
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Catalytic alkaline hydrolysis of PET and BPA-PC waste in minutes at atmospheric pressure without microwaves or organic solvents†
Anshul
Jain
 and
Stephen J.
Connon
*
and
Stephen J.
Connon
*
School of Chemistry, Trinity Biomedical Sciences Institute, Trinity College Dublin, 152-160 Pearse St., Dublin 2, Ireland. E-mail: connons@tcd.ie
First published on 11th April 2025
Abstract
Rapid hydrolysis of poly(ethylene terephthalate) (PET) waste usually requires organic cosolvents, high pressures or microwave irradiation, which can increase the environmental impact/expense/operational complexity of an emerging enabling technology for more sustainable plastic recycling. Using a combination of solute-derived boiling point elevation and phase transfer catalysis, operationally facile, rapid alkaline hydrolysis of PET and poly(bisphenol A carbonate) (BPA-PC) waste – from beverage bottles/textiles and compact discs respectively – is achievable in minutes (≤5 min for PET and 20 min for BPA-PC) at atmospheric pressure without the need for either microwaves or organic cosolvents. Dimethyldialkylammonium halides were found to be optimal catalysts at low loadings. The rapid, one-pot catalytic hydrolysis of a waste stream of both plastics followed by ready isolation of the terephthalic acid and bis-phenol A monomer units in excellent yields (without decomposition) is possible by selective protonolysis.
Green foundation1. A catalytic methodology for the chemical recycling of PET and BPA-PC plastic waste by alkaline hydrolysis. This occurs on timescales comparable with the most efficient methodologies in the literature/industry, without requiring organic co-solvents, microwaves or high-pressure apparatus. This is possible through the combination of a simple and efficient (1 mol%) catalyst with boiling point elevation. The first hydrolytic rapid recycling of mixed streams of the two plastic wastes is possible – again without organic solvents.2. The ability to quantitatively depolymerise PET in 4–5 min in minimal volumes of aqueous NaOH at atmospheric pressure without organic solvents reduces both solvent waste and the hazards associated with the containment of high-pressure reactions at scale. The short timescales at reaction temperatures <150 °C also reduce energy usage. 3. Efforts to improve catalysis and reduce the loading of NaOH closer to 2 equivalents could reduce waste further. |
Poly(ethylene terephthalate) (PET) is a fossil-derived condensation polymer thermoplastic constituent of beverage bottles, food packaging and textiles. It has been reported that in 2021, the production of this material totaled 24.2 billion kg (ref. 1) and PET disposal comprised 12% of worldwide solid waste.2 Despite the increasing environmental concern, if current trends continue the OECD estimates that the use of plastics is set to almost triple from 2019 to 2060.3 Alongside investment in recycling infrastructure and more stringent regulatory frameworks (e.g. the EU setting minimum future levels of recycled plastic in beverage bottles4), the rapid improvement and expansion of humanity's arsenal of recycling technologies are therefore of some urgency.
Non-destructive PET recycling methodologies can be classified as being either mechanical or chemical in nature. Mechanical recycling broadly involves cutting, washing and melting waste materials to form recycled PET flakes/pellets.5 This is the most common form of PET recycling and can lead to lower quality products due to contamination and/or hydrolytic/thermal degradation (requiring the utilisation of remedial, more resource-intensive ‘superclean’ methodologies) and ultimate downcycling.5–7 Chemical PET recycling is an emerging technology that involves solvolysis with various nucleophiles, i.e. aminolysis, alcoholysis, glycolysis and hydrolysis (possible under neutral, acidic or alkaline conditions).8 Solvolytic PET recycling is more resource intensive and requires efficient chemistry at scale – but can regenerate pure monomers which can be either upcycled or repolymerised to virgin PET, thereby increasing circularity and reducing global dependence on petroleum-based plastic feedstocks.5,8
The scale of the PET recycling challenge is daunting in both volume and time domains – for instance, in 2021 on average >700 kg of PET was synthesised every second,1 while the total greenhouse gas emissions associated with the PET production supply chain are the highest of the major plastic types.3,9 It follows that even if an international agreement aimed at slowing production is reached, it is not difficult to foresee that future sustainability from a plastics perspective will rely in part on recycling technologies that are (a) rapid, (b) circular, (c) involving minimal waste, (d) operationally straightforward and (e) applicable in both developed and developing nations alike.
Nascent industrial processes have largely focused on catalytic methanolysis and (in particular) glycolysis.5,8l,10,11 The high boiling point of ethylene glycol allows glycolysis at temperatures up to 190 °C to occur at atmospheric pressure – which is advantageous in terms of rate (PET is a hydrophobic material insoluble in water and alcohol) and operational simplicity (i.e. avoidance of high-pressure vessels) at the process scale. The product is directly polymerisable to PET; however, an equilibrium between the product and oligomers/precursors diminishes the yield and complicates purification.11
PET hydrolysis offers advantages in terms of the use of water as a solvent and the formation of water-insoluble terephthalic acid (1) – a starting material for the production of PET in the petrochemical industry. Acidic-,12 neutral-13 and alkaline14 hydrolysis methodologies are known. The reactions are (in a practical sense) irreversible, with outstanding product yields possible. Alkaline hydrolysis has been generally preferred in the small number of industrial processes using hydrolytic technology,11a as it can be conducted at lower temperatures, with less EG degradation and shorter reaction times – at the cost of the use of NaOH, together with mineral acids to precipitate the product. The main challenge associated with alkaline hydrolysis is the aforementioned hydrophobicity of PET, and researchers in industry and academia have attempted to circumvent this through the use of (either alone or in combination) high temperatures/pressures,15 phase transfer catalysts,16 organic co-solvents,17 microwaves,17k,18 UV-irradiation,19 ultrasound20 and extruders21/mechanochemistry.22 All except the use of phase transfer catalysts would increase the operational complexity of the process, especially at scale.11
Representative recent advances are depicted in Fig. 1. The application of high temperatures/pressures by Karayannidis et al.15 resulted in complete PET degradation to monomer 1 at 200 °C after 60 min (Fig. 1A). Gutiérrez-Ortiz and coworkers16d (Fig. 1B) reported the alkaline hydrolysis of PET flakes using a phosphonium ion-based phase transfer catalyst (12.5 mol%) – which allowed the reaction to proceed to a considerable extent at 80 °C over 90 min. A study from Xu's laboratory17j demonstrated efficient alkaline hydrolysis at just 35 °C in 10 min (Fig. 1C). While this level of reactivity is impressive, the methodology relies on a dichloromethane-based solvent system likely to be of toxicological23 and environmental24 concern at scale. Gr3n has achieved similarly rapid alkaline hydrolysis using a combination of ethylene glycol cosolvent and microwave irradiation at a high temperature and pressure (Fig. 1D).11a,18c The technology is currently at the pilot plant (60 kg PET h−1) stage; however, an agreement to build a 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 ton plant operational in 2027 has recently been announced.25
000 ton plant operational in 2027 has recently been announced.25
 | ||
| Fig. 1 Representative strategies for the alkaline hydrolysis of PET. | ||
Given that it seems likely that future plastic recycling will have to be accomplished in the developing as well as the developed world, we were interested in the design of an operationally simple, minimal laboratory process for PET alkaline hydrolysis which still occurred on rapid timescales – ideally at atmospheric pressure without irradiation, cosolvents or mechanochemical assistance. We recently evaluated the performance of over 50 phase transfer catalysts in the hydrolysis of PET by NaOH at 90 °C.16k Catalyst-design guidelines were developed and dimethyl dialkylammonium halides emerged as an exceptional class of phase transfer agents for this process. Reactions however were prohibitively slow (1.5–3 h) for commercial use. Herein we report the unique combination of ad hoc-designed phase transfer catalysts (at low loadings) with alkaline aqueous solution boiling point elevation, which allows the complete hydrolysis of bottle waste PET in 4 min at 138 °C under atmospheric pressure using standard laboratory apparatus without any cosolvent (Fig. 1E). Extension to poly-BPA-carbonate plastic from compact disc (CD) waste and the selective isolation of monomers from an unprecedented hydrolysis of a PET/poly-(BPA carbonate) mixed-waste stream are also demonstrated.
Since it is well known that solutes increase the boiling point of aqueous solutions (sodium hydroxide is a particularly effective salt for this purpose26), the dearth of reports involving the exploitation of this phenomenon in PET alkaline hydrolysis is surprising.18e We posited that if sufficient boiling point elevation could be coupled with compatible and powerful phase-transfer catalysis, significantly accelerated depolymerisation could occur without the need for either cosolvents or high-pressure reactors. In preliminary studies we investigated the hydrolysis of PET flakes cut from waste beverage bottles purchased from a local supermarket (5 mm × 5 mm) using the same loading of NaOH as used previously (1 g NaOH/g PET, 4.8 eq.16k) but at a considerably higher concentration (i.e. from the more standard 10% w/v16k to ∼50% w/v, Scheme 1) in the absence of catalysis.
 | ||
| Scheme 1 Uncatalysed, atmospheric pressure PET alkaline hydrolysis using concentrated NaOH (50% w/v): influence of temperature. | ||
At 90 °C after 15 min TPA (1) could be isolated after acidification in a low yield. However, at atmospheric pressure (non-sealed carousel glass vessel) the high hydroxide concentrations allowed hydrolysis to occur at temperatures considerably beyond 100 °C (see ESI, Table S1†). At the maximum we recorded under these conditions of 138 °C (note: the boiling temperature decreases from the maximum as the reaction progresses due to sodium hydroxide consumption and concomitant disodium terephthalate precipitation), formation of 1 was possible in 72% yield (Scheme 1, Table S1†).
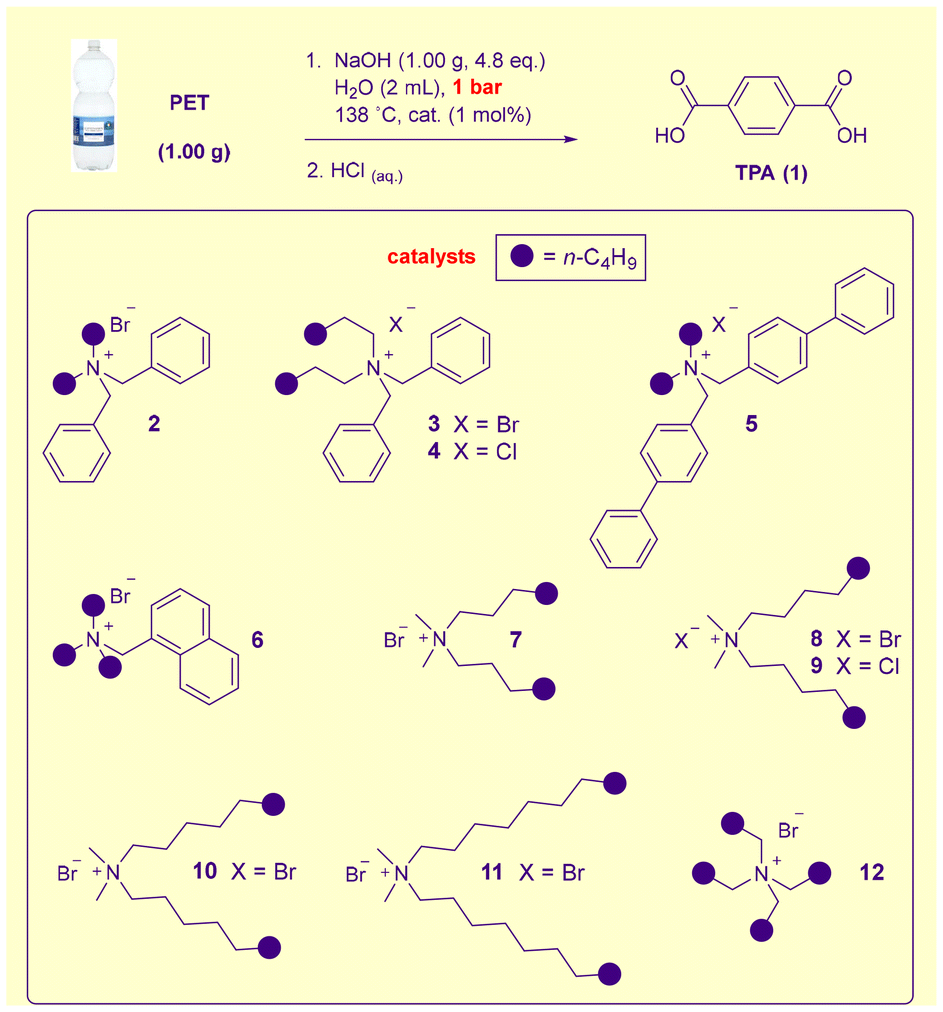
Our attention next turned to catalysis. Benzylated- and dimethyldialkylammonium halides have been identified as superior phase transfer catalyst classes for PET hydrolysis,16k so the most active members of these classes were evaluated under the reaction conditions outlined in Scheme 1 at 1 mol% loading. To both challenge the catalysts and facilitate comparisons, the reaction time was reduced to 10 min – on par with timescales associated with a microwave- and cosolvent-mediated, high-pressure current industrial process.11a,18c In the absence of a catalyst, ca. 50% yield is obtained (entry 1). The soluble dibutyldibenzyl ammonium salt 2 and its dihexyl analogue 3 (a highly efficacious catalyst at 90 °C under more dilute conditions16k) were largely ineffectual (entries 2 and 3). While the exchange of the bromide counterion for chloride (i.e.4, entry 4) made little difference, the presence of an extended p-system (i.e.5–6, entries 5 and 6) led to appreciable improvements in activity. Dimethyldialkylammonium salts, which are marginally superior to isomeric phase transfer catalysts under dilute conditions,16k distinguish themselves in ∼50% NaOH. The diheptyl system 7 (entry 7) proved capable of significant activity, while its immediate homologues 8 and 9 mediated hydrolysis to either near or full completion (entries 8 and 9). Previously we had found 8 to be the most powerful promoter in 10% NaOH and that once higher homologues became insoluble in the reaction medium, progressively slower catalysis resulted as the catalyst chain size was increased.16k Intriguingly here 8–9 are insoluble, yet superior to the soluble 7. Given that under these conditions catalyst insolubility was no longer a predictor of reduced activity, the higher homologues 10 and 11 were evaluated. The former species proved to be an active catalyst (entry 10), while the depolymerisation was clearly promoted more slowly by the larger 11 (entry 11). It is worth noting that the most active previously identified16k member of the symmetrical tetraalkylammonium halide catalyst family (i.e.12, entry 12) remains inferior to 8–10 here despite being a soluble, constitutional isomer of 10.
To allow the relative performance of 8–10 to be more easily assessed, depolymerisation involving these catalysts was repeated with a 5 min reaction time (and an increase in the stirring speed to 1000 rpm to prevent excessive adherence of 1 to the PET flakes) – which allowed the superiority of 10 to be clearly identified (entries 13–15). Further experimentation revealed that a 4 min (note – this includes the ramp time required to heat from ambient temperature to 138 °C) reaction time is sufficient for essentially quantitative generation of 1 (entries 16 and 17). This is an unprecedented depolymerisation rate for a simple alkaline PET hydrolysis process without pressure, cosolvent or microwave irradiation. Without a catalyst, <30% yield of the product is obtained after 4 min (entry 18); however, the use of a 60% w/v NaOH solution (without altering the hydroxide loading) allows hydrolysis at 145 °C – under these conditions after 13 min the uncatalysed recycling is complete (entry 19). At these higher temperatures/concentrations, phase transfer catalysis is less effective (see ESI, Table S2†).
The superiority of the partially soluble 10 over the soluble 7 merits comment. Under dilute conditions, lipophilic soluble catalysts outperform insoluble homologues;16k however, the faster hydrolysis catalysed by 10 is ascribable to the physical contact with the PET flakes in the reaction vessel. PET is denser than water (1.4 g mL−1), and the phase transfer catalysts generally have densities <1 g mL−1. Under dilute conditions, insoluble phase transfer catalysts tend to accumulate at the top of the solvent, while PET sinks to the bottom, leading to reduced contact between the two even with vigorous stirring. The depolymerisation reported here occurs in 2 mL H2O per g PET. The plastic floats more easily on the denser ∼50% NaOH solution alongside the insoluble catalyst, leading to improved surface contact. In addition, the solvent generally just covers the insoluble PET in the reaction vessel, so that any insoluble catalyst floating at the top of the solution remains in contact with the PET, especially at low conversions.
The depolymerisation was also carried out on a multigram scale using standard laboratory apparatus (Scheme 2). Heat transfer to a flask in an open oil bath was inferior to that associated with the carousel reactor, leading to increased ramp times. To obviate this problem (unlikely to be an issue using heated industrial reactors), PET was added to preheated aqueous NaOH. To avoid complications stemming from PET addition at reflux, a marginally lower reaction temperature of 135 °C was selected. Otherwise, conditions were identical to those listed in Table 1. A 95% isolated yield of 1 was possible after 5 min. A drawback associated with this technology would be the requirement for lined reactors to avoid corrosion by the high-concentration alkaline solution. High concentration NaOH-mediated industrial PET hydrolysis processes have been described in the patent literature.11,18e
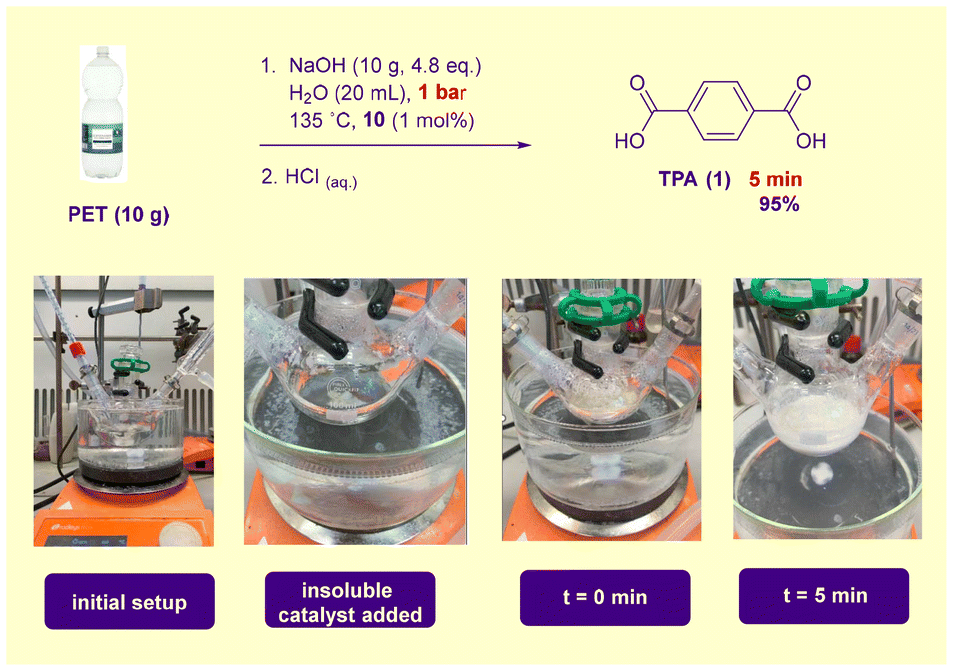 | ||
| Scheme 2 Multigram scale PET (bottle) waste hydrolysis. | ||
|
|
|||||
|---|---|---|---|---|---|
| Entry | Cat. | Time (min) | Stirringa (rpm) | Cat. solubilityb | Yieldc (%) |
| a Revolutions per minute. b Refers to solubility under the reaction conditions at 138 °C. c Isolated yield. d 60% NaOH (w/v), 145 °C (the initial temperature at low conversion – the solution boiling point reduces as the hydroxide reacts due to precipitation of Na2TPA). | |||||
| 1 | None | 10 | 500 | — | 51.4 |
| 2 | 2 | 10 | 500 | Soluble | 55.4 |
| 3 | 3 | 10 | 500 | Soluble | 57.8 |
| 4 | 4 | 10 | 500 | Soluble | 59.4 |
| 5 | 5 | 10 | 500 | Insoluble solid | 70.4 |
| 6 | 6 | 10 | 500 | Insoluble oil | 69.2 |
| 7 | 7 | 10 | 500 | Soluble | 89.2 |
| 8 | 8 | 10 | 500 | Insoluble oil | 99.9 |
| 9 | 9 | 10 | 500 | Insoluble oil | 98.1 |
| 10 | 10 | 10 | 500 | Insoluble oil | 98.5 |
| 11 | 11 | 10 | 500 | Insoluble oil | 58.6 |
| 12 | 12 | 10 | 500 | Soluble | 90.4 |
| 13 | 8 | 5 | 1000 | Insoluble oil | 90.2 |
| 14 | 9 | 5 | 1000 | Insoluble oil | 89.5 |
| 15 | 10 | 5 | 1000 | Insoluble oil | 99.9 |
| 16 | 10 | 4 | 1000 | Insoluble oil | 99.9 |
| 17 | 10 | 3.5 | 1000 | Insoluble oil | 96.7 |
| 18 | None | 4 | 1000 | — | 29.5 |
| 19d | None | 13 | 1000 | — | 99.9 |
Catalyst recovery and reuse is possible in principle. After PET hydrolysis but before neutralisation, water was added to dissolve all disodium-TPA, followed by extraction of the catalyst with dichloromethane. The 1H NMR spectrum of the recovered catalyst (99%) indicated that no decomposition had occurred – most likely due to the short reaction time. Reuse of the recovered catalyst in a subsequent PET hydrolysis allowed the isolation of TPA in 99.4% yield after 4 min (see the ESI†). It is clear that to be of use in an industrial process, the advantages associated with catalyst recovery would have to be balanced against the use of an organic extraction solvent (which itself would have to be recycled) – however, catalyst recovery and reuse do appear possible in principle at this juncture.
We have not recovered the ethylene glycol generated by the hydrolysis reaction by distillation – however, glycol and water do not form an azeotrope so recovery of this valuable (often overlooked) product from PET alkaline hydrolysis reaction is not problematic on an industrial scale.11b,18c,e
In terms of green chemistry metrics, the E factor (E), energy economy coefficient (ε), and environmental energy impact factor (ξ) were calculated to be 2.09, 0.00179 C−1 min−1 and 1167.6 °C min respectively (see ESI†), comparable with the most sustainable values from the alkaline PET hydrolysis literature.8k,18d It is worth noting that in the calculation of these metrics, we did not factor NaCl electrolysis back to NaOH, ethylene glycol distillation or catalyst recovery – all of which are possible on a larger scale and would lead to superior process metrics overall.
Compared to the alkaline hydrolysis of PET from bottle waste, comparatively little is known regarding the corresponding depolymerisation of PET derived from textiles, despite textile production traditionally being the largest consumer of PET.27 For example, a recent study reported >90% textile PET degradation in 3 h at 90 °C (10% NaOH), with full degradation in 24 h.28 Under the previously established catalytic hydrolysis conditions (Table 2, entry 1), complete depolymerisation of PET textile waste was observed after 15 min. The longer reaction time is related to a physical issue: the more voluminous textile waste was not completely covered by the concentrated aqueous solvent. If faster reaction times are required, the use of double the solvent provides quantitative yields in 4 min (entries 2 and 3).

We were next interested in the possibility of the rapid hydrolysis of a more challenging polymer. Poly(bisphenol A carbonate) (BPA-PC) is a high-strength hydrophobic thermoplastic used in (inter alia) safety goggles, headlights, computer casings, compact disks (CDs), window panes and safety paraphernalia.28 Although controversial, there is concern regarding BPA-PC/BPA-PC-waste focused on the monomer constituent bis-phenol A (13) as an endocrine disrupting agent.29 Advances in the chemical recycling of BPA-PC have proceeded along similar lines to those associated with PET,29 with a major difference: BPA-PC is considerably more susceptible to the action of organic co-solvents. PET has been blended with BPA-PC to improve its resistance to organic solvents, while blending with BPA-PC can improve the impact strength and dimensional stability of PET.30 Perhaps unsurprisingly, there has been a considerable focus on solvent-assisted hydrolytic depolymerisation of BPA-PC in the literature, where often completely recalcitrant base-mediated hydrolysis in water proceeds upon the addition of an organic cosolvent.31 In the absence of cosolvents, hydrolysis of BPA-PC is difficult and, in addition, the product monomer 13 is prone to decomposition at high temperatures.28,32 Achillas utilised microwave radiation to degrade waste BPA-PC in the presence of 1-hexadecyltrimethylammonium bromide as a phase transfer catalyst via alkaline hydrolysis (NaOH, 10% w/v). After 10 min at 150 °C (pressure = 4 bar), the polymer was 30% degraded, which increased to 95% degradation at 160 °C. No yield/characterisation of 13 was recorded and methanol was required to solubilise 13 and its degradation products isopropenyl phenol/t-butyl phenol/phenol so they could be separated from unreacted BPA-PC.33
Waste polycarbonate from CDs was pre-treated to remove the aluminum layer33 cut into 2.5 mm squares and subjected to the standard hydrolysis conditions at 135 °C (Table 3). In the absence of a catalyst, BPA-PC is remarkably resistant to hydrolysis after 15 min of reaction time (entry 1). This would be attributable to the greater hydrophobicity and higher Tg (>140 °C) associated with this polymer relative to PET. Appreciable degradation occurs in the presence of 1 mol% of one of the catalysts 7, 8 or 10, with the dioctyl variant 8 proving superior (entries 2–4). Further optimisation allowed the formation of 13 in >90% yield after 30 min of reaction time using a 65% (w/v) NaOH solution (entries 5–8). The more concentrated solution permits the reaction at 145 °C – here depolymerisation with >90% yield requires only 20 min (entry 9).
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Cat. | [NaOH] (% w/v) | Soln vol.a (mL) | Time (min) | Temp.b (°C) | Conversionc (%) | Yieldd (%) |
| a Refers to the volume of H2O used in the sodium hydroxide solution. b Refers to the initial temperature at low conversion – the solution boiling point reduces as the hydroxide reacts due to precipitation of Na2BPA. c Calculated based on unreacted BPA-PC (see the ESI†). d Isolated yield. The value in parentheses is determined by 1H NMR spectroscopy using 4-iodoanisole as an internal standard (see the ESI†). | |||||||
| 1 | None | 50 | 2 | 15 | 135 | 14.7 | 15.0 (14.8) |
| 2 | 7 | 50 | 2 | 15 | 135 | 46.7 | 46.7 (46.6) |
| 3 | 8 | 50 | 2 | 15 | 135 | 53.2 | 53.1 (53.2) |
| 4 | 10 | 50 | 2 | 15 | 135 | 33.4 | 33.5 (33.4) |
| 5 | 8 | 50 | 2 | 30 | 135 | 72.5 | 72.5 (72.3) |
| 6 | 8 | 65 | 2 | 20 | 135 | 80.2 | 80.3 (80.0) |
| 7 | 8 | 65 | 3 | 20 | 135 | 86.7 | 86.8 (86.5) |
| 8 | 8 | 65 | 3 | 30 | 135 | 91.8 | 92.0 (91.6) |
| 9 | 8 | 65 | 3 | 20 | 145 | 90.4 | 90.2 (90.2) |

Real-world waste streams usually comprise mixed plastic, which necessitates sorting by various means, including by density (PET = 1.4 g cm−3, BPA-PC = 1.2 g cm−3). There is therefore burgeoning interest in the chemolytic selective depolymerisation of mixed waste streams34 involving the separation of 2 or more plastic types based on their relative rates of depolymerisation under a given set of conditions. Reports concerning glycolysis,35 methanolysis36 and reductions37 have appeared. For instance, Dove et al.35a reported an elegant organocatalytic solution for the selective depolymerisation of BPA-PC in the presence of PET and a triol at 130 °C in under 1 h. The unreacted PET could then be removed by filtration and depolymerised at a higher temperature. Wang and coworkers36b developed an efficient ‘one pot’ depolymerisation of BPA-PC-PET by Zn(HMDS)2-catalysed methanolysis for 15 h at 100 °C involving complete destruction of both plastics followed by selective monomer isolation: washing with cold methanol allowed 13 (97%) to be separated from the soluble dimethyl terephthalate PET-derived methanolysed product (90%, which could be concentrated in vacuo) by filtration.
To the best of our knowledge, no hydrolytic methodology for the depolymerisation of PET-based mixed streams has been reported. Given the smooth degradation of both polymers under the concentrated conditions described above, we envisaged a ‘one pot’ catalytic alkaline hydrolysis process involving rapid depolymerisation of a BPA-PC/PET mixture to Na2–TPA and Na2–BPA, followed by pH-driven selective protonolysis to allow the sequential precipitation and isolation of the hydrophobic monomers 13 and 1.
Accordingly, equimolar amounts of PET bottle and BPA-PC CD waste were hydrolysed catalysed by 8 (1 mol%) at 145 °C using 65% (w/v) NaOH at atmospheric pressure (Scheme 3). Cleavage of all ester and carbonate moieties was complete in 30 min. In contrast to methods involving organic solvents – where PC-PBA is the more reactive polymer35 – here PET is hydrolysed first in a matter of minutes, followed by degradation of BPA-PC. Upon completion of the reaction, the addition of water and adjustment of the pH to 7.9 resulted in the precipitation of pure 13 in 90% yield, which was removed by filtration. Notably, no decomposition of 13 was observed. The filtrate was subsequently acidified to pH 2.0, allowing the precipitation of 1 in near quantitative yields. The TPA monomer contained 0.1% 13. If required, this can be removed by washing with a small amount of ethanol (recoverable by distillation).
 | ||
| Scheme 3 Mixed stream PET/BPA-PC waste hydrolysis and selective precipitation. | ||
The relative reactivity of PET and BPA-PC waste under these conditions is intriguing in light of the known superior susceptibility of BPA-PC to glycolysis.35a We suspected that the switch in the order of reactivity towards alkaline hydrolysis was related to the hydrophobicity of BPA-PC in the absence of organic solvents. To test this hypothesis, both PET and BPA-PC waste were separately hydrolysed in the presence of minimal hydroxide in a 4:1 THF:H2O medium (Scheme 4). In this predominantly organic solvent, the reactivity order one would anticipate from Sardon and Dove's study35a was restored: BPA-PC was completely hydrolysed after 30 min at 60 °C, whereas PET was largely resistant.
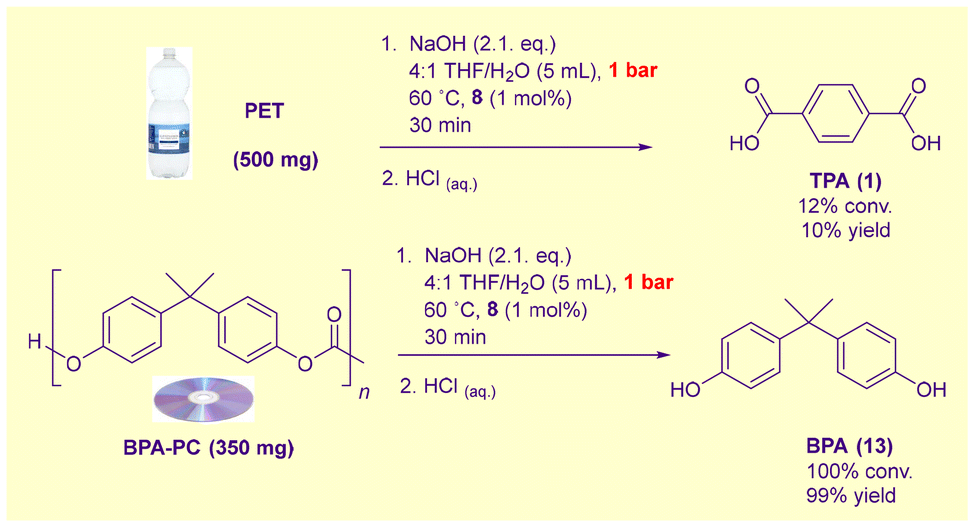 | ||
| Scheme 4 Hydrolysis of PET and BPA-PC in a predominantly organic medium. | ||
In summary, the rapid, operationally facile alkaline hydrolysis of PET and BPA-PC waste is possible at atmospheric pressure. The power of phase transfer catalysis has been used to magnify rate accelerations due to boiling point elevation in highly concentrated NaOH solutions of 50–65% w/v to allow the complete depolymerisation of PET in ≤5 min and BPA-PC in 20 min – timescales either similar to or shorter than those more usually associated with reactions involving either microwave irradiation or the use of organic cosolvents. The phase transfer catalysts were selected from the recently identified lipophilic dimethyldialkylammonium halide class of particular utility in chaperoning anions to the polymer surface.16k,38 It is worth noting that this has been accomplished without increasing the loading of NaOH (and hence saline waste after neutralisation) relative to typical PET hydrolysis processes in the literature. In this regard, the recent electrochemical recovery of NaOH from the generated saline by Gr3N in their industrial process is an interesting avenue for future development. The first hydrolytic recycling of a mixed stream of PET and BPA-PC could be accomplished in 30 min without the involvement of bulk organic solvents in either the depolymerisation or product isolation steps. It is hoped that this operationally facile proof-of-concept hydrolysis methodology, which is devoid of complications stemming from expensive/environmentally impacting solvents, microwave irradiation or high pressures at scale, could enable the future development of globally applicable chemical recycling of hydrolytically susceptible polymer waste.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This publication has emanated from research conducted with the financial support from Taighde Éireann – Research Ireland under Grant Number 19/FFP/6527. We are grateful to Prof. Ramesh Padamati for the gift of waste PET textile. For the purpose of Open Access, the author has applied a CC BY public copyright license to any Author Accepted Manuscript version arising from this submission.References
- https://plasticseurope.org/knowledge-hub/plastics-the-facts-2022/ (accessed 23/01/25).
- P. Benyathiar, P. Kumar, G. Carpenter, J. Brace and D. K. Mishra, Polymers, 2022, 14, 2366 CrossRef CAS PubMed.
- OECD , Global plastics outlook: policy scenarios to 2060: https://www.oecd.org/en/events/2022/06/Global-plastics-outlook-policy-scenarios-to-2060.html (accessed 23/01/25).
- Commission Implementing Decision (EU) 2023/2683: https://eur-lex.europa.eu/eli/dec_impl/2023/2683/oj (accessed 23/01/25).
- H. Li, H. A. Aguirre-Villegas, R. D. Allen, X. Bai, C. H. Benson, G. T. Beckham, S. L. Bradshaw, J. L. Brown, R. C. Brown, V. S. Cecon, J. B. Curley, G. W. Curtzwiler, S. Dong, S. Gaddameedi, J. E. García, I. Hermans, M. S. Kim, J. Ma, L. O. Mark, M. Mavrikakis, O. O. Olafasakin, T. A. Osswald, K. G. Papanikolaou, H. Radhakrishnan, M. A. Sanchez Castillo, K. L. Sánchez-Rivera, K. N. Tumu, R. C. Van Lehn, K. L. Vorsth, M. M. Wright, J. Wu, V. M. Zavala, P. Zhou and G. W. Huber, Green Chem., 2022, 24, 8899 RSC.
- (a) R. Assadi and X. Verdu, Polymer, 2004, 45, 4403 CrossRef CAS; (b) F. Awaja and D. Pavel, Eur. Polym. J., 2005, 41, 1453 CrossRef CAS; (c) L. Incarnato, L. Scarfalo, L. Maio and D. Acierno, Polymer, 2000, 41, 6825 CrossRef CAS; (d) L. K. Nait-Ali, X. Colin and A. Bergeret, Polymer, 2011, 96, 236 CAS.
- A. H. Tullow, Plastic has a problem; is chemical recycling the solution?, Chem. Eng. News, 2019, 39 Search PubMed , https://cen.acs.org/environment/recycling/Plastic-problem-chemical-recycling-solution/97/i39 (accessed 23/01/25).
- Selected reviews: (a) T. M. Joseph, S. Azat, Z. Ahmadi, O. M. Jazani, A. Esmaeili, E. Kianfar, J. Haponiuk and S. Thomas, Case Stud. Chem. Environ. Eng., 2024, 9, 100673 CrossRef; (b) S. Conroy and X. Zhang, Polym. Degrad. Stab., 2024, 223, 110729 CrossRef CAS; (c) M. Muszyński, J. Nowicki, M. Zygadło and G. Dudek, Molecules, 2023, 28, 6385 CrossRef PubMed; (d) H. Abedsoltan, Polym. Eng. Sci., 2023, 63, 2651 CrossRef CAS; (e) V. Tournier, S. Duquesne, F. Guillamot, H. Cramail, D. Taton, A. Marty and I. André, Chem. Rev., 2023, 123, 5612 CrossRef CAS PubMed; (f) K. Ghosal and C. Nayak, Mater. Adv., 2022, 3, 1974 RSC; (g) F. Cao, L. Wang, R. Zheng, L. Guo, Y. Chen and X. Qian, RSC Adv., 2022, 12, 31564 RSC; (h) J. Xin, Q. Zhang, J. Huang, R. Huang, Q. Z. Jaffery, D. Yan, Q. Zhou, J. Xu and X. Lu, J. Environ. Manage., 2021, 296, 113267 CrossRef CAS PubMed; (i) S. C. Kosloski-Oh, Z. A. Wood, Y. Manjarrez, J. P. de los Rios and M. E. Fieser, Mater. Horiz., 2021, 8, 1084 RSC; (j) J. Payne and M. D. Jones, ChemSusChem, 2021, 14, 4041 CrossRef CAS PubMed; (k) E. Barnard, J. J. Rubio Arias and W. Thielemans, Green Chem., 2021, 23, 3765 RSC; (l) I. Vollmer, M. J. G. P. van der Laan, F. Meirer, J. T. F. Keurentjes and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2020, 59, 15402 CrossRef CAS PubMed; (m) C. Jehanno, M. M. Pérez-Madrigal, J. Demarteau, H. Sardon and A. P. Dove, Polym. Chem., 2019, 10, 172 RSC.
- S. R. Nicholson, N. A. Rorrer, A. C. Carpenter and G. T. Beckham, Joule, 2021, 5, 673 CrossRef CAS.
- Damayanti and H.-S. Wu, Polymers, 2021, 13, 147 CrossRef PubMed.
- (a) A. McNeeley and Y. A. Liu, Ind. Eng. Chem. Res., 2024, 63, 3355 CrossRef CAS; (b) A. McNeeley and Y. A. Liu, Ind. Eng. Chem. Res., 2024, 63, 3400 CrossRef CAS.
- Representative references: (a) T. Yoshioka, T. Sato and A. Okuwaki, J. Appl. Polym. Sci., 1994, 52, 1353 CrossRef CAS; (b) T. Yoshioka, N. Okayama and A. Okuwaki, Ind. Eng. Chem. Res., 1998, 37, 336 CrossRef CAS; (c) F. Liu, X. Cui, S. Yu, Z. Li and X. Ge, J. Appl. Polym. Sci., 2009, 114, 3561 CrossRef CAS; (d) W. Yang, R. Liu, C. Li, Y. Song and C. Hu, Waste Manage., 2021, 135, 267 CrossRef CAS PubMed; (e) W. Yang, J. Wang, L. Jiao, Y. Song, C. Li and C. Hu, Green Chem., 2022, 24, 1362 RSC; (f) P. Pereira, P. E. Savage and C. W. Pester, Green Chem., 2024, 26, 1964 RSC.
- Representative references: (a) J. R. Campanelli, D. G. Cooper and M. R. Kamal, J. Appl. Polym. Sci., 1994, 53, 985 CrossRef CAS; (b) C. Y. Kao, B. Z. Wan and W. H. Cheng, Ind. Eng. Chem. Res., 1998, 37, 1228 CrossRef CAS; (c) S. D. Mancini and M. Zanin, Prog. Rubber, Plast. Recycl. Technol., 2004, 20, 117 CrossRef CAS; (d) V. S. Zope and S. Mishra, J. Appl. Polym. Sci., 2008, 110, 2179 CrossRef CAS; (e) O. Sato, Y. Masuda, N. Hiyoshi, A. Yamaguchi and M. Shirai, J. Chem. Eng., 2010, 43, 313 CrossRef CAS; (f) Y. Liu, M. Wang and Z. Pan, J. Supercrit. Fluids, 2012, 62, 226 CrossRef CAS; (g) S. D. Mancini, A. R. Nogueira, E. C. Rangel and N. C. da Cruz, J. Appl. Polym. Sci., 2013, 127, 1989 CrossRef CAS; (h) L. Zhang, J. Gao, J. Zou and F. Yi, J. Appl. Polym. Sci., 2013, 130, 2790 CrossRef CAS; (i) L. Zhang, Eur. Polym. J., 2014, 60, 1 CrossRef CAS; (j) A. Căta, M. Miclău, I. Ienascu, D. Ursu, C. Tănasie and M. N. Stefănuţ, Rev. Roum. Chim., 2015, 60, 579 Search PubMed; (k) D. Stanica-Ezeanu and D. Matei, Sci. Rep., 2021, 11, 4431 CrossRef CAS PubMed; (l) C. N. Onwucha, C. O. Ehi-Eromosele, S. O. Ajayi, M. Schaefer, S. Indris and H. Ehrenberg, Ind. Eng. Chem. Res., 2023, 62, 6378 CrossRef CAS; (m) P. Pereira, P. E. Savage and C. W. Pester, ACS Sustainable Chem. Eng., 2023, 11, 7203 CrossRef CAS; (n) V. Tournier, S. Duquesne, F. Guillamot, H. Cramail, D. Taton, A. Marty and I. André, Chem. Rev., 2023, 123, 5612 CrossRef CAS PubMed; (o) P. Pereira, W. Slear, A. Testa, K. Reasons, P. Guirguis, P. E. Savage and C. W. Pester, RSC Sustainability, 2024, 2, 1508 RSC; (p) I. L. Martin, L. B. Anderson, D. A. McAdams, C. Molloy, P. W. Dunne and S. J. Connon, Chem. Commun., 2025, 61, 2750 RSC.
- J. Pitat, V. Holcik and M. Bacak, GB Patent 822834, 1959.
- G. P. Karayannidis, A. P. Chatziavgoustis and D. S. Achilias, Adv. Polym. Technol., 2002, 21, 250 CrossRef CAS.
- (a) M. Polk, L. Leboeuf, M. Shah, C.-Y. Won, X. Hu and W. Ding, Polym.-Plast. Technol. Eng., 1999, 38, 459 CrossRef CAS; (b) V. A. Kosmidis, D. S. Achilias and G. P. Karayannidis, Macromol. Mater. Eng., 2001, 286, 640 CrossRef CAS; (c) J. Das, A. B. Halgeri, V. Sahu and P. A. Parikh, Indian J. Chem. Technol., 2007, 14, 173 CAS; (d) R. López-Fonseca, M. P. González-Marcos, J. R. González-Velasco and J. I. Gutiérrez-Ortiz, WIT Trans. Ecol. Environ., 2008, 109, 511 Search PubMed; (e) R. López-Fonseca, J. R. González-Velasco and J. I. Gutiérrez-Ortiz, Chem. Eng. J., 2009, 146, 287 CrossRef; (f) D. Spaseska and M. Civkaroska, J. Univ. Chem. Technol. Metall., 2010, 45, 379 CAS; (g) A. Palme, A. Peterson, H. de la Motte, H. Theliander and H. Brelid, Text. Cloth. Sustain., 2017, 3, 4 CrossRef; (h) I. Čorak, A. Tarbuk, D. Đorđević, K. Višić and L. Botteri, Materials, 2022, 15, 1530 CrossRef PubMed; (i) Y. Wang, H. Wang, H. Chen and H. Liu, Chin. J. Chem. Eng., 2022, 51, 53 CrossRef CAS; (j) A. Barredo, A. Asueta, I. Amundarain, J. Leivar, R. Miguel-Fernández, S. Arnaiz, E. Epelde, R. López-Fonseca and J. I. Gutiérrez-Ortiz, J. Environ. Chem. Eng., 2023, 11, 109823 CrossRef CAS; (k) L. B. Anderson, C. Molloy, L. Pedrini, I. L. Martin and S. J. Connon, Green Chem., 2024, 26, 11125 RSC.
- (a) A. Oku, L.-C. Hu and E. Yamada, J. Appl. Polym. Sci., 1997, 63, 595 CrossRef CAS; (b) L.-C. Hu, A. Oku, E. Yamada and K. Tomari, Polym. J., 1997, 29, 708 CrossRef CAS; (c) H. Essaddam, US9550713B1, 2015; (d) S. Ügdüler, K. M. Van Geem, R. Denolf, M. Roosen, N. Mys, K. Ragaert and S. De Meester, Green Chem., 2020, 22, 5376 RSC; (e) W. Chen, Y. Yang, X. Lan, B. Zhang, X. Zhang and T. Mu, Green Chem., 2021, 23, 4065 RSC; (f) J. J. Rubio Arias and W. Thielemans, Green Chem., 2021, 23, 9945 RSC; (g) H. Yu, Y. Wang, L. Chen, C. Wei, T. Mu and Z. Xue, Green Chem., 2023, 25, 7807 RSC; (h) X.-L. Wang, W.-L. An, R. Du, F. Tian, Y. Yang, X. Zhao, S. Xu and Y.-Z. Wang, J. Environ. Chem. Eng., 2023, 11, 109434 CrossRef CAS; (i) H. Chen and H. Hu, Ind. Eng. Chem. Res., 2023, 62, 12925 CrossRef CAS; (j) S. Zhang, W. Xu, R. Du, W. An, X. Liu, S. Xu and Y.-Z. Wang, J. Chem. Eng., 2023, 470, 144032 CrossRef CAS; (k) S. Zhang, Y. Xue, Y. Wu, Y.-X. Zhang, T. Tan and Z. Niu, Chem. Sci., 2023, 14, 6558 RSC; (l) M. Azeem, O. A. Attallah, C. Erdinc Tas and M. Brennan Fournet, J. Polym. Environ., 2024, 32, 303 CrossRef CAS; (m) F. Pedroso de Lima, C. Alves, R. Gomes-Dias, M. Fernandes, B. Vieira, R. Rodrigues, J. Padrão and A. Zille, J. Polym. Environ., 2025, 33, 1847 CrossRef.
- (a) H. I. Khalaf and O. A. Hasan, Chem. Eng. J., 2012, 192, 45 CrossRef CAS; (b) M. N. Siddiqui, D. S. Achilias, H. H. Redhwi, D. N. Bikiaris, K.-A. G. Katsogiannis and G. P. Karayannidis, Macromol. Mater. Eng., 2010, 295, 575 CrossRef CAS; (c) M. Parravicini, M. Crippa and M. V. Bertele, WO2013014650A1, 2012; (d) A. Câta, N. Ş. Mariana, M. Ioana, C. Tânasie and M. Miclâu, Rev. Roum. Chim., 2017, 62, 531 Search PubMed; (e) J. Sramek, M. Trzewiczek and K. Travnicek, WO2019174656A1, 2019; (f) M. Crippa and B. Morico, in Studies in Surface, Science and Catalysis, ed. A. Basile, G. Centi, M. D. Falco and G. Iaquaniello, Elsevier, 2020, vol. 179, ch. 12, pp. 215–229 Search PubMed; (g) M. Crippa, US20240317666A1, 2022.
- S. Anderson, C. Ireland, B. Smit and B. K. Stylianou, WO2020173961A1, 2020.
- N. R. Paliwal and A. K. Mungray, Polym. Degrad. Stab., 2013, 98, 2094 CrossRef CAS.
- L. Biermann, E. Brepohl, C. Eichert, M. Paschetag, M. Watts and S. Scholl, Green Process. Synth., 2021, 10, 361 CrossRef CAS.
- (a) L. Biermann, D. Quast, E. Brepohl, C. Eichert and S. Scholl, Chem. Eng. Technol., 2021, 44, 2300 CrossRef CAS; (b) V. Štrukil, ChemSusChem, 2021, 14, 330 CrossRef PubMed; (c) Y. Y. Li, D. G. Yao and M. Q. Ge, eXPRESS Polym. Lett., 2021, 15, 153 CrossRef CAS; (d) A. W. Tricker, A. A. Osibo, Y. Chang, J. X. Kang, A. Ganesan, E. Anglou, F. Boukouvala, S. Nair, C. W. Jones and C. Sievers, ACS Sustainable Chem. Eng., 2022, 10, 11338 CrossRef CAS; (e) S. Cai, Y. Li, Y. Wang, Z. Guo, B. Liu, L. Huang, J. Beiyuan, D. Liu, R. Cha and W. Yuan, Chem. Eng. J., 2024, 500, 157131 CrossRef CAS.
- (a) Agency for Toxic Substances and Disease Registry (ATSDR), Toxicological profile for Methylene Chloride, U.S. Department of Health and Human Services, Public Health Service, Atlanta, GA, 2000. https://www.epa.gov/sites/default/files/2014-04/documents/methylene_chloride_toxicology_profile_tp14_3v.pdf, (accessed 31/01/05) Search PubMed; (b) World Health Organization , International Labor Organization, International Chemical Safety Cards (ICSCs). ICSC 0058 – Dichloromethane, https://www.ilo.org/dyn/icsc/showcard.display?p_card_id=0058#:~:text=ICSC0058%2DDICHLOROMETHANE&text=Flammableunderspecific (accessed 31/01/05).
- G. S. Cooper, A. S. Bale and P. Schlosser, Toxicological Review of Dichloromethane, U.S. EPA, 2011, https://www.epa.gov/iris/toxreviews/0070tr.pdf (accessed 31/01/25) Search PubMed.
- https://blog.gr3n-recycling.com/2023/07/24/gr3n-to-build-in-spain-the-first-of-a-kind-manufacturing-plant-for-microwave-assisted-depolymerization-of-pet-in-partnership-with-intecsa-industrial/ (accessed 31/01/25).
- M. Bialik, P. Sedin and H. Theliander, Ind. Eng. Chem. Res., 2008, 47, 1283 CrossRef CAS.
- Recent reviews: (a) U. Nshokano Ndagano, L. Cahill, C. Smullen, J. Gaughran and S. M. Kelleher, Molecules, 2025, 30, 299 CrossRef PubMed; (b) T. El Darai, A. Ter-Halle, M. Blanzat, G. Despras, V. Sartor, G. Bordeau, A. Lattes, S. Franceschi, S. Cassel, N. Chouini-Lalanne, E. Perez, C. Déjugnat and J. C. Garrigues, Green Chem., 2024, 26, 6857 RSC; (c) C. Moreno-Marrodán, F. Brandi, P. Barbaro and F. Liguori, Green Chem., 2024, 26, 11832 RSC.
- Reviews: (a) E. V. Antonakou and D. S. Achilias, Waste Biomass Valorization, 2013, 4, 9 CrossRef CAS; (b) J. G. Kim, Polym. Chem., 2020, 11, 4830 RSC; (c) E. A. Gilbert, M. L. Polo, J. M. Maffi, J. F. Guastavino, S. E. Vaillard and D. A. Estenoz, J. Polym. Sci., 2022, 60, 3284 CrossRef CAS.
- C. M. Metz, Workplace Health Saf., 2016, 64, 28 CrossRef PubMed.
- (a) S. S. Pesetskii, B. Jurkowski and V. N. Koval, J. Appl. Polym. Sci., 2002, 84, 1277 CrossRef CAS; (b) S. S. Pesetskii, B. Jurkowski, O. V. Filimonov, V. N. Koval and V. V. Golubovich, J. Appl. Polym. Sci., 2011, 119, 225 CrossRef CAS.
- (a) F. Liu, Z. Li, S.-T. Yu, X. Cui, C.-X. Xie and X.-P. Ge, J. Polym. Environ., 2009, 17, 208 CrossRef CAS; (b) N. Deirrama and A. R. Rahmat, APCBEE Proc., 2012, 3, 172 CrossRef; (c) X. Song, F. Liu, L. Li, X. Yang, S. Yu and X. Ge, J. Hazard. Mater., 2013, 244, 204 CrossRef PubMed.
- A. Ikeda, K. Katoh and H. Tagaya, J. Mater. Sci., 2008, 43, 2437 CrossRef CAS.
- (a) G. P. Tsintzou, E. V. Antonakou and D. S. Achilias, J. Hazard. Mater., 2012, 241, 137 CrossRef PubMed; (b) G. P. Tsintzou and D. S. Achilias, Waste Biomass Valorization, 2013, 4, 3 CrossRef CAS. For a recent review.
- Y. Shi, X. Diao, N. Ji, H. Ding, Z. Ya, D. Xu, R. Wei, K. Cao and S. Zhang, ACS Catal., 2025, 15, 841 CrossRef CAS.
- (a) C. Jehanno, J. Demarteau, D. Mantione, M. C. Arno, F. Ruipérez, J. L. Hedrick, A. P. Dove and H. Sardon, Angew. Chem., Int. Ed., 2021, 60, 6710 CrossRef CAS PubMed; (b) A. J. Spicer, A. Brandolese and A. P. Dove, ACS Macro Lett., 2024, 13, 189 CrossRef CAS PubMed.
- (a) A. Carne Sanchez and S. R. Collinson, Eur. Polym. J., 2011, 47, 1970 CrossRef CAS; (b) R. Yang, G. Xu, B. Dong, X. Guo and Q. Wang, ACS Sustainable Chem. Eng., 2022, 10, 9860 CrossRef CAS.
- (a) S. Westhues, J. Idel and J. Klankermayer, Sci. Adv., 2018, 4, eaat9669 CrossRef CAS PubMed; (b) L. Monsigny, J.-C. Berthet and T. Cantat, ACS Sustainable Chem. Eng., 2018, 6, 10481 CrossRef CAS; (c) B. F. S. Nunes, M. C. Oliveira and A. C. Fernandes, Green Chem., 2020, 22, 2419 RSC.
- (a) L. Pedrini, C. Zappelli and S. J. Connon, ACS Sustainable Chem. Eng., 2025, 13, 1424 CrossRef CAS PubMed; (b) D. Bura, L. Pedrini, C. Trujillo and S. J. Connon, RSC Sustainability, 2023, 1, 2197 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5gc01183c |
| This journal is © The Royal Society of Chemistry 2025 |