
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Streamlining squaramide synthesis using a sustainable and versatile paper-based platform†
Antonella Ilenia
Alfano‡
 a,
Panagiota M.
Kalligosfyri‡
b,
Valerio
Baia
a,
Margherita
Brindisi§
*a and
Stefano
Cinti§
*b
a,
Panagiota M.
Kalligosfyri‡
b,
Valerio
Baia
a,
Margherita
Brindisi§
*a and
Stefano
Cinti§
*b
aSPOTS-Lab, Department of Pharmacy (DoE 2023-2027), University of Naples Federico II, via D. Montesano 49, 80131, Naples, Italy. E-mail: margherita.brindisi@unina.it
bUniNanobiosensors, Department of Pharmacy (DoE 2023-2027), University of Naples Federico II, via D. Montesano 49, 80131, Naples, Italy. E-mail: stefano.cinti@unina.it
First published on 9th April 2025
Abstract
Green sustainable synthesis minimizes environmental impact by reducing waste, energy use, and hazardous materials, thus promoting safer and efficient methods aligned with ecological principles. Building on this, we herein disclose a sustainable methodology for the synthesis of squaramide-based compounds, key structural templates for medicinal and organic chemistry applications, via an innovative eco-friendly protocol. This innovative approach leverages the benefits of filter paper as a suitable reaction platform for water-based solvent systems, thus triggering a significant advance in environmentally friendly practices in organic synthesis. Our newly conceived protocol guarantees high reaction yields (up to 99.5%) and excellent green metrics (up to 89.5/100), unequivocally demonstrating the suitability and versatility of a simple material like filter paper to not only streamline the overall process but also enhance the efficiency of the reaction. Specifically, the adoption of a paper-based reaction platform for synthesizing squaramide-based compounds showcases clear advantages, such as enabling the reaction to proceed without the complications of water removal and avoiding the need for complex equipment or time-consuming purification steps. This approach represents a promising starting point that could be exploited for further applications in chemical synthesis, thus fostering and empowering a sustainable vision in organic and medicinal chemistry.
Green foundation1. In this work, we disclose a sustainable methodology for the synthesis of squaramide-based compounds, employing an eco-friendly protocol. Our innovative approach leverages the benefits of filter paper as a suitable reaction platform for water-based solvent systems, thus triggering a significant advance in environmentally friendly practices in organic synthesis.2. Our newly conceived protocol guarantees high reaction yields (up to 99.5%) and excellent green metrics (ecoscale up to 89.5/100). The adoption of a paper-based platform allows the reaction to proceed without the complication of water removal and avoids the need for complex equipment or time-consuming purification steps. 3. The study demonstrates the versatility of the platform towards both small- and large-scale reaction settings. This approach represents a promising starting point that could be exploited for further applications in chemical synthesis, thus fostering and empowering a sustainable vision in organic and medicinal chemistry. |
Introduction
Squaramides are organic compounds formally derived from squaric acid, in which the hydroxyl groups are replaced by amino groups.1–3 Squarate is a 2e− aromatic oxocarbon dianion characterized by a cyclic structure and two C–O double bonds, and thus exhibits a quasi-aromatic structure. Squarate acts a precursor for both squaramides and squaric esters (Fig. 1a). Structurally, squaramide is considered a member of the vinylogous amide family, as it contains a C–N bond with partial double-bond character due to the delocalization of the nitrogen ion pair electrons. Although squaramides have been known since 1950, only in the past 10 years has this structural template received broader attention from the research community. Accordingly, a steady increase in the application of squaramides has been observed in various fields, such as organocatalysis,4 materials chemistry,5 biosensors,6 bioconjugate linkers,7 anion sensing polymers,8 dyes,9 and the development of novel drug candidates.3 | ||
| Fig. 1 (a) Squarate family, (b) squaramide-containing drugs/drug candidates, and (c) squaramide-based catalysts. | ||
In the context of medicinal chemistry, squaramides are known as bioisosteres of various functional groups, such as ureas, thioureas, guanidines, and cyanoguanidines.1 More recently, squaramides have also increasingly been applied as bioisosteres of carboxylic acids, amides, phosphates, sulfonamides, hydroxamic acids and carbamates.10 The importance of squaramides in drug discovery is mainly ascribable to their unique potential to interrogate the hydrogen bonding ability in several drug targets through the presence of multiple acceptor–donor functionalities in such a small cyclic template, in addition to their intrinsic nature as an optimal appendage for diverse structural moieties in a symmetrical or unsymmetrical fashion.
Accordingly, a variety of medicinally relevant compounds incorporating a squaramide moiety can be found, including H2 antagonists,11 an NMDA antagonist,12 and a CXCR1/2 antagonist13,14 (Fig. 1b).
Squaramides have also emerged as effective bifunctional organocatalysts (Fig. 1c) due to their ability to stabilize transition states, which makes them important in several chemical reactions.15–17
Consequently, the efficient integration of squaramide moieties into simple or more complex molecular frameworks is a compelling need within the synthetic organic and pharmaceutical chemistry communities. Although several synthetic strategies have been developed in this context, most of them to date suffer from various issues that considerably limit their broad-spectrum applicability. The main drawbacks include (i) the need to employ high-boiling-point solvents such as DMF or DMSO for their synthesis, (ii) long reaction times (24–48 h), (iii) the need for a catalyst for some substrates, i.e., Zn(OTf)2 as Lewis acid to coordinate the squarate, (iv) lengthy and/or ineffective purification procedures, (v) limited substrate scope due to the limited access to different monomers, and (vi) high operating temperatures (110–120 °C).18–21 Hence, the development of an effective synthetic strategy for squaramide-based structural templates, especially one that is more eco-friendly and favorable in terms of atom economy, is a highly desirable task in the context of modern chemical synthesis.
With the aim of addressing the challenges of traditional methods, i.e., high-boiling-point solvents, the need for catalysts, long reaction times, harsh purification steps, etc., we sought to develop a reliable and versatile platform for performing squaramide synthesis. A key input towards this task was the driving idea of merging the potential of a filter-paper-based platform with the use of an eco-friendly solvent system, namely, a mixture of ethanol and water (EtOH/H2O). Filter-paper-based platforms have been widely employed in several applications, including the isolation and extraction of chemical compounds, reagent storage or immobilization, filtration, and purification.22 In particular, filter paper is commonly used in organic synthesis for chromatography, water removal from solvents and filtration/purification of chemical products of various types.23–25 Recent works have also revealed that paper-based platforms can be functionalized with metal nanoparticles and cellulose composites that serve as catalysts,26 as well as being used for catalyst storage and subsequent reactant filtration via conventional glassware setups,27,28 an approach that is also known as catalytic filtration.
In the context of sustainability, this manuscript highlights the innovative application of filter paper in organic synthesis, emphasizing its inherent simplicity and eco-friendliness. For the first time, filter paper has been adopted as a unique platform for successfully synthesizing 36 squaramide-based compounds. This approach allows for efficient mixing of reagents and drying processes, which significantly reduced the reaction time, usage of toxic solvents and purification steps.
The intrinsic physical properties of paper make it an ideal platform for the synthesis of compounds without employing complex experimental setups. Moreover, filter paper can be easily customized to various sizes, facilitating scalability and adaptability for a wide range of experimental scale requirements. Additive manufacturing has also been employed in the present work; a 3D-printed housing was fabricated for the paper-based platform to ensure the stability of the reaction setup. To the best of our knowledge, this study is the first to integrate the numerous benefits of filter paper into organic synthesis as an alternative reaction platform. It highlights the way in which this simple yet effective material can be exploited to simplify and replace complex laboratory procedures while at the same time promoting environmentally friendly practices in the field of medicinal chemistry. The synthesis of molecules on paper-based platforms not only enables faster reactions but also maintains high reaction yields compared to the standard routes employed to date for the preparation of the selected chemical template. These preliminary results confirm the bright future of the use of paper-based devices to streamline synthetic protocols while prioritizing sustainability in organic synthesis.
Experimental section
Optimization of the reaction
Ethanol has been reported as a crucial solvent in the synthesis of squaramide-based compounds due to several important factors. As a polar protic solvent, ethanol enhances the solubility of reactants, thus enabling proper mixing and influencing the reaction rate. Additionally, ethanol is advantageous during purification steps because of its ease of handling; its employment has also been shown to improve reaction yields.29,30 In this study, batch reactions were performed in ethanol consistent with previously reported methods18,20 for the optimization of the reaction. However, to enable the use of a filter paper disc as a reaction platform, water was added. The ethanol-to-water ratio was optimized to ensure sufficient ethanol content for the reaction, maintain compatibility with the filter paper, and implement a more sustainable approach towards the synthesis of squaramide-based compounds. Water plays a pivotal role in our reaction through enhancing the coordination with the squaramide carbonyl oxygens. Additionally, the use of paper as a reaction platform avoids the need for lengthy work-up procedures to remove water from the crude mixture, which are typically required in batch protocols. An EtOH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O ratio of 1:1 was selected as the optimal solvent mixture, as outlined in Fig. S2 and further detailed in the ESI.† The interaction of the reaction components with the filter was assessed by characterizing the filter paper before and after running the reaction on it, i.e., after the removal of the product. Microscopic characterization and image analysis, via ImageJ software, showed no significant changes (Fig. S3†), highlighting the reliability of the paper-based platform under the designed reaction conditions.
:H2O ratio of 1:1 was selected as the optimal solvent mixture, as outlined in Fig. S2 and further detailed in the ESI.† The interaction of the reaction components with the filter was assessed by characterizing the filter paper before and after running the reaction on it, i.e., after the removal of the product. Microscopic characterization and image analysis, via ImageJ software, showed no significant changes (Fig. S3†), highlighting the reliability of the paper-based platform under the designed reaction conditions.
Optimization of the paper-based synthesis protocol
The capacity of paper-based supports to be loaded with various amounts of sample has been demonstrated in previous works; in fact, paper-based platforms have been tested for pre-concentration of ca. 10 drops of aqueous solution.31 The volume and the initial filter disc size/shape were selected accordingly. Before establishing the optimal conditions for the paper-based reaction, several parameters were tested and assessed based on the yield of the desired product and comparison with previously developed protocols. Key factors included the use of strong bases, the concentration of the reactants and the diameter of the employed filter disc. The use of an alkaline reaction environment was evaluated with both an organic base (namely DIPEA) and NaOH (entries 2 and 3, Table 1). The selectivity ratio was higher in the case of NaOH, reaching 95:5 for products 3a and 3a′. DIPEA affected the viscosity of the reaction mixture, causing it to spread excessively on the paper-based platform, leading to the loss of reactants/solvent and thus reducing the final reaction ratio. In stark contrast with most of the reported procedures in the literature,15,16 the base is not essential for our protocol, as demonstrated in entry 4. Decreasing the number of equivalents of 2a from 2.5 to 2 did not affect the reaction outcome or the ratio between 3a and 3a′, as shown in entry 5. Another important parameter, i.e., the size of the filter disc, was also investigated. It was observed that increasing the reaction concentration while keeping the filter disc diameter constant resulted in a decrease in the product ratio (entry 6). Conversely, increasing the diameter of the filter disc increased the reaction area, which caused the reaction mixture to spread out uniformly, forming a thinner drop layer. This effect can be advantageous, as a larger area can improve reagent interaction and reaction efficiency. Moreover, a thinner liquid layer enhances interaction with the paper platform and facilitates solvent evaporation, aiding in the recovery of the dry product after the reaction. A 10-fold increase in the scale from 0.014 mmol to 0.14 mmol gave slightly different results compared to the small-scale reaction (entries 7 and 8). Thus, we explored different paper sizes and dilution of the reaction mixture from 1 M to 0.5 M. Among the tested sizes, the 2 cm diameter was the most effective (see also Table S2† for filter disc size optimizations). The final optimized reaction conditions employed dimethyl squarate 1, 2 equiv. of benzylamine 2a, 1:1 EtOH/H2O as the solvent, 2 cm of W1 paper, and a 5 min reaction time at room temperature, which furnished a 3a:3a′ ratio of 97:3 and a 97% yield of compound 3a, as obtained after weighing the product on an analytical balance. Finally, when a batch reaction was performed in a polyester vial using EtOH following previously reported batch conditions,1,32 a 42:58 ratio was obtained, instead a higher ratio of 88:12 was registered when utilizing the paper-based conditions (entries 9 and 10, respectively). In fact, as shown in the optimization in Table S1,† the reaction is completed within 20 min in a polyester vial.
|
|
||
|---|---|---|
| Entry | Deviation from the standard conditions |
3a:3a′ ratiob |
| W1: Whatman chromatographic paper no.1.a Reaction conditions: 1 (0.14 mmol), 2a (2 equiv.), EtOH/H2O 1:1 (0.5 M), 2.0 cm W1, 25 °C, 1 min.b Calculated by UPLC analysis through integrating the area of the peaks.c 3,4-Diethoxy-3-cyclobutene-1,2-dione was used.d 0.014 mmol scale (2 mg).e Same ratio using 3,4-dimethoxy-3-cyclobutene-1,2-dione. |
||
| 1 | None | 97:3 |
| 2c,d | 2.5 equiv. of 2a, 1.5 equiv. of DIPEA, 0.8 cm paper | 50:50 |
| 3c,d | 2.5 equiv. of 2a, 2 equiv. of NaOH 0.5 M, 0.8 cm paper | 95:5 |
| 4c,d | 2.5 equiv. of 2a, 0.8 cm paper | 96:4 |
| 5c,d | 0.8 cm paper | 95:5 |
| 6c,d | 1 M, 0.8 cm paper | 90:10e |
| 7 | 1 M, 1.0 cm paper | 68:32 |
| 8 | 4 M, 0.8 cm paper | 71:29 |
| 9 | polyester vial, 1 min, EtOH 0.1 M | 42:58 |
| 10 | polyester vial, 5 min | 88:12 |

Scale-up and scope of the reaction protocol
Leveraging the robustness of the newly developed methodology, the small-scale setup was adapted into a larger-scale version, as shown in Fig. 2. | ||
| Fig. 2 Images of (A) the experimental setup for the general procedure, i.e., 0.14 mmol scale and the (B) large-scale, i.e., 1.4 mmol scale, experimental setup. The filter paper disc diameter is scalable to (A) 2 cm or (B) 6 cm according to the experimental needs. A 3D-printed device was employed as a housing for the paper-based platform to ensure stability throughout the entire procedure (from mixing of reagents to drying in a fume hood). | ||
Briefly, the synthesis was performed at a 1.4 mmol scale (large-scale) in 10 min with a similar isolated yield to that obtained at the 0.14 mmol scale (small-scale). In the scale-up experiment, a 10-fold proportional increase in the reaction volume was used with respect to the small-scale counterpart. Accordingly, a 10-fold increase in the surface area of the filter disc was applied by increasing the diameter of the filter disc from 2 cm to 6 cm, demonstrating the scalability of our proposed system in all aspects. Once the functional protocol using benzylamine as a model substrate had been established, the reaction scope was broadened by applying the developed methodology to several readily available sterically and electronically diverse benzylamine derivatives in order to investigate the robustness of the paper-based reaction protocol, as detailed in Fig. 3.
 | ||
| Fig. 3 Scope of symmetric dimers with different benzylamine derivatives. | ||
The presence of one or two strongly electron-donating methoxy substituents did not appear to dramatically perturb the performance of our synthesis, with products 3b and 3c being obtained in 84% and 78% yield, respectively. Benzylamines bearing electron-withdrawing substituents, such as trifluoromethyl and nitro substituents, were successfully coupled with dimethyl squarate 1 to give the target products with high yields in both cases, 97% and 81%, respectively (3d and 3e). Similarly, halogen substituents on the aromatic ring, which are useful synthetic handles for further chemical transformations, were well tolerated under our conditions, with compounds 3f and 3g being obtained in 93% and 88% yields, respectively. Notably, a methyl group at the α position on the benzylamine led to the formation of the desired dimer in excellent yield, without any interference being observed during the synthesis and with the preservation of the integrity of the chiral center, giving compound 3h in 93% yield. Conversely, the use of a secondary benzylamine caused a significant drop in the yield, with product 3i being obtained in 51% yield. Low monomer-to-dimer conversion was observed even when the number of equivalents of the starting benzylamine was increased (data not shown). Gratifyingly, even a more complex and Boc-protected isoindoline derivative exhibited complete compatibility with our protocol, yielding the final compound 3j in excellent yield (87%) and confirming the integrity of moderately stable protecting groups in the reaction medium. This observation also highlights the potential of our proposed methodology as a starting point for further functionalization of the generated compounds. In most scenarios, it is worth underlining that the symmetric dimers did not require any additional purification processes, which significantly contributes to further enhancing the eco-friendliness of the developed methodology.
To further showcase the synthetic potential of our method, we investigated the formation of unsymmetrical derivatives employing a small set of benzylamine and aniline derivatives, as detailed in Fig. 4.
 | ||
| Fig. 4 Scope of asymmetric dimers with different benzylamine and aniline derivatives. | ||
First, we analyzed the formation of monomer 5a′ following the procedures reported in the literature, which usually require a long reaction time, toxic solvents, and high temperature.33–37 Due to the poor nucleophilicity of aniline compared to benzylamine, a Lewis acid catalyst like Zn(OTf)2 is usually added to the reaction to promote coordination with the carbonyl oxygens, which renders the compounds more susceptible to the substitution. Employing 1 equiv. of aniline as the amine partner for dimethyl squarate 1 in EtOH as the solvent in the presence of 10 mol% of Zn(OTf)2 at room temperature for 16 h, we achieved a 95% yield of compound 5a′. We then sought to investigate the same reaction under our developed conditions using the EtOH/H2O solvent mixture and 1 equiv. of aniline. Gratifyingly, within only 5 min, at room temperature, and without the need for a catalyst, we were able to successfully achieve the product. This highlights the pivotal role of water in our reaction, as it likely enhances the coordination with the carbonyl oxygens. Additionally, the use of paper as a platform avoids the necessity for a work-up procedure to remove water from the crude mixture, which is typically required in batch protocols. With the aniline monomer in hand, we turned our attention to accessing the asymmetric derivatives through the addition of commercially available benzylamines. The addition of unsubstituted benzylamine led to compound 5a in quantitative yield. Notably, the presence of electron-donating substituents, such as methoxy groups, on the aromatic ring of benzylamine, did not affect the performance of the protocol. Indeed, quantitative yields were observed with one or two methoxy groups (5b and 5c, 97% and 95% yield, respectively). Analogously, the presence of electron-withdrawing groups such as trifluoromethyl and nitro functionalities did not adversely affect the reaction outcome, with compounds 5d and 5e being obtained in 91% and 88% yields, respectively. Comparable yields of 95% and 85% were achieved with two different halogen substituents (5f, 5g), as well as with the introduction of a methyl group at the α position (5h, 99%). In stark contrast to the results observed for the generation of the symmetric dimer, the monomer with 1-(2-bromophenyl)-N-methylmethanamine provided the corresponding unsymmetrical squaramide derivative in quantitative yield (5i, 82%). The protocol was also compatible with an N-Boc derivative, furnishing compound 5j in 84% yield. Then, we shifted our focus to broadening the scope of the aniline counterpart while keeping the benzylamine component unchanged. The introduction of aliphatic substituents such as isopropyl and methyl groups was well tolerated (5k, 5l), resulting in yields of 82% and 97% respectively, as was the addition of a halogen at the 3-position (5m, 52%). Our method was also amenable to an electron-donating substituent (5n) as well as fused heterocycles such as benzo[d][1,3]dioxole (5o) and quinoline (5p), which gave yields of 98%, 60% and 98%, respectively.
To further investigate the versatility of our paper-based protocol, we extended the scope to unsymmetrical squaramide derivatives derived from both primary and secondary aliphatic amines and an aniline counterpart (Fig. 5).
 | ||
| Fig. 5 Scope of unsymmetrical dimers with different aliphatic amines and aniline. | ||
The cyclic primary amines cyclohexylmethanamine and cyclobutylmethanamine were successfully coupled with the aniline monomer in excellent yields (7a and 7b, 99% and 88%). When a double bond functionality was added, low conversion was observed, and consequently, a yield of 40% was obtained for compound 7c. The reaction effectiveness was not limited to the use of primary amines; application of the protocol to 6-membered secondary amines, namely, piperidine and piperazine, bearing a Boc protecting group provided 7d and 7e in high yields (96% and 65%). Slightly different behavior was observed for the 5-membered secondary amines pyrrolidine and L-proline, which gave compounds 7f and 7g in slightly lower yields of 55% and 72%, respectively. This outcome can be associated with the low conversion of monomer to dimer. The reaction involving two fused secondary amines bearing a hydroxy or ether group yielded favorable results, furnishing compounds 7h and 7j in good yields (91% and 64%). It is particularly noteworthy to highlight that the presence of a free hydroxyl group does not adversely affect the overall reaction efficiency.
For completeness, we have also included a list of unsuccessful compounds in the ESI (Fig. S4†). Most failures were linked to solubility issues of the employed amine derivatives. It is important to underscore that all our compounds, with the exception of 6 derivatives, are new chemical entities or at least compounds that have not been reported and characterized in the literature previously. We have therefore contributed the synthesis and characterization of 30 new chemical entities, further demonstrating the versatility of the protocol for further research and application in various fields.
Green metrics
Finally, green metrics were evaluated for three model compounds, a symmetric dimer, a monomer and an unsymmetrical dimer, to measure the environmental impact, effectiveness and sustainability of our paper-based protocol. The parameters contributing to the green metrics of the compound synthesis were calculated as demonstrated in Fig. 6 for the model compound dimer 3a.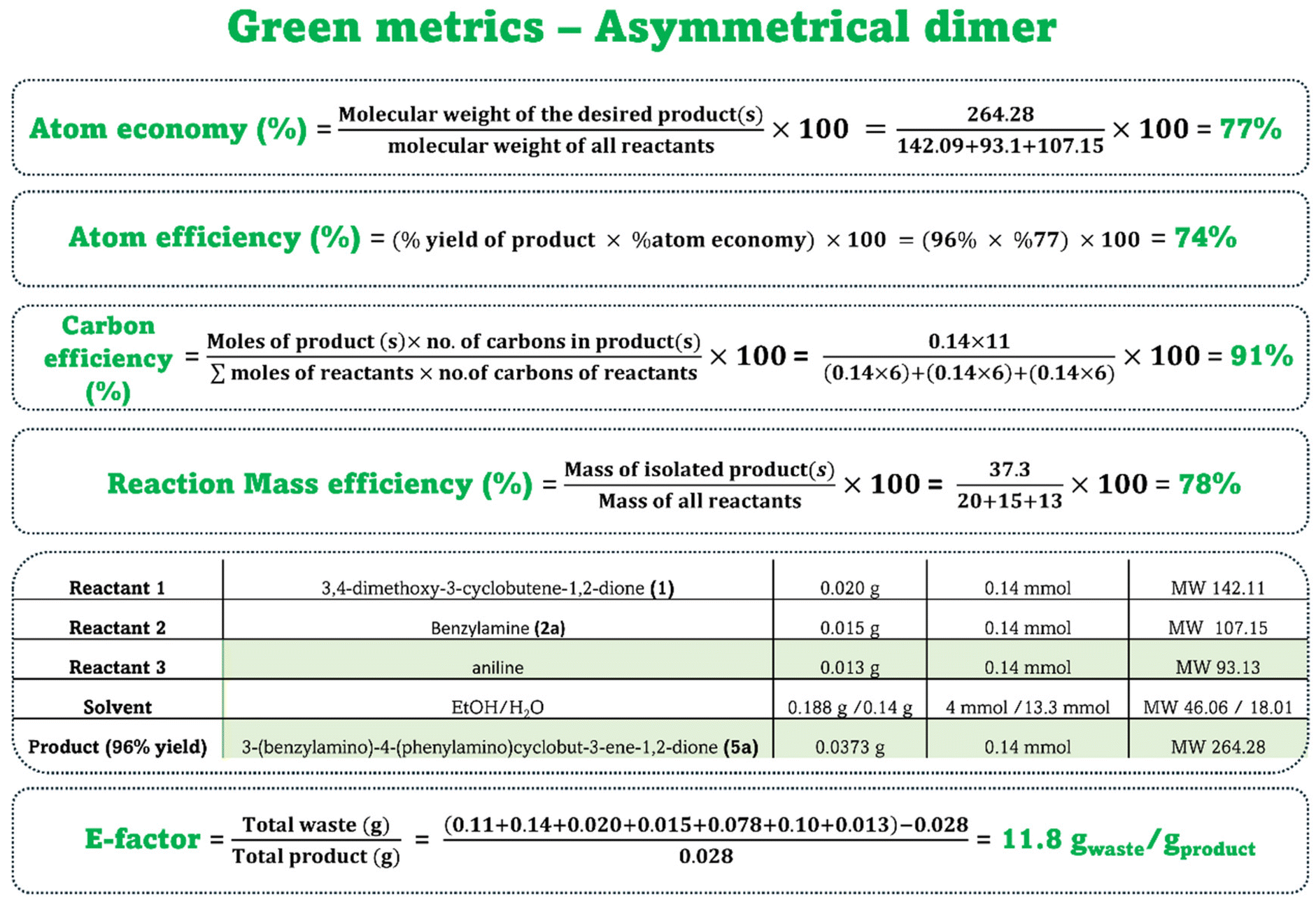 | ||
| Fig. 6 Detailed calculation of the green metrics components for the model compound 3a. | ||
All the metrics were found to be excellent. We calculated the E-factor, which is defined as the weight of total waste (in grams) relative to the weight of the product (in grams) and compared it with those of various procedures previously reported in the literature. Our method showed a very low E-factor, highlighting its advantages. Significantly, two crucial green chemistry parameters, i.e., atom economy, which is calculated as the molecular weight of the desired product(s) divided by molecular weight of all reactants, and atom efficiency, which is determined as the product yield multiplied by the atom economy percentage, were found to be favorable as well. The carbon efficiency, which is calculated as moles of product multiplied by the number of carbons present in the product divided by moles of reactants multiplied by the number of carbons in the reactants, and reaction mass efficiency, which is defined as the mass of isolated product divided by mass of all reactants, approached 100% across all processes.
These calculations were performed for the monomer (using aniline as the reactant) and the asymmetrical dimer; detailed results are provided in Fig. S5 and S6 of the ESI.† For all three compounds, the E-factors related to waste generation were up to ∼85 times lower than those previously reported for similar synthesized compounds (Table S4†). This is a result of the eco-friendly solvent and the paper-based reaction platform, which enable fast reaction at room temperature without requiring long isolation and purification steps. Moreover, an essential component of green metrics is the final EcoScale, which was calculated following the previously reported methodology.38 The individual penalties contributing to the EcoScore of the model compound 3a are detailed in the ESI† (green metrics calculation section). Notably, for the symmetric dimer, the EcoScale, which assesses organic safety, economic and ecological features, was calculated to be an impressive 88.5 (>75, excellent; >50, acceptable; and <50, inadequate), which established the excellence of our process.
Conclusions
The study incorporated a chromatographic paper-based platform in the field of organic synthesis, highlighting its ability to simplify laboratory procedures while promoting environmentally friendly practices. The proposed paper-based reaction platform demonstrated greater efficiency than batch solutions and offered important advantages. Its porous, high-surface-area structure facilitates capillary-driven flow,22 ensuring uniform reagent distribution and enhancing reaction kinetics. In contrast, batch solutions often experience localized over-concentration of reactants, increasing unwanted side reactions, especially when mixing is not employed. Additionally, the porous filter paper enables continuous solvent evaporation,22 shifting reaction equilibria toward product formation. This passive solvent removal is significantly more efficient than in batch systems, in which excess solvent slows reaction progress. The faster drying on filter paper accelerates reaction completion and simplifies product isolation, reducing the need for extensive purification or solvent removal steps. Here, we have successfully applied this synergistic and interdisciplinary approach to the generation of a large library of squaramide-based templates, which have great relevance in medicinal and organic chemistry. Paper-based platforms were exploited as reactors, and their robustness was confirmed through scale-up experiments. The model reaction yielded consistent results across different scales, underscoring the scalability and reliability of the brand-new platform. Unlike conventional methods that rely on bulky and costly equipment, this system offers a low-cost and streamlined solution. Furthermore, the excellent green metric values confirm the sustainability of this approach. By bridging medicinal chemistry and sustainable materials, this work introduced a novel approach that led to the straightforward synthesis of 36 squaramide-based compounds, 30 of which are newly characterized chemical entities. The study demonstrates the versatility of the platform for both small- and large-scale reaction settings while avoiding the need for extensive purification steps and expensive equipment. The combination of high reaction yields and excellent green metrics firmly establishes this as a sustainable and efficient solution and paves the way for advancing environmentally friendly practices in medicinal and organic chemistry.Data availability
The data underlying this article are included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the Department of Excellence of Pharmacy (2023-2027) at the University of Naples Federico II. MB acknowledges financial support by the Italian Ministry for University and Research (PRIN project n. 202295845T) and EU funding within the MUR PNRR Extended Partnership initiative on Emerging Infectious Diseases (project no. PE00000007, INF-ACT).References
- R. I. Storer, C. Aciro and L. H. Jones, Squaramides: Physical Properties, Synthesis and Applications, Chem. Soc. Rev., 2011, 40(5), 2330, 10.1039/c0cs00200c.
- L. A. Marchetti, L. K. Kumawat, N. Mao, J. C. Stephens and R. B. P. Elmes, The Versatility of Squaramides: From Supramolecular Chemistry to Chemical Biology, Chem, 2019, 5(6), 1398–1485, DOI:10.1016/j.chempr.2019.02.027.
- K. A. Agnew-Francis and C. M. Williams, Squaramides as Bioisosteres in Contemporary Drug Design, Chem. Rev., 2020, 120(20), 11616–11650, DOI:10.1021/acs.chemrev.0c00416.
- A. Rouf and C. Tanyeli, Squaramide Based Organocatalysts in Organic Transformations, Curr. Org. Chem., 2016, 20(28), 2996–3013, DOI:10.2174/1385272820666160805113749.
- D. Basak, C. Versek, D. T. Toscano, S. Christensen, M. T. Tuominen and D. Venkataraman, Anhydrous Proton Conductivities of Squaric Acid Derivatives, Chem. Commun., 2012, 48(47), 5922, 10.1039/c2cc31283b.
- W. Lai, D. Tang, L. Fu, X. Que, J. Zhuang and G. Chen, A Squaric Acid-Stimulated Electrocatalytic Reaction for Sensing Biomolecules with Cycling Signal Amplification, Chem. Commun., 2013, 49(42), 4761, 10.1039/c3cc41708e.
- F. R. Wurm and H.-A. Klok, Be Squared: Expanding the Horizon of Squaric Acid-Mediated Conjugations, Chem. Soc. Rev., 2013, 42(21), 8220, 10.1039/c3cs60153f.
- A. Rostami, G. Guérin and M. S. Taylor, Structure–Activity Relationships for Anion-Responsive Poly(Squaramides): Support for an Analyte-Induced Noncovalent Polymer Cross-Linking Mechanism, Macromolecules, 2013, 46(16), 6439–6450, DOI:10.1021/ma401263q.
- G. Xia and H. Wang, Squaraine Dyes: The Hierarchical Synthesis and Its Application in Optical Detection, J. Photochem. Photobiol., C, 2017, 31, 84–113, DOI:10.1016/j.jphotochemrev.2017.03.001.
- K. A. Agnew-Francis and C. M. Williams, Squaramides as Bioisosteres in Contemporary Drug Design, Chem. Rev., 2020, 120(20), 11616–11650, DOI:10.1021/acs.chemrev.0c00416.
- C. R. Ganellin and R. C. Young, Pharmacologically Active Cyclo Butenediones, U.S. Patent4062863, 1977 Search PubMed.
- A. A. Algieri and R. R. Crenshaw, 1,2-Diaminocyclobutene-3,4- Diones and a Pharmaceutical Composition Containing Them, FR 2505835A1, 1982.
- D. F. Jones and T. O. Yellin, Guanidino Imidazoles and Thiazoles, U.S. Patent4165377, 1979 Search PubMed.
- T. H. Brown and R. C. Young, Dioxocyclobutene Compounds, U.S. Patent4521625, 1985 Search PubMed.
- F. Spiaggia, G. Uccello Barretta, A. Iuliano, C. Baldassari, F. Aiello and F. Balzano, A Squaramide-Based Organocatalyst as a Novel Versatile Chiral Solvating Agent for Carboxylic Acids, Molecules, 2024, 29(10), 2389, DOI:10.3390/molecules29102389.
- S. Sopeña, E. Martin, E. C. Escudero-Adán and A. W. Kleij, Pushing the Limits with Squaramide-Based Organocatalysts in Cyclic Carbonate Synthesis, ACS Catal., 2017, 7(5), 3532–3539, DOI:10.1021/acscatal.7b00475.
- P. Chauhan, S. Mahajan, U. Kaya, D. Hack and D. Enders, Bifunctional Amine–Squaramides: Powerful Hydrogen–Bonding Organocatalysts for Asymmetric Domino/Cascade Reactions, Adv. Synth. Catal., 2015, 357(2–3), 253–281, DOI:10.1002/adsc.201401003.
- G. Printz, D. Ryzhakov, C. Gourlaouen, B. Jacques, S. Messaoudi, F. Dumas, F. Le Bideau and S. Dagorne, First Use of Thiosquaramides as Polymerization Catalysts: Controlled ROP of Lactide Implicating Key Secondary Interactions for Optimal Performance, ChemCatChem, 2024, 16(1), e202301207 CrossRef CAS.
- D. Givaudan, B. Biletskyi, A. Recupido, V. Héran, O. Chuzel, T. Constantieux, J. Parrain and X. Bugaut, Bifunctional Iodoazolium Salts: Searching for Cooperation Between Halogen Bonding and Hydrogen Bonding, Eur. J. Org. Chem., 2024, 27(15), e202300261 CrossRef CAS.
- V. E. Zwicker, K. K. Y. Yuen, D. G. Smith, J. Ho, L. Qin, P. Turner and K. A. Jolliffe, Deltamides and Croconamides: Expanding the Range of Dual H–bond Donors for Selective Anion Recognition, Chem. – Eur. J., 2018, 24(5), 1140–1150, DOI:10.1002/chem.201704388.
- G. Dong, M. Bao, X. Xie, S. Jia, W. Hu and X. Xu, Asymmetric Allylation by Chiral Organocatalyst–Promoted Formal Hetero–Ene Reactions of Alkylgold Intermediates, Angew. Chem., Int. Ed., 2021, 60(4), 1992–1999, DOI:10.1002/anie.202012678.
- P. M. Kalligosfyri and S. Cinti, Paper-Based Materials for Diagnostics, ACS Mater. Lett., 2024, 1184–1198, DOI:10.1021/acsmaterialslett.3c01474.
- R. H. Kang and D. Kim, Thermally Induced Silane Dehydrocoupling: Hydrophobic and Oleophilic Filter Paper Preparation for Water Separation and Removal from Organic Solvents, Materials, 2021, 14(19), 5775, DOI:10.3390/ma14195775.
- G. S. Shinde, P. S. Rao, R. S. Jadhav, P. Kolhe and D. Athare, A Review on Chromatography and Advancement in Paper Chromatography Technique, Asian J. Pharm. Anal. Med. Chem., 2021, 11(1), 45–48, DOI:10.5958/2231-5675.2021.00009.0.
- R. M. Lemmon, W. Tarpey and K. G. Scott, Paper Chromatography in Synthetic Organic Chemistry. Microgram Scale Syntheses of Labeled Monoiodotyrosine, Diiodotyrosine and Thyroxine, J. Am. Chem. Soc., 1950, 72(2), 758–761, DOI:10.1021/ja01158a030.
- S. Kamel and T. A. Khattab, Recent Advances in Cellulose Supported Metal Nanoparticles as Green and Sustainable Catalysis for Organic Synthesis, Cellulose, 2021, 28(8), 4545–4574, DOI:10.1007/s10570-021-03839-1.
- I. Raya, S. Danshina, A. T. Jalil, W. Suksatan, M. Z. Mahmoud, A. B. Roomi, Y. F. Mustafa and M. Kazemnejadi, Catalytic Filtration: Efficient C-C Cross-Coupling Using Pd (II) -Salen Complex-Embedded Cellulose Filter Paper as a Portable Catalyst, RSC Adv., 2022, 12(31), 20156–20173, 10.1039/D2RA03440A.
- D. O. Bokov, M. Z. Mahmoud, G. Widjaja, W. Suksatan, S. Chupradit, U. S. Altimari, H. A. Hussein, Y. F. Mustafa and M. Kazemnejadi, Transfer Hydrogenation of Nitroarenes Using Cellulose Filter Paper-Supported Pd/C by Filtration as Well as Sealed Methods, RSC Adv., 2022, 12(18), 10933–10949, 10.1039/D2RA01151D.
- L. A. Marchetti, T. Krämer and R. B. P. Elmes, Amidosquaramides – a New Anion Binding Motif with PH Sensitive Anion Transport Properties, Org. Biomol. Chem., 2022, 20(35), 7056–7066, 10.1039/D2OB01176J.
- G. Carullo, L. Bottoni, S. Pasquini, A. Papa, C. Contri, S. Brogi, V. Calderone, M. Orlandini, S. Gemma, K. Varani, S. Butini, F. Galvagni, F. Vincenzi and G. Campiani, Synthesis of Unsymmetrical Squaramides as Allosteric GSK–3β Inhibitors Promoting B–Catenin–Mediated Transcription of TCF/LEF in Retinal Pigment Epithelial Cells, ChemMedChem, 2022, 17(24), e202200456 CrossRef CAS PubMed.
- P. M. Kalligosfyri and S. Cinti, 3D Paper-Based Origami Device for Programmable Multifold Analyte Preconcentration, Anal. Chem., 2024, 96(24), 9773–9779, DOI:10.1021/acs.analchem.4c02032.
- A. E. Aydin and S. Culha, Enantioselective Conjugate Addition of Pyrazolones to Nitroalkenes Catalyzed by Chiral Squaramide Organocatalyst, Chirality, 2021, 33(3), 106–114, DOI:10.1002/chir.23295.
- A. Rostami, A. Colin, X. Y. Li, M. G. Chudzinski, A. J. Lough and M. S. Taylor, N, N′ -Diarylsquaramides: General, High-Yielding Synthesis and Applications in Colorimetric Anion Sensing, J. Org. Chem., 2010, 75(12), 3983–3992, DOI:10.1021/jo100104g.
- S. Bujosa, E. Castellanos, A. Frontera, C. Rotger, A. Costa and B. Soberats, Self-Assembly of Amphiphilic Aryl-Squaramides in Water Driven by Dipolar π–π Interactions, Org. Biomol. Chem., 2020, 18(5), 888–894, 10.1039/C9OB02085C.
- N. Long, A. Le Gresley, A. Solomonsz, A. Wozniak, S. Brough and S. P. Wren, Synthesis of Squaric Acid Monoamides as Building Blocks for Drug Discovery, SynOpen, 2023, 07(03), 401–407, DOI:10.1055/a-2148-9518.
- C. Jin, M. Zhang, L. Wu, Y. Guan, Y. Pan, J. Jiang, C. Lin and L. Wang, Squaramide-Based Tripodal Receptors for Selective Recognition of Sulfate Anion, Chem. Commun., 2013, 49(20), 2025, 10.1039/c3cc00196b.
- C. Jin, M. Zhang, C. Deng, Y. Guan, J. Gong, D. Zhu, Y. Pan, J. Jiang and L. Wang, Novel Calix[4]Arene-Based Receptors with Bis-Squaramide Moieties for Colorimetric Sensing of Anions via Two Different Interaction Modes, Tetrahedron Lett., 2013, 54(8), 796–801, DOI:10.1016/j.tetlet.2012.11.117.
- K. Van Aken, L. Strekowski and L. Patiny, EcoScale, a Semi-Quantitative Tool to Select an Organic Preparation Based on Economical and Ecological Parameters, Beilstein J. Org. Chem., 2006, 2(3) DOI:10.1186/1860-5397-2-3.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5gc00535c |
| ‡ Co-first authors. |
| § Co-last authors. |
| This journal is © The Royal Society of Chemistry 2025 |