
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA fundamental study of lignin reactions with formaldehyde and glyoxal†
Mohsen
Siahkamari‡
*a,
Debkumar
Debnath‡
b,
Tuo
Wang
 *b and
Mojgan
Nejad
*ac
*b and
Mojgan
Nejad
*ac
aDepartment of Forestry, Michigan State University, 480 Wilson Rd, East Lansing, MI 48824, USA. E-mail: nejad@msu.edu
bDepartment of Chemistry, Michigan State University, 578 S. Shaw Lane, East Lansing, MI 48224, USA. E-mail: wangtuo1@msu.edu
cDepartment of Chemical Engineering and Materials Science, 428 S. Shaw Lane, Michigan State University, East Lansing, MI 48224, USA
First published on 3rd February 2025
Abstract
This study investigates the synthesis and characterization of lignin-formaldehyde (LF) and fully biobased lignin-glyoxal (LG) resins as alternatives to the phenol-formaldehyde (PF) adhesive currently used in the manufacturing of plywood and oriented strand boards (OSB). In this process, phenol was entirely replaced by a commercially available kraft-softwood lignin, while formaldehyde was substituted with glyoxal (a biobased dialdehyde) in the LG resins. Additionally, lignin monomers were used as model compounds to better understand the behavior of lignin in LF and LG resins. The reactions of phenol, lignin monomers, and commercial lignin with formaldehyde and glyoxal were investigated through Fourier transform infrared (FT-IR) and nuclear magnetic resonance (NMR) spectroscopy in both solution and solid states. The results confirmed the successful integration of formaldehyde and glyoxal into phenolic structures, leading to the formation of methylene and glyoxylene linkages during resin synthesis. This process created a robust three-dimensional network, as evidenced by 2D 13C–13C correlation solid-state NMR spectra and FT-IR analyses, which are crucial in studying the structural properties of the cured thermoset solid resins that are insoluble in NMR solvents. These findings highlight the innovative potential of lignin as a renewable alternative to petroleum-based phenol and emphasize the practical applications of a fully biobased lignin-glyoxal adhesive in the production of greener, more sustainable, and formaldehyde-free plywood and OSB wood panels commonly used in building construction.
Green foundation1. A fully biobased wood adhesive was developed by replacing phenol with kraft-softwood lignin and formaldehyde with glyoxal (a biobased dialdehyde).2. Solid-state NMR was employed to confirm the formation of glyoxylene linkages in the cured lignin-glyoxal resins. 3. Future studies can focus on the recyclability and reusability of the green plywood made with this biobased resin. |
Introduction
Adhesives have played a pivotal role in enhancing the efficient utilization of wood resources.1 A wood adhesive can interact with the wood's surface through physical and/or chemical bonding. This interaction effectively transfers stresses between bonded components, ensuring the adhesive creates strong bonds with wood without detachment or rupture.2 Formaldehyde-based adhesives, categorized as thermosetting polymers, have significantly contributed to the advancement and utilization of various wood-based products.3 Phenol-formaldehyde (PF) polymers, developed in the early 20th century, are among the earliest synthetic polymers.4–6 Their widespread use in laminations and composites is due to their exceptional durability, strong adhesion to wood, robust polymer strength, and chemical stability.5 PF adhesives meet most wood bonding needs when cost and hot curing time are not primary concerns.6–8 These adhesives are synthesized by reacting phenol with formaldehyde or a formaldehyde precursor under specific conditions, resulting in a resin that can further polymerize during the setting phase. There are two primary categories of pre-polymers: novolacs produced under acidic conditions, with a formaldehyde/phenol (F/P) ratio below 1, and resole resins, produced under basic conditions, with F/P ratios above 1.9–11 In wood adhesive applications, resole resins are preferred due to their solubility, excellent wood penetration, and ability to delay curing until activated by heat, allowing ample time for assembly.6Resole resins are produced using alkali hydroxides, an F/P ratio of 1–3, and a pH between 7–13. Fig. S1† shows the chemical reaction of base-activated phenol with formaldehyde. In basic conditions, formaldehyde adds to phenol quickly, but hydroxymethyl derivatives transform into oligomers slowly. This allows higher formaldehyde concentrations without forming the final polymer until sufficient heat is applied during press time.6,7
Although resole resins are widely used for their durability and excellent water resistance properties, concerns are raised due to the toxicity of phenol and formaldehyde and the fact that both are derived from petrochemicals.12–14 Replacing petrochemicals with renewable alternatives can address these challenges.8,15 Our previous study used two bio-based raw materials to replace phenol and formaldehyde and developed a fully bio-based adhesive using lignin and glyoxal.16 This study focuses on investigating the reactions of lignin with formaldehyde and glyoxal in liquid—and solid-cured resins.16
Lignin is the most abundant natural aromatic biopolymer, polymerized and synthesized from three hydroxycinnamyl alcohol monomers: p-coumaryl alcohol (H), coniferyl alcohol (G), and sinapyl alcohol (S), as illustrated in Fig. S2.†17–21 This results in a disordered, three-dimensional polymer network coexisting with various polysaccharides within the secondary plant cell wall.22–26 Lignin composition varies among plant species, tissues, and cell types.27–30 Ecological factors like tree age, climate, nutrients, and sunlight influence this diversity.31,32 Hardwood lignin mainly contains G and S units, with few H units. Softwood lignin is primarily G units with fewer H units.27,33 Annual crops have similar G and S units and a higher proportion of H units than softwoods and hardwoods.27,34,35
Glyoxal, the simplest dialdehyde, is recognized for its non-volatility and biodegradability, making it a promising formaldehyde substitute in phenolic adhesives.16 Glyoxal is known to have much lower toxicity compared to formaldehyde.36 Formaldehyde is classified as a carcinogen, mutagen, and reprotoxic substance by the European Chemicals Agency (ECHA) and has an LD50 value ranging from 500 to 800 mg kg−1,37 significantly lower amount than glyoxal, which has an LD50 between 2960 and 8979 mg kg−1.38 Glyoxal is typically found in a 40 wt% aqueous solution and naturally exists in many foods.36,39,40 It acts as a cross-linker or precursor in paper, textiles, and leather applications.36,39,40 Glyoxal's water solubility allows it to equilibrate between mono- and di-hydrated states and form dimers and small oligomers through reversible condensation, depending on concentration, as illustrated in Fig. S3.†36
While lignin and glyoxal may initially seem to have similar active functional groups to phenol and formaldehyde, they have significant differences. Lignin is solid, requiring a solvent for dissolution, and has lower reactivity due to its complex structure and significantly fewer reactive sites than phenol. In addition, glyoxal is less reactive and more prone to side reactions than formaldehyde.16 These differences significantly impact resin synthesis and properties.16 Therefore, a comprehensive structural investigation of the resulting resins using advanced analytical techniques is essential to better understand the reaction of lignin with glyoxal, compared with the reaction of lignin with formaldehyde.
The synthesis of alkali-catalyzed resole PF resin involves a two-stage reaction. Initially, phenolate/enolate intermediates form, creating resonance-stabilized ions with high negative charge density at the 2-, 4-, and 6-positions of phenol. Subsequently, methylol formation occurs through the nucleophilic attack of the anion on the formaldehyde carbonyl. In the second stage, increased temperature causes resoles to condense, forming methylene bridges through a quinone methide intermediate, as illustrated in Fig. S4.†41
A key difference between lignin and phenol in producing phenolic adhesives is lignin's side chain at the para position of the phenolic hydroxyl group, hindering methylol or methylene bridge formation. This prevents reactions at the para position, which is usually more reactive than the ortho sites.4 Furthermore, the substitution of formaldehyde with glyoxal changes the resulting compounds. During the methylolation process, formaldehyde is introduced into an alkaline lignin solution, leading to three distinct reactions (Fig. 1). The primary reaction involves the Lederer–Manasse reaction, where hydroxymethyl groups (–CH2OH) become part of the C5 position of the aromatic rings in lignin.42 However, concurrent side reactions may occur, such as the Cannizzaro reaction, in which two formaldehyde molecules react together, forming formic acid and methanol. Another side reaction, known as the Tollens reaction, replaces lignin's side chains with aliphatic methylol groups.36 Glyoxalation involves introducing glyoxal into lignin's ortho positions, glyoxal's Cannizzaro reaction, and substituting glyoxal at lignin's side chains.36,43
 | ||
| Fig. 1 Reactions occurring during methylolation (left) and glyoxalation (right) of Lignin. (a) Linder-Manasse reaction, (b) Self-Cannizzaro reaction, (c) Tollen's reaction. | ||
A crucial element in the synthesis of phenolic resole resins involves preventing aldehyde side reactions, with a particular focus on avoiding the Cannizzaro reaction. Aldehydes lacking alpha hydrogen tend to undergo a disproportionation reaction in an alkaline setting, forming alcohol and carboxylic acid compounds.44 Glyoxal, with its two neighboring carbonyl groups, readily undergoes an intramolecular Cannizzaro reaction in an alkaline solution, as depicted in Fig. 2b.36 This reaction can occur even under relatively mild alkaline conditions, leading to the production of glycolic acid.36 Since glyoxal can undergo the Cannizzaro reaction within a single molecule without the need for two aldehyde molecules, the potential for glyoxal's side reaction exceeds that of formaldehyde. Additionally, the proximity of the two carbonyl groups with partial positive charges makes glyoxal more susceptible to this side reaction.16 The production of glycolic acid leads to a decline in the resin's pH level.16 If the resin's alkalinity is not adequately controlled, this pH decrease can interfere with the electrophilic substitution reactions between lignin and glyoxal during the synthesis of phenolic resole resin. Conversely, excessive alkalinity can adversely affect the adhesive's adhesion performance, especially when exposed to moisture. Therefore, it becomes crucial to control the pH of the reaction media to achieve optimal performance.45
 | ||
| Fig. 2 Proposed mechanism of the self-Cannizzaro reaction of (a) Formaldehyde, (b) Glyoxal.16 | ||
Despite extensive research aimed at elucidating the reaction mechanism of lignin with formaldehyde and glyoxal, particularly formaldehyde, there is a gap in the systematic investigation into the reactions of lignin and its monomers with formaldehyde and glyoxal, especially post-curing (solid resin).
This study aimed to investigate the reaction mechanisms of commercial kraft softwood lignin with formaldehyde and glyoxal to determine the potential formation of methylene and glyoxylene linkages during the reaction. For this purpose, phenol-formaldehyde and phenol-glyoxal resins were synthesized, and phenol was substituted with commercial lignin. Formaldehyde and glyoxal resins were also prepared using H and G lignin monomers as intermediate models between phenol and lignin to provide insights into lignin's behavior. The analysis was planned for all three monomers. However, due to the very low solubility of syringyl (S) monomers, even at high solvent concentrations, the study focused on H and G monomers. Liquid and solid-state NMR, along with FT-IR, were employed to examine the reactions of phenol, H and G lignin monomers, and commercial K-SW lignin with 13C-formaldehyde and glyoxal in resole resins. As the majority of methylene and glyoxylene linkages were formed after curing and due to the low solubility of cured resins in organic solvents, solid-state NMR was used to address this challenge.
Materials and methods
Ingevity generously supplied the kraft softwood (K-SW) lignin (Indulin-AT) sample. Glyoxal (40 wt% in water), 13C-formaldehyde, standard formaldehyde (37 wt% in water), and all additional reagents were procured from Sigma-Aldrich and Fisher Scientific, Inc., and utilized without further modification.Ash content of lignin
The ash content of lignin was assessed following the TAPPI-T 211 om-93 test method.46 Ceramic crucibles were dried and weighed. Then two grams of oven-dried lignin were added to each crucible (in triplicate). The samples were heated in a muffle furnace to 525 °C at a rate of 5 °C min−1. After 4 hours, they were cooled to 100 °C and transferred to a desiccator. Once at room temperature, the samples were weighed, and the ash mass fraction was calculated by dividing the weight of the ash by the weight of the oven-dried lignin.Lignin molecular weight assessment
The number average molecular weight (Mn), weight average molecular weight (Mw), and polydispersity index (PDI) of the lignin sample were determined using gel permeation chromatography (GPC). To ensure solubility in tetrahydrofuran (THF), lignin samples were acetylated.47 This process involved adding 1 g of lignin to a 40 mL mixture of pyridine (20 mL) and acetic anhydride (20 mL), and stirring at room temperature for 24 hours. The acetylated lignin was precipitated with 0.1 M HCl, separated by vacuum filtration, and washed three times with 0.05 M HCl and deionized water. The sample was then dried in a vacuum oven at 40 °C for 16 hours.47 The acetylated sample was dissolved in HPLC grade THF (5 mg mL−1), filtered through a PTFE syringe filter (0.45 μm), and analyzed by GPC. The GPC system (Waters e2695 Separation Module) was equipped with Styragel HR 4 THF, Styragel HR 3 THF, and Ultrastyragel THF columns. Calibration standards of monodisperse polystyrene (162–16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 200 Da) were used. The instrument injected 25 μL of the filtrate, and detection was performed with a 2414 refractive index detector at 35 °C. Data were analyzed using Empower GPC Software.
200 Da) were used. The instrument injected 25 μL of the filtrate, and detection was performed with a 2414 refractive index detector at 35 °C. Data were analyzed using Empower GPC Software.
Lignin hydroxyl content estimated through 31P NMR
The determination of the phenolic and aliphatic hydroxyl content in K-SW lignin involved using 31P NMR, following the method outlined by Asgari and Argyropoulos47 with some modifications. A solution was prepared by combining 325 μL of anhydrous pyridine/deuterated chloroform (1.6:1 v/v) and 300 μL of anhydrous dimethylformamide (DMF). Subsequently, 40 mg of oven-dried lignin was dissolved in the solution. To this mixture, 100 μL of cyclohexanol (22 mg mL−1 in anhydrous pyridine and deuterated chloroform at a ratio of 1.6:1.0 v/v) was added, followed by the addition of 50 μL of chromium(III) acetylacetonate solution (5.6 mg mL−1 concentration in the mixture of 1.6:1.0 v/v anhydrous pyridine and deuterated chloroform). Cyclohexanol served as the internal standard, and chromium(III) acetylacetonate functioned as a relaxation reagent. Finally, 100 μL of the phosphitylating reagent (2-chloro-4,4,5,5-tetramethyl-1,3,2-dioxaphospholane or TMDP) was introduced to the mixture. The samples were analyzed using an Agilent DDR2 500 MHz NMR spectrometer with 7600AS, running VnmrJ 3.2A, a relaxation delay of 5 s, and 128 scans. Various hydroxyl groups were calculated based on the ratio of the area under the internal standard peak to the integrated areas corresponding to different hydroxyl groups.
Solution NMR spectroscopy
13C NMR and 1H–13C-gradient heteronuclear single-quantum coherence (HSQC) spectra were recorded at room temperature on a 600 MHz Bruker NMR spectrometer equipped with a 5 mm iProbe. For the NMR experiments, 100 mg of the sample (based on the dry weight of the resin or raw materials) was dissolved in 600 microliters of deuterated dimethyl sulfoxide (d-DMSO), followed by the addition of 2.5 mg of chromium(III) acetylacetonate as the relaxation agent. In the 13C NMR test, 100 microliters of a standard solution (1,3,5-trioxane in d-DMSO at 100 mg mL−1) were also added.Solid-state NMR
Cured resins were packed into Magic-Angle Spinning (MAS) rotors for measurements on 400 MHz (9.4 Tesla) Bruker Avance Neo spectrometer using a 3.2 mm HCN MAS probe. All experiments were collected under 15 kHz MAS at 298 K unless otherwise stated. 13C chemical shifts were referenced to the tetramethyl silane (TMS) scale and externally referenced to the methyl Cδ of a model peptide N-formyl-Met-Leu-Phe-OH (MLF) at 14.0 ppm. The radiofrequency field strengths were 83.3 kHz for 1H decoupling, 62.5 kHz for 1H cross-polarization (CP) contact pulse, and 50–62.5 kHz for 13C. The analysis and plotting of NMR data were conducted using TopSpin, Microsoft Excel, OriginPro, and Adobe Illustrator. After curing, complex linkages of resins were achieved by the 2D 13C–13C correlation experiments. A 100 ms dipolar-assisted rotational resonance (DARR) experiment were measured for analysis of 13C–13C spatial proximity by analyzing the well-resolved cross-peaks.48Fourier transform infrared spectroscopy (FT-IR)
FT-IR was employed to record the spectrum of each oven-dried raw material or cured sample. A PerkinElmer Spectrum II was utilized in attenuated total reflectance (ATR) mode, covering a wavenumber range of 4000–400 cm−1, with a resolution set at 4 cm−1, and 64 scans in total.Resin preparation
The formulation of lignin-based resins followed a slightly modified procedure as outlined by Siahkamari et al.16 The consistent molar ratio of total phenolic content to formaldehyde or glyoxal was maintained at 1:2, signifying two moles of aldehyde per one mole of the phenolic hydroxyl group in lignin, lignin monomers, or phenol, across all resin variations. Initially, 0.5 g of phenolic material (including phenol, lignin monomers, or lignin) was dissolved in 2 mL of 1 M NaOH (or 2 M NaOH for glyoxal). The temperature was then set at 65 °C using a water bath, lasting for 60 min with continuous stirring at 600 rpm using a digital hot-plate stirrer. Subsequently, 1 M NaOH solution (or 1 mL of 2 M NaOH for glyoxal) was added to the vial, followed by an increase in temperature to 90 °C, maintained for 2 hours. The resulting resin was then cooled to room temperature and stored in a freezer to prevent further polymerization.
Results and discussion
Lignin characterization
Table 1 summarizes the chemical characteristics of commercially available K-SW lignin. Data from 31P-NMR analysis is essential for determining the total phenolic hydroxyl content of lignin. The 31P-NMR method is employed following the reactions of hydroxyl groups in lignin, providing a precise way to quantify different hydroxyl groups using phosphorus-containing reagents.47 This information calculates the amount of formaldehyde or glyoxal needed to achieve a 2:1 molar ratio with lignin, based on its phenolic OH content. Formaldehyde (or glyoxal) reacts at the ortho positions adjacent to the phenolic hydroxyl group in lignin. Phenolic units with more vacant ortho positions offer more active sites for this reaction. In this case, the aldehyde reacts with p-hydroxyphenyl and guaiacyl units, which have two and one reactive sites, respectively.49 According to 31P-NMR analysis, K-SW lignin contains 0.27 mmol g−1 of p-hydroxyphenyl and 1.76 mmol g−1 of guaiacyl units, making it suitable for reactions with formaldehyde and glyoxal.
| Techniques | K-SW lignin | |
|---|---|---|
| Ash content (%) | 4.34 ± 0.22 | |
| GPC analysis results | M n (Da) | 1900 |
| M w (Da) | 6230 | |
| PDI | 3.3 | |
| Hydroxyl functional groups of kraft softwood lignin 31P NMR Analysis result (mmol g−1) | Aliphatic hydroxyl | 1.98 |
| Syringyl | — | |
| Guaiacyl | 1.76 | |
| Condensed phenolic | 1.24 | |
| p-Hydroxyphenyl | 0.27 | |
| Carboxylic acid | 0.39 | |
| Total phenolic | 3.66 | |
| Total hydroxyl content | 5.64 |
Characterization of formaldehyde resins
In the synthesis of resole resin, methylolphenols are formed through the Lederer–Manasse reaction (Fig. 3a), which involves aromatic hydroxylation, attaching a –CH2OH group to a phenolic ring.50 These methylolphenols can self-condense, creating binuclear or polynuclear oligomers by forming methylene bridges between aromatic rings. When phenol reacts with o-methylolphenols (I), it primarily forms ortho–ortho (III) intermediates, while p-methylolphenols produce ortho–para (IV) and para–para (V) dimers (Fig. 3b and c). These reactions increase molecular weight, leading to branching and crosslinking in thermosetting resins. To avoid solidification, the reaction is halted by cooling, yielding resole resin with low polymerization. This intermediate remains soluble in alkaline conditions and needs heating to complete curing.51 | ||
| Fig. 3 Reaction of phenol with formaldehyde. (a) ortho and para-methylolphenols formation (I and II), (b) ortho–ortho methylene linkage formation (III), (c) ortho–para and para–para methylene linkage formation (IV and V). Here, linkages coming from formaldehyde are represented in red color, and labeled carbons are brown circles. | ||
In resole resin synthesis, the condensation step is the rate-limiting step, so the reaction rate is initially slow for further oligomer reactions. However, at elevated temperatures (>100 °C), hydroxymethylphenols (compound VI) react with unreacted phenols to continue polymerization, as shown in Fig. 4a.52 Alternative pathways include combining two hydroxymethyl groups to form a dibenzyl ether linkage (Ar–CH2–O–CH2–Ar), depicted in Fig. 4b (VIII). Phenolic hydroxyl groups can also form Ar–O–CH2–Ar ethers with hydroxymethyl groups (Fig. 4c) (IX). Additionally, methylene linkages can react with formaldehyde to yield bis(hydroxymethyl)phenol (X), as shown in Fig. 4d. Formic acid, a principal product of the self-Cannizzaro reaction, can react with hydroxymethyl and phenolic hydroxyl groups to produce formate esters, XI and XII, as illustrated in Fig. 4e. To control the self-Cannizzaro reaction, formaldehyde was gradually added to the flask. Moreover, during the addition step, when formaldehyde interacts with the phenolic ring, both temperature and pH were maintained at low levels to prevent the Cannizzaro reaction. Then, during the subsequent condensation step, the temperature was raised, and a second charge of the alkaline catalyst was introduced. These adjustments not only facilitated the condensation necessary for developing the prepolymer (when methylol and glyoxylene bridges are formed before hot-press curing) but also helped reduce the free formaldehyde content by promoting the Cannizzaro reaction at this stage.16
 | ||
| Fig. 4 Different condensation reactions of PF resin. (a) Formation of tetramer (VII) from dimer (VII); (b) formation of dibenzyl ether through two CH2OH (VIII); (c) formation of phenyl benzyl ether (IX); (d) reaction of methylene linkage with formaldehyde forms compound X; (e) formation of formate esters. | ||
The PF resin, synthesized using 13C-formaldehyde, was analyzed using 13C-NMR experiments to confirm the successful incorporation of formaldehyde into the phenolic ring and the development of desirable linkages. 13C-formaldehyde was utilized in the synthesis because 13C is a naturally less abundant isotope (about 1.1% of carbon). Using 13C-labeled formaldehyde increases the sensitivity and detection capability of the NMR experiment for the carbon atoms in formaldehyde. Additionally, 13C-labeling allows precise tracking of the formaldehyde carbon throughout the chemical reactions, providing detailed insights into reaction mechanisms and product formation.53
In Fig. 5a, the presence of CH2OH groups was identified by peaks ranging from 61–64 ppm, referenced by TMS.54 At temperatures below 100 °C, minimal methylene linkages were formed, shown around 34 ppm in the liquid PF resin spectra. Conversely, the crosslinked PF resin, which was obtained after curing for 30 minutes at 130 °C, exhibited significantly increased methylene linkages (Fig. 5b). Additional peaks at 155 ppm and 170 ppm correspond to phenolic carbon 1 and carbonyl carbon, respectively.55,56 The carbonyl peak is attributed to the self-Cannizzaro reaction of formaldehyde (Fig. 2a).56 Additionally, HSQC experiments have shown the direct bonding of the proton to the carbon. Specifically, the aromatic protons linked to carbons are significantly shifted downfield compared to the aliphatic –CH2– protons. In ESIFig. 5,† the 1H peak at 3.7 ppm corresponds with methylene linkages at 35 ppm, whereas the methylol (CH2OH) protons are observed around 4.5 ppm.57 Additionally, proton peaks at approximately 5–8 ppm are associated with aromatic carbons.
 | ||
| Fig. 5 (a) Comparison of liquid 13C NMR spectra of liquid PF resin with the 13C-CP MAS spectra of cured PF resin (CH2OH at 63.2 ppm and methylene linkage at 34.7 ppm). (b) Dimer fragment of the polymeric cured PF resin, which consists of both CH2OH group and methylene linkage (blue circles identify conversion of CH2OH to CH2). Blue asterisk shows the solvent peak of DMSO-d6 at 39.5 ppm. | ||
When the 1D solution 13C-NMR spectra was compared with the 13C-CP MAS spectra, a significant change after curing was observed. The solid-state NMR spectra appeared broad due to residual dipolar coupling and incomplete removal of chemical shift anisotropy by magic angle spinning.58–60 A decrease in intensity at 64 ppm and an increase at 34 ppm indicated the crucial role of curing in resin synthesis. The dominance of the band around 34 ppm suggested that most methylol groups participated in condensation to form methylene linkages. To further analyze linkages in cured PF resin, 2D 13C–13C correlation experiments were conducted. Diagonal peaks in dipolar-assisted rotational resonance (DARR) signify carbon self-correlation,48 while cross-diagonal peaks indicate correlations between different carbons in space.61,62 It should be noted that observing these 13C–13C cross-peaks can be technically challenging since only one carbon site is 13C-enriched, while the other one involved in these cross-peaks relies on natural isotopic abundance (1.1% for 13C). The correlation of methylene bridges with various carbons illustrated that linkage A (Fig. 6) was predominant. The slight interaction between 64 ppm and 70 ppm suggested the formation of –CH2–O–CH2– and –Ar–O–CH2– ether linkages, introducing linkages B and C, respectively. The absence of correlation with other carbons for the 170 ppm peak indicated its origin from the self-Cannizaro reaction of formaldehyde (Fig. 2a).
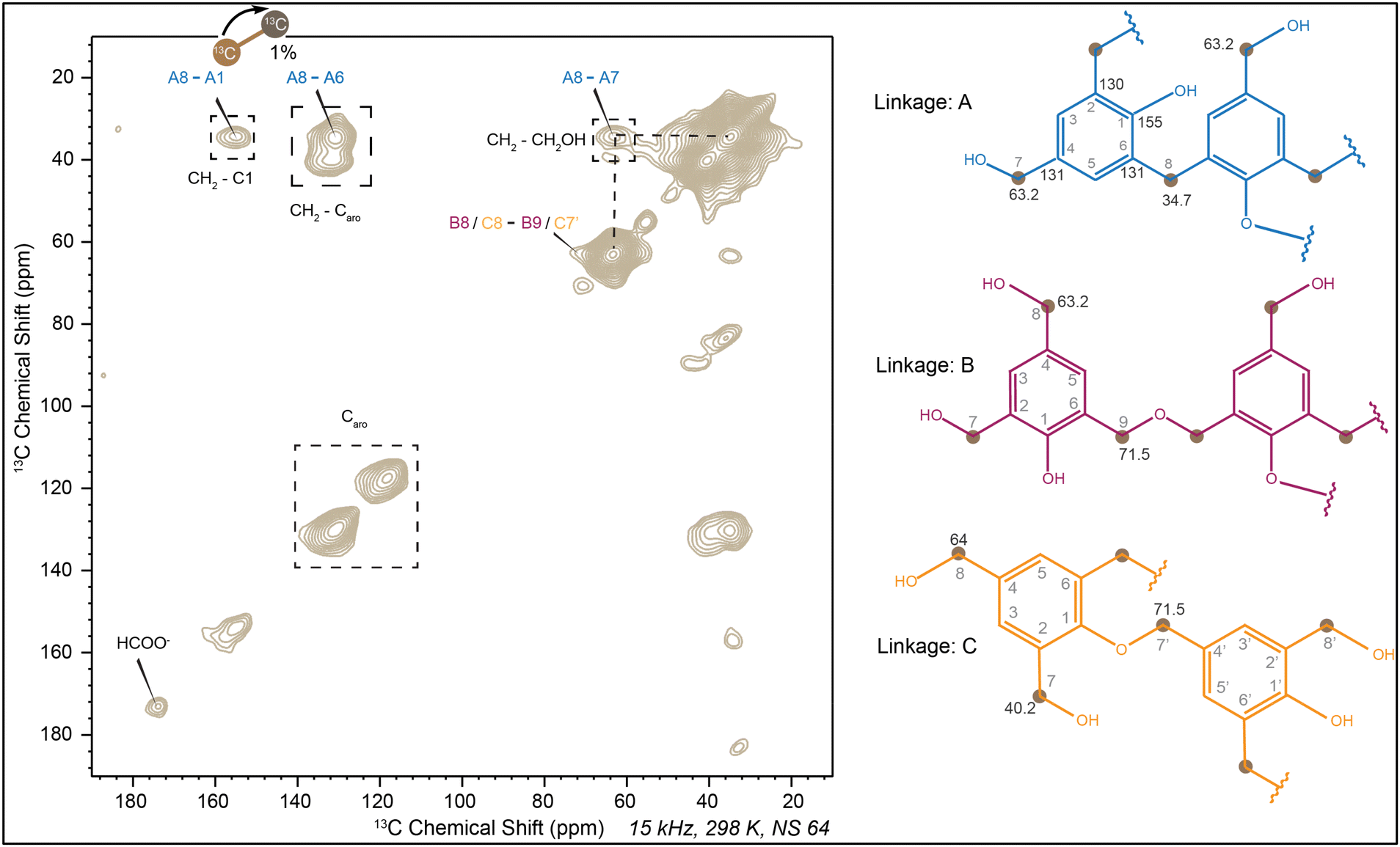 | ||
| Fig. 6 13C–13C 2D dipolar assisted rotational resonance spectra (DARR) of cured PF resin. Linkage A: interaction of methylene linkage with CH2OH, linkage B: formation of –CH2–O–CH2– ether, linkage C: formation of –Ar–O–CH2– ether (labeled carbons are pointed out as brown dots). Note that for each cross peak, only one of the two carbon sites involved was 13C-enriched, while the other site remained at a natural abundance of 1.1% for 13C. | ||
Before delving into commercial lignin, the reaction of lignin monomers with formaldehyde was studied using both liquid and solid-state NMR techniques (Fig. 7). The reaction of H-monomer and G-monomer with formaldehyde resulted in adducts similar to PF resin, which indicated CH2OH, methylene linkages, ether, and formate ester formation. Peaks at approximately 63 ppm and 34 ppm indicated CH2OH and methylene linkages, respectively.54,55,57 HSQC spectra for HF and GF resins were obtained (referenced in Fig. S5b and c†) to examine the proton-carbon correlations, which further support the structural properties of the resins.
 | ||
| Fig. 7 Comparison between the liquid NMR of liquid formaldehyde resins with solid-state NMR of cured formaldehyde resins. (a) NMR spectra of HF liquid resin (blue) and cured HF resin (purple) (chemical shifts of CH2OH and methylene linkage are respectively 63.3 ppm and 34.3 ppm); (b) dimer form of cured HF resin; (c) NMR spectra of GF liquid resin (pink) and cured GF resin (brown) (chemical shifts of CH2OH and methylene linkage are respectively 62.9 ppm and 34.5 ppm); (d) dimer form of cured GF resin (methylene linkages in dimers are represented in dark blue color with a yellow circle representing 13C labeled formaldehyde carbon). | ||
DARR experiments (Fig. 8) on cured resins provided further insights into the fact that CH2OH and methylene linkages belong to the same moiety, as shown by linkage A (methylene linkage) due to the cross peak A7–A8. Similar interactions between 64 and 70 ppm carbons indicated linkages C (–Ar–O–CH2–) and D (–CH2–O–CH2–). New cross-peaks from the interaction between CH2OH and the carbonyl group at 171 ppm validated the transformation of uncondensed CH2OH into formate esters, formed by the self-Cannizaro reaction of formaldehyde, illustrated by linkage B (HCOOAr) and B′ (HCOOCH2), depicting ester formation. Due to the presence of the meta-OCH3 group and the limited reaction sites in the G-monomer (only one free ortho position), leads to the noticeable appearance of unreacted formaldehyde (as paraformaldehyde) around 80–100 ppm, as shown in both Fig. 7c and 8b. In Fig. 8b, the cross peak between 80–60 ppm confirms that the CH2OH group is a form of formaldehyde.
 | ||
| Fig. 8 13C–13C 2D dipolar assisted rotational resonance spectra (DARR) of cured resins. (a) DARR of cured HF resin with linkage A (methylene), B (HCOOAr), B′ (HCOOCH2–), C (–Ar–O–CH2–) and D (–CH2–O–CH2–); (b) DARR of cured GF resin with linkage A (methylene), B (HCOOAr), B′ (HCOOCH2-), C (–Ar–O–CH2–) and D (–CH2–O–CH2–). Each off-diagonal cross peak had one 13C-enriched carbon site, with the other at 1.1% natural abundance. | ||
The analysis of PF resin, along with the structural similarities observed in HF and GF resins, suggests that similar linkage information applies to lignin-formaldehyde (LF) resin (Fig. 9a). The limited availability of reactive sites in commercial lignin, compared to lignin monomers, significantly reduces the possibility of reactions.5 In a liquid medium, the predominant reaction pathway is the self-Cannizzaro reaction of formaldehyde, producing peaks at 172 ppm (carbonyl) and 50 ppm (CH3OH),55,56 which further reduces the signal intensity of the aromatic regions. Since formaldehyde is the only labeled reagent in the reaction medium, and due to the longer 13C-T1 relaxation time of the aromatic carbons, the signal intensity of the aromatic region in the liquid NMR spectra is not observable; however, it can be clearly seen in the solid-state NMR spectrum. The self-Cannizzaro reaction, depicted in Fig. 9d, involves one unit oxidized to form formic acid (172 ppm) and another reduced to form a CH3OH group (50 ppm) (detailed mechanism depicted in Fig. 2a).56 However, after curing, 13C-CP MAS analysis of cured LF resin shows a significant increase in peaks at 64.7 ppm (CH2OH) and 34.5 ppm (CH2) (Fig. 9b). Comparing the relative intensity of the peak at 34.5 ppm to that at 64.7 ppm reveals an approximately sixfold increase in methylene linkage formation after curing (Fig. 9c). The peak around 172 ppm indicates the potential formation of formate ester with CH2OH groups.63
 | ||
| Fig. 9 (a) Comparison between 13C NMR spectra of lignin with the liquid LF resin, which shows self-Cannizaro reaction of formaldehyde that form formic acid (172 ppm) and methanol (50 ppm); (b) comparison of liquid NMR of liquid LF resin (green) with 13C-CP MAS NMR spectra of cured LF resin (orange) (CH2OH at 64.7 ppm and methylene linkage at 34.5 ppm) (missing of methanol peak at 50 ppm); (c) table of relative intensity study of figure (b) with respect to CH2OH band (as it is the primary product of addition reaction); (d) self-Cannizaro reaction of formaldehyde which for formic acid (172 ppm) and methanol (50 ppm), which ultimately form methyl formate. | ||
The use of only 1D spectra cannot fully explain the course of the reaction. To clarify the initial formation of methylene linkages and formate esters, HSQC spectra were obtained from liquid LF resin, which clearly indicates the presence of formate, CH2OH, and methylene groups (Fig. S5d†). Due to the significant difference between the solid-state NMR and the liquid NMR spectra of LF resin, the DARR experiment will provide important information about the different linkages formed after curing. Similar to other formaldehyde resins, the cross peak between 64.7 ppm and 34.5 ppm shows that the CH2OH group and methylene linkages belong to the same moiety as depicted by linkage A in Fig. 10. Along with that the interaction of carbonyl carbon with CH2OH groups is found to be more pronounced in cured LF resin than cured monomer-formaldehyde resins due to its different reaction pathways. While the interaction of carbonyl carbon with aromatic carbon should theoretically exist in every possible linkage, Fig. 10 highlights this interaction with linkage E. Additionally, a peak at 70 ppm (CHOH or ether) correlated with 23 ppm (carbon β), which can only be explained by linkage F. Cross peaks from linkages B, B′, C and D shows the formation of formate esters and ethers, which have already been observed in other cured resins. It is important to note that only the formaldehyde is 13C-labeled, while lignin remains unlabeled (with 99% 12C instead of 13C). Consequently, lignin aromatic carbons do not produce any signals. As a result, the cross peaks primarily originate from the labeled (13C) formaldehyde and its correlations with nearby carbons located in close spatial proximity.
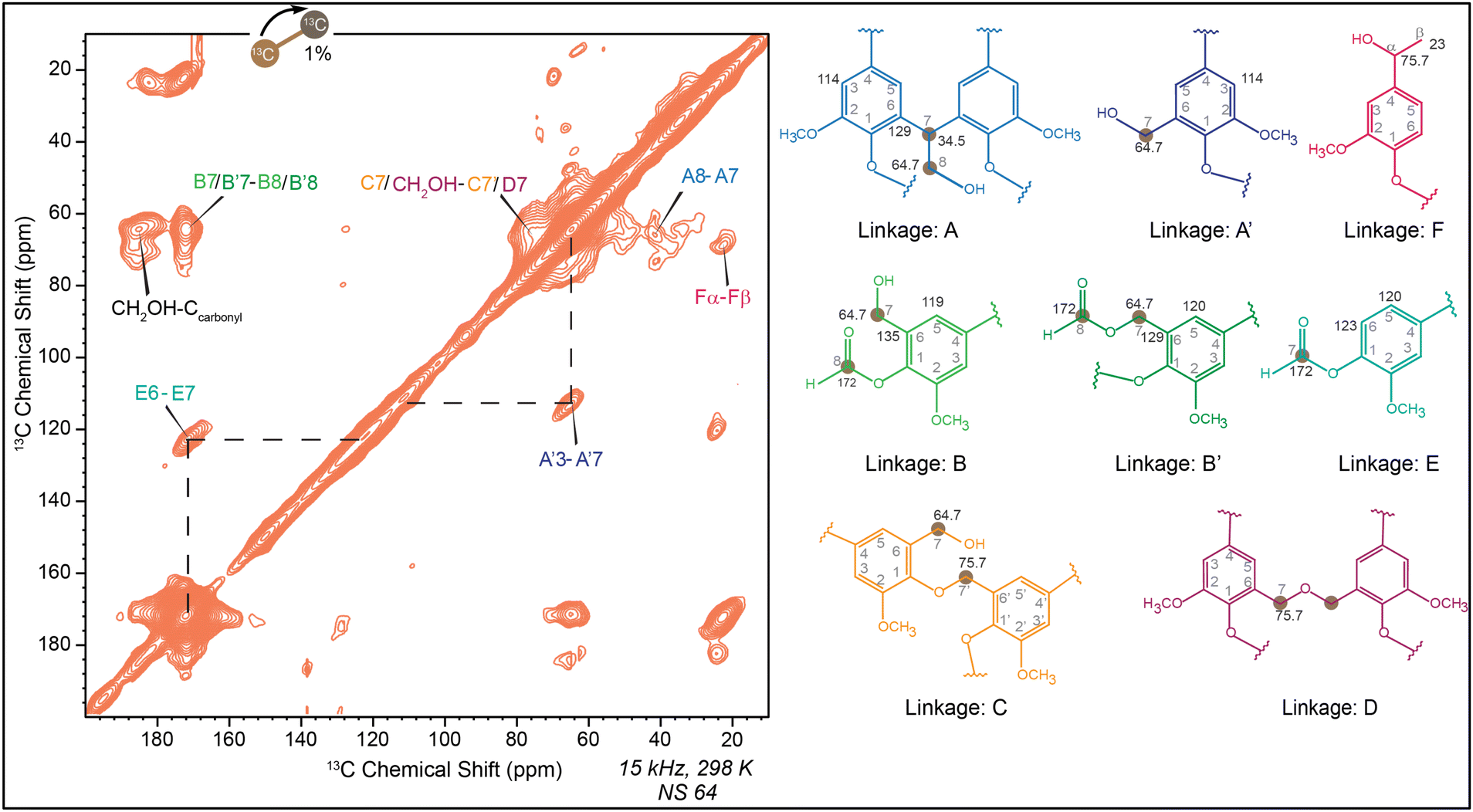 | ||
| Fig. 10 13C–13C 2D dipolar assisted rotational resonance spectra (DARR) of cured LF resin. Linkage A: methylene linkage, linkage A′: interaction between CH2OH and unsubstituted aromatic carbon, linkage B: HCOOAr formate ester, linkage B′: HCOOCH2– formate ester, linkage C: formation of –Ar–O–CH2– ether, linkage D: formation of –CH2–O–CH2– ether, linkage E: interaction of formate ester and aromatic carbon, linkage F: reduced Cα and Cβ interaction. | ||
Characterization of glyoxal resins
In the reaction between lignin and glyoxal, various condensates can be produced. Therefore, simple phenol was used as a model compound to identify possible reactions and products before investigating lignin monomers and commercially available K-SW lignin. Adjusting the pH significantly influences the stability of the condensates. Similarly, modifying the reaction temperature at various stages tends to inhibit the Cannizzaro reaction, which is crucial for forming desirable compounds. Additionally, adjusting the reaction time results in an increased molecular weight of the final resin, affecting its performance as an adhesive.16 These parameters are meticulously optimized to enhance the adhesion performance of the lignin-glyoxal resins. When phenol reacts with glyoxal in an alkaline environment, it initially forms compound XIV (depicted in Fig. 11b) through the addition step.36,64 This addition occurs at the ortho or para positions, with –CH(OH)–CHO added to either position. For simplicity, compound XIV is illustrated as an ortho-substituted intermediate. This ortho-substituted compound then undergoes a further Cannizzaro reaction with another glyoxal unit, yielding compound XV and oxidized glyoxal (XVI) (Fig. 11b).36,64 Following glyoxalation, further condensation by unreacted phenolic units leads to the formation of compound XVII (Fig. 11c).36,64 | ||
| Fig. 11 Reaction of phenol and glyoxal. (a) Self-Cannizaro reaction of glyoxal forms glycolic acid (XI); (b) ortho substitution of phenol forms –CH(OH)–CHO (XII) and –CH(OH)––CH2OH (XIII). Also oxidized form of glyoxal (XIV); (c) formation of dibenzyl derivative with CHCH2OH (XV); (d) formation of compound XVIII and compound XIX (enol ester). Compound XIX is stabilized by hydrogen bonding. | ||
In contrast to formaldehyde, glyoxal can undergo a self-Cannizzaro reaction to produce glycolic acid (Fig. 11a). Even under mild basic conditions, glyoxal readily converts into glycolic acid.16 This transformation influences the structure of Phenol-glyoxal (PG) resin, similar to what was reported by Ramires et al.65,66 through 31P, 1H, and 13C NMR techniques. They detected residual glycolic acid and glycolic ester derivatives in PG resin samples. Their findings showed that the glycolic enol ester derivative (XXI) (depicted in Fig. 11d) forms through the esterification of residual glycolic acid with the enolate structure of compound XX, created via keto–enol tautomerism. The glycolic enol ester is also stabilized through hydrogen bonding, resulting in a six-membered ring, as illustrated in Fig. 11d.
The reaction between lignin and glyoxal is more complex than that of LF resin. Functional groups potentially associated with condensates are evident in the 13C CP-MAS NMR spectra of glyoxylated lignin. In Fig. 12a, the CH2OH group in PG resin is indicated by a peak at approximately 61.9 ppm,64,67,68 with an additional band around 73.2 ppm, signifying the presence of the CHOH group.69 Furthermore, a notable band between 180–190 ppm is linked to the formation of a quinone-stabilized structure and glyoxylate, which results from the self-Cannizzaro reaction of glyoxal (Fig. 11a) in a basic environment.65 In the basic environment provided during the preparation of resole resin, the two adjacent carbonyl groups on the glyoxal can participate in a Cannizzaro reaction,16,36 leading to the formation of the glyoxylate structure, as has been previously documented in the literature.65
 | ||
| Fig. 12 (a) Comparison of liquid 13C NMR spectra of liquid PG resin with the 13C-CP MAS spectra of cured PG resin (CH2OH at 61.9 ppm and methylene linkage at 34.9 ppm) (b) dimer fragment of the polymeric cured PG resin consists of both CH2OH group and methylene linkage. | ||
In HG resin (Fig. 13a and b), the peak at 62.2 ppm corresponds to CH2OH,64,67,68 and an additional band at 71.5 ppm indicates the presence of CHOH groups.69 The range of peaks from 62–72 ppm is attributed to CH2OH and CHOH groups due to structural complexities. After curing, there is a significant increase in peaks at 71.5 ppm (CHOH) and 32.4 ppm (CH2), suggesting further progress toward condensation reaction.64 In the HSQC of HG resin (Fig. S6b†), a 1H peak around 3.8 ppm correlates with 13C peaks between 60–70 ppm, representing CH2OH and CHOH groups.65 Furthermore, the 8.3 ppm peak correlates with 178 ppm, indicating the presence of glyoxylate.65
 | ||
| Fig. 13 Comparison of the liquid NMR of liquid glyoxal resin with the solid NMR of cured glyoxal resin (In cured resin there are distinct band of CH2OH (60–72 ppm), CHOH and ethers (70–72 ppm), methylene linkages (30–40 ppm), quinone and glyoxylate (170–190 ppm)). (a) Overlapped spectra of HG liquid and cured resin; (b) dimer fragment of cured HG resin; (c) overlapped spectra of GG liquid and cured resin; (d) dimer fragment of cured GG resin; (e) overlapped spectra of LG liquid and cured resin; (f) dimer fragment of cured LG resin. | ||
In the cured GG resin (Fig. 13c and d), a broad band around 37.2 ppm confirms the formation of CH2 linkages, while the band at approximately 71.6 ppm indicates CHOH groups.64 The hindered ortho positions, evidenced by the sharp OCH3 band at 54.1 ppm in the 1D CP-MAS spectra (Fig. 13c), result in a lower rate of condensation in GG resin.70–72 Additionally, the peak at 182.2 ppm signifies the carbonyl carbon of quinone formed before glyoxalation. Another band around 175–177 ppm suggests glyoxylate ester formation in both cured HG and GG resins.65 Moreover, the significant increase in the intensity of the peaks at 71.6 ppm and 32–35 ppm indicates condensation and CH2 linkage formation, along with CHOH groups.64 The HSQC spectra of GG resin (Fig. S6c†) exhibit a peak at 3.8 ppm, which signifies the presence of CH2OH which has a 13C chemical shift near about 63 ppm.65
The broad peak spanning 171–184 ppm indicates glyoxal and lignin oxidation, potentially leading to various linkages (Fig. 13b, d and f). The 13C CP-MAS spectra of cured LG resin (Fig. 13e) appear broader due to the complex linkages formed by the reaction of lignin with glyoxal. After curing, CH2 linkage formation results in a band in the 30–40 ppm region, while a band around 62–75 ppm depicts CHOH and CH2OH groups.64 Similar to GG resin, LG resin shows a distinct OCH3 peak at approximately 55 ppm.73,74
Fourier-transform infrared analysis of resins
Fourier-transform infrared spectroscopy is particularly valuable for detecting changes in specific functional groups and monitoring chemical reactions in various materials.75 Here FT-IR spectra of formaldehyde and glyoxal resins showed the systemic changes in the spectral region which helps to identify the incorporation of methylene linkage. The sharp O–H stretches around 3300–3250 cm−1 in pure phenol (Fig. 14a) changes to a broad O–H stretches (3500–2700 cm−1) after curing PF resin (Fig. 14b). The broadening in the O–H stretch region indicates increased hydroxyl group incorporation, potentially from unreacted CH2OH groups, suggesting both intramolecular and intermolecular interactions.76 A sharp peak increase in the 1150–1050 cm−1 region suggests the incorporation of aliphatic C–O stretches. Additionally, significant changes in the 750–730 cm−1 range represent aromatic C–H bending, which reduces after the formation of methylene linkages as aromatic C–H bonds are substituted by methylene groups.76 | ||
| Fig. 14 (a) FT-IR spectra of Phenol (contain sharp O–H stretch) (phenol structure in black color); (b) FT-IR spectra of PF cured resin (blue). O–H stretch becomes broad due to presence of intra and inter-molecular bonding and incorporation of CH2OH. Incorporation of aliphatic O–H bending and C–O stretch at 145–1310 cm−1 (red circle and red arrow) and 1150–1050 cm−1, respectively. Decrease in C–H bending in the fingerprint region (750–730 cm−1) (PF dimer in blue color); (c) FT-IR spectra of PG cured resin (red). (The red box usually shows the changes in the regions.) | ||
The IR spectra of PG resin (Fig. 14c) are quite similar to that of PF resin. Due to glyoxalation, more OH groups are introduced into the cured resins, resulting in a much broader O–H stretch region than in PF resin. The aromatic C–H bending (750–730 cm−1) in PG resin decreases significantly due to the association of CH(OH)CH2OH and CHCH2OH linkages.77–80
Similar changes were observed for the reaction of various lignin monomers with formaldehyde. However, LF resin (Fig. 15b) exhibits a notable increase in the 1120 cm−1 peak, indicating the formation of C–O bonds. This bond formation may arise from the incorporation of CH2OH or ether groups.76 The presence of methylene linkages is evidenced by the alkane C–H stretch in the 3000–2700 cm−1 region. A decrease in peaks within the 860–680 cm−1 range also suggests substitution reactions leading to CH2OH or methylene linkages.76 Additionally, the cured LG resin (Fig. 15c) displays FT-IR spectra similar to those of LF cured resin.
 | ||
| Fig. 15 (a) FT-IR spectra of commercial lignin which contains broad O–H stretching (green spectra); (b) FT-IR spectra of LF cured resin which contain sharp C–O stretch at 1120 cm−1 and significant changes in O–H and fingerprint region (blue spectra) (dimerization in LF resin in blue); (c) FT-IR spectra of LG cured resin which contain sharp C–O stretch at 1120 cm−1 and significant changes in O–H and fingerprint region (red spectra) (dimerization in LG resin in orange). | ||
This systemic analysis of the FTIR spectra provided insights into the incorporation of methylol and glyoxal groups into the resins. The notable differences in the spectral patterns between phenolic and lignin resins, arise from their distinct reaction pathways and structural moieties. However, both types of resins exhibit common spectral changes in the fingerprint region, indicating alterations in the aromatic C–H bending. These changes suggest that the aromatic rings react with formaldehyde and glyoxal.
Conclusions
This study presents a comprehensive evaluation that covers the development and structural characterization of eco-friendly phenolic resins, where lignin serves as a bio-based phenol substitute. At the same time, glyoxal is utilized as a greener cross-linker to replace formaldehyde in the formulation of phenolic adhesives. This investigation is the first to systematically analyze lignin monomers and commercial lignin reactions with formaldehyde and glyoxal, not just in liquid resins but also in their cured solid structures, confirming the linkages formed during the cross-linking (curing) steps. FT-IR, liquid, and solid-state NMR analyses conducted at various stages of resin synthesis confirmed the successful integration of formaldehyde and glyoxal onto the phenolic rings. They verified the development of critical linkages essential for enhancing adhesive performance. The formation of a large number of methylene and glyoxylene linkages with lignin confirmed lignin's suitability to fully replace phenol. Lignin, unlike phenol, requires less extensive degrees of crosslinking during the curing step, as it is a natural glue in the wood and already a prepolymer, making it an inherently valuable substitute. The creation of methylene and glyoxylene bridges between phenolic units, as observed in the solid-state NMR analysis, highlighted the cross-linked structure of lignin-based resins. These findings align with the fundamental principles of sustainable chemistry, aiming to reduce reliance on non-renewable resources and hazardous chemicals. This study contributes to developing bio-based adhesives that offer significant opportunities for manufacturing more sustainable wood panels. Future research should focus on evaluating alternative solvents for lignin that exhibit lower reactivity with glyoxal. This approach aims to reduce undesired side reactions and enhance resin quality. Additionally, studying commercial lignin samples with higher reactivity toward aldehydes would be beneficial, as this could potentially improve the overall reactivity of resin systems for industrial use.Author contributions
MS: data curation, formal analysis, methodology, writing – original draft; DD: solid-State NMR Data curation, formal analysis, visualization, writing – original draft; TW: supervision solid-state NMR, resources, data validation, writing – review & editing, funding acquisition (DOE grant); and MN: conceptualization, investigation, project administration, resources, supervision, validation, writing – review & editing. Funding acquisition (WBC Grant).Data availability
The data supporting this study's findings are available from the corresponding authors, Mojgan Nejad and Tuo Wang, upon reasonable request.Conflicts of interest
There is no conflict of interest.Acknowledgements
The authors are grateful to technical advisors from Willamette Valley, Hexion, Oxiquim, Roseburg, Fortum, West Fraser, and Arclin for their invaluable technical advice throughout the project. We appreciate the funding support provided by the Wood-Based Composites Center, a National Science Foundation Industry/University Cooperative Research Center (Award 1624599-IIP), and funding from the Center for Research and Innovation in Bioeconomy (CRIBE). D. D. and T. W. acknowledge the support from the U.S. Department of Energy, Office of Science, Basic Energy Sciences under grant no. DE-SC0023702 for solid-state NMR analysis.References
- C. R. Frihart, For. Prod. J., 2015, 65, 4–8 Search PubMed.
- A. H. Conner, Encycl. Mater. Sci. Technol., 2001, 9583–9599 Search PubMed.
- L. Nasseri, C. Rosenfeld, P. Solt, M. Mihalic, A. Kandelbauer, J. Konnerth and H. W. G. van Herwijnen, ACS Appl. Polym. Mater., 2023, 5, 6354–6363 CrossRef CAS.
- R. N. Kumar and A. Pizzi, Adhesives for Wood and Lignocellulosic Materials, John Wiley & Sons, Hoboken, New Jersey, 2019 Search PubMed.
- B. K. Saulnier, M. Siahkamari, S. K. Singh, M. Nejad and D. B. Hodge, J. Agric. Food Chem., 2022, 71, 592–602 CrossRef PubMed.
- C. R. Frihart, Handb. wood Chem. wood Compos., 2005, pp. 215–278 Search PubMed.
- A. Pizzi and C. C. Ibeh, in Handbook of thermoset plastics, Elsevier, 2014, pp. 13–44 Search PubMed.
- W. Tian, X. Wang, Y. Ye, W. Wu, Y. Wang, S. Jiang, J. Wang and X. Han, Green Chem., 2023, 10304–10337 RSC.
- A. Gardziella, L. A. Pilato, A. Knop, A. Gardziella, L. A. Pilato and A. Knop, Phenolic Resins Chem. Appl. Stand. Saf. Ecol., 2000, 24–82 Search PubMed.
- P. Georgiou, E. Kyriakopoulou and L. Zoumpoulakis, Polym. Int., 2024, 73, 658–672 CrossRef CAS.
- G. Özbay, E. S. Kökten and A. Özçifçi, Wood Res., 2021, 66, 161–170 Search PubMed.
- I. Calvez, R. Garcia, A. Koubaa, V. Landry and A. Cloutier, Curr. For. Rep., 2024, 1–15 Search PubMed.
- B. Zhan, L. Zhang, Y. Deng and L. Yan, Green Chem., 2023, 25, 10061–10071 RSC.
- W. Tian, H. Feng, X. Wang, W. Wu, S. Zeng, Y. Zhu, J. Wang, Y. Ye, C. Mei and Y. Liu, J. Appl. Polym. Sci., 2024, e56548 Search PubMed.
- X. Han, L. Ding, Z. Tian, Y. Song, R. Xiong, C. Zhang, J. Han and S. Jiang, Int. J. Biol. Macromol., 2023, 224, 1236–1243 CrossRef CAS PubMed.
- M. Siahkamari, S. Emmanuel, D. B. Hodge and M. Nejad, ACS Sustainable Chem. Eng., 2022, 10, 3430–3441 CrossRef CAS.
- R. Whetten and R. Sederoff, Plant Cell, 1995, 7, 1001 CrossRef CAS.
- Y.-Q. Gao, J.-Q. Huang, G. Reyt, T. Song, A. Love, D. Tiemessen, P.-Y. Xue, W.-K. Wu, M. W. George and X.-Y. Chen, Science, 2023, 382, 464–471 CrossRef CAS PubMed.
- R. Vanholme, B. Demedts, K. Morreel, J. Ralph and W. Boerjan, Plant Physiol., 2010, 153, 895–905 CrossRef CAS PubMed.
- M. Perkins, R. A. Smith and L. Samuels, Curr. Opin. Biotechnol., 2019, 56, 69–74 CrossRef CAS PubMed.
- X. Wang, W. Tian, Y. Ye, Y. Chen, W. Wu, S. Jiang, Y. Wang and X. Han, Adv. Colloid Interface Sci., 2024, 103142 CrossRef CAS PubMed.
- N. Terashima, M. Yoshida, J. Hafrén, K. Fukushima and U. Westermark, Holzforschung, 2012, 66, 907–915 CrossRef CAS.
- P. Langan, L. Petridis, H. M. O'Neill, S. V. Pingali, M. Foston, Y. Nishiyama, R. Schulz, B. Lindner, B. L. Hanson and S. Harton, Green Chem., 2014, 16, 63–68 RSC.
- X. Kang, A. Kirui, M. C. Dickwella Widanage, F. Mentink-Vigier, D. J. Cosgrove and T. Wang, Nat. Commun., 2019, 10, 347 CrossRef CAS PubMed.
- O. M. Terrett, J. J. Lyczakowski, L. Yu, D. Iuga, W. T. Franks, S. P. Brown, R. Dupree and P. Dupree, Nat. Commun., 2019, 10, 4978 CrossRef CAS PubMed.
- A. Kirui, W. Zhao, F. Deligey, H. Yang, X. Kang, F. Mentink-Vigier and T. Wang, Nat. Commun., 2022, 13, 538 CrossRef CAS PubMed.
- W. Boerjan, J. Ralph and M. Baucher, Annu. Rev. Plant Biol., 2003, 54, 519–546 CrossRef CAS PubMed.
- J. Gui, P. Y. Lam, Y. Tobimatsu, J. Sun, C. Huang, S. Cao, Y. Zhong, T. Umezawa and L. Li, New Phytol., 2020, 226, 1074–1087 CrossRef CAS PubMed.
- K. Morreel, J. Ralph, H. Kim, F. Lu, G. Goeminne, S. Ralph, E. Messens and W. Boerjan, Plant Physiol., 2004, 136, 3537–3549 CrossRef CAS PubMed.
- R. A. Dixon and J. Barros, Open Biol., 2019, 9, 190215 CrossRef PubMed.
- C. Li, X. Zhao, A. Wang, G. W. Huber and T. Zhang, Chem. Rev., 2015, 115, 11559–11624 CrossRef CAS PubMed.
- M. M. Abu-Omar, K. Barta, G. T. Beckham, J. S. Luterbacher, J. Ralph and R. Rinaldi, Energy Environ. Sci., 2021, 14, 262–292 RSC.
- R. Vanholme, B. De Meester, J. Ralph and W. Boerjan, Curr. Opin. Biotechnol., 2019, 56, 230–239 CrossRef CAS PubMed.
- J. Ralph, K. Lundquist, G. Brunow, F. Lu, H. Kim, P. F. Schatz, J. M. Marita, R. D. Hatfield, S. A. Ralph and J. H. Christensen, Phytochem. Rev., 2004, 3, 29–60 CrossRef CAS.
- F. Chen and R. A. Dixon, Nat. Biotechnol., 2007, 25, 759–761 CrossRef CAS PubMed.
- I. Van Nieuwenhove, T. Renders, J. Lauwaert, T. De Roo, J. De Clercq and A. Verberckmoes, ACS Sustainable Chem. Eng., 2020, 8, 18789–18809 CrossRef CAS.
- K. Tsuchiya, Y. Hayashi, M. Onodera and T. Hasegawa, Keio J. Med., 1975, 24, 19–37 CrossRef CAS PubMed.
- J. Kielhorn, C. Pohlenz-Michel, S. Schmidt and I. Mangelsdorf, World Health Organ., 2004, pp. 1–41 Search PubMed.
- P. R. Sarika, P. Nancarrow, A. Khansaheb and T. Ibrahim, Polymers, 2020, 12, 2237 CrossRef CAS PubMed.
- G. Mattioda, B. Metivier and J. P. Guette, Chemtech, 1983, 13, 478–481 CAS.
- D. J. Yelle and J. Ralph, Int. J. Adhes. Adhes., 2016, 70, 26–36 CrossRef CAS.
- L. Hu, H. Pan, Y. Zhou and M. Zhang, BioResources, 2011, 6, 3515–3525 CAS.
- J. O'Brien, The Maillard reaction in foods and medicine, Woodhead Publishing, 1998, vol. 223 Search PubMed.
- C. G. Swain, A. L. Powell, W. A. Sheppard and C. R. Morgan, J. Am. Chem. Soc., 1979, 101, 3576–3583 CrossRef CAS.
- Y. Zhang, X. M. Wang, R. Casilla, P. Cooper, Z. Huang and X. Wang, J. Appl. Polym. Sci., 2010, 117, 2888–2898 CrossRef CAS.
- TAPPI - Technical Association Of The Pulp And Paper Industry. 1993. T 211 om-93: Ash in wood, pulp, paper and paperboard: combustion at 525 °C. Atlanta., 1993.
- F. Asgari and D. S. Argyropoulos, Artic. Can. J. Chem., 1998, 76, 1606–1615 CAS.
- K. Takegoshi, S. Nakamura and T. Terao, Chem. Phys. Lett., 2001, 344, 631–637 CrossRef CAS.
- M. Ghorbani, F. Liebner, H. W. G. van Herwijnen, L. Pfungen, M. Krahofer, E. Budjav and J. Konnerth, BioResources, 2016, 11, 6727–6741 CrossRef CAS.
- G. E. Maciel, I. S. Chuang and L. Gollob, Macromolecules, 1984, 17, 1081–1087 CrossRef CAS.
- L. Uyigue and E. O. Kubiangha, Int. J. Eng. Mod. Technol., 2018, 4, 25–39 Search PubMed.
- I. S. Chuang and G. E. Maciel, Macromolecules, 1991, 24, 1025–1032 CrossRef CAS.
- D. D. Werstler, Polymer, 1986, 27, 750–756 CrossRef CAS.
- T. Holopainen, L. Alvila, J. Rainio and T. T. Pakkanen, J. Appl. Polym. Sci., 1997, 66, 1183–1193 CrossRef CAS.
- J. Li, J. Zhang, S. Zhang, Q. Gao, J. Li and W. Zhang, Polymers, 2017, 9, 428 CrossRef PubMed.
- H. Paananen and T. T. Pakkanen, Holzforschung, 2020, 74, 663–672 CrossRef CAS.
- H. Paananen, L. Alvila and T. T. Pakkanen, Sustainable Chem. Pharm., 2021, 20, 100376 CrossRef CAS.
- B. Reif, S. E. Ashbrook, L. Emsley and M. Hong, Nat. Rev. Methods Prime, 2021, 1, 2 CrossRef CAS PubMed.
- J. Schaefer and E. O. Stejskal, J. Am. Chem. Soc., 1976, 98, 1031–1032 CrossRef CAS.
- E. R. Andrew, A. Bradbury and R. G. Eades, Nature, 1958, 182, 1659 CrossRef CAS.
- M. Veshtort and R. G. Griffin, J. Chem. Phys., 2011, 135, 134509 CrossRef PubMed.
- G. Hou, S. Yan, J. Trebosc, J.-P. Amoureux and T. Polenova, J. Magn. Reson., 2013, 232, 18–30 CrossRef CAS PubMed.
- X. Duan, X. Wang, A. Huang, G. Liu and Y. Liu, Molecules, 2022, 27, 2905 CrossRef CAS PubMed.
- P. Navarrete, A. Pizzi, H. Pasch and L. Delmotte, J. Adhes. Sci. Technol., 2012, 26, 1069–1082 CrossRef CAS.
- E. C. Ramires, J. D. Megiatto, C. Gardrat, A. Castellan and E. Frollini, Bioresour. Technol., 2010, 101, 1998–2006 CrossRef CAS PubMed.
- E. C. Ramires, D. Jackson Jr, C. Gardrat, A. Castellan and E. Frollini, Polim.:Cienc. Tecnol., 2010, 20, 126–133 CrossRef CAS.
- M. H. Hussin, N. A. Samad, N. H. A. Latif, N. A. Rozuli, S. B. Yusoff, F. Gambier and N. Brosse, Int. J. Biol. Macromol., 2018, 113, 1266–1272 CrossRef PubMed.
- H. I. Huzyan, A. A. Aziz and M. H. Hussin, BioResources, 2021, 16, 4106–4125 CAS.
- N. E. El Mansouri, A. Pizzi and J. Salvado, J. Appl. Polym. Sci., 2007, 103, 1690–1699 CrossRef CAS.
- L. Zhao, B. F. Griggs, C.-L. Chen, J. S. Gratzl and C.-Y. Hse, J. Wood Chem. Technol., 1994, 14, 127–145 CrossRef CAS.
- H. Paananen, E. Eronen, M. Mäkinen, J. Jänis, M. Suvanto and T. T. Pakkanen, Ind. Crops Prod., 2020, 152, 112473 CrossRef CAS.
- W. Zhang, Y. Ma, C. Wang, S. Li, M. Zhang and F. Chu, Ind. Crops Prod., 2013, 43, 326–333 CrossRef CAS.
- N. A. Aziz, A. F. A. Latip, L. C. Peng, N. H. Abd Latif, N. Brosse, R. Hashim and M. H. Hussin, Int. J. Biol. Macromol., 2019, 141, 185–196 CrossRef CAS PubMed.
- M. Hazwan Hussin, A. A. Aziz, A. Iqbal, M. N. M. Ibrahim and N. H. A. Latif, Int. J. Biol. Macromol., 2019, 122, 713–722 CrossRef CAS PubMed.
- K. M. Kelani, M. R. Rezk, H. H. Monir, M. S. ElSherbiny and S. M. Eid, Anal. Methods, 2020, 12, 5893–5907 RSC.
- I. Poljansek and M. Krajnc, Acta Chim. Slov., 2005, 52, 238 Search PubMed.
- L. Chupin, B. Charrier, A. Pizzi, A. Perdomo and F. Charrier-El Bouhtoury, J. Therm. Anal. Calorim., 2015, 119, 1577–1585 CrossRef CAS.
- S. Wang, Y. Yu and M. Di, Polymers, 2018, 10, 631 CrossRef PubMed.
- P. L. de Hoyos-Martínez, E. Robles, A. Khoukh, F. Charrier-El Bouhtoury and J. Labidi, Biomacromolecules, 2019, 20, 3535–3546 CrossRef PubMed.
- N.-E. El Mansouri, Q. L. Yuan and F. Huang, Mater. Sci., 2011, 6, 4523–4536 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4gc05695g |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |