
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence1,2-trans and regioselective glycosylation of multihydroxy sugars via a simple yet synergistic combination of BF3·Et2O in THF†
Zhimei
Wang‡
 a,
Longwei
Gao‡
a,
Yingjie
Wang
b,
Fuzhu
Yang
b,
Jinpeng
Sang
a,
Shuheng
Pan
a,
Xin
Huang
a,
Pan
Zhang
a,
Weijia
Xie
a,
Xiaoxing
Wu
a,
Biao
Yu
b,
Peng
Xu
b,
Xiaheng
Zhang
c,
Zhaolun
Zhang
*a and
Wei
Li
*a
a,
Longwei
Gao‡
a,
Yingjie
Wang
b,
Fuzhu
Yang
b,
Jinpeng
Sang
a,
Shuheng
Pan
a,
Xin
Huang
a,
Pan
Zhang
a,
Weijia
Xie
a,
Xiaoxing
Wu
a,
Biao
Yu
b,
Peng
Xu
b,
Xiaheng
Zhang
c,
Zhaolun
Zhang
*a and
Wei
Li
*a
aDepartment of Medicinal Chemistry, School of Pharmacy, China Pharmaceutical University, 639 Longmian Avenue, Nanjing, Jiangsu 211198, China. E-mail: wli@cpu.edu.cn; zhangzl_cn@outlook.com
bState Key Laboratory of Chemical Biology, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Lingling Road, Shanghai 200032, China
cSchool of Chemistry and Materials Science, Hangzhou Institute for Advanced Study, University of Chinese Academy of Sciences, 1 Sub-lane Xiangshan, Hangzhou 310024, China
First published on 21st January 2025
Abstract
Carbohydrates play pivotal roles in numerous biological processes. The precise synthesis of structurally defined and pure carbohydrates is of paramount importance in pathological research and drug development. However, achieving stereo- and regioselective glycosylation during carbohydrate synthesis is often a tedious process that exhibits low atom economy. Herein, we present a surprisingly simple yet synergistic combination of BF3·Et2O in THF as a green solution to shorten the synthetic procedures, utilizing readily accessible imidate donor mixtures, regardless of their anomeric configuration. Glycosylation selectively occurs on the more nucleophilic hydroxyl group, giving 1,2-trans glycosides across a broad substrate scope in a highly stereo- and regioselective manner. This strategy is easy to apply and scale up, as demonstrated by an atom-economical synthetic route to achieve an oligosaccharide framework related to the Enterococcus faecalis antigen. Variable-temperature (VT) NMR studies revealed the formation of BF3·ROH complexes, suggesting their roles as the true promoters and acceptors during glycosylation. Density functional theory (DFT) calculations suggested that 1,2-trans selectivity arises from the energy discrepancy between putative transition states involving [BF3OR]− and the oxocarbenium–solvent complex.
Green foundation1. This work allows for 1,2-trans and regioselective glycosylation of multihydroxy sugars with minimal protection in an environmentally friendly manner. It can significantly shorten the synthetic procedures required for obtaining complex carbohydrates and reduces the reliance on protecting and directing groups.2. The advantages of this strategy in terms of green chemistry include the use of readily available donors and acceptors, a metal-free system, short reaction time, good scalability, inexpensive reagents, and halogen-free solvents. 3. Future research endeavors can focus on refining reagent design to minimize the amount of promoters utilized and enable selective glycosylation reactions at room temperatures. |
Introduction
Carbohydrates serve as the primary energy source and play a crucial role in numerous essential biological processes, including cell recognition and interaction.1,2 Studies on carbohydrates provide valuable insights into their pathomechanism and facilitate drug development for combating diseases, such as cancers, viral infections, and diabetes.3,4 Currently, access to structurally precise oligosaccharides, glycoconjugates, and glycans5–10 heavily relies on chemical synthesis via crucial glycosylation reactions.11–15 In order to achieve high stereo- and regioselectivity in glycosylation during complex carbohydrate synthesis, directing/protecting groups are frequently manipulated on both donors and acceptors,13,16–21 such as in our recent studies on preparing challenging β/α-2-deoxyglycosides, β-rhamnosides, α-glucosides, and β/α-2′-deoxynucleosides.22–26 However, these tactics usually suffer from a poor atom economy, long synthetic route, and low overall efficiency due to the tedious introduction, replacement, and cleavage of directing/protecting groups. To make carbohydrate synthesis greener, many reagent-controlled strategies for glycosylation have emerged over the past decade, primarily focusing on stereoselectivity,27–41 with a limited number also offering simultaneous regioselectivity toward multihydroxy sugars bearing few protecting groups (Fig. 1a).42–52 These include the effective utilization of borinic acid, boronic acid, noble metal chloride, and Ca(OTf)2 with α-configured donors (Fig. 1b).43–45,47–51,53 Recent studies have reported precisely designed catalysts bearing multiple functional sites as stereocontrol and regiocontrol factors, such as synthetic bis-thioureas for (1→2)-selective β-galactosylation with α-configured donors and aminoboronic acid for 1,2-cis-glycosylation (Fig. 1b).42,52 | ||
| Fig. 1 Stereo- and regioselective glycosylation. | ||
Despite these innovative achievements, the synthesis of complex carbohydrates still calls for a greener and more atom-economical strategy favoring sustainable development. While pursuing high yields and selectivity in glycosylation, several aspects are also noteworthy. For example, simple and cheap reagents are preferred for achieving a better atom economy and easier preparation of donors, acceptors, and promoters. Some strategies depend on donor anomeric configuration and only employ specific anomers as donors. However, obtaining such single α- or β-specific donors can be difficult during complex carbohydrate synthesis, particularly for donors bearing multiple functional groups or oligosaccharide donors. This would require careful separation and result in wastage of other undesired anomers.40 Furthermore, the recent ban by the US Environmental Protection Agency (EPA) on most uses of CH2Cl2, a widely used solvent in glycosylation, poses additional challenges for finding more eco-friendly solvents. Scale-up, operation, functional group tolerance, and reaction time are also of great concern. Therefore, it is still a highly desirable but challenging goal to develop a green and efficient strategy for glycosylation to synthesize complex carbohydrates.
Herein, we present a solution to utilize a simple combination of BF3·Et2O in ether solvent (Fig. 1c) to enable the highly 1,2-trans and regioselective glycosylation of multihydroxy acceptors with readily accessible imidate donors, regardless of the donor anomeric configuration. BF3·Et2O, a cheap Lewis acid widely employed in organic synthesis, was recommended by Yu and co-workers as a better catalyst for phenol O-glycosylation to suppress the formation of side-products during neighbouring group participation.54 However, BF3·Et2O itself is generally considered a poor factor for stereocontrol;55,56 ether solvents, on the other hand, are typically expected to lead to the reverse 1,2-cis α-selectivity.57–62 Neither has been known as a regiocontrol factor to date. However, their combination resulted in high 1,2-trans and regioselectivity (Fig. 1c). Furthermore, ether solvents, such as THF in our protocol, are more eco-friendly than regularly used halogen solvents (e.g., CH2Cl2 and DCE) in glycosylation, providing an alternative to comply with the ban on CH2Cl2 by the EPA. We consider this strategy greener and highly practical due to the simple and eco-friendly reagents/promoters/solvents utilized, readily accessible donors/acceptors, ease of handling, broad substrate scope, easy scale-up, mild conditions, reasonable reaction time, and low cost.
Results and discussion
Reaction discovery and validation
As control reactions during our pursuit of new reagents for stereoselective glycosylation using regular imidate donors, we were surprised to discover that a simple combination of BF3·Et2O (stored or freshly distilled) in ether solvent (Table S1†), without any additives, resulted in high β-selectivity in glucosylation with the benzylated imidate donor 1 (a mixture of anomers, α/β = 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). Despite the general perception that β-glucosylation has already been addressed by the 2-O-acyl directing group-controlled strategy, we perceived this finding still holds high value in consideration of a practical reagent-controlled strategy. It promises more straightforward access to base-labile glycosides and 2-O-glycosylated glycosides.63 In addition, it can circumvent the issues associated with the 2-O-acyl strategy,64 including orthoester formation, selective installation/cleavage of the 2-O-acyl groups, and diminished reactivity due to the disarming effect of acyl groups. Further optimization showed that the standard conditions were 0.4 eq. of BF3·Et2O in THF at −50 °C (Table 1, entry 1), which led to the desired disaccharide 4 with complete β-selectivity and a high 91% yield. Although THF is generally avoided in glycosylation due to concerns about sluggish reactions and potential side reactions, such as polymerization,65 this reaction proceeded smoothly and could be easily scaled up (entry 2).
:1). Despite the general perception that β-glucosylation has already been addressed by the 2-O-acyl directing group-controlled strategy, we perceived this finding still holds high value in consideration of a practical reagent-controlled strategy. It promises more straightforward access to base-labile glycosides and 2-O-glycosylated glycosides.63 In addition, it can circumvent the issues associated with the 2-O-acyl strategy,64 including orthoester formation, selective installation/cleavage of the 2-O-acyl groups, and diminished reactivity due to the disarming effect of acyl groups. Further optimization showed that the standard conditions were 0.4 eq. of BF3·Et2O in THF at −50 °C (Table 1, entry 1), which led to the desired disaccharide 4 with complete β-selectivity and a high 91% yield. Although THF is generally avoided in glycosylation due to concerns about sluggish reactions and potential side reactions, such as polymerization,65 this reaction proceeded smoothly and could be easily scaled up (entry 2).
|
|
||
|---|---|---|
| Entry | Deviation | Yielda (β/α ratio)b |
| a Products were isolated via chromatography. b Ratios were determined through 1H NMR. | ||
| 1 | None (donor 1) | 91% (β only) |
| 2 | Gram scale | 99% (β only) |
| 3 | Pure β-donor (2) | 92% (β only) |
| 4 | TMSOTf (0.1 eq.), CH2Cl2 | 85% (1.3:1) |
| 5 | CH2Cl2 | 70% (5.1:1) |
| 6 | Pure β-donor (2), CH2Cl2 | 80% (3.5:1) |
| 7 | TMSOTf | 86% (2.5:1) |
| 8 | Et2O | 71% (20:1) |
| 9 | TMSOTf, Et2O | 68% (1:1.2) |
| 10 | −25 °C | 70% (β only) |
| 11 | Et2O, −25 °C | 66% (3:1) |

Schmidt and co-workers reported BF3·Et2O-directed β/α ratios of up to 6:1 by using pure α-imidate donors in CH2Cl2via a presumed intramolecular SN2 attack on the α-donors.55,56 However, this could not rationalize the complete β-selectivity we obtained from a mixture of α- and β-donors (α/β = 7:1) in THF. For further verification, we prepared glucosyl imidate 2 as a pure β-donor and then evaluated it under the standard conditions of 0.4 eq. of BF3·Et2O in THF at −50 °C (Table 1, entry 3). Gratifyingly, we still obtained complete β-selectivity with a similar 92% yield, indicating the presence of other strong β-directing factors independent of the anomeric configuration of the imidate donors. To gain further insights into the β-directing effect of BF3·Et2O and THF, we conducted four control reactions (entries 4–7) by replacing them with TMSOTf and CH2Cl2, respectively. The condensation of 1 and 3 with TMSOTf in CH2Cl2 (entry 4) was found to be almost non-stereoselective (β/α = 1.3:1) at −50 °C. Utilizing BF3·Et2O in CH2Cl2 (entry 5) increased the β/α ratio to 5.1:1, which was in agreement with Schmidt's report.56 Replacement of donor 1 (predominantly α-donor) with β-donor 2 in CH2Cl2 (entry 6) also led to β-selectivity (β/α = 3.5:1), reaffirming the presence of alternative β-directing pathways distinct from the direct SN2 attack on α-donors.
On the other hand, the β-selectivity (β/α = 2.5:1, entry 7) that we achieved with TMSOTf in THF was not expected. It is well-known that ether solvents, such as Et2O and 1,4-dixoane, are experimentally α-directing in glucosylation.57–62 However, by comparing the β/α ratios in THF and CH2Cl2 (2.5:1 in entry 7 vs. 1.3:1 in entry 4), we found that THF could actually be β-directing at −50 °C. Although the β-directing effects of BF3·Et2O (entries 5 and 6) and THF (entry 7) did not seem significant individually, it seemed their combination could, in some way, provide a strong synergistic effect to give complete β-selectivity (entries 1–3) in glucosylation. In fact, the combination of BF3·Et2O in Et2O also offered a high β/α ratio of 20:1 at −50 °C (entry 8), compared to the poor β/α ratio of 1:1.2 with TMSOTf in Et2O (entry 9). Additionally, we also achieved high β-selectivity (β/α > 20:1 or β only) with BF3·Et2O in MTBE, 2-methyl-THF, and 2,5-dimethyl-THF (Table S1†), suggesting that this synergistic effect with BF3·Et2O may be common among ether solvents.
A low temperature of −50 °C was found necessary to ensure the high 91% yield for the glycosylation in THF (entry 1), while increasing the reaction temperature to −25 °C maintained the β-specificity but reduced the yield to 70% (entry 10) due to the formation of glucosyl fluoride. In comparison, other ether solvents, such as Et2O (entry 11), at −25 °C decreased the β/α ratio down to 3:1 (Table S1†). Nevertheless, the results in Table 1 indicate that the present high stereoselectivity could primarily be attributed to the synergistic effect of BF3·Et2O and THF rather than to the donors being α- or β-configured. Low temperatures, such as −50 °C, could further increase the β-selectivity and yields.
Scope for stereoselectivity
To further understand the unexpected stereoselectivity induced by BF3·Et2O and THF, we investigated 11 pyranosyl and furanosyl imidate donors (5–15) varying in sugar types and protecting groups (Fig. 2b). These donors were prepared from their corresponding hemiacetals using the routine protocol (NCCCl3 and DBU in CH2Cl2) without deliberately separating the anomers, leading to α-donors (e.g., 5), β-donors (e.g., 13), and α/β mixtures (e.g., 9). Regardless of their anomeric configuration, they were employed to glycosylate alcohol 3 under the standard conditions (0.4 eq. of BF3·Et2O, THF, −50 °C) to evaluate the stereoselectivity and donor scope (Fig. 2a). To our delight, the results in Fig. 2b clearly show that the glycosylation was 1,2-trans selective rather than axial- or equatorial-selective for both pyranoses and furanoses, giving the corresponding β-glucosides 22–26, β-galactoside 19, α-mannosides 20/27, α-rhamnoside 21, α-arabinofuranoside 28, and β-ribofuranoside 29. Most reactions exhibited complete stereoselectivity with yields exceeding 90%, indicating a broad substrate scope and robust compatibility with various functional groups. Taking glucosylation as an example, an α-directing 6-OAc group (donor 10) or a bulky 6-OTIPS group (donor 9) did not diminish the β-selectivity. Donor 8 with a benzylidene-restrained conformation also resulted in complete β-selectivity for the disaccharide 22 with a yield of 98%. Even the glucuronic donor 12 bearing the electron-withdrawing methyl ester group, which is usually considered to be a poor donor due to its tendency toward elimination and epimerization, was smoothly converted into the disaccharide 26 with a high 91% yield and complete β-selectivity. Arabinosyl imidate 14 and ribosyl imidate 15 as representative furanosyl donors also led to the desired α-arabinoside 28 and β-riboside 29 with complete stereoselectivity and high yields.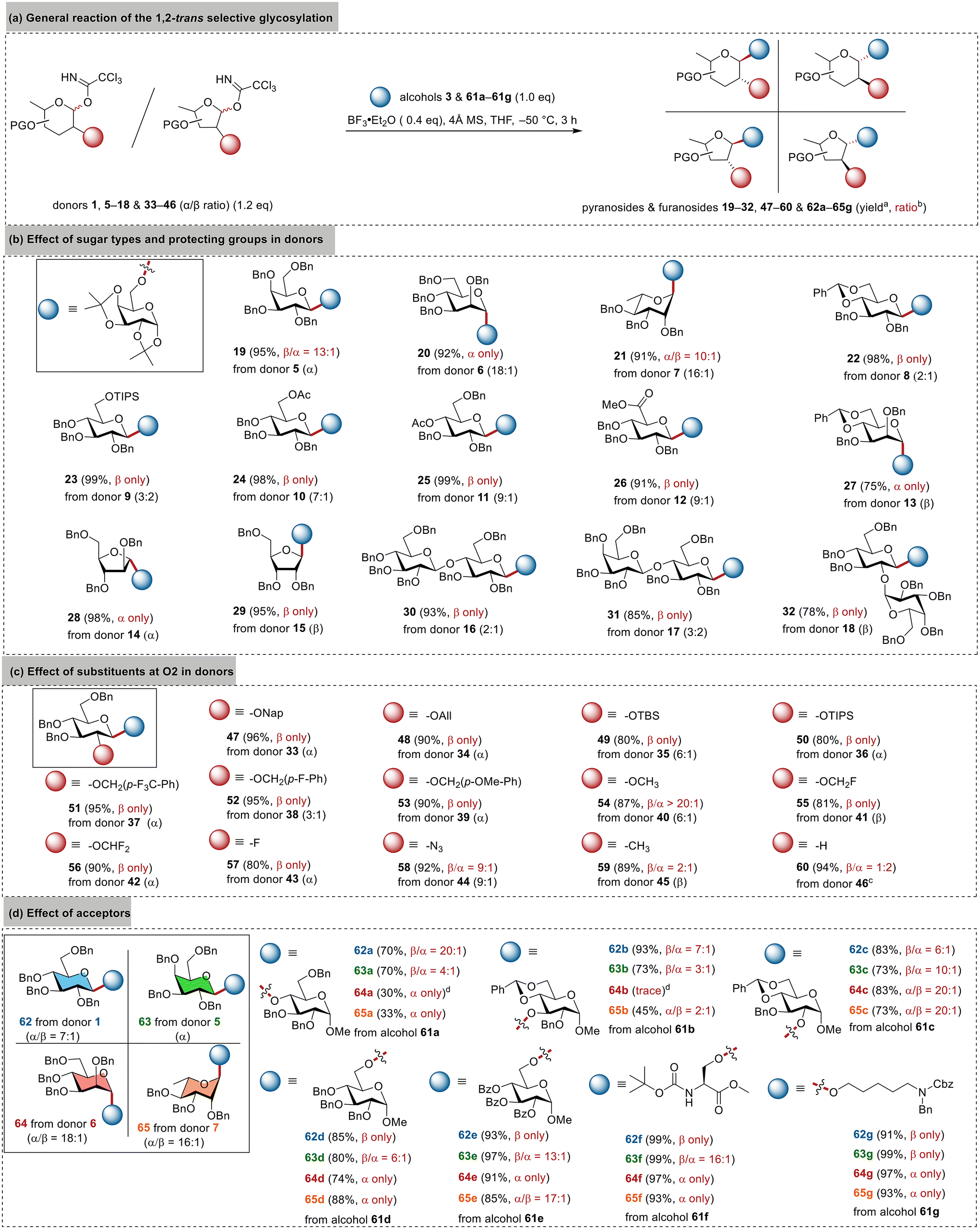 | ||
| Fig. 2 Scope for stereoselectivity. aProducts were isolated using chromatography. bRatios were determined via1H NMR. cα/β ratio of donor 46 was not determined due to its lability. dReactions were sluggish. | ||
Compared to monosaccharide donors, achieving stereocontrol with oligosaccharide donors is more challenging yet crucial for implementing a convergent strategy for the efficient synthesis of complex carbohydrates.22–24 Thus, we prepared cellobiosyl imidate 16, lactosyl imidate 17, and the 2-O-glycosylated disaccharide donor 18 for further evaluation (Fig. 2b). Gratifyingly, all these donors resulted in complete 1,2-trans selectivity to give the desired trisaccharides 30–32. These results highlight the valuable potential of BF3·Et2O/THF glycosylation in the convergent synthesis of complex carbohydrates, particularly in cases involving the use of 2-O-glycosylated donors,63 which cannot adopt the conventional 2-O-acyl directing strategy for stereocontrol.
All the donors in Fig. 2b share a benzyl group at O2. We were curious about whether this could be replaced with other groups while maintaining high 1,2-trans selectivity. Thus, 10 additional glucosyl imidate donors (33–42) bearing diverse groups at the O2 position were prepared and evaluated. As depicted in Fig. 2c, all the reactions were 1,2-trans β-specific, giving the corresponding disaccharides 47–56 in high yields. These results indicated the broad compatibility with various groups at O2. Notably, the stereoselectivity was barely affected by the electron density (e.g., 54vs.56) or steric hindrance (e.g., 54vs.50) of the substituents at O2, suggesting that direct repulsion was unlikely the primary factor for the stereocontrol. Additionally, we prepared four 2-deoxy donors (43–46) bearing different substituents at C2. The donor with a 2-fluoro group or a 2-azide group (43 or 44) still exhibited satisfactory β-selectivity in glycosylation, while the 2-methyl donor 45 resulted in a poor β/α ratio of 2:1, and the 2-deoxy donor 46 even led to a reversed β/α ratio of 1:2. These findings underscore the necessity of a heteroatom at C2 for maintaining high stereoselectivity.
Meanwhile, we conducted studies on the acceptor scope for the BF3·Et2O/THF glycosylation (Fig. 2d). In addition to glucosyl imidate 1, we also employed galactosyl, mannosyl, and rhamnosyl imidates 5–7 to glycosylate seven representative acceptors (61a–61g) varying in electron density, steric hindrance, and structural rigidity. These acceptors included five glucosides with 2-OH, 3-OH, 4-OH, and 6-OH, among which the 4,6-O-benzylidene-restrained 3-OH glucoside 61b was one of the poorest acceptors in terms of stereocontrol.22,24,66 As depicted in Fig. 2d, primary alcohols generally led to both high yields and 1,2-trans selectivity, giving β-configured glucosides/galactosides and α-configured mannosides/rhamnosides. This protocol was also found to be highly efficient for coupling the fatty alcohol 61g, a prevalent linker for preparing carbohydrate vaccines.67,68 On the other hand, secondary alcohols resulted in decreased stereoselectivity due to the increased hindrance and rigidity.66,69,70 Their yields also decreased due to the formation of glycosyl fluorides that were inert under these conditions, particularly in the mannosylation and rhamnosylation of hindered alcohols 61a and 61b. The reactions were also found to be sluggish to further reduce the yields. Among these four donors, glucosyl donor 1 proved to be the most effective one in terms of both yield and stereoselectivity. Even with the poorest acceptor 61b, donor 1 led to a satisfactory β/α ratio of 7:1 for 62b, compared to 2:1 in our previous studies.22
Scope for regioselectivity and synthetic application
The results in Fig. 2d suggest a valuable sensitivity to the nucleophilicity of the acceptors in glycosylation, inspiring us to pursue regioselective glycosylation under the conditions of BF3·Et2O in THF. Thus, 1.0 eq. of glucosyl donor 1 was utilized to glycosylate 1.0 eq. of multihydroxy acceptors (Fig. 3a). First, we evaluated benzylated glucoside 66a and benzoylated glucoside 66b as diol acceptors possessing a primary 6-OH and a secondary 4-OH but different electron densities. Their glycosylation with donor 1 proceeded smoothly, resulting in 6-O-glycosylated disaccharides 67a (83%) and 67b (98%) with complete β-selectivity, indicating the excellent regioselectivity of the primary 6-OH. For the secondary diols 66c and 66d bearing an equatorial 3-OH and an axial 2-OH or 4-OH, only the equatorial 3-OH was glycosylated to give 67c and 67d in over 95% yields. The glycosylated hydroxyl group was confirmed based on the downfield-shifted δH related to the unglycosylated one after acetylation. However, we also observed minor components in the 1H NMR spectra of both 67c and 67d, which were assigned to the α-anomers rather than the regioisomers based on their δC1′ values being less than 100 ppm. An acid-labile glucal 66e, with its 2-OH and 3-OH both in the equatorial orientation, was also found to be effective, providing 3-O-glycosylated 67e with complete β-selectivity. | ||
| Fig. 3 Scope for regioselectivity and synthetic applications. aReactions of diols were conducted at −50 °C. bReactions of triols were conducted at −60 °C. cProducts were isolated by chromatography. dRatios were determined by 1H NMR. e0.1 eq. of BF3·Et2O was used for 67e to obtain β only. f67i was contaminated with 3-O-(β-glucosyl)-galactoside in a 10% yield. | ||
The high regio- and stereoselectivity toward diols encouraged us to extend the studies to more challenging secondary triol acceptors (Fig. 3a).42,43,48,49,52,71 Using THF as the solvent ensures good solubility for these polar and hydrophilic triols. Thus, seven easily accessible glycoside 2,3,4-triols, i.e., glucosides 66f/66g, 6-deoxyglucoside 66h, galactosides 66i/66j, mannoside 66k, and rhamnoside 66l, were utilized with variations in the protecting group at C6/C1 and the anomeric configuration. Gratifyingly, upon treatment with 1.0 eq. of glucosyl donor 1, these 2,3,4-triols were glycosylated at the relatively more nucleophilic hydroxyl group in a highly regioselective manner, i.e., 2-OH on α-glucosides/α-galactosides or 3-OH on β-galactosides/α-mannosides/α-rhamnosides. The corresponding products 67f–67l were successfully isolated with both satisfactory yields (74%–97%) and β-selectivity (β/α = 7:1 to β only). Compared to the protected acceptor 61c with 2-OH (Fig. 2d), glucoside triol 66f and 66g even provided improved yields and β-selectivity for 2-O-glycosylated 67f (97%, β/α = 7:1) and 67g (87%, β/α = 9:1). Removal of the substituent at C6 led to 6-deoxyglucoside 66h and further increased the β/α ratio to 17:1 for 2-O-glycosylated 67h in a high 93% yield.
It is noteworthy that the glycosylation of α-galactoside 66i resulted in 2-O-glycosylated 67i in an 80% yield with complete β-selectivity, while β-galactoside 66j led to the regioisomeric 3-O-glycosylated 67j in a 74% yield, also with complete β-selectivity. These results suggest a practical manipulation of the regioselectivity between O2 and O3 was possible by using α- or β-configured galactoside acceptors. On the other hand, both the mannoside and rhanmnoside triols 66k and 66l were converted into 3-O-glycosylated 67k and 67l with good yields and β-selectivity.
This 1,2-trans and regioselective glycosylation can enable concise access to an oligosaccharide antigen (Fig. 3b) related to Enterococcus faecalis,72 a common nosocomial pathogen that causes severe urinary tract infections, surgical wound infections, pneumonia, bacteremia, etc.73,74 A conventional synthesis from precursor 68 would require at least four chemical steps, including a selective protection of 3-OH with the toxic organotin reagent and a β-selective glucosylation reaction of 3-OH with 2-O-acyl donors. However, previous reports with such disarmed 2-O-acyl donors and similar acceptors raised our concern about the undesired orthoesters that would arise as the predominant products,75,76 and so an alternative β-(1→3)-selective approach with an armed 2-OBn donor would be highly preferred. Thus, we subjected diol 68 to condensation with benzylated imidate 1 under BF3·Et2O (0.2 eq.) in THF at −50 °C. To our delight, the reaction was highly β- and O3-selective, giving the desired 3-O-glucosylated galactoside in a 76% yield (β only, scaled up to 940 mg). The remaining 4-OH was then glycosylated with donor 69 to provide the branched oligosaccharide 70 in an 81% yield. Compared to the conventional four-step route, this two-step route was straightforward, with a considerable decrease in E factor (see the SI† for the detailed calculations), and eliminated concerns about organotin toxicity, orthoester formation, and the need for the additional removal of acyl groups.
Mechanism studies
It was intriguing to explore the intricate mechanism underlying this stereo- and regioselective glycosylation driven by such a simple combination of BF3·Et2O in THF. In fact, our current findings appear to challenge or even contradict the previously proposed stereocontrol mechanisms associated with ether solvents or BF3·Et2O. For instance, ether solvents are generally expected to induce α-selectivity in glucosylation,57–62 which is contrary to the β-selectivity we observed. The model of direct SN2-type attack to α-donors55 could not justify the formation of β-glucosides from β-donors, since no isomerization from β-donors to α-donors was observed during our variable-temperature (VT) NMR studies (Fig. S1†). Some reports suggest glycosyl fluorides are important intermediates or that there is an involvement of in situ HF or SiF4.77–80 However, we found glycosyl fluorides were stable with BF3·Et2O in THF, and our VT NMR studies did not detect any signals for HF or SiF4. These disparities indicate that the BF3·Et2O/THF glycosylation proceeded via a different pathway from the regular BF3·Et2O-promoted glycosylation.Nevertheless, we conducted multiple VT NMR studies to seek out any meaningful intermediates. To minimize interference from other stereocontrol pathways, such as direct SN2 attack on glucosyl α-imidates, we preferred to use glucosyl β-imidates because the resulting β-selectivity should rely more on this unknown mechanism. In terms of stability and accessibility, we chose the β-anomer of the benzylidene-restrained donor 8, i.e., 8β. Although our VT NMR studies did not reveal intermediates that were directly responsible for the selectivity, they provided valuable information, particularly on the formation and transformation of fluoroboron species. We were aware that the promoter BF3·Et2O is unlikely to survive in THF solvent due to the replacement of the coordinated Et2O with THF. Indeed, δH was upfield-shifted to 3.37 (CH2) and 1.12 (CH3) ppm (Fig. S3†) after the addition of BF3·Et2O to deuterated THF (THF-d8) at −70 °C, compared to the reported δH at around 4.2 and 1.5 ppm for BF3·Et2O in CDCl3,81,82 indicating an instant and complete release of Et2O from BF3·Et2O. In the corresponding 19F NMR spectrum, two sets of two singlets, corresponding to 10B and 11B, were observed at −151.6 and −156.5 ppm at −70 °C (Fig. 4b), assigned to BF3·H2O (Fig. S3 and S4†) and BF3·THF (Fig. S5†), respectively.
 | ||
| Fig. 4 Mechanistic studies of highly 1,2-trans and regioselective glycosylation. DFT calculations were performed at the M06-2X/6-311+G(d,p)/PCM(THF)//M06-2X/6-31G(d,p)/PCM(THF) level of theory. See ESI† for mannosylation results and additional details. CIP, contact ion pair; SSIP, solvent-separated ion pair; Int, intermediate; Glc-TS-Hα, transition state in 4H3 conformation for α-attack on the glucosyl cation; Glc-TS-Hβ, transition state in 4H3 conformation for β-attack on the glucosyl cation; Glc-TS-Bα, transition state in B2,5 conformation for α-attack on the glucosyl cation; Glc-TS-Bβ, transition state in B2,5 conformation for β-attack on the glucosyl cation. | ||
Interestingly, these signals of BF3·THF vanished upon the addition of MeOH (0.2 M) as an acceptor to the solution of BF3·Et2O (0.02 M) in THF-d8 at −70 °C (Fig. 4b). Instead, a new set of two singlets was observed at −157.0 ppm, assigned to BF3·MeOH as a minor component (Fig. S6†). Increasing the amount of BF3·Et2O provided BF3·THF in coexistence with BF3·H2O and BF3·MeOH (Fig. S6†). Consequently, the coordination ability order with BF3 appeared to be in the order: H2O > MeOH > THF. Moreover, the signals of BF3·MeOH were broadened and slightly downfield-shifted in the presence of the imidate donor 8β (Fig. 4b), suggesting an interaction between BF3·MeOH and the imidate. This leads to an assumption that the coordinated complexes of BF3 with acceptors (i.e., BF3·ROH) might be the true promoters acting as Brønsted acids, releasing anions [BF3OR]− as nucleophiles for glycosylation. This assumption aligns with an interesting finding in the VT NMR studies, wherein donor 8β remained intact at −50 °C in the absence of acceptors (Fig. S1†), suggesting the involvement of acceptors in initiating the glycosylation. Furthermore, this assumption was experimentally supported by the similar glycosylation results obtained from the control reactions using either commercially available BF3·MeOH or BF3·Et2O/MeOH (Fig. 4a).
We propose that BF3·ROH complexes act as Brønsted acids to activate imidate donors and release anions [BF3OR]− (Fig. 4c). Under this assumption, the resulting anions [BF3OR]− are not only more reactive but also closer to the reaction center than regular alcohols, thereby rationalizing the high regioselectivity in Fig. 3a. Given the similar reaction results with α- or β-imidate donors, the selective outcomes were not associated with the departure of the imidate group. Thereupon the post-activation reaction system presumably consisted of a glycosyl cation, anion [BF3OR]−, inactive trichloroacetamide, and solvent before the stereoselective formation of glycosidic bonds. Therefore, we propose a putative mechanism involving nucleophilic attack of [BF3OR]− on the oxocarbenium–solvent complex Int-2 (Fig. 4c),11 wherein THF can stabilize the oxocarbenium.
Next, we conducted density functional theory (DFT) calculations on the glucosylation and mannosylation of methanol as models (Fig. 4c and Fig. S7†). To simplify the calculations, we employed donors (e.g., 72) with a restrained conformation, replacing benzyl and benzylidene groups with methyl and methylene groups, respectively. Specifically, we focused on the energy differences of the reaction pathways with the 4H3 and B2,5 conformations based on previous reports and conformational analysis.83–85 Calculations at the M06-2X/6-31G**/PCM(THF) quantum chemical level for structure optimization and frequency, with single-point energy correction at the higher M06-2X/6-311+G**/PCM(THF) level, were performed. The computed results for the glycosylation process are depicted in Fig. 4d. Notably, the glucosyl cation favored the 4H3 conformation while the mannosyl cation preferred the B2,5 conformation (Fig. S7†), with both desirable transition states featuring the pseudoequatorial C-2 substituent. The free energy barrier for the β-attack on the glucosyl cation was significantly lower than that for α-attack by 5.1 kcal mol−1. This causes the β-side attack preference and predominate outcome of Int-5β, resulting in the β-glucoside 73β (Fig. 4c) after the release of BF3. Conversely, α-attack on the mannosyl cation led to a more stable transition state than in the case of the β-attack (Fig. S7†). As a result, mannosylation under the same condition primarily afforded the α-product.
Conclusions
In conclusion, we discovered a straightforward approach for 1,2-trans and regioselective glycosylation enabled by a simple yet synergistic combination of BF3·Et2O in THF. Ether solvents, including THF, are typically regarded as promoting 1,2-cis α-selectivity, while BF3·Et2O is considered to be a poor stereocontrol factor. However, their combination permits the construction of multiple 1,2-trans glycosides with remarkable stereo-outcomes, irrespective of whether using α- or β-imidate donors. This ensures facile access to both pyranosides and furanosides. This protocol is applicable to oligosaccharide donors and demonstrates broad compatibility with variations of the protecting groups, including those at O2. Furthermore, the glycosylation of diols or triols under these conditions exhibits exceptional regioselectivity by amplifying the reactivity differences among hydroxyl groups, favoring the one with a relatively higher nucleophilicity. We utilized this approach, avoiding laborious protection and deprotection on the central GalpNAc, to synthesize an oligosaccharide antigen precursor related to Enterococcus faecalis. In addition, experimental studies on the mechanism revealed an unconventional process promoted by the BF3–acceptor complex BF3·ROH, leading to a putative pathway involving nucleophilic attack of the anion [BF3OR]− on the THF-stabilized anomeric center. DFT calculations rationalized the stereoselectivity through comparing the energy barriers during the process.We consider this a green and practical strategy for 1,2-trans glycosides construction. With its high stereo- and regioselectivity, this strategy utilizes a concise, cheap, and eco-friendly protocol to shorten the synthetic routes to obtain complex carbohydrates and reduces the reliance on the protecting groups, thereby enhancing the flexibility of carbohydrate synthesis. Atom-economical imidate donors, especially a mixture of anomers, can be utilized avoiding extra separation and waste of the undesired anomer. Furthermore, the need for noble metals and halogenated solvents is circumvented, making these transformations sustainable. This study demonstrates that a longstanding challenge in chemical synthesis can be addressed using a simple yet often overlooked system.
Author contributions
W. L. and Z. Z. designed the research and wrote the manuscript. Z. W., L. G., J. S., S. P., X. H., and P. Z. conducted the synthetic work. Y. W. and F. Y. conducted VT NMR studies. Z. Z. contributed computational calculations. W. X., X. W., B. Y., P. X., and X. Z. revised the manuscript and fully engaged in discussion.Data availability
All data related to this study, including the characterization data for new chemical compounds, are included within the paper and its ESI† and are available from the authors upon reasonable request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (92478108, 22477136, 21977115 and 21877128), the National Key R&D Program of China (2022YFA1303803 and 2022YFE0105400), the Natural Science Foundation of Zhejiang Province (LDQ23B020001), and the Hangzhou Leading Innovation and Entrepreneurship Team project (TD2022002). We thank Prof. Zhe Wang for the helpful discussion, and Dr Hui-Min Xu from the Public Laboratory Platform in China Pharmaceutical University for NMR assistance.References
- A. Varki, Glycobiology, 2017, 27, 3–49 CrossRef CAS PubMed.
- S. Wisnovsky and C. R. Bertozzi, Curr. Opin. Struct. Biol., 2022, 75, 102395 CrossRef CAS PubMed.
- B. A. H. Smith and C. R. Bertozzi, Nat. Rev. Drug Discovery, 2021, 20, 217–243 CrossRef CAS PubMed.
- X. Cao, X. Du, H. Jiao, Q. An, R. Chen, P. Fang, J. Wang and B. Yu, Acta Pharm. Sin. B, 2022, 12, 3783–3821 CrossRef CAS PubMed.
- W. Yao, D.-C. Xiong, Y. Yang, C. Geng, Z. Cong, F. Li, B.-H. Li, X. Qin, L.-N. Wang, W.-Y. Xue, N. Yu, H. Zhang, X. Wu, M. Liu and X.-S. Ye, Nat. Synth., 2022, 1, 854–863 CrossRef CAS.
- Q. Zhu, Z. Shen, F. Chiodo, S. Nicolardi, A. Molinaro, A. Silipo and B. Yu, Nat. Commun., 2020, 11, 4142 CrossRef CAS PubMed.
- A. A. Joseph, A. Pardo-Vargas and P. H. Seeberger, J. Am. Chem. Soc., 2020, 142, 8561–8564 CrossRef CAS PubMed.
- T. J. Boltje, T. Buskas and G.-J. Boons, Nat. Chem., 2009, 1, 611–622 CrossRef CAS PubMed.
- Y. Ma, Y. Zhang, Y. Huang, Z. Chen, Q. Xian, R. Su, Q. Jiang, X. Wang and G. Xiao, J. Am. Chem. Soc., 2024, 146, 4112–4122 CrossRef CAS PubMed.
- M. Guberman and P. H. Seeberger, J. Am. Chem. Soc., 2019, 141, 5581–5592 CrossRef CAS PubMed.
- P. O. Adero, H. Amarasekara, P. Wen, L. Bohé and D. Crich, Chem. Rev., 2018, 118, 8242–8284 CrossRef CAS PubMed.
- M. M. Nielsen and C. M. Pedersen, Chem. Rev., 2018, 118, 8285–8358 CrossRef CAS PubMed.
- A. V. Demchenko, Handbook of chemical glycosylation, Wiley-VCH, 2008 Search PubMed.
- W. Li and B. Yu, Chem. Soc. Rev., 2018, 47, 7954–7984 RSC.
- D. Crich, J. Am. Chem. Soc., 2021, 143, 17–34 CrossRef CAS PubMed.
- W.-L. Leng, H. Yao, J.-X. He and X.-W. Liu, Acc. Chem. Res., 2018, 51, 628–639 CrossRef CAS PubMed.
- C. S. Bennett and M. C. Galan, Chem. Rev., 2018, 118, 7931–7985 CrossRef CAS PubMed.
- A. I. Tokatly, D. Z. Vinnitskiy, N. E. Ustuzhanina and N. E. Nifantiev, Russ. J. Bioorg. Chem., 2021, 47, 53–70 CrossRef CAS.
- A. G. Volbeda, G. A. van der Marel and J. D. C. Codée, in Protecting Groups: Strategies and Applications in Carbohydrate Chemistry, ed. S. Vidal, Wiley–VCH Verlag GmbH & Co. KGaA, Weinheim, 2018, pp. 1–27. DOI:10.1002/9783527697014.ch1.
- B. S. Komarova, N. E. Ustyuzhanina, Y. E. Tsvetkov and N. E. Nifantiev, in Modern Synthetic Methods in Carbohydrate Chemistry: From Monosaccharides to Complex Glycoconjugates, ed. D. B. Werz and S. Vidal, Wiley-VCH, 2014, pp. 125–159. DOI:10.1002/9783527658947.ch5.
- J. Guo and X.-S. Ye, Molecules, 2010, 15, 7235–7265 CrossRef CAS PubMed.
- X. Liu, Y. Lin, A. Liu, Q. Sun, H. Sun, P. Xu, G. Li, Y. Song, W. Xie, H. Sun, B. Yu and W. Li, Chin. J. Chem., 2022, 40, 443–452 CrossRef CAS.
- X. Liu, Y. Song, A. Liu, Y. Zhou, Q. Zhu, Y. Lin, H. Sun, K. Zhu, W. Liu, N. Ding, W. Xie, H. Sun, B. Yu, P. Xu and W. Li, Angew. Chem., Int. Ed., 2022, 61, e202201510 CrossRef CAS PubMed.
- X. Liu, Y. Lin, W. Peng, Z. Zhang, L. Gao, Y. Zhou, Z. Song, Y. Wang, P. Xu, B. Yu, H. Sun, W. Xie and W. Li, Angew. Chem., Int. Ed., 2022, 61, e202206128 CrossRef CAS PubMed.
- X. Tang, Y. Zhou, Y. Wang, Y. Lin, S. Pan, Q. Che, J. Sang, Z. Gao, W. Zhang, Y. Wang, G. Li, L. Gao, Z. Wang, X. Yang, A. Liu, S. Wang, B. Yu, P. Xu, Z. Wang, Z. Zhang, P. Yang, W. Xie, H. Sun and W. Li, J. Am. Chem. Soc., 2024, 146, 8768–8779 CrossRef CAS PubMed.
- A. Liu, L. Gao, X. Tang, X. Yang, X. Liu, W. Xie, J. Qi and W. Li, Chem. – Eur. J., 2024, 30, e202400946 CrossRef CAS PubMed.
- H. Xu, R. N. Schaugaard, J. Li, H. B. Schlegel and H. M. Nguyen, J. Am. Chem. Soc., 2022, 144, 7441–7456 CrossRef CAS PubMed.
- J. Zeng, R. Wang, S. Zhang, J. Fang, S. Liu, G. Sun, B. Xu, Y. Xiao, D. Fu, W. Zhang, Y. Hu and Q. Wan, J. Am. Chem. Soc., 2019, 141, 8509–8515 CrossRef CAS PubMed.
- L. Wang, H. S. Overkleeft, G. A. van der Marel and J. D. C. Codée, J. Am. Chem. Soc., 2018, 140, 4632–4638 CrossRef CAS PubMed.
- S.-R. Lu, Y.-H. Lai, J. H. Chen, C.-Y. Liu and K.-K. T. Mong, Angew. Chem., Int. Ed., 2011, 50, 7315–7320 CrossRef CAS PubMed.
- M. Liu, K.-M. Liu, D.-C. Xiong, H. Zhang, T. Li, B. Li, X. Qin, J. Bai and X.-S. Ye, Angew. Chem., Int. Ed., 2020, 59, 15204–15208 CrossRef CAS PubMed.
- M. M. Nielsen, T. Holmstrom and C. M. Pedersen, Angew. Chem., Int. Ed., 2022, 61, e202115394 CrossRef CAS PubMed.
- J. P. Issa and C. S. Bennett, J. Am. Chem. Soc., 2014, 136, 5740–5744 CrossRef CAS PubMed.
- D. Zhu, K. N. Baryal, S. Adhikari and J. Zhu, J. Am. Chem. Soc., 2014, 136, 3172–3175 CrossRef CAS PubMed.
- Y. Park, K. C. Harper, N. Kuhl, E. E. Kwan, R. Y. Liu and E. N. Jacobsen, Science, 2017, 355, 162–166 CrossRef CAS PubMed.
- L.-F. Deng, Y. Wang, S. Xu, A. Shen, H. Zhu, S. Zhang, X. Zhang and D. Niu, Science, 2023, 382, 928–935 CrossRef CAS PubMed.
- C. Palo-Nieto, A. Sau and M. C. Galan, J. Am. Chem. Soc., 2017, 139, 14041–14044 CrossRef CAS PubMed.
- H. Yao, M. D. Vu and X. W. Liu, Carbohydr. Res., 2019, 473, 72–81 CrossRef CAS PubMed.
- J. Ling and C. S. Bennett, Asian J. Org. Chem., 2019, 8, 802–813 CrossRef CAS PubMed.
- C. C. J. Loh, Nat. Rev. Chem., 2021, 5, 792–815 CrossRef CAS PubMed.
- F. Yu, J. Li, P. M. DeMent, Y. J. Tu, H. B. Schlegel and H. M. Nguyen, Angew. Chem., Int. Ed., 2019, 58, 6957–6961 CrossRef CAS PubMed.
- Q. Li, S. M. Levi, C. C. Wagen, A. E. Wendlandt and E. N. Jacobsen, Nature, 2022, 608, 74–79 CrossRef CAS PubMed.
- T. Li, T. Li, H. Zhuang, F. Wang, R. R. Schmidt and P. Peng, ACS Catal., 2021, 11, 10279–10287 CrossRef CAS.
- G. Pelletier, A. Zwicker, C. L. Allen, A. Schepartz and S. J. Miller, J. Am. Chem. Soc., 2016, 138, 3175–3182 CrossRef CAS PubMed.
- S. O. Bajaj, E. U. Sharif, N. G. Akhmedov and G. A. O'Doherty, Chem. Sci., 2014, 5, 2230–2234 RSC.
- K. B. Pal, A. Guo, M. Das, G. Báti and X.-W. Liu, ACS Catal., 2020, 10, 6707–6715 CrossRef CAS.
- S. P. Desai, G. Yatzoglou, J. A. Turner and M. S. Taylor, J. Am. Chem. Soc., 2024, 146, 4973–4984 CrossRef CAS PubMed.
- K. A. D'Angelo and M. S. Taylor, J. Am. Chem. Soc., 2016, 138, 11058–11066 CrossRef PubMed.
- M. Tanaka, A. Nakagawa, N. Nishi, K. Iijima, R. Sawa, D. Takahashi and K. Toshima, J. Am. Chem. Soc., 2018, 140, 3644–3651 CrossRef CAS PubMed.
- K. Inaba, Y. Naito, M. Tachibana, K. Toshima and D. Takahashi, Angew. Chem., Int. Ed., 2023, 62, e202307015 CrossRef CAS PubMed.
- A. Nakagawa, M. Tanaka, S. Hanamura, D. Takahashi and K. Toshima, Angew. Chem., Int. Ed., 2015, 54, 10935–10939 CrossRef CAS PubMed.
- Q.-D. Dang, Y.-H. Deng, T.-Y. Sun, Y. Zhang, J. Li, X. Zhang, Y.-D. Wu and D. Niu, Nature, 2024, 632, 313–319 CrossRef CAS PubMed.
- T. J. Wadzinski, A. Steinauer, L. Hie, G. Pelletier, A. Schepartz and S. J. Miller, Nat. Chem., 2018, 10, 644–652 CrossRef CAS PubMed.
- Y. Li, H. Mo, G. Lian and B. Yu, Carbohydr. Res., 2012, 363, 14–22 CrossRef CAS PubMed.
- P. Peng and R. R. Schmidt, Acc. Chem. Res., 2017, 50, 1171–1183 CrossRef CAS PubMed.
- A. Kumar, V. Kumar, R. T. Dere and R. R. Schmidt, Org. Lett., 2011, 13, 3612–3615 CrossRef CAS PubMed.
- S. Chatterjee, S. Moon, F. Hentschel, K. Gilmore and P. H. Seeberger, J. Am. Chem. Soc., 2018, 140, 11942–11953 CrossRef CAS PubMed.
- K.-K. T. Mong, T. Nokami, N. T. T. Tran and P. B. Nhi, in Selective Glycosylations: Synthetic Methods and Catalysts: Synthetic Methods and Catalysts, Wiley, 2017, pp. 59–77. DOI:10.1002/9783527696239.ch3.
- E. C. Lourenco and M. R. Ventura, Carbohydr. Res., 2011, 346, 163–168 CrossRef CAS PubMed.
- H. Satoh, H. S. Hansen, S. Manabe, W. F. van Gunsteren and P. H. Hunenberger, J. Chem. Theory Comput., 2010, 6, 1783–1797 CrossRef CAS PubMed.
- S. S. Nigudkar and A. V. Demchenko, Chem. Sci., 2015, 6, 2687–2704 RSC.
- K. M. Dorst, O. Engström, T. Angles d'Ortoli, H. Mobarak, A. Ebrahemi, U. Fagerberg, D. M. Whitfield and G. Widmalm, Carbohydr. Res., 2024, 535, 109010 CrossRef CAS PubMed.
- D. Zhu, M. Geng, F. Yang and B. Yu, J. Carbohydr. Chem., 2019, 38, 494–508 CrossRef CAS.
- H. M. Christensen, S. Oscarson and H. H. Jensen, Carbohydr. Res., 2015, 408, 51–95 CrossRef CAS PubMed.
- Y. Li and B. Yu, Chem. Commun., 2010, 46, 6060–6062 RSC.
- C.-W. Chang, M.-H. Lin, C.-K. Chan, K.-Y. Su, C.-H. Wu, W.-C. Lo, S. Lam, Y.-T. Cheng, P.-H. Liao, C.-H. Wong and C.-C. Wang, Angew. Chem., Int. Ed., 2021, 60, 12413–12423 CrossRef CAS PubMed.
- M. Emmadi, N. Khan, L. Lykke, K. Reppe, S. G. Parameswarappa, M. P. Lisboa, S.-M. Wienhold, M. Witzenrath, C. L. Pereira and P. H. Seeberger, J. Am. Chem. Soc., 2017, 139, 14783–14791 CrossRef CAS PubMed.
- X. Zou, J. Hu, M. Zhao, C. Qin, Y. Zhu, G. Tian, J. Cai, P. H. Seeberger and J. Yin, J. Am. Chem. Soc., 2022, 144, 14535–14547 CrossRef CAS PubMed.
- S. van der Vorm, J. M. A. van Hengst, M. Bakker, H. S. Overkleeft, G. A. van der Marel and J. D. C. Codée, Angew. Chem., Int. Ed., 2018, 57, 8240–8244 CrossRef CAS PubMed.
- S. van der Vorm, T. Hansen, J. M. A. van Hengst, H. S. Overkleeft, G. A. van der Marel and J. D. C. Codée, Chem. Soc. Rev., 2019, 48, 4688–4706 RSC.
- C. Gouliaras, D. Lee, L. Chan and M. S. Taylor, J. Am. Chem. Soc., 2011, 133, 13926–13929 CrossRef CAS PubMed.
- S. Geiss-Liebisch, S. H. M. Rooijakkers, A. Beczala, P. Sanchez-Carballo, K. Kruszynska, C. Repp, T. Sakinc, E. Vinogradov, O. Holst, J. Huebner and C. Theilacker, J. Biol. Chem., 2012, 287, 37769–37777 CrossRef CAS PubMed.
- A. L. Flores-Mireles, J. N. Walker, M. Caparon and S. J. Hultgren, Nat. Rev. Microbiol., 2015, 13, 269–284 CrossRef CAS PubMed.
- J.-L. Vincent, J. Rello, J. Marshall, E. Silva, A. Anzueto, C. D. Martin, R. Moreno, J. Lipman, C. Gomersall, Y. Sakr and K. Reinhart, J. Am. Med. Assoc., 2009, 302, 2323–2329 CrossRef CAS PubMed.
- S. Dziadek, D. Kowalczyk and H. Kunz, Angew. Chem., Int. Ed., 2005, 44, 7624–7630 CrossRef CAS PubMed.
- D. J. Keith and S. D. Townsend, J. Am. Chem. Soc., 2019, 141, 12939–12945 CrossRef CAS PubMed.
- Y. Manabe, T. Matsumoto, Y. Ikinaga, Y. Tsutsui, S. Sasaya, Y. Kadonaga, A. Konishi, M. Yasuda, T. Uto, C. Dai, K. Yano, A. Shimoyama, A. Matsuda and K. Fukase, Org. Lett., 2021, 24, 6–10 CrossRef PubMed.
- M. M. Nielsen, B. A. Stougaard, M. Bols, E. Glibstrup and C. M. Pedersen, Eur. J. Org. Chem., 2017, 1281–1284 CrossRef CAS.
- M. M. Nielsen, Y. Qiao, Y. Wang and C. M. Pedersen, Eur. J. Org. Chem., 2020, 140–144 CrossRef CAS.
- Q. Long, J. Gao, N. Yan, P. Wang and M. Li, Org. Chem. Front., 2021, 8, 3332–3341 RSC.
- R. J. Gillespie and J. S. Hartman, Can. J. Chem., 1967, 45, 859–863 CrossRef CAS.
- E. L. Myers, C. P. Butts and V. K. Aggarwal, Chem. Commun., 2006, 28, 4434–4436 RSC.
- T. Hosoya, P. Kosma and T. Rosenau, Carbohydr. Res., 2015, 401, 127–131 CrossRef CAS PubMed.
- M. Huang, G. E. Garrett, N. Birlirakis, L. Bohe, D. A. Pratt and D. Crich, Nat. Chem., 2012, 4, 663–667 CrossRef CAS PubMed.
- T. Hansen, L. Lebedel, W. A. Remmerswaal, S. van der Vorm, D. P. A. Wander, M. Somers, H. S. Overkleeft, D. V. Filippov, J. Desire, A. Mingot, Y. Bleriot, G. A. van der Marel, S. Thibaudeau and J. D. C. Codee, ACS Cent. Sci., 2019, 5, 781–788 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4gc04572f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |