhysChem, 2011, 12, 1413–1418 CrossRef CAS PubMed.
- P. P. Pescarmona, Cyclic carbonates synthesised from CO2: Applications, challenges and recent research trends, Curr. Opin. Green Sustainable Chem., 2021, 29, 100457 CrossRef CAS.
- P. Yingcharoen, C. Kongtes, S. Arayachukiat, K. Suvarnapunya, S. V. C. Vummaleti, S. Wannakao, L. Cavallo, A. Poater and V. D'Elia, Assessing the pKa-dependent activity of hydroxyl hydrogen bond donors in the organocatalyzed cycloaddition of carbon dioxide to epoxides: Experimental and theoretical study, Adv. Synth. Catal., 2019, 361, 366–373 CrossRef CAS.
- W. Natongchai, S. Posada-Pérez, Ch. Phungpanya, J. A. Luque Urrutia, M. Solà, V. D'Elia and A. Poater, Enhancing the catalytic performance of group I, II metal halides in the cycloaddition of CO2 to epoxides under atmospheric conditions by cooperation with homogeneous and heterogeneous highly nucleophilic aminopyridines: Experimental and theoretical study, J. Org. Chem., 2022, 87, 2873–2886 CrossRef CAS PubMed.
- V. Aomchad, S. Del Globo, A. Poater and V. D'Elia, Exploring the potential of Group III salen complexes for the conversion of CO2 under ambient conditions, Catal. Today, 2021, 375, 324–334 CrossRef CAS.
- G. S. Hammond, A correlation of reaction rates, J. Am. Chem. Soc., 1955, 77, 334–338 CrossRef CAS.
- L. Falivene, Z. Cao, A. Petta, L. Serra, A. Poater, R. Oliva, V. Scarano and L. Cavallo, Towards the online computer-aided design of catalytic pockets, Nat. Chem., 2019, 11, 872–879 CrossRef CAS PubMed.
- A. Poater, B. Cosenza, A. Correa, S. Giudice, F. Ragone, V. Scarano and L. Cavallo, SambVca: A web application for the calculation of buried volumes of N-heterocyclic carbene ligands, Eur. J. Inorg. Chem., 2009, 2009, 1759–1766 CrossRef.
- S. Escayola, N. Bahri-Laleh and A. Poater, %VBur index and steric maps: From predictive catalysis to machine learning, Chem. Soc. Rev., 2024, 53, 853–882 RSC.
- L. Falivene, R. Credendino, A. Poater, A. Petta, L. Serra, R. Oliva, V. Scarano and L. Cavallo, SambVca 2. A web tool for analyzing catalytic pockets with topographic steric maps, Organometallics, 2016, 35, 2286–2293 CrossRef CAS.
- Y. A. Alassmy and P. P. Pescarmona, The role of water revisited and enhanced: A sustainable catalytic system for the conversion of CO2 into cyclic carbonates under mild condition, ChemSusChem, 2019, 12, 3856–3863 CrossRef CAS PubMed.
- J. V. Alegre-Requena, S. V. S. Sowndarya, R. Pérez-Soto, T. M. Alturaifi and R. S. Paton, AQME v1.7.1: Automated quantum mechanical environments for researchers and educators, WIREs Comput. Mol. Sci., 2023, 13, e1663663 Search PubMed.
- C. Bannwarth, E. Caldeweyher, S. Ehlert, A. Hansen, P. Pracht, J. Seibert, S. Spicher and S. Grimme, Extended tight-binding quantum chemistry methods, WIREs Comput. Mol. Sci., 2021, 11, e1493 CrossRef CAS.
- MORFEUS, https://github.com/kjelljorner/morfeus/, (accessed April 2025) Search PubMed.
- RDKit: Open-Source Cheminformatics 2010, https://www.rdkit.org, (accessed April 2025) Search PubMed.
- D. Dalmau and J. V. Alegre-Requena, ROBERT v1.3.0: Bridging the gap between machine learning and chemistry, Wiley Interdiscip. Rev.:Comput. Mol. Sci., 2024, 14, e1733 CAS.
- For a more detailed explanation of the three “flawed” models used, see https://robert.readthedocs.io/en/latest/Modules/verify.html, (accessed April 2025).
- For a more detailed explanation of the ROBERT score, see https://robert.readthedocs.io/en/latest/Report/score.html, (accessed April 2025).
- D. Dalmau and J. V. Alegre-Requena, Integrating digital chemistry within the broader chemistry community, Trends Chem., 2024, 6, 459–469 CrossRef CAS.
- A. D. Becke, Density-functional exchange-energy approximation with correct asymptotic behavior, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098–3100 CrossRef CAS PubMed.
- J. P. Perdew, Density-functional approximation for the correlation energy of the inhomogeneous electron gas, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 8822–8824 CrossRef PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford, CT, 2016 Search PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- A. Schäfer, H. Horn and R. Ahlrichs, Fully optimized contracted Gaussian basis sets for atoms Li to Kr, J. Chem. Phys., 1992, 97, 2571–2577 CrossRef.
- A. Bergner, M. Dolg, W. Küchle, H. Stoll and H. Preuß, Ab initio energy-adjusted pseudopotentials for elements of groups 13–17, Mol. Phys., 2006, 80, 1431–1441 CrossRef.
- A. D. Becke, Density-functional thermochemistry. III. The role of exact Exchange, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- R. A. Kendall, T. H. Dunning and R. J. Harrison, Electron affinities of the first-row atoms revisited. Systematic basis sets and wave functions, J. Chem. Phys., 1992, 96, 6796–6806 CrossRef CAS.
- J. Tomasi and M. Persico, Molecular interactions in solution: An overview of methods based on continuous distributions of the solvent, Chem. Rev., 1994, 94, 2027–2094 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: xyz coordinates and absolute energies. See DOI: https://doi.org/10.1039/d5im00037h |
| This journal is © Institute of Process Engineering of CAS 2025 |