
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTwo-step continuous flow aerobic oxidation of cannabidiol to cannabinoquinone derivatives†
Manuel
Zielke
 ab,
Christof
Aellig
c,
Dominique M.
Roberge
*c,
Christopher A.
Hone
*ab and
C. Oliver
Kappe
*ab
ab,
Christof
Aellig
c,
Dominique M.
Roberge
*c,
Christopher A.
Hone
*ab and
C. Oliver
Kappe
*ab
aCenter for Continuous Flow Synthesis and Processing (CCFLOW), Research Center Pharmaceutical Engineering GmbH (RCPE), Inffeldgasse 13, 8010 Graz, Austria
bInstitute of Chemistry, University of Graz, NAWI Graz, Heinrichstrasse 28, 8010 Graz, Austria. E-mail: christopher.hone@rcpe.at; oliver.kappe@uni-graz.at
cAdvanced Chemistry Technologies, Lonza AG, CH-3930 Visp, Switzerland. E-mail: dominique.roberge@lonza.com
First published on 15th May 2025
Abstract
A two-step continuous flow oxidation procedure is presented for the preparation of Etrinabdione, a cannabidiol quinone derivative. Etrinabdione is currently undergoing clinical trials for the treatment of peripheral artery disease. Both steps described herein utilize molecular oxygen as a green and inexpensive stoichiometric oxidant. Initially, the two-steps were optimized separately. The first step is a very fast oxidation process to form quinonoid derivative HU-331 and includes an in situ quench for the safe handling of peroxy intermediates. The second step is a segmented flow oxidative amination to afford Etrinabdione. The two steps can be run either separately or telescoped by incorporating a gravity-based continuous separation. The two steps were telescoped to afford Etrinabdione in 98% yield. The process mass intensity (PMI) was significantly reduced when compared to an existing batch protocol. The second step was also adapted for the preparation of two additional cannabidiol quinone analogs. The described protocol significantly improves the efficiency, safety and scalability when compared to existing batch protocols.
Green foundation1. We develop a two-step continuous-flow synthesis of cannabinoquinone derivatives from cannabidiol. O2 is used as a sustainable and efficient oxidant source and the use of flow facilitates its safe handling.2. Calculation of the PMI for the newly defined procedure and previous literature batch protocols. The yield is very high and the PMI for the telescoped sequence corresponds to a reduction in the PMI of 97% from existing reported procedures. 3. Future research would examine the associated challenges with scaling-up to larger flow equipment. |
Introduction
Compounds bearing the quinone motif have been widely investigated and are currently used in a number of active pharmaceutical ingredients (APIs).1,2 In general, these compounds display high biological activity, and have found applications as antibiotics and for the treatment of certain cancers.1 However, compounds bearing this quinone motif often have considerable side effects, such as heart toxicity, which limits their use.3,4 Recent research has focused on identifying derivatives that display promising therapeutic properties along with minimal side effects.The quinone motif is found in semisynthetic compounds, see Fig. 1b, which can be prepared from naturally occurring cannabinoids. Natural products, such as cannabidiol (CBD, 1), Δ8-tetrahydrocannabinol (Δ8-THC) and cannabinol (CBN), can be extracted from the cannabis plant (Fig. 1a). One of the semisynthetic compounds, HU-331 (2), exhibits potent antineoplastic activity in vivo, and is currently being investigated for its anticancer activity.5 In addition, HU-331 (2) was also found to inhibit topoisomerase II catalytically, without causing damage to DNA strains or producing a reactive oxygen species.6 This mode of action avoids many of the side effects observed for other quinone-based compounds.
 | ||
| Fig. 1 (a) Natural products extracted from the cannabis plant; (b) semisynthetic cannabinoids. | ||
In 2007, O'Sullivan reported that phytocannabinoids, such as THC and CBD (1), bind and activate peroxisome proliferator-activated receptors (PPARs).7 PPAR-γ are known to be involved in inflammatory processes, and these receptors have been shown to be activated by natural and synthetic cannabinoids. Thus, PPAR-γ are an ideal target for the treatment of neuroinflammatory diseases.8 In 2012 Muñoz and co-workers reported an increase in binding activity to PPAR-γ for the oxidation products, HU-331 (2) and cannabigeroquinone (CBGQ), over their non-oxidized counterparts, CBD (1) and cannabigerol, respectively.9 Unfortunately, HU-331 (2) proved to be thiophilic and rather unstable towards oxidative dimerization, which completely removed any activity in vivo. To render HU-331 (2) more stable, the group of Muñoz utilized its high electrophilicity and introduced a range of different amines onto the unsubstituted position in the quinone.10 The fully substituted amine derivatives (VCE-004.1-14) were demonstrated not to dimerize and did not exhibit any thiol trapping activity as shown for HU-331 (2) in a cysteamine-recovery assay.11 Etrinabdione, VCE-004.8 (3a), the benzylamine derivative, was shown to be the most promising compound for activity toward PPAR-γ and cannabinoid receptor type 2 (CB2) pathways. Recent studies have demonstrated that Etrinabdione activates the AMPK/Sirt1/eNOS pathway in a PP2A/B55α-dependent manner, effectively preventing endothelial cell damage and senescence.12 Additionally, it promotes arteriogenesis and angiogenesis, highlighting its potential for vascular protection and regeneration.
Etrinabdione (3a) has been investigated for its ability to prevent diet-induced obesity,13 sclerodermia14 and multiple sclerosis (MS),15 adult and neonatal stroke,16,17 traumatic brain injury,18 and critical limb ischemia.12 Etrinabdione is currently in phase 2a clinical trial in Spain for the treatment against peripheral artery disease.19 Thus, there is considerable interest in developing a scalable synthetic route for the synthesis of Etrinabdione (3a) via HU-331 (2) from the natural product CBD (1).
HU-331 (2) was first synthesised via batch aerobic oxidation of CBD (1) in the presence of KOH in alcoholic media, either ethanol (EtOH) or methanol (MeOH), with air as the oxidant source (Scheme 1a).5 The purple color produced is used to test for the presence of cannabinoids, and is commonly known as the Beam test.20 The reaction proceeds in relatively poor yield (20%) due to the reactive electrophilic position at the unsubstituted carbon undergoing side reactions. The reaction of CBD (1) to form HU-331 (2) was further improved to 74% yield by using potassium tert-butoxide (KOtBu) as a non-nucleophilic base instead of an aqueous KOH solution (Scheme 1b).10 The use of KOtBu was shown to be superior to KOH because no side products were formed. This approach, however; suffers from the low solubility of the KOtBu in toluene (PhMe) resulting in very high dilution of CBD 1 (0.044 M in PhMe). The batch protocols required purification by column chromatography to provide the necessary product purity.
 | ||
| Scheme 1 (a) First reported synthesis of HU-331 (2) via aerobic oxidation of CBD (1) in the presence of KOH; (b) improved batch synthesis of HU-331 (2) from CBD (1) using KOtBu. | ||
The conversion of HU-331 (2) to Etrinabdione (3a) was achieved via treatment of 2 with a very high excess of benzylamine (40 equiv.) and subsequent oxidation with air to afford 3a in 66% yield after purification by column chromatography (Scheme 2).10
 | ||
| Scheme 2 Synthesis of Etrinabdione (3) via treatment of 2 with a large excess of benzylamine and subsequent aerobic oxidation with air. | ||
The batch protocols described above rely on vigorous stirring of the reaction solutions within an air-opened flask. This mode of operation is impractical at larger scales due to safety and scalability challenges. Historically, the pharmaceutical sector have been reliant on multipurpose batch reactors for manufacture of APIs.21 Multiphase batch reactions often suffer from mass transfer limitations due to the reduction in surface-to-volume ratio with increase in batch reactor size.22 Studies have successfully demonstrated that mass transfer can be enhanced within small scale flow devices due to the high interfacial area between the gas and liquid phases providing a high surface-to-volume ratio.22,23 Henry's law states that the solubility of a gas increases as a function of pressure, thus increasing the pressure is often used to increase the concentration of gas in the liquid phase. A standard commercial batch reactor can typically operate only up to 6 bar. If a higher pressure is needed, then a more specialized and expensive batch vessel is required.
Over recent years, there has been a paradigm shift to continuous flow technologies with their use for API manufacture encouraged by the US Food and Drug Administration.24 This shift is driven by the increased need for improved sustainability, higher product quality, improved safety and more cost effective API manufacture.25 Continuous flow reactors have been demonstrated to provide precise parameter control, improved mixing, and safer handling of hazardous chemistry.26 The safer handling of hazardous reagents and intermediates is possible in flow because a small inventory is processed at any one time.27 Continuous processing has been demonstrated to improve the process efficiency, in terms of mass, energy, space, and time – due to the enhanced heat and mass transfer, precise residence time control, shorter process times, better product quality, smaller footprint and facile scalability.28–30 Vilé and co-workers performed techno-economic analyses (TEA) and life-cycle assessments (LCA) for seven industrially relevant APIs. The results showed that continuous-flow processes were significantly more sustainable than batch with improvements in energy efficiency, water consumption, and waste reduction.31
Oxygen is perhaps one of the greenest and inexpensive reagents available to the organic chemist, but it is underutilized within API manufacture due safety concerns.32 Concentrations of O2 in N2 below 10% are generally used in batch pharmaceutical manufacturing to prevent the risk of combustions in the presence of flammable organic solvents (“limiting oxygen concentration”).33 However, higher concentrations have been demonstrated to be inherently safe through the application of flow technologies.34 Recently, there have been examples presented where pure oxygen was safely used instead of synthetic air for API synthesis, with significant improvements in product yield and process efficiency obtained.35 In addition, potentially explosive peroxy radicals are often formed in oxidation reactions, within a continuous flow environment these can be formed in very small quantities at any one time and quenched in situ.
Results and discussion
One of the limitations of the batch oxidation of CBD (1) to HU-331 (2) reported by Muñoz and co-workers was the low solubility of the KOtBu in PhMe, thus causing a high dilution of CBD (1) to be necessary (0.044 M).10 This high dilution results in a very high process mass intensity (PMI) (kg raw materials per kg product) of 787. To circumvent this problem, we commenced our investigation by considering a more appropriate solvent system for this reaction type by considering the nucleophilicity, solubility of the base, stability against oxidation and inherent greenness of the solvent. KOtBu displays high solubility in tetrahydrofuran (THF) and tert-butanol (tBuOH).36 Unfortunately, the use of THF as a solvent is discouraged due to the high energy requirements for its production.37 In addition, the use of THF is avoided for reactions employing oxidants because THF readily forms explosive peroxides. tBuOH is generally considered a green solvent,37 however; its high melting point of ∼26 °C can make it difficult to handle due to it freezing to form a solid at room temperature (e.g., tBuOH can freeze inside pumps). In 2013, Ley and co-worker reported a continuous flow oxidation process using a solvent mixture of tBuOH and PhMe (6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 = v:v), which ensured that the solvent remained as a liquid.38 Based on this report we evaluated the solubility of KOtBu and CBD (1) within this solvent mixture (Table 1). Gratifyingly, both KOtBu and CBD (1) were soluble within tBuOH/PhMe (6:1) at concentrations above 1 M, therefore this solvent mixture was used for all experiments described herein.
:1 = v:v), which ensured that the solvent remained as a liquid.38 Based on this report we evaluated the solubility of KOtBu and CBD (1) within this solvent mixture (Table 1). Gratifyingly, both KOtBu and CBD (1) were soluble within tBuOH/PhMe (6:1) at concentrations above 1 M, therefore this solvent mixture was used for all experiments described herein.
Initially, we attempted a 1 mmol scale batch oxidation of CBD (1) to HU-331 (2) by using a 1 M solution of 1 in tBuOH/PhMe (6:1) and 2.7 equiv. of KOtBu within a vigorously stirred air-opened flask at room temperature. To our delight, quantitative conversion of 1 and 89% yield was achieved within 40 min reaction time. The conversion of 1 dropped to 50% when a 1 min reaction time was used. With these initial results in hand, we envisioned that the utilization of continuous flow technology could improve the inherent safety, control over the process parameters and scalability of the oxidation. When using pure O2 reaction rate is substantially enhanced, since O2 is not competing with N2 for dissolution in the liquid phase and the process is less likely to be mass-transfer limited.34 A further benefit of using pure O2 in flow is that smaller reactor unit volume is possible because there is a lower gas contribution within the flow channel, thus providing improved space–time yields (within diluted mixtures the majority of gas present is N2 and it is not consumed), thus improving process efficiency.
For initial screening studies, we assembled the Modular MicroReaction System (MMRS), equipped with a FlowPlate™ Lab reactor, see Table 2 and Fig. S1,† manufactured by Ehrfeld Mikrotechnik.39 The system has been designed so that it can be reconfigured depending on the particular application and scaled in a facile manner.40 The feed solutions were introduced by using sample loops and the liquid phases were pumped by two HPLC pumps (Uniqsis FlowSyn). Unless otherwise stated, the feed solutions were comprised of: (1) 0.5 M solution of CBD (1) in t-BuOH/PhMe (6:1), and (2) 1 M solution of KOtBu in t-BuOH/PhMe (6:1). The two liquid feeds were pre-mixed within a T-piece prior to entering the reactor. The T-piece also contained a pressure sensor for monitoring the system pressure. A FlowPlate™ reactor with a LL mixing structure to facilitate enhanced mixing between the two phases,41 with a 0.24 mL internal volume, 0.2 mm minimum channel width and 2 mm average channel width, was used. A mass flow controller (Bronkhorst, EL-FLOW) was used for the introduction of oxygen. Pure oxygen was introduced at an inlet within the FlowPlate. Initially, a perfluoroalkoxy (PFA) tubular reactor, with 40 mL internal volume and 1 mm internal diameter, was installed after the LL mixing plate to enable access to longer residence times. The system pressure was maintained by a back pressure regulator (BPR, Zaiput) set to 7 bar. The effluent was quenched into a flask containing an aqueous 0.5 M solution of HCl.
|
|
||||
|---|---|---|---|---|
| Entry | Reactor volume [mL] | Flow rate [mL min−1] | Residence time | Conv. 1 [%]f |
|
a Unless otherwise stated, conditions: Feed 1: 0.5 M CBD in tBuOH/toluene (6:1); Feed 2: 1 M KOtBu in tBuOH/toluene (6:1) with flow rate ratio for Feed 1/Feed 2 = 1:1.05; 1.05 equiv. O2 introduced by a mass flow controller. The effluent was quenched into a vessel containing 0.5 M HCl.
b 0.25 M CBD and 0.625 M KOtBu used.
c 0.90 equiv. O2 used.
d No PFA coil used.
e T-piece used instead of micromixer.
f Analyzed by HPLC UV/VIS at 215 nm. 7 bar backpressure was applied.
|
||||
| 1b | 40 | 1 | 41 min | >99 |
| 2 | 40 | 2 | 20.5 min | >99 |
| 3 | 40 | 4 | 10.2 min | >99 |
| 4c | 40 | 4 | 10.2 min | 93 |
| 5d | 0.8 | 4 | 12 s | >99 |
| 6d | 0.8 | 8 | 6 s | >99 |
| 7d,e | 0.2 | 2 | 6 s | 83 |

The results from the preliminary flow screening are shown in Table 2. The residence time could be reduced from 41 min to 10.2 min without any loss in conversion (Table 2, entries 1 to 3). The reaction did not reach full completion when oxygen was used in substoichiometric amounts (Table 2, entry 4), which demonstrated the need for at least equimolar amounts of oxygen in the system. We observed that the oxygen fully dissolved in the liquid phase within only a few seconds, which indicated the rapid consumption of oxygen in the reaction. Thus, the PFA tubular reactor was removed to enable access to shorter residence times. The residence time could be successfully reduced to ∼6 seconds within the LL mixing plate without any loss in conversion (Table 2, entry 6). We were also interested in assessing the mixing characteristics of the system, a drop in conversion to 83% was observed when the LL mixing plate was replaced with a T-piece and the same volume of tubing (entry 7), which indicated the poor mixing properties of the T-piece and that the reaction rate was limited by mass transfer in this instance. The flow optimization was conducted with only a small excess of oxygen (1.05 equiv.), so the reaction becomes starved of oxygen towards the end of the reactor. Therefore, at the beginning of the reactor there is a higher oxygen concentration present while towards the end the concentration is very low, which improved the inherent process safety at the outlet. In addition, the collection vessel was kept under a stream N2 and cooled within an ice batch to operate below the flash point.
After the initial screening, we modified the flow setup to the configuration shown in Scheme 3 and Fig. S2.† Temperature and pressure probes were placed immediately before and after the FlowPlate for inline process monitoring. The feed solutions were introduced directly through the HPLC pumps. The T-piece which had been used for pre-mixing the two liquid feeds was replaced with a cascade mixer, which mixes the two feeds by a split and recombine principle. There were two different configurations used within the FlowPlate, either the reaction solution was collected after passing through 7 mixing elements (0.026 mL internal volume) at outlet 3, or the reaction solution was collected after passing through the entire FlowPlate, corresponding to 70 mixing elements (0.24 mL internal volume) at outlet 7 (Fig. 2). The effluent was quenched into a flask containing 0.5 M aqueous HCl after passing through a coil with a volume of 1.2 mL.
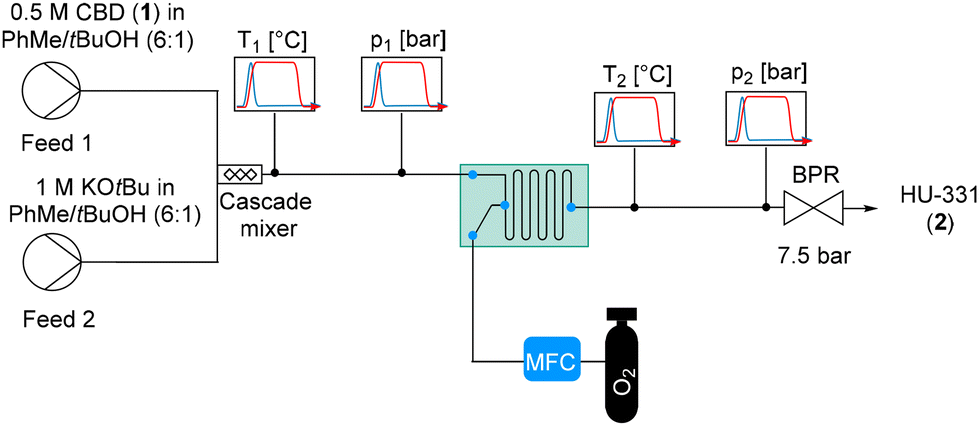 | ||
| Scheme 3 Continuous flow setup for the oxidation of CBD (1) to HU-331 (2). Conditions: Feed 1: 0.5 M CBD (1) in tBuOH/PhMe (6:1); Feed 2: 1 M KOtBu in tBuOH/PhMe with flow rates for Feed 1/Feed 2 = 2:2.1 mL min−1; 1.05 equiv. O2 introduced through a mass flow controller. 7.5 bar backpressure was applied. The effluent was quenched into a vessel containing 0.5 M HCl. | ||
 | ||
| Fig. 2 Liquid/liquid (LL) mixing structures (representative image). Temperature was measured before inlet 1 and after outlet 3 or 7. Oxygen was introduced via inlet 2. | ||
We were interested in investigating the exothermic nature of the transformation in a steady state flow experiment using the conditions shown in Scheme 3, either using outlet 3 or outlet 7. We observed an average temperature difference of 6.2 °C when outlet 3 was used. This rapid increase clearly demonstrates the highly exothermic nature of the reaction. In contrast, when outlet 7 was used an average temperature difference of 1.5 °C was observed. The smaller temperature difference at outlet 7 can be explained by heat dissipation occurring over the entire surface of the FlowPlate. Furthermore, a gradual increase in temperature over operation time was observed (see Fig. S3 and S4†). The conversion dropped to 93% when 7 mixing elements were used (outlet 3). Multiphase reactions that occur in such a short time are typically highly mass transfer limited under certain conditions which can make them difficult to scale.23 A scale-out experiment was successfully conducted over 19.4 min operation time by using the same conditions as shown in Scheme 3 and through utilization of the full length of the flow plate (70 mixing elements, outlet 7) to afford HU-331 (2) in 99.4% isolated yield.
Our hypothesis for the mechanism of this transformation is through the formation of dipotassium salt 4 by the deprotonation of 1 by KOtBu, and subsequent addition of oxygen to give the peroxy intermediate 5 (Scheme 4). The reactive intermediate 5 is then quenched by acid to form the desired product 2. This mechanism is supported by investigations conducted by Musso on similar compounds.42 Peroxy compounds need to be handled with care, and if possible quenched in situ, because of their potential to undergo explosive decomposition.
 | ||
| Scheme 4 Proposed mechanism for the oxidation of CBD (1) to 2 using KOtBu as base. | ||
To address the issue associated with the accumulation of the hazardous peroxy intermediate 5, we selected to incorporate an inline quench consisting of aqueous HCl. The quench stream was introduced by a syringe pump (HighTec Zhang Syrdos) at inlet 5 within the LL mixing structure (Fig. 2 and 3). This resulted in a reduction in reactor volume available for the oxidation step (165 μL), thus the flow rates were lowered accordingly to allow for a sufficient residence time for satisfactory conversion values resulting in a slight reduction throughput. We also selected to increase the applied back pressure to 10 bar to ensure complete dissolution of oxygen within the liquid phase. The amount of base displayed no apparent influence on the conversion under the range explored (Table 3, entries 1–3). The oxidation reaction provided >99% conversion within 4.4 s residence time (Table 3, entry 5). The quenching of the reaction solution could be accomplished with 2.5 equiv. of aqueous HCl with a concentration of either 1 M, or 0.5 M HCl at double the flow rate. 0.5 M aqueous HCl was preferred because the subsequent phase separation was superior. The quenching of the peroxy intermediate 5 was observed via a color change from deep purple to orange (Fig. 3).
 | ||
| Fig. 3 Image of the continuous fow oxidation of CBD (1) to HU-331 (2) with an inline quench by aqueous HCl solution. The first part of the reaction was a purple color. The color change from purple to orange indicated quenching of reactive intermediate 5. | ||
| Entry | Feed 1 low rate [mL min−1] | Feed 2 flow rate [mL min−1] | KOtBu [equiv.] | Residence time [s] | Conv. 1 [%] |
|---|---|---|---|---|---|
|
a Conditions: Feed 1: 0.5 M CBD in tBuOH/toluene (6:1); Feed 2: 1 M KOtBu in tBuOH/toluene (6:1); Feed 3: 1 M HCl with a flow rate of 2.5 mL min−1; 1.05 equiv. O2 introduced through a mass flow controller. Conversion was measured by HPLC UV/VIS at 215 nm. Isolated yield given in parentheses.
b 0.5 M HCl was introduced into the microreactor with a flow rate of 2.7 mL min−1.
|
|||||
| 1 | 1.95 | 2.05 | 2.1 | 2.5 | 83 |
| 2 | 1.90 | 2.10 | 2.2 | 2.5 | 87 |
| 3 | 1.80 | 2.20 | 2.44 | 2.5 | 85 |
| 4 | 1.35 | 1.40 | 2.04 | 3.6 | 93 |
| 5b | 1.10 | 1.15 | 2.09 | 4.4 | >99 (>99) |
The identified optimized conditions were used in a long run for 3 h operation time to afford HU-331 (2) in 99% isolated yield (31.5 g) after simple phase extraction and solvent evaporation, which corresponded to 94.4 mmol of product over the 3 h collection period (31.6 mmol h−1 or 10.5 g h−1). This resulted in a calculated space–time yield of 43.8 kg (L h)−1, based on the combined volume of 0.24 mL for the reaction and quench steps.
Subsequently, we turned our attention to investigate the second step, the oxidative amination of HU-331 (2) with benzylamine to form Etrinabdione (3a). The published batch protocol involves the treatment of 2 (0.5 M in EtOH) with 45 equiv. of benzylamine vigorously stirred within an air-opened flask for 18 h (Scheme 2). We were able to reproduce the batch protocol to synthesize 3a in 89% yield. The mechanism for the formation of 3a most likely proceeds via the nucleophilic addition of benzylamine to the unsubstituted position of 2 followed by reoxidization of the resulting enol using air to the para-quinone 3a. The nucleophilic addition is probably the rate-determining step. For the optimization of the reaction, we assembled a flow system similar as for the first step described above. The feed solutions were comprised of: (feed 1) 0.5 M solution of HU-331 (2) in EtOH, and (feed 2) neat benzylamine (9.15 M). Initially, 2 equiv. of oxygen were used.
We commenced our screening by evaluating the influence of the equivalents of benzylamine on conversion. Surprisingly, by using only 10 equiv. of benzylamine at 20 °C was sufficient to achieve a conversion of 32% within 1.8 min of residence time. Increasing to 40 equiv. of benzylamine resulted in 58% conversion (Chart 1) within the same residence time. To minimize waste, we selected to optimize the transformation with 10 equiv. of benzylamine. The residence time was extended by increasing the volume of the reactor by adding a capillary reactor (8 mL) after the FlowPlate™ to provide a total volume of 9.6 mL, corresponding to a residence time of 10.6 min. Under these conditions, a side reaction was identified, corresponding to the oxidation of benzylamine to benzylimine, which then subsequently hydrolyzed to benzaldehyde within the quench (Scheme 5). The highest conversion of 2 was achieved when 1.5 equiv. of oxygen was used, but the equivalents of oxygen appeared to have minimal influence over benzaldehyde formation (Chart 2).
 | ||
| Chart 1 Influence of equivalents of benzylamine used on conversion of 2. Conditions: Feed 1: 0.5 M HU-331 (2) in EtOH; Feed 2: 9.15 M benzylamine with a total flow rate of Feed 1 + Feed 2 = 0.9 mL min−1; reactor volume = 1.6 mL; 2.0 equiv. O2 introduced by a mass flow controller. 13 bar back pressure were applied. Conversion measured by 1H NMR and HPLC UV/VIS at 250 nm. | ||
 | ||
| Scheme 5 Oxidation of benzylamine with molecular oxygen and subsequent hydrolysis to benzaldehyde with diluted HCl. | ||
 | ||
| Chart 2 Influence of equivalents of oxygen used on conversion of 2 and benzaldehyde formation. Conditions: Feed 1: 0.5 M HU-331 in EtOH; Feed 2: 9.15 M benzylamine with flow rates for Feed 1/Feed 2 = 0.58:0.32 mL min−1; reactor volume = 9.6 mL; O2 introduced through a mass flow controller. 13 bar back pressure were applied. Conversion measured by HPLC UV/VIS at 250 nm. | ||
Since our goal was to telescope the first and the second reaction step without isolating in-between the stages, we attempted the transformation with tBuOH/toluene (6:1) as a solvent replacement for EtOH. Gratifyingly, >99% conversion was observed, and the formation of benzaldehyde was almost completely suppressed (<1area%). Decreasing to 5 equiv. of benzylamine resulted in a drop in conversion to 87% (Table 4, entry 1). Through further increase of the residence time and temperature, we were able to further increase the conversion (Table 4, entries 2 and 3). We could successfully operate this optimized flow procedure (Table 4, entry 4) over a 2 h operation time to afford Etrinabdione (3a) in 99% isolated yield (19.4 g, 45.0 mmol, 9.7 g h−1). This result corresponds to a space–time yield of 0.836 kg (L h)−1, based on the volume of 11.6 mL. Furthermore, we demonstrated that the protocol could be successfully applied for different amine nucleophiles with the methyl derivative 3b and propyl derivative 3c both isolated in 95% yield.
|
|
||||
|---|---|---|---|---|
| Entry | Reactor volume [mL] | Residence time [min] | Temperature [°C] | Conv. 2 [%] |
| Conditions: Feed 1: 0.5 M HU-331 in EtOH; Feed 2: 9.15 M benzylamine with flow rates for Feed 1/Feed 2 = 0.58:0.32 mL min−1; 1.5 equiv. of O2 introduced through a mass flow controller. 13 bar back pressure were applied. The effluent was collected into a vessel containing H2O. Conversion measured by HPLC UV/VIS at 250 nm. Isolated yields given in parentheses.a Feed 1/Feed 2 = 0.75:0.41 mL min−1.b Feed 1: 0.5 M HU-331 in tBuOH/toluene; Feed 1/Feed 2 = 0.75:0.41 mL min−1.c Feed 1: 0.5 M HU-331 in tBuOH/toluene; Feed 1/Feed 2 = 0.95:0.25 mL min−1. |
||||
| 1 | 9.6 | 10.6 | 20 | 87 |
| 2 | 11.6 | 12.9 | 20 | 90 |
| 3 | 11.6 | 12.9 | 30 | >99 |
| 4a | 11.6 | 10 | 30 | >99 (99) |
| 5b | 11.6 | 10 | 30 | >99 (95) |
| 6c | 11.6 | 10 | 30 | >97 (95) |

Subsequently, we examined the possibility of telescoping the two reaction steps into one reaction sequence without isolation or purification in-between. Initially, we evaluated the possibility of using a membrane-based liquid–liquid separator for separation of the organic and aqueous feed after the first step.43 However, we found that a membrane separation resulted in very poor separation of the phases, because tBuOH acts as a surfactant between the aqueous and organic phases. Thus, we opted for a gravity-based continuous separation approach by adapting Dean–Stark apparatus, see Scheme 6 for details. The concentration of HU-331 (2) in the organic phase was determined to be 0.21 M. The decrease in concentration from the expected 0.24 M was accounted due to some water partitioning into the organic phase. Thus, the flow procedure was adapted for the oxidative amination step to account for the lower concentration. The second step for the telescoped sequence was operated over 30 min. The presence of water within the second step did not have a significant influence the reaction outcome with Etrinabdione (3a), which was isolated in 98% yield after phase extraction and solvent evaporation.
 | ||
| Scheme 6 Continuous separation of phases using a Dean–Stark apparatus for telescoping the preparation Etrinabdione (3) from CBD (1). | ||
Finally, we were interested in assessing the greenness of our flow approach compared with the original batch procedure by Muñoz. Both the batch protocols required purification by column chromatography, however; in the case of the flow protocols only a simple solvent extraction was necessary. The product yields obtained were improved for the first and second reaction steps from 74% and 56% respectively for the batch protocol by Muñoz to virtually quantitative yields obtained for the flow protocols. We compared the process mass intensity (PMI) from the patented procedure by Muñoz10 (Table 5, entries 1 and 2) with our flow processes (Table 5, entries 3–5).44 For simplicity we did not include the solvents used in the column chromatography in the batch protocols for the calculation of the PMIs, thus our calculations were conservative when considering the entire processing.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Process | Yield [%] | Reaction PMI | Workup PMI | Total PMI |
| 1 | HU-331-batch | 74 | 224 | 563 | 787 |
| 2 | Etrinabdione-batch | 56 | 133 | 1723 | 1856 |
| 3 | HU-331-flow | >99 | 10.1 | 27.9 | 37.9 |
| 4 | Etrinabdione-flow | >99 | 16.6 | 45.6 | 62.2 |
| 5 | Etrinabdione-tele | 98 | 37.3 | 48.0 | 85.3 |

The PMI was significantly reduced from 787 to 37.9 for the oxidation of CBD (1) to HU-331 (2), and from 1856 to 62.2 in the case of the oxidation of HU-331 (2) to Etrinabdione (3). Moreover, the PMI of the telescoped approach was lower than for the two separate flow stages, a PMI of 85.3 was achieved by carrying forward the organic layer straight into the oxidative amination. These results demonstrate that the established flow protocols are substantially more sustainable and efficient than the published batch protocols.
Conclusions
A two-step gas–liquid continuous flow aerobic oxidation protocol for the preparation of Etrinabdione from CBD was reported. Etrinabdione is an API currently in clinical phase 2 trials for the treatment of peripheral artery disease. The utilization of continuous flow technology enabled safer and more scalable handling of pure oxygen when compared to using an air-opened flask. In particular, the rapid and exothermic oxidation of CBD to HU-331 could be carefully controlled within a microreactor platform. PhMe previously used for the oxidation of CBD was changed to a solvent mixture of tBuOH/PhMe (6:1) allowing for a higher substrate and base concentration to be used. The second oxidation step was also developed to use the same solvent mixture to allow for efficient reaction telescoping. Purification by column chromatography was no longer necessary for the flow protocols because the products were obtained in very high yield and purity after a simple extraction procedure. The PMI for the telescoped sequence corresponded to a reduction in the PMI of 97%. The new two-step flow protocol represents a significant improvement in terms of safety, scalability, sustainability, reagent cost and atom economy. The developed process will be particularly important into the future as Etrinabdione becomes increasingly used in the clinic.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The CCFLOW project (Austrian Research Promotion Agency FFG No. 862766) is funded through the Austrian COMET Program by the Austrian Federal Ministry of Transport, Innovation and Technology (BMVIT), the Austrian Federal Ministry of Digital and Economic Affairs (BMDW) and by the State of Styria (Styrian Funding Agency SFG). The CBD used in this study was supplied by Lonza AG.References
- A. Begleiter, Front. Biosci., 2000, 5, 153–171 CrossRef.
- K.-H. Lee, Med. Res. Rev., 1999, 19, 569–596 CrossRef CAS.
- R. Zucchi and R. Danesi, Curr. Med. Chem. Anticancer Agents, 2003, 3, 151–171 CrossRef CAS PubMed.
- X. Thomas, Q. Le and D. Fiere, Ann. Hematol., 2002, 81, 504–507 CrossRef CAS PubMed.
- N. M. Kogan, R. Rabinowitz, P. Levi, D. Gibson, P. Sandor, M. Schlesinger and R. Mechoulam, J. Med. Chem., 2004, 47, 3800–3806 CrossRef CAS PubMed.
- N. M. Kogan, M. Schlesinger, E. Priel, R. Rabinowitz, E. Berenshtein, M. Chevion and R. Mechoulam, Mol. Cancer Ther., 2007, 6, 173–183 CrossRef CAS PubMed.
- S. E. O'Sullivan, Br. J. Pharmacol., 2009, 152, 576–582 CrossRef PubMed.
- S. E. O'Sullivan, D. A. Kendall and M. D. Randall, PPAR Res., 2009, 2009, 425289 Search PubMed.
- A. G. Granja, F. Carrillo-Salinas, A. Pagani, M. Gómez-Cañas, R. Negri, C. Navarrete, M. Mecha, L. Mestre, B. L. Fiebich, I. Cantarero, M. A. Calzado, M. L. Bellido, J. Fernandez-Ruiz, G. Appendino, C. Guaza and E. Muñoz, J. Neuroimmune Pharmacol., 2012, 7, 1002–1016 CrossRef PubMed.
- G. Appendino, M. L. Bellido and E. Muñoz, Novel Cannabidiol Quinone Derivatives, World Patent, 2015/158381, 2015 Search PubMed.
- C. Del Río, C. Navarrete, J. A. Collado, M. L. Bellido, M. Gómez-Cañas, M. R. Pazos, J. Fernández-Ruiz, F. Pollastro, G. Appendino, M. A. Calzado, I. Cantarero and E. Muñoz, Sci. Rep., 2016, 6, 1–14 Search PubMed.
- A. García-Martín, M. E. Prados, I. Lastres-Cubillo, F. J. Ponce-Diaz, L. Cerero, M. Garrido-Rodríguez, C. Navarrete, R. Pineda, A. B. Rodríguez, I. Muñoz, J. Moya, A. Medeot, J. A. Moreno, A. Chacón, J. García-Revillo and E. Muñoz, J. Transl. Med., 2024, 22, 1003 CrossRef.
- B. Palomares, F. Ruiz-Pino, C. Navarrete, I. Velasco, M. A. Sánchez-Garrido, C. Jimenez-Jimenez, C. Pavicic, M. J. Vazquez, G. Appendino, M. L. Bellido, M. A. Calzado, M. Tena-Sempere and E. Muñoz, Sci. Rep., 2018, 8, 1–15 CAS.
- A. García-Martín, M. Garrido-Rodríguez, C. Navarrete, C. del Río, M. L. Bellido, G. Appendino, M. A. Calzado and E. Muñoz, Biochem. Pharmacol., 2018, 157, 304–313 CrossRef PubMed.
- C. Navarrete, F. Carrillo-Salinas, B. Palomares, M. Mecha, C. Jiménez-Jiménez, L. Mestre, A. Feliú, M. L. Bellido, B. L. Fiebich, G. Appendino, M. A. Calzado, C. Guaza and E. Muñoz, J. Neuroinflammation, 2018, 15, 1–19 Search PubMed.
- B. P. Lavayen, C. Yang, J. Larochelle, L. Liu, R. J. Tishko, A. C. Pinheiro de Oliveira, E. Muñoz and E. Candelario-Jalil, Neurochem. Int., 2023, 165, 105508 Search PubMed.
- M. Villa, M. Martínez-Vega, L. Silva, A. Romero, M. de Hoz-Rivera, M. E. Prados, E. Muñoz and J. Martínez-Orgado, Eur. J. Pharmacol., 2024, 972, 176554 CrossRef CAS PubMed.
- C. Navarrete, A. García-Martín, A. Correa-Sáez, M. E. Prados, F. Fernández, R. Pineda, M. Mazzone, M. Álvarez-Benito, M. A. Calzado and E. Muñoz, J. Neuroinflammation, 2022, 19, 177 Search PubMed.
- ClinicalTrials.gov ID: NCT06774040.
- R. Mechoulam, Z. Ben-Zvi and Y. Gaoni, Tetrahedron, 1968, 24, 5615–5624 CrossRef CAS PubMed.
- E. H. Stitt, Chem. Eng. J., 2002, 90, 47–60 Search PubMed.
- (a) C. J. Mallia and I. R. Baxendale, Org. Process Res. Dev., 2016, 20, 327–360 CrossRef CAS; (b) A. A. H. Laporte and T. M. Noël, Angew. Chem., Int. Ed., 2024, 63, e202316108 Search PubMed.
- A. Günther and K. F. Jensen, Lab Chip, 2006, 6, 1487–1503 RSC.
- S. L. Lee, T. F. O'Connor, X. Yang, C. N. Cruz, S. Chatterjee, R. D. Madurawe, C. M. V. Moore, L. X. Yu and J. Woodcock, J. Pharm. Innov., 2015, 10, 191–199 CrossRef.
- (a) C. Jiménez-González, P. Poechlauer, Q. B. Broxterman, B. S. Yang, D. Am Ende, J. Baird, C. Bertsch, R. E. Hannah, P. Dell'Orco, H. Noorman, S. Yee, R. Reintjens, A. Wells, V. Massonneau and J. Manley, Org. Process Res. Dev., 2011, 15, 900–911 CrossRef; (b) P. Poechlauer, J. Colberg, E. Fisher, M. Jansen, M. D. Johnson, S. G. Koenig, M. Lawler, T. Laporte, J. Manley, B. Martin and A. O'Kearney-McMullan, Org. Process Res. Dev., 2013, 17, 1472–1478 CrossRef CAS; (c) J. C. McWilliams, A. D. Allian, S. M. Opalka, S. A. May, M. Journet and T. M. Braden, Org. Process Res. Dev., 2018, 22, 1143–1166 Search PubMed; (d) D. Dallinger and C. O. Kappe, Curr. Opin. Green Sustainable Chem., 2017, 7, 6–12 Search PubMed; (e) A. J. Blacker, J. R. Breen, R. A. Bourne and C. A. Hone, The Growing Impact of Continuous Flow Methods on the Twelve Principles of Green Chemistry, in Green and Sustainable Medicinal Chemistry: Methods, Tools and Strategies for the 21st Century Pharmaceutical Industry, Royal Society of Chemistry, Cambridge, U.K., 2016, ch. 12, pp. 140–155 Search PubMed.
- (a) M. B. Plutschack, P. Bartholomäus, K. Gilmore and P. H. Seeberger, Chem. Rev., 2017, 117, 11796–11893 Search PubMed; (b) B. Gutmann, D. Cantillo and C. O. Kappe, Angew. Chem., Int. Ed., 2015, 54, 6688–6728 Search PubMed; (c) R. Gérardy, N. Emmanuel, T. Toupy, V. Kassin, N. N. Tshibalonza, M. Schmitz and J. M. Monbaliu, Eur. J. Org. Chem., 2018, 2301–2351 CrossRef; (d) M. Baumann and I. R. Baxendale, Beilstein J. Org. Chem., 2015, 11, 1194–1219 CrossRef CAS PubMed; (e) C. A. Hone and C. O. Kappe, Chem.:Methods, 2021, 1, 454–467 CAS.
- (a) M. Movsisyan, E. I. P. Delbeke, J. K. E. T. Berton, C. Battilocchio, S. V. Ley and C. V. Stevens, Chem. Soc. Rev., 2016, 45, 4892–4928 RSC; (b) B. Gutmann and C. O. Kappe, J. Flow Chem., 2017, 7, 65–71 CrossRef CAS.
- M. Baumann, T. S. Moody, M. Smyth and S. Wharry, Synthesis, 2021, 3963–3976 Search PubMed.
- P. Natho and R. Luisi, Tetrahedron Green Chem., 2023, 2, 100015 CrossRef.
- L. Roger and K. F. Jensen, Green Chem., 2019, 21, 3481–3498 RSC.
- M. C. Ince, B. Benyahia and G. Vilé, ACS Sustainable Chem. Eng., 2025, 13, 2864–2874 CrossRef CAS PubMed.
- S. Caron, R. W. Dugger, S. G. Ruggeri, J. A. Ragan and D. H. B. Ripin, Chem. Rev., 2006, 106, 2943–2989 CrossRef CAS PubMed.
- P. M. Osterberg, J. K. Niemeier, C. J. Welch, J. M. Hawkins, J. R. Martinelli, T. E. Johnson, T. W. Root and S. S. Stahl, Org. Process Res. Dev., 2015, 19, 1537–1543 CrossRef CAS PubMed . 26.
- (a) H. P. L. Gemoets, Y. Su, M. Shang, V. Hessel, R. Luque and T. Noël, Chem. Soc. Rev., 2016, 45, 83–117 Search PubMed; (b) A. Gavriilidis, A. Constantinou, K. Hellgardt, K. K. M. Hii, G. J. Hutchings, G. L. Brett, S. Kuhn and S. P. Marsden, React. Chem. Eng., 2016, 1, 595–612 RSC; (c) C. A. Hone, D. M. Roberge and C. O. Kappe, ChemSusChem, 2017, 10, 32–41 CrossRef CAS PubMed; (d) C. A. Hone and C. O. Kappe, Top. Curr. Chem., 2019, 377, 2 CrossRef PubMed.
- For selected examples of API synthesis in flow using oxygen, see: (a) V.-E. H. Kassin, R. Gérardy, T. Toupy, D. Collin, E. Salvadeo, F. Toussaint, K. Van Hecke and J.-C. M. Monbaliu, Green Chem., 2019, 21, 2952–2966 RSC; (b) T. L. Laporte, M. Hamedi, J. S. Depue, L. Shen, D. Watson and D. Hsieh, Org. Process Res. Dev., 2008, 12, 956–966 CrossRef CAS; (c) S. De Angelis, C. A. Hone, L. Degennaro, P. Celestini, R. Luisi and C. O. Kappe, J. Flow Chem., 2018, 8, 109–116 CrossRef CAS; (d) B. Gutmann, P. Elsner, D. P. Cox, U. Weigl, D. M. Roberge and C. O. Kappe, ACS Sustainable Chem. Eng., 2016, 4, 6048–6061 CrossRef CAS; (e) B. Gutmann, U. Weigl, D. P. Cox and C. O. Kappe, Chem. – Eur. J., 2016, 22, 10393–10398 CrossRef CAS PubMed; (f) F. Lévesque and P. H. Seeberger, Angew. Chem., Int. Ed., 2012, 51, 1706–1709 CrossRef PubMed; (g) D. Kopetzki, F. Lévesque and P. H. Seeberger, Chem. – Eur. J., 2013, 19, 5450–5456 CrossRef CAS PubMed.
- D. Caine, Potassium tert–Butoxide, in Encyclopedia of Reagents for Organic Synthesis. John Wiley & Sons, Ltd, Chichester, 2006, vol. 8 Search PubMed.
- D. Prat, A. Wells, J. Hayler, H. Sneddon, C. R. McElroy, S. Abou-Shehada and P. J. Dunn, Green Chem., 2015, 18, 288–296 RSC.
- S. L. Bourne and S. V. Ley, Adv. Synth. Catal., 2013, 355, 1905–1910 CrossRef CAS.
- For an overview of the MMRS with multiple analysis tools, see: P. Sagmeister, J. D. Williams, C. A. Hone and C. O. Kappe, React. Chem. Eng., 2019, 4, 1571–1578 RSC.
- (a) D. M. Roberge, M. Gottsponer, M. Eyholzer and N. Kockmann, Chim. Oggi, 2009, 27, 8–11 CAS; (b) N. Kockmann, M. Gottsponer, B. Zimmermann and D. M. Roberge, Chem. – Eur. J., 2008, 14, 7470–7477 Search PubMed.
- (a) E. Mielke, D. M. Roberge and A. Macchi, J. Flow. Chem., 2016, 6, 279–287 CrossRef CAS; (b) A. Macchi, P. Plouffe, G. S. Patience and D. M. Roberge, Can. J. Chem. Eng., 2019, 97, 2578–2587 CrossRef CAS; (c) M. Köckinger, B. Wyler, C. Aellig, D. M. Roberge, C. A. Hone and C. O. Kappe, Org. Process Res. Dev., 2020, 24, 2217–2227 CrossRef.
- H. Musso, Angew. Chem., Int. Ed. Engl., 1963, 2, 723–735 CrossRef.
- A. Adamo, P. L. Heider, N. Weeranoppanant and K. F. Jensen, Ind. Eng. Chem. Res., 2013, 52, 10802–10808 CrossRef CAS.
- We used the calculation toolbox outlined here: (a) C. R. McElroy, A. Constantinou, L. C. Jones, L. Summerton and J. H. Clark, Green Chem., 2015, 17, 3111–3121 RSC for a previous example of its application to a flow process, see: M. Prieschl, J. García-Lacuna, R. Munday, K. Leslie, A. O'Kearney-McMullan, C. A. Hone and C. O. Kappe, Green Chem., 2020, 22, 5762–5770 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, further optimization data and characterization of all compounds. For full experimental details, description and images of the continuous flow set-up, 1H NMR and 13C NMR spectra of all isolated products. See DOI: https://doi.org/10.1039/d5gc01304f |
| This journal is © The Royal Society of Chemistry 2025 |