
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Chemical looping hydrogen production from ammonia and water: materials and technoeconomics†
Amirmohammad
Arjomand Kermani
a,
Kyle
Shank
a and
Shang
Zhai
 *ab
*ab
aDepartment of Mechanical and Aerospace Engineering, The Ohio State University, 201 West 19th Ave, Columbus, OH 43210, USA. E-mail: zhai.218@osu.edu
bSchool of Earth Sciences, The Ohio State University, 125 South Oval Mall, Columbus, OH 43210, USA
First published on 28th May 2025
Abstract
Ammonia (NH3) is a promising hydrogen carrier due to high hydrogen density and established infrastructure. We present a novel chemical looping process to produce H2 from NH3 oxidation and decomposition and from water splitting, integrating thermochemical redox looping and catalytic reaction. Unlike single-step catalytic NH3 decomposition, the looping configuration produces high purity H2 from the water splitting that significantly lowers separation energy and cost. FeOx/YSZ and Fe0.5Co0.5Ox/YSZ were shown as durable dual-functional oxygen carriers and catalysts, achieving 95% and 99% NH3 conversion and 39% and 25% water splitting conversion, respectively. The materials’ redox capacities were explained by simultaneous Fe and Co redox reactions and solid-state phase transition between metal (alloy) and spinel. From 450 to 600 °C, Fe redox capacity increased, while Co redox capacity decreased. Kinetic limitations hindered full reduction of FeOx/YSZ to metallic Fe at 450 °C due to lack of the effective Co catalyst, while thermodynamic limitations prevented complete oxidation of Co metal in Fe0.5Co0.5Ox/YSZ. Techno-economic analysis showed the looping process achieves 52% to 86% lower energy and equipment costs than single-step catalytic NH3 decomposition with different H2 separation methods.
Green foundation1. This paper presents a novel process of chemical looping ammonia oxidation and decomposition integrated with water splitting (CLAOD-WS). Compared to single-step catalytic NH3 decomposition, CLAOD-WS gives high purity H2 to significantly lower separation energy and cost, enabling NH3 as a promising hydrogen carrier.2. FeOx/YSZ and Fe0.5Co0.5Ox/YSZ were shown as durable dual-functional oxygen carriers and catalysts. The materials’ redox capacities were explained by simultaneous Fe and Co redox reactions and solid-state phase transition between metal (alloy) and spinel. The looping process achieves 52% to 86% lower energy and equipment costs than single-step catalytic NH3 decomposition. 3. Future research can include reactor design and materials chemistry to co-optimize oxygen exchange capacity and catalytic reactivity, so that almost all H2 is produced from water splitting step rather than NH3 conversion step, further lowing H2 separation energy and cost. |
1. Introduction
Addressing the Gigaton-scale greenhouse gas (GHG) emissions challenge is critical for mitigating global warming.1 Clean hydrogen (H2) produced without GHG emissions offers significant potential to support carbon neutrality, especially in hard-to-decarbonize sectors including industry and transportation.2,3 Its applications span chemical feedstock, steel production, fuel for heavy-duty transport, industrial/domestic heating, and renewable energy storage.2,3 To expand clean H2 utilization, the issue of energy-intensive and costly H2 delivery needs to be overcome.1,4 The current cost to produce GHG-free H2 with water electrolysis is about $4–5 per kg,5 while the average cost of transporting and dispensing H2 can range from $8.17–$9.46 per kg for gaseous tube trailers and $8.31–$11.35 per kg for liquid tankers,6 mainly because H2 compression and liquefaction are expensive and energy-intensive.7–11 Chemical hydrogen carriers have the potential to help lower energy demand and cost for H2 delivery.1,4,12NH3 proves to be an excellent hydrogen carrier. Firstly, it possesses a high volumetric and gravimetric hydrogen density compared to several well-known hydrogen carriers (Fig. 1a).1,12,13 At 298 K and 1 MPa, the volumetric hydrogen density of NH3 is 60 times that of H2, and the storage cost of NH3 is only 3% of H2.1 Secondly, NH3 is vastly utilized as a fertilizer in agriculture; consequently, well-established infrastructure exists in many countries.14 NH3 has been easily and safely transported via pipelines, trucks, and rail cargos for many decades.1,15,16
 | ||
| Fig. 1 (a) Comparison among chemical hydrogen carriers for volumetric and gravimetric hydrogen densities at 0.1 MPa and 240 K. Adapted from references.1,12,13,17–20 (b) Chemical looping ammonia oxidation and decomposition integrated with water splitting (CLAOD-WS) process schematic. MOx is a redox active metal oxide, and M* is a metallic catalyst. Compositions of M and M* may change between the reduced and oxidized states. | ||
Catalytic NH3 decomposition to H2 has been extensively studied and catalysts such as noble metals of ruthenium (Ru) and platinum (Pt) or transition metal-based catalysts of nickel (Ni), cobalt (Co), and iron (Fe) have been investigated to give high conversions (>95%) at moderate temperatures (450–600 °C).4,21–24 In a study on noble metal catalysts by Yao et al., high NH3 conversion of 96% was achieved at the moderate temperature of 500 °C using nanoparticles of Ru on silica (SiO2) support.24 In a comprehensive study by Gu et al., Co, Ni, and Fe were synthesized on alumina (Al2O3) support, with catalytic NH3 decomposition at 600 °C revealing conversion rates of 100%, 93%, and 86% for Co, Ni, and Fe catalysts, respectively.23 Xie et al. tuned CoMoFeNiCu high entropy alloy catalyst nanoparticles dispersed on carbon nanofibers which reached 100% conversion of NH3 at 525 °C for Co25Mo45Fe10Ni10Cu10. This was compared to bimetallic Co–Mo and monometallic Ru which only reached 46% and 73% conversion, respectively, at 600 °C.22 Tabassum et al. present a Ru-free catalyst composed of a CoNi alloy supported on a K-promoted MgO–CeO2–SrO mixed oxide, achieving nearly complete NH3 conversion at 450 °C. The catalyst demonstrates high stability and hydrogen production rates comparable to Ru-based catalysts, highlighting the role of metal–oxide interfaces and promoter effects in enhancing activity and stability.21
Oxygen carrier metal oxides have been researched for NH3 oxidation to nitrogen (N2) and nitrogen oxides (NOx). Normann et al. introduced a chemical looping scheme in which NH3 was oxidized using Fe, manganese (Mn), and Ni-based oxygen carriers; afterwards, the oxygen carriers were re-oxidized by oxygen.25 This work focused on the production of nitric oxide (NO) and H2O from NH3 oxidation and concluded that Mn-based oxide demonstrates the best performance in NO formation. In another study, Cheng et al. investigated NH3 oxidation to NOx and N2 using Ilmenite (iron–titanium oxide).26 It was discovered that decreasing NH3 concentration decreases the selectivity of NH3 decomposition toward N2 and increases that toward NO formation. Ruan et al. investigated a chemical looping scheme in which NH3 was selectively oxidized to form NO using a vanadium pentoxide (V2O5) oxygen carrier and then the oxygen carrier was re-oxidized using oxygen gas (O2).27 This configuration achieved over 95% NH3 conversion and 100% NO selectivity at 650 °C.
Oxygen carriers have also been studied in thermochemical redox cycles for H2 production by water splitting.28–43 The oxygen capacity of the carrier materials comes from either solid phase transformation or oxygen vacancy. One category of the redox cycles uses high temperature to thermally reduce the oxygen carrier. Zhai et al. utilized mixed metal oxides called poly-cation oxides (PCOs) which consist of transition metals Fe, Mg, Co, and Ni. Fe redox in the PCOs provided the redox capacity, while the other redox inactive metals helped tune the oxide phase transformation to lower the reduction reaction temperature for a lower system cost.28 Tran et al. examined the performance of iron aluminate ((Fe0.33Al0.67)3−δO4) for H2O and CO2 splitting reactions. Thermal reduction at 1400 °C was followed by oxidation at elevated pressures (1–10 atm), resulting in enhanced equilibrium extent and reaction kinetics.38 Wexler et al. presented a newly developed perovskite material, Ca2/3Ce1/3Ti1/3Mn2/3O3, which exhibits high hydrogen production capacity and stable redox performance. The authors identified that the A-site Ce4+ reduction is the key to its enhanced water splitting capabilities.43 Tran et al. reviewed the potential of perovskite oxides as redox materials for CO2 splitting using solar thermochemical methods. This work highlights their lower reduction temperatures, tunable properties, and challenges such as sintering and cycle stability for solar-driven fuel production.44 The other category of redox cycles uses chemical reduction for the oxygen carrier. We refer to review articles and book in this area.45–47
Building on the previous work, we present chemical looping ammonia oxidation and decomposition integrated with water splitting (CLAOD-WS), a novel two-step cyclic process involving redox looping and catalytic reactions for H2 production from NH3 and H2O (Fig. 1b). Here NH3 is first partially oxidized by a solid oxygen carrier and meanwhile decomposed by a catalyst, to produce H2, N2, and H2O (eqn (1)); secondly, H2O re-oxidizes the oxygen carrier to produce H2 (eqn (2)).
 | (1) |
| δH2O + MOx−δ → δH2 + MOx | (2) |
Over CLAOD-WS cycles, the net reaction is the same as NH3 decomposition producing N2 and H2, but looping presents advantages that will translate to significant energy and cost savings shown by this paper. Firstly, it gives two H2 streams at different purity levels suitable for different applications. On the one hand, the H2 produced by water splitting step is intrinsically separated from N2 and NH3. This H2 has only unreacted H2O as impurity and is thus more suitable for high-purity H2 applications such as fuel cells.48–50 If necessary, this stream of H2 mixed with unreacted H2O can be further purified by removing H2O, which is simpler than H2 separation from N2 and unreacted NH3 in direct NH3 decomposition. On the other hand, the low-purity H2 from the NH3 conversion step is mixed with N2, H2O, and unreacted NH3, and can be suitable for industrial heating and power generation. Secondly, according to Le Chatelier's principle, chemical looping facilitates NH3 conversion thanks to the removal of H2 by the chemical reduction of the oxygen carrier.
Previous literature on system energy and technoeconomic assessments provides information for our analysis of CLAOD-WS in comparison to direct NH3 decomposition in terms of energy consumption, H2 production, and process costs. Yun et al. analyzed catalytic NH3 decomposition plants equipped with various types of H2 separation systems including pressure swing absorption (PSA) and Pd membranes, where the levelized cost of H2 (LCOH) was reported between $5–7.5 per kg-H2.51 Nordio et al. proposed a H2 separation system integrated with multiple membrane modules and conducted a comprehensive technoeonomic analysis. This study concluded that LCOH ranges between €3.56 and €12.63 per kg-H2.52 Makhloufi et al. studied a large-scale NH3 cracking system for clean H2 production. This plant showed 68.5% thermal efficiency and €4.83 per kg-H2.53 In another study by Devkota et al., technoeconomic and environmental assessment of an NH3 decomposition plant equipped with a packed bed reactor concluded that LCOH was $6.05 per kg-H2.54
This study leverages catalytic and redox activities of FeOx/YSZ and Fe0.5Co0.5Ox/YSZ for CLAOD-WS. We identified the different redox and catalytic properties of Fe and Co. Also, we showed that kinetic limitation and thermodynamic limitation occurred for different materials under different reaction conditions. Stable 50-cycle operation at 600 °C showed the materials’ durability. Our energy and technoeconomic analyses compared CLAOD-WS with direct catalytic NH3 decomposition systems.
2. Materials and methods
2.1. YSZ supported materials synthesis
Iron oxide and mixed iron–cobalt oxide were synthesized using a modified wet impregnation method. For the iron oxide, Fe(NO3)3·9H2O (Sigma-Aldrich) was mixed with ethanol, and 8% yttria-stabilized zirconia (YSZ8%) ((ZrO2)0.92(Y2O3)0.08) (Inframat Advanced Materials) was supplemented to the mixture as the oxygen carrier's support material. The mass of the support material was calculated to be 30% of the total mass of metal oxide and support. The mixture was stirred on a hotplate at 300 rpm for three hours at ambient temperature, followed by heating to 100 °C and stirring for two more hours. At this point, a thick paste was formed which was moved to a box furnace (MTI KSL-1200X) and dried at 200 °C for seven hours (heating and cooling ramp rates were 5 °C min−1). The resulting dry material was ground with a mortar and pestle to obtain a fine powder. Lastly, calcination was carried out under air at 600 °C for three hours (heating and cooling ramp rates were 15 and 5 °C min−1 respectively) in the box furnace. The final product was further ground by mortar and pestle. This sample is denoted as FeOx/YSZ. For the mixed iron–cobalt oxide, Fe(NO3)3·9H2O and Co(NO3)2·6H2O (Sigma-Aldrich) were mixed with the support following the same procedure. This sample is denoted as Fe0.5Co0.5Ox/YSZ.2.2. Materials looping performance testing
CLAOD-WS experiments were conducted in a U-shaped quartz tube (O.D. = 0.5 in) in a Micromeritics tube furnace. 0.01 g material was uniformly placed inside the tube between quartz wool to form a fixed bed reactor. The reactor was programmed for isothermal reaction temperatures of 450, 500, 550, and 600 °C, with a 15 °C min−1 ramp rate, and three different space velocity values of 6, 18, and 30 LNH3 (gsolid h)−1. During heating up, 100 sccm of argon (Ar) flowed over the sample. At the determined reaction temperature, the first step began by flowing NH3 gas balanced with Ar for 12 minutes unless otherwise noted, and consequently, H2, N2, and H2O were produced. Different concentrations of NH3 (1%, 3%, and 5%) were chosen for space velocities of 6, 18, and 30 LNH3 (gsolid h)−1, respectively (eqn (S1)†). In the second step, 2% H2O vapor diluted in Ar flowed over the reduced material for 3 minutes unless otherwise noted; H2O step space velocity was equal to 12 LH2O (gsolid h)−1 in all experiments (eqn (S2)†). After each step, 100 sccm of Ar flowed for 5 minutes to purge the reactor and gas lines. A mass spectrometer (Hiden Analytical HPR-20 R&D) and gas chromatograph (Inficon Micro GC Fusion) were used in parallel to detect and quantify the gas mixture in the exhaust line. Potential NOx was detected at a small amount in the NH3 step of the first CLAOD-WS cycle for several samples, and no NOx was detected beyond the first cycle (Fig. S4†).2.3. Material characterization using XRD
X-ray diffraction (XRD) characterizations were performed using a Rigaku SmartLab X-ray diffractometer at ambient pressure and temperature. The data collection range was 2θ from 20° to 90° with a 0.02° step size and 1.3° min−1 rate. XRD measurements were conducted on NH3-reduced and steam-oxidized samples. To prepare the NH3-reduced samples, 0.07 g material was reduced by NH3 gas for 60 minutes and was carefully protected in a glove box filled with Ar due to their sensitivity to air. To prepare the steam-oxidized samples, the material was first reduced by NH3 for 60 minutes and then re-oxidized by H2O for 30 minutes and protected in Ar glove box. To keep the sample shielded from air oxidation, Kapton tape was used to protect the samples when taking the sample from the glove box to diffractometer.We used 0.07 g sample for XRD and XANES measurements rather than the 0.01 g used for looping performance experiments because 0.01 g sample gave too low signal-to-noise ratio in the characterization results. Also, the 60-minute duration of the reduction step gave stable concentrations of products (N2 and H2) for at least 10 minutes in the NH3 step, meaning the reduction of oxygen carrier was completed and the gas products were exclusively from NH3 decomposition. Similarly, the 30-minute duration of the re-oxidation step gave zero H2 product concentration for at least 10 minutes so that steam oxidation of the oxygen carrier was completed.
Refinement and Reference Intensity Ratio (RIR) methods in PDXL software were used to analyze the XRD data to quantify the phase compositions. Using chemical stoichiometry and molar mass of different solid phases, the molar ratio of oxygen to Fe plus Co can be calculated for each material at reduced and re-oxidized states. The difference in this ratio between reduced and re-oxidized samples shows the sample's oxygen exchange capacity normalized by the redox active metals (Fe and Co) molar amount (eqn (3)).
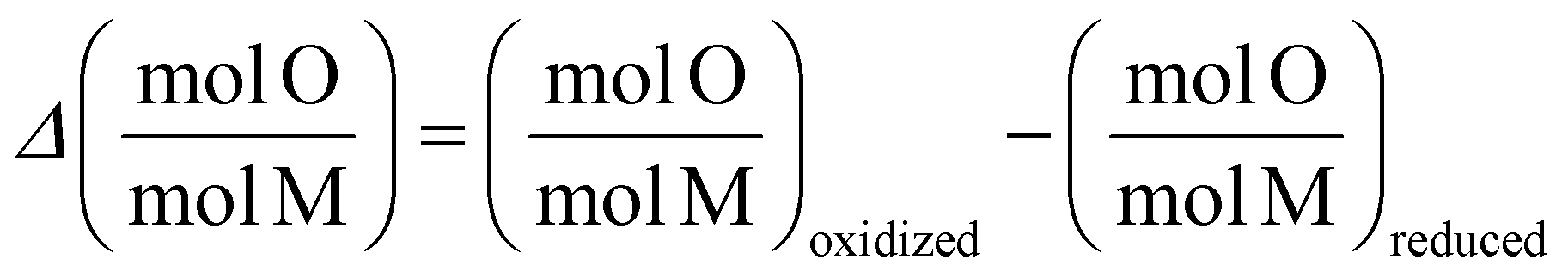 | (3) |
2.4. Material characterization using XANES
X-ray absorption near edge structure (XANES) was conducted by easyXAFS300+ to measure the change in the oxidation state of the Fe and Co. The X-ray source was set at 36 kV and 20 mA to acquire energy absorption spectra ranging from 7000 eV to 7600 eV for Fe K edge and from 7600 eV to 7900 eV for Co K edge. NH3-reduced and steam-oxidized samples (FeOx/YSZ and Fe0.5Co0.5Ox/YSZ) and standard materials (Co, CoO, Co3O4, Fe, Fe3O4, and CoFe2O4) were protected against air oxidation using Kapton tape. FeOx/YSZ and Fe0.5Co0.5Ox/YSZ samples were prepared in the same way as XRD samples. X-ray energy value at 0.5 normalized absorption was defined as the edge energy, and the edge energy of the reduced and re-oxidized samples relative to standard materials allowed us to determine the average oxidation states of Fe and Co in our samples. XANES data was analyzed by Athena software to give normalized absorption (μ) vs. absorption energy.552.5. Material characterization using SEM
Scanning Electron Microscopy (SEM) images were acquired using the Everhart–Thornley Detector (ETD) of a Thermo Fisher Scientific Apreo Scanning Electron Microscope set at 5 kV and 3.2 nA in the Secondary Electron (SE) mode. As-synthesized or cycled FeOx/YSZ and Fe0.5Co0.5Ox/YSZ were mixed in deionized water and sonicated at ambient temperature (25 °C) for 5 minutes, a 10 μL droplet of the mixture was placed on an SEM stub covered with carbon tape; afterwards, the stub was dried for 5 hours at 80 °C on a hot plate to remove water.3. Results and discussion
We conducted five-cycle CLAOD-WS experiments using 0.01 g FeOx/YSZ or Fe0.5Co0.5Ox/YSZ at 450, 500, 550, and 600 °C. Fig. 2a shows reactor outlet gas mixture profile during a 600 °C experiment using FeOx/YSZ at 6 LNH3 (gsolid h)−1. Initially, NH3 was mostly oxidized into H2O, and then when oxygen carrier capacity was used up, NH3 was only catalytically decomposed to H2 and N2. In water splitting, H2O re-oxidizes the oxygen carrier to produce additional H2 until the re-oxidation is complete. | ||
| Fig. 2 CLAOD-WS performance for different materials at different temperatures. Reactor outlet gas profiles using FeOx/YSZ at 600 °C with different step durations of (a) ΔtNH3 = 12 min and ΔtH2O = 3 min, and (b) ΔtNH3 = 5 min and ΔtH2O = 5 min. (c) NH3 and (d) H2O conversions dependence on temperature for Fe0.5Co0.5Ox/YSZ and FeOx/YSZ. “max” refers to equilibrium and “exp” refers to average over five-cycle CLAOD-WS experiments. Experiments in (a–d) had space velocities 6 LNH3 (gsolid h)−1 and 12 LH2O (gsolid h)−1. | ||
Space velocities and step durations can be adjusted to regulate the relative ratio of H2 produced from the two steps. As in Fig. 2a, the ratio of H2 from the NH3 and H2O steps was 7.43![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, while in Fig. 2b with shorter NH3 step and longer H2O step, this ratio became 2.13:1. Future research can involve materials chemistry adjustments and process optimizations to enhance high-purity H2 production from H2O in the CLAOD-WS system. Also, increasing the space velocity of the NH3 step from 6 to 30 LNH3 (gsolid h)−1 only led to minor decay in NH3 conversion from 99% to 95% for Fe0.5Co0.5Ox/YSZ and from 95% to 84% for FeOx/YSZ at 600 °C (Fig. S1 and S2†). Therefore, sufficiently high conversions were achieved at space velocities suitable for commercial use conditions (≥7.8 LNH3 (gsolid h)−1).56
:1, while in Fig. 2b with shorter NH3 step and longer H2O step, this ratio became 2.13:1. Future research can involve materials chemistry adjustments and process optimizations to enhance high-purity H2 production from H2O in the CLAOD-WS system. Also, increasing the space velocity of the NH3 step from 6 to 30 LNH3 (gsolid h)−1 only led to minor decay in NH3 conversion from 99% to 95% for Fe0.5Co0.5Ox/YSZ and from 95% to 84% for FeOx/YSZ at 600 °C (Fig. S1 and S2†). Therefore, sufficiently high conversions were achieved at space velocities suitable for commercial use conditions (≥7.8 LNH3 (gsolid h)−1).56
Experimental results from different temperatures are demonstrated in Fig. 2c, d, S1, and S2.† The solid lines indicate experimental results in Fig. 2c and d, while the dashed lines show thermodynamic equilibrium simulation results via FactSage software, giving the maximum possible conversions of NH3 and H2O. Fig. 2c shows an increasing trend in experimental NH3 conversion as the temperature increases due to kinetics enhancement. Meanwhile, water splitting is improved at higher temperatures (Fig. 2d), specifically in FeOx/YSZ, because both reaction kinetics and equilibrium H2O conversion are improved at elevated temperatures.
To measure the materials’ durability, 50-cycle CLAOD-WS were conducted at 600 °C and 30 LNH3 (gsolid h)−1 space velocity, the most demanding reaction condition in this paper. Over the 50 cycles, FeOx/YSZ had only slight decays of 1.3% and 0.9% in NH3 and H2O conversions, respectively (Fig. 3a). Similarly, in Fig. 3d, Fe0.5Co0.5Ox/YSZ gave slight decays of 1.6% and 0.6% in NH3 and H2O conversions, respectively. Scanning electron microscope (SEM), showed surface topology and particle size did not change after the 50-cycle experiments (Fig. 3b–f), indicating negligible agglomeration and sintering. Stability assessment of these materials in commercial reactors requires longer duration which is out of the scope of this study.
 | ||
| Fig. 3 CLAOD-WS durability and performance comparison with NH3 decomposition and thermochemical water splitting. (a) 50-cycle NH3 and H2O conversions using FeOx/YSZ at 600 °C, with 30 LNH3 (gsolid h)−1 for 12 min, 12 LH2O (gsolid h)−1 for 3 min. SEM images of (b) as-synthesized FeOx/YSZ, and (c) 50-cycled FeOx/YSZ. (d–f) are Fe0.5Co0.5Ox/YSZ results corresponding to (a–c). (g) H2 yield from NH3 in our 600 °C CLAOD-WS experiments (red star, only counting H2 from the NH3 step), compared to direct decomposition catalysts in literature ranging 450 °C to 700 °C (blue circle: Fe-based,23,57–69 black square: Co-based,66,70,71 orange triangle: Ni-based,72,73 brown triangle: mixed transition metals,21,22 green triangle: noble metal-based74–77). (h) H2 production in our 600 °C CLAOD-WS experiments (green, only counting H2 from the H2O step), compared to thermochemical water splitting in literature (orange: chemical reduction ranging 650 °C to 900 °C,36,78,79 red: thermal reduction28–30,33,37,80–82). | ||
Fig. 3g compares H2 yield from NH3 between chemical looping (only counting H2 from the NH3 step) and catalytic NH3 decomposition in literature. The looping scheme shows high H2 yields from the NH3 at a range of space velocities. Fig. 3h compares thermochemical water splitting in literature with our CLAOD-WS experiments in millimoles H2 production from H2O per gram of oxygen carrier (OC). FeOx/YSZ and Fe0.5Co0.5Ox/YSZ are among the best-performing water splitting oxygen carriers.
Other oxygen carriers and catalysts FeOx/Al2O3, Fe0.5Co0.5Ox/Al2O3, CoOx/YSZ, NiOx/YSZ, and Fe0.5Ni0.5Ox/YSZ were also synthesized and tested for CLAOD-WS (Fig. S3 and Table S1†). We found that Al2O3 is not as effective a support as YSZ. Also, Co and Ni both showed minimal water splitting performance due to limited redox capacity. Additionally, Co is a better NH3 conversion catalyst than Ni.
Material solid-phase transformation was studied by X-ray diffraction. At 450 °C, Fe0.5Co0.5Ox/YSZ was fully reduced by NH3 to iron–cobalt metallic alloy (Fig. 4a), and then oxidized by H2O to spinel phase while a portion of Co remained metallic (Fig. 4b), so the steam oxidation limited the redox capacity. With the same reaction conditions, FeOx/YSZ was partially reduced by NH3 to metallic iron with a spinel oxide phase remaining (Fig. 4c), and then oxidized by H2O to complete spinel (Fig. 4d), so the reduction reaction limited the redox capacity. At 600 °C, FeOx/YSZ was fully reduced by NH3 to metallic Fe (Fig. 4g); and then fully oxidized by H2O to spinel (Fig. 4h), giving complete phase transformation and thus high redox capacity. However, the Fe0.5Co0.5Ox/YSZ exhibited full reduction to metallic phase but still partial oxidation to spinel phase at 600 °C (Fig. 4e and f), similar to its behavior at 450 °C. Therefore, the elevated temperature more significantly enhanced the redox phase transformation of FeOx/YSZ than Fe0.5Co0.5Ox/YSZ, explaining their looping performance trend in Fig. 2d. Kapton tape was used to minimize XRD samples’ oxidation by air, so amorphous peaks at 2θ of 20° to 30° showed up.
 | ||
| Fig. 4 Solid-state phase transformation of materials for CLAOD-WS. XRD patterns of NH3-reduced and steam-oxidized samples: (a) reduced Fe0.5Co0.5Ox/YSZ, (b) re-oxidized Fe0.5Co0.5Ox/YSZ, (c) reduced FeOx/YSZ, and (d) re-oxidized FeOx/YSZ at 450 °C; (e–h) are results at 600 °C corresponding to (a–d). (i) Fe–O system, and (j) Fe–Co–O system phase diagrams, where red arrows indicate the equilibrium pO2 ranges between reduced and re-oxidized states according to simulation. (k) Redox ranges of FeOx/YSZ (blue) and Fe0.5Co0.5Ox/YSZ (red) at 450 °C and 600 °C, concluded from XRD refinement. | ||
The red arrows in Fig. 4i and j indicate the phase transition range at equilibrium conditions of NH3 reduction and H2O re-oxidation steps. For Fe–O system, at lower temperatures, the equilibrium phase transition is directly from Fe to Fe3O4, whereas at 600 °C the phase transition becomes Fe → FeO → Fe3O4. Fig. 4c showed that FeOx/YSZ was not fully reduced by NH3 to the metallic phase at 450 °C. Therefore, kinetic limitation prevented FeOx/YSZ from achieving a fully reduced state at 450 °C. For the Fe–Co–O system, after oxidation at both temperatures, a portion of Co remains in the metallic phase due to thermodynamic limitation, consistent with XRD results. Therefore, thermodynamic limitation prevents metallic Co from being completely oxidized by H2O and thus limits the redox capacity of Fe0.5Co0.5Ox/YSZ compared to FeOx/YSZ.
By XRD refinement, the oxygen-to-metal molar ratios were calculated for 450 °C and 600 °C looping, and are shown in Fig. 4k. At 450 °C, owing to the full reduction of Fe0.5Co0.5Ox/YSZ and its oxidation to mostly spinel phase, the redox capacity of Fe0.5Co0.5Ox/YSZ is significantly higher than that of FeOx/YSZ. At 600 °C, full reduction of FeOx/YSZ to metallic phase followed by full oxidation to spinel phase resulted in its higher oxygen capacity than Fe0.5Co0.5Ox/YSZ. These are consistent with their water splitting capacity performance (Fig. 2d). The redox capacity of Fe0.5Co0.5Ox/YSZ is not significantly different between 450 °C and 600 °C based on XRD refinement, so X-ray Absorption Near Edge Structure (XANES) was utilized to further interpret each element's redox behavior.
XANES was conducted to analyze oxidation states of Fe and Co. Fig. 5a and b demonstrate Co K edge XANES spectra of NH3-reduced and steam-oxidized Fe0.5Co0.5Ox/YSZ at 450 °C and 600 °C, along with standard materials Co, CoO, and Co3O4. Comparing the edge energy shifts at the normalized absorption of 0.5 gave that at 450 °C, Co underwent an oxidation state variation of 1.49; whereas at 600 °C, Co underwent a much lower oxidation state change of 0.71, proving that Co becomes less redox active at the higher temperature of 600 °C. Fig. 5c and d show the Fe edge XANES results of reduced and re-oxidized Fe0.5Co0.5Ox/YSZ along with standard materials of Fe, Fe3O4, and CoFe2O4. We found that at 450 °C Fe underwent a smaller oxidation state variation than at 600 °C. The oxidation state changes of Co and Fe (Tables S2, S3, and S4, and Fig. S6†) were then weight-averaged based on their mole fractions to result in the overall metal redox capacity of Fe0.5Co0.5Ox/YSZ at 450 °C and 600 °C in Table 1. Additionally, Fig. 5e and f show the Fe redox of FeOx/YSZ. The order of overall oxidation state variation in Table 1 agreed with those of water splitting capacity (Fig. 2d) and oxygen capacity derived from XRD phase transformation (Fig. 4k). At 450 °C, Fe0.5Co0.5Ox/YSZ is a more redox-active material, while at 600 °C FeOx/YSZ becomes more redox-active.
 | ||
| Fig. 5 Simultaneous redox reactions of Fe and Co for CLAOD-WS. XANES results for Co and Fe between NH3-reduced and steam-oxidized states: Co K edge of Fe0.5Co0.5Ox/YSZ at (a) 450 °C and (b) 600 °C, Fe K edge of Fe0.5Co0.5Ox/YSZ at (c) 450 °C and (d) 600 °C, and Fe K edge of FeOx/YSZ at (e) 450 °C and (f) 600 °C. | ||
| T [°C] | Overall averaged oxidation state variation per mol of active metals (Fe & Co) | |
|---|---|---|
| FeOx/YSZ | Fe0.5Co0.5Ox/YSZ | |
| 450 | 1.31 | 1.59 |
| 600 | 2.86 | 1.86 |
As discussed earlier, the materials for CLAOD-WS play the dual-role of oxygen carriers and catalysts. XRD and XANES characterizations of NH3-reduced and H2O-oxidized samples illustrate their catalytic activity. For 600 °C experiments, XRD results in Fig. 4e and g show that the FeOx/YSZ and Fe0.5Co0.5Ox/YSZ are fully reduced in NH3 step to metal phases of Fe and Fe–Co alloy, while the support material YSZ remained unchanged in crystal structure. The XANES experiments (Fig. 5b, d, and f) also show the fully reduced metal phases. Combining the results of XRD and XANES, the metal phases are the active sites for NH3 decomposition that continues beyond the NH3 oxidation reaction. High conversions of NH3 at 600 °C (99% for Fe0.5Co0.5Ox/YSZ and 95% for FeOx/YSZ) show that metal phases are highly catalytically active for NH3 decomposition at 600 °C. Additionally, we tested as-synthesized and H2-reduced samples for NH3 conversion and observed that H2-reduced samples reach their maximum NH3 conversion faster than as-synthesized oxides (Fig. S5†), indicating that after reduction of oxides to metal phases, NH3 conversion is significantly enhanced due to catalytic activity of these active metal sites.
In comparison, at 450 °C, Fig. 4a shows that Fe0.5Co0.5Ox/YSZ is fully reduced to Fe–Co metal phase and this phase is responsible for catalytic NH3 decomposition; however, due to the lower temperature at 450 °C, NH3 conversion is relatively low (20%). Fig. 4c shows that FeOx/YSZ is partly reduced to metal Fe phase mixed with Fe3O4 spinel phase, resulting in a significantly lower NH3 conversion of 5% at 450 °C. The XANES experiments for 450 °C samples also agreed with the XRD results. For FeOx/YSZ, Fig. 5e showed that the oxidation state of Fe is very close to that of Fe3O4. For Fe0.5Co0.5Ox/YSZ sample, Fig. 5a (Co edge) and Fig. 5c (Fe edge) show that Co and Fe are both reduced to metal phases which are the active phases for NH3 conversion.
Energy and techno-economic analyses were conducted to assess two systems: (1) Chemical looping ammonia oxidation and decomposition integrated with water splitting (CLAOD-WS) (Fig. 6a), and (2) Catalytic ammonia decomposition (CAD) (Fig. 6b). Major assumptions for both scenarios are: NH3 conversion is 90%, steam conversion is 40%, reaction temperature is 600 °C, total pressure is 1 atm (except for the PSA where the compressor increases the pressure to 700 kPa), heat exchanger effectiveness is 95%, total H2 production rate is 10 metric tons per day, and electricity serves as the energy input and is converted to heat when needed and the cost of electricity is assumed to be $0.053 per kWhe (following utility-scale PV-plus-battery system, class 5, moderate case, market, PTC + ITC, 2030).83 For CLAOD-WS, 5 tons per day of H2 comes out of each of NH3 reaction and steam reaction. For CAD, 5 tons per day of H2 is purified in the separation step, where two types of H2 separation technologies were analyzed: PSA and Pd-Ag membrane supported on ceramic material. Base case membrane separation has a Second Law efficiency of 20%. The redox or catalyst material is assumed to be replaced 4 times per year,84 and we find that the replacement frequency varying from once per year to once per two weeks only changes LCOH by less than 2% for all cases in this work.
 | ||
| Fig. 6 Process schematics with major model parameters of (a) chemical looping ammonia oxidation and decomposition integrated with water splitting (CLAOD-WS), and (b) catalytic ammonia decomposition (CAD). MTPH: metric ton per hour. | ||
In CLAOD-WS, the NH3 step is the main thermal energy consumer (2.965 MWth). In CAD, although the chemical reactor is less energy intensive (2.207 MWth), the H2 separation requires an energy input of 0.237 MWe for the membrane and 2.190 MWe for the PSA. Overall, the CLAOD-WS system is 22.5% more energy-consuming than the membrane-based CAD system and 29.1% less energy-consuming than the PSA-based CAD system.
As Fig. 7a shows, notably, CLAOD-WS process achieves cost savings of 52% and 86% in energy and equipment expenses compared to CAD plants utilizing PSA and membrane systems, respectively. The CLAOD-WS plant requires an annual cost of $8.64 M, 51.8% lower than the CAD plant equipped with membrane separation and 14.8% lower than the CAD plant equipped with PSA separation. Accordingly, the LCOH is $2.5 per kg-H2 in CLAOD-WS, $5.1 per kg-H2 in CAD with membrane, and $2.9 per kg-H2 in CAD with PSA. LCOH refers to the cost per kilogram of H2 production from both NH3 and H2O steps in CLAOD-WS. In addition, thermal energy release in CLAOD-WS can be used to produce hot water byproduct, whose revenue is not included in our economic analysis. Fig. 7b compares the LCOH and total energy consumption rate in the CAD as a function of the membrane's H2 separation Second Law efficiency, showing the impact of membrane performance.
 | ||
| Fig. 7 (a) Annual cost and levelized cost of hydrogen (LCOH) for CLAOD-WS and for CAD equipped with pressure swing absorption (PSA) or Pd-Ag/ceramic membrane separation. (b) Dependence of LCOH and total system energy consumption rate on H2 separation membrane Second Law efficiency in CAD process with Pd-Ag/ceramic membrane separation. | ||
4. Conclusions
We have proposed and studied integrated chemical looping NH3 oxidation and decomposition with water splitting (CLAOD-WS) as a promising novel pathway toward H2 production from NH3 and H2O. FeOx/YSZ and Fe0.5Co0.5Ox/YSZ each played the dual roles of oxygen carrier and catalyst. Looping experiments at 450 to 600 °C and space velocities of 6, 18, and 30 LNH3 (gsolid h)−1 and 12 LH2O (gsolid h)−1 were conducted. XRD coupled with phase diagrams illustrated that FeOx/YSZ is kinetically limited from achieving full reduction at 450 °C, while Fe0.5Co0.5Ox/YSZ is thermodynamically limited from complete oxidation under H2O. Moreover, XRD refinements and XANES results showed the Fe and Co redox activities that explain the overall redox capacity of Fe0.5Co0.5Ox/YSZ and FeOx/YSZ at different temperatures, consistent with the experimental water splitting capacity. Durability of the materials was shown by 50-cycle CLAOD-WS experiments at 600 °C and 30 LNH3 (gsolid h)−1. Lastly, energy and techno-economic analysis of the CLAOD-WS and catalytic NH3 decomposition systems indicated that the looping scheme can significantly reduce energy consumption and cost mainly due to the intrinsic H2 purification in the looping scheme.Data availability
Further information on materials and code used in the work are available from the lead contact upon reasonable request. Crystallographic data used for XRD data analysis has been deposited at the ICSD and can be obtained from https://icsd.products.fiz-karlsruhe.de/en. Additional data used in this study is available in the ESI.†Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The authors acknowledge Center for Electron Microscopy and Analysis (CEMAS) and Surface Analysis Laboratory at The Ohio State University (OSU). The research is partly supported by OSU President's Research Excellence Program. A. Arjomand is partly supported by US Department of Energy DE-EE0010733. K. Shank is supported by Distinguished University Fellowship and ENGIE-Axium Fellowship at OSU.References
- S. Ristig, M. Poschmann, J. Folke, O. Gómez-Cápiro, Z. Chen, N. Sanchez-Bastardo, R. Schlögl, S. Heumann and H. Ruland, Chem. Ing. Tech., 2022, 94, 1413–1425 CrossRef CAS.
- B. Modu, M. P. Abdullah, A. L. Bukar and M. F. Hamza, Int. J. Hydrogen Energy, 2023, 48, 38354–38373 CrossRef CAS.
- A. Arjomand Kermani and E. Houshfar, Int. J. Hydrogen Energy, 2024, 52, 177–189 CrossRef.
- I. Lucentini, X. Garcia, X. Vendrell and J. Llorca, Ind. Eng. Chem. Res., 2021, 18560–18611 CrossRef CAS.
- A. Pal, S. Kakran, A. Kumar, A. B. Youssef, U. P. Singh and A. Sidhu, Int. J. Hydrogen Energy, 2024, 49, 16–41 CrossRef CAS.
- M. Koleva and N. Rustagi, Hydrogen Delivery and Dispensing Cost – DOE Hydrogen and Fuel Cells Program Record.
- J. W. Sheffield, K. B. Martin and R. Folkson, in Alternative Fuels and Advanced Vehicle Technologies for Improved Environmental Performance, ed. R. Folkson, Woodhead Publishing, 2014, pp. 117–137 Search PubMed.
- Physical Hydrogen Storage – Hydrogen and Fuel Cell Technologies Office, https://energy.gov/eere/fuelcells/physical-hydrogen-storage.
- L. Mulky, S. Srivastava, T. Lakshmi, E. R. Sandadi, S. Gour, N. A. Thomas, S. Shanmuga Priya and K. Sudhakar, Mater. Chem. Phys., 2024, 325, 129710 CrossRef CAS.
- F. Dawood, M. Anda and G. M. Shafiullah, Int. J. Hydrogen Energy, 2020, 45, 3847–3869 CrossRef CAS.
- M. F. Platzer and N. Sarigul-Klijn, in The Green Energy Ship Concept: Renewable Energy from Wind Over Water, ed. M. F. Platzer and N. Sarigul-Klijn, Springer International Publishing, Cham, 2021, pp. 71–72 Search PubMed.
- A. Valera-Medina, H. Xiao, M. Owen-Jones, W. I. F. David and P. J. Bowen, Prog. Energy Combust. Sci., 2018, 69, 63–102 CrossRef.
- A. Roy, S. Sen Gupta, A. Samanta, P. V. S. Sai Likhith and S. K. Das, Int. J. Hydrogen Energy, 2024, 71, 131–142 CrossRef CAS.
- N. Othman, H. Salleh and H. Purwanto, Procedia Chem., 2016, 119–124 CrossRef CAS.
- R. Lan, J. T. S. Irvine and S. Tao, Int. J. Hydrogen Energy, 2012, 37, 1482–1494 CrossRef CAS.
- J. Bartels, A Feasibility Study of Implementing an Ammonia Economy, Iowa State University, 2008 Search PubMed.
- L. Zhai, S. Liu and Z. Xiang, Ind. Chem. Mater., 2023, 1, 332–342 RSC.
- O. Ojelade and S. Zaman, Chem. Pap., 2020, 57–65 Search PubMed.
- F. Schuth, R. Palkovits, R. Schlogl and D. S. Su, Energy Environ. Sci., 2011, 6278–6289 Search PubMed.
- Z. Hu, C. Weng, C. Chen and Z. Yuan, Appl. Catal., A, 2018, 49–57 CrossRef CAS.
- H. Tabassum, S. Mukherjee, J. Chen, D. Holiharimanana, S. Karakalos, X. Yang, S. Hwang, T. Zhang, B. Lu, M. Chen, Z. Tang, E. A. Kyriakidou, Q. Ge and G. Wu, Energy Environ. Sci., 2022, 15, 4190–4200 RSC.
- P. Xie, Y. Yao, Z. Huang, Z. Liu, J. Zhang, T. Li, G. Wang, R. Shahbazian-Yassar, L. Hu and C. Wang, Nat. Commun., 2019, 10, 4011 CrossRef PubMed.
- Y.-Q. Gu, Z. Jin, H. Zhang, R.-J. Xu, M.-J. Zheng, Y.-M. Guo, Q.-S. Song and C.-J. Jia, J. Mater. Chem. A, 2015, 3, 17172–17180 RSC.
- L. Yao, T. Shi, Y. Li, J. Zhao, W. Ji and C.-T. Au, Catal. Today, 2011, 164, 112–118 CrossRef CAS.
- F. Normann, A. O. Wismer, C. R. Müller and H. Leion, Fuel, 2019, 57–63 CrossRef CAS.
- M. Cheng, F. Normann, D. Zhao, Z. Li, N. Cai and H. Leion, Energy Fuels, 2015, 8126–8134 CrossRef CAS.
- C. Ruan, X. Wang, C. Wang, L. Zheng, L. Li, J. Lin, X. Liu, F. Li and X. Wang, Nat. Commun., 2022, 1–12 Search PubMed.
- S. Zhai, J. Rojas, N. Ahlborg, K. Lim, M. F. Toney, H. Jin, W. C. Chueh and A. Majumdar, Energy Environ. Sci., 2018, 2172–2178 RSC.
- X. Qian, J. He, E. Mastronardo, B. Baldassarri, C. Wolverton and S. M. Haile, Chem. Mater., 2020, 32, 9335–9346 CrossRef CAS.
- Y. Mao, Y. Gao, W. Dong, H. Wu, Z. Song, X. Zhao, J. Sun and W. Wang, Appl. Energy, 2020, 267, 114860 CrossRef CAS.
- A. Pérez, M. Orfila, M. Linares, R. Sanz, J. Marugán, R. Molina and J. A. Botas, Catal. Today, 2022, 390–391, 22–33 CrossRef.
- X. Li, X. Sun, Q. Song, Z. Yang, H. Wang and Y. Duan, Int. J. Hydrogen Energy, 2022, 47, 33619–33642 CrossRef CAS.
- C. Bu, T. Gu, S. Cen, D. Liu, J. Meng, C. Liu, X. Wang, H. Xie, J. Zhang and G. Piao, Int. J. Hydrogen Energy, 2023, 48, 12227–12239 CrossRef CAS.
- V. K. Budama, J. P. Rincon Duarte, M. Roeb and C. Sattler, Sol. Energy, 2023, 249, 353–366 CrossRef CAS.
- B. Ghorbani, S. Zendehboudi, Y. Zhang, H. Zarrin and I. Chatzis, Energy Convers. Manage., 2023, 297, 117599 CrossRef CAS.
- Y. Liu, T. Gu, C. Bu, D. Liu and G. Piao, Int. J. Hydrogen Energy, 2024, 62, 1077–1088 CrossRef CAS.
- A. M. Huerta-Flores, F. Torre, M. Taeño, R. C. Barreno, E. Palomo Del Barrio and S. Doppiu, Int. J. Hydrogen Energy, 2024, 71, 1293–1302 CrossRef CAS.
- J. T. Tran, K. J. Warren, D. Mejic, R. L. Anderson, L. Jones, D. S. Hauschulz, C. Wilson and A. W. Weimer, Joule, 2023, 7, 1759–1768 CrossRef CAS.
- E. Gager, M. Frye, D. McCord, J. Scheffe and J. C. Nino, Int. J. Hydrogen Energy, 2022, 47, 31152–31164 CrossRef CAS.
- D. Zhang, H. A. De Santiago, B. Xu, C. Liu, J. A. Trindell, W. Li, J. Park, M. A. Rodriguez, E. N. Coker, J. D. Sugar, A. H. McDaniel, S. Lany, L. Ma, Y. Wang, G. Collins, H. Tian, W. Li, Y. Qi, X. Liu and J. Luo, Chem. Mater., 2023, 35, 1901–1915 CrossRef CAS.
- G. L. Schieber, E. B. Stechel, A. Ambrosini, J. E. Miller and P. G. Loutzenhiser, Int. J. Hydrogen Energy, 2017, 42, 18785–18793 CrossRef CAS.
- B. Bulfin, M. Miranda and A. Steinfeld, Front. Energy Res., 2021, 9, 677980 CrossRef.
- R. B. Wexler, G. Sai Gautam, R. T. Bell, S. Shulda, N. A. Strange, J. A. Trindell, J. D. Sugar, E. Nygren, S. Sainio, A. H. McDaniel, D. Ginley, E. A. Carter and E. B. Stechel, Energy Environ. Sci., 2023, 16, 2550–2560 RSC.
- H. N. N. Tran, W. Li and X. Liu, Chem. Eng. J., 2024, 500, 156613 CrossRef CAS.
- L.-S. Fan and F. Li, Ind. Eng. Chem. Res., 2010, 49, 10200–10211 CrossRef CAS.
- L.-S. Fan, Chemical looping partial oxidation: gasification, reforming, and chemical syntheses, Cambridge University Press, 2017 Search PubMed.
- X. Zhu, Q. Imtiaz, F. Donat, C. R. Müller and F. Li, Energy Environ. Sci., 2020, 13, 772–804 RSC.
- Z. Du, C. Liu, J. Zhai, X. Guo, Y. Xiong, W. Su and G. He, Catalysts, 2021, 11, 393 CrossRef CAS.
- I. Staffell, D. Scamman, A. Velazquez Abad, P. Balcombe, P. E. Dodds, P. Ekins, N. Shah and K. R. Ward, Energy Environ. Sci., 2019, 12, 463–491 RSC.
- K. Wróbel, J. Wróbel, W. Tokarz, J. Lach, K. Podsadni and A. Czerwiński, Energies, 2022, 15, 8937 CrossRef.
- S. Yun, J. Im, J. Kim, H. Cho and J. Lee, Chem. Eng. J., 2024, 491, 151875 CrossRef CAS.
- M. Nordio, S. A. Wassie, M. Van Sint Annaland, D. A. Pacheco Tanaka, J. L. Viviente Sole and F. Gallucci, Int. J. Hydrogen Energy, 2021, 46, 23417–23435 CrossRef CAS.
- C. Makhloufi and N. Kezibri, Int. J. Hydrogen Energy, 2021, 46, 34777–34787 CrossRef CAS.
- S. Devkota, J.-Y. Cha, B.-J. Shin, J.-H. Mun, H. C. Yoon, S. A. Mazari and J.-H. Moon, Appl. Energy, 2024, 358, 122605 CrossRef CAS.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed.
- H. F. Rase and J. R. Holmes, Chemical reactor design for process plants, Wiley, New York, 1977 Search PubMed.
- X. Duan, G. Qian, X. Zhou, Z. Sui, D. Chen and W. Yuan, Appl. Catal., B, 2011, 101, 189–196 CrossRef CAS.
- A.-H. Lu, J.-J. Nitz, M. Comotti, C. Weidenthaler, K. Schlichte, C. W. Lehmann, O. Terasaki and F. Schüth, J. Am. Chem. Soc., 2010, 132, 14152–14162 CrossRef CAS PubMed.
- M. Itoh, M. Masuda and K.-I. Machida, Mater. Trans., 2002, 43, 2763–2767 CrossRef CAS.
- T. Otremba, N. Frenzel, M. Lerch, T. Ressler and R. Schomäcker, Appl. Catal., A, 2011, 392, 103–110 CrossRef CAS.
- Z.-P. Hu, L. Chen, C. Chen and Z.-Y. Yuan, Mol. Catal., 2018, 455, 14–22 CrossRef CAS.
- X.-C. Hu, W.-W. Wang, Z. Jin, X. Wang, R. Si and C.-J. Jia, J. Energy Chem., 2019, 38, 41–49 CrossRef.
- H. Tüysüz, F. Schüth, L. Zhi, K. Müllen and M. Comotti, ChemCatChem, 2015, 7, 1453–1459 CrossRef.
- B. Lorenzut, T. Montini, M. Bevilacqua and P. Fornasiero, Appl. Catal., B, 2012, 125, 409–417 CrossRef CAS.
- W. Arabczyk and J. Zamlynny, Catal. Lett., 1999, 60, 167–171 CrossRef CAS.
- H. Zhang, Y. A. Alhamed, Y. Kojima, A. A. Al-Zahrani and L. A. Petrov, Proceedings of the Bulgarian Academy of Sciences, 2013, 519–524 Search PubMed.
- K. F. Ortega, D. Rein, C. Lüttmann, J. Heese, F. Özcan, M. Heidelmann, J. Folke, K. Kähler, R. Schlögl and M. Behrens, ChemCatChem, 2017, 9, 659–671 CrossRef CAS.
- L. Li, Q. Meng, W. Ji, J. Shao, Q. Xu and J. Yan, Mol. Catal., 2017, 442, 147–153 CrossRef CAS.
- Y. Yuan, Science, 2022, 889–893 CrossRef PubMed.
- C. Huang, Y. Yu, X. Tang, Z. Liu, J. Zhang, C. Ye, Y. Ye and R. Zhang, Appl. Surf. Sci., 2020, 532, 147335 CrossRef CAS.
- S. Podila, H. Driss, S. F. Zaman, Y. A. Alhamed, A. A. AlZahrani, M. A. Daous and L. A. Petrov, J. Mol. Catal. A: Chem., 2016, 414, 130–139 CrossRef CAS.
- K. Okura, K. Miyazaki, H. Muroyama, T. Matsui and K. Eguchi, RSC Adv., 2018, 8, 32102–32110 RSC.
- H. Liu, H. Wang, J. Shen, Y. Sun and Z. Liu, Appl. Catal., A, 2008, 337, 138–147 CrossRef CAS.
- S.-F. Yin, B.-Q. Xu, C.-F. Ng and C.-T. Au, Appl. Catal., B, 2004, 48, 237–241 CrossRef CAS.
- P. F. Ng, L. Li, S. Wang, Z. Zhu, G. Lu and Z. Yan, Environ. Sci. Technol., 2007, 3758–3762 CrossRef CAS PubMed.
- R. Antunes, R. Steiner, L. Marot and E. Meyer, Int. J. Hydrogen Energy, 2022, 47, 14130–14140 CrossRef CAS.
- B. Bahrami, V. G. Komvokis, M. S. Ziebarth, O. S. Alexeev and M. D. Amiridis, Appl. Catal., B, 2013, 130–131, 25–35 CrossRef CAS.
- Y. Qiu, S. Zhang, D. Cui, M. Li, J. Zeng, D. Zeng and R. Xiao, Appl. Energy, 2019, 252, 113454 CrossRef CAS.
- Y. Zheng, K. Li, H. Wang, X. Zhu, Y. Wei, M. Zheng and Y. Wang, Energy Fuels, 2016, 30, 638–647 CrossRef CAS.
- K. Zheng, Z. Yu, S.-C. Tan, T. Liu and H. Kong, Energy Convers. Manage., 2024, 303, 118116 CrossRef CAS.
- A. Haeussler, S. Abanades, A. Julbe, J. Jouannaux, M. Drobek, A. Ayral and B. Cartoixa, Chem. Eng. Res. Des., 2020, 156, 311–323 CrossRef CAS.
- M. Orfila, D. Sanz, M. Linares, R. Molina, R. Sanz, J. Marugán and J.Á Botas, Int. J. Hydrogen Energy, 2021, 46, 17458–17471 CrossRef CAS.
- Annual Technology Baseline – Utility Scale PV plus Battery, https://atb.nrel.gov/electricity/2024/utility-scale_pv-plus-battery.
- S. Zhai, Iron-based Oxides for Thermochemical Splitting of Water and Carbon Dioxide, Stanford University, 2020 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental methods, data analysis methods and results, additional looping performance data, equilibrium simulation method, energy and technoeconomic analysis methods. See DOI: https://doi.org/10.1039/d5gc00236b |
| This journal is © The Royal Society of Chemistry 2025 |