
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Identification of catalyst optimization trends for electrocatalytic CO(2) reduction to ethylene†
Stefan J.
Raaijman
a,
Maarten P.
Schellekens
ac,
Yoon Jun
Son
b,
Marc T. M.
Koper
*c and
Paul J.
Corbett
 *a
*a
aEnergy Transition Campus Amsterdam, Shell Global Solutions International B.V., Grasweg 31, 1031 HW Amsterdam, The Netherlands. E-mail: paul.corbett@shell.com
bShell International Exploration and Production Inc., Houston, Texas 77079, USA
cLeiden Institute of Chemistry, Leiden University, PO Box 9502, 2300 RA Leiden, The Netherlands. E-mail: m.koper@lic.leidenuniv.nl
First published on 27th March 2025
Abstract
In this perspective we analyze copper and copper-based electrocatalysts with high ethylene selectivities from the literature to identify global catalyst formulation trends that allow for making catalysts with improved ethylene performance for industrial application. From our analysis, we identified six trends that can aid researchers in creating novel, high selectivity electrocatalysts for the electroreduction of CO(2) to ethylene. These trends were as follows. (i) Tandem-type and (ii) supported-type catalysts perform relatively more poorly than other types of systems. Engineering the nanoenvironment through implementing nanoconfining morphologies (iii) or via the addition of polymeric additives (iv) brings about significant C2H4 selectivity enhancements. (v) Catalyst heterogeneity is an important driver for improving C2H4 selectivity. (vi) Both CO2 and CO can serve as feedstock with little impact on maximum achievable C2H4 selectivity. As we identified during our study that the field lacks reproducibility of catalyst performance and independent reproduction of results, we propose several strategies on how to improve. Finally, we discuss changes that authors can implement to improve the industrial relevancy of their work.
Broader contextOverall, these trends act as a framework for designing catalyst systems with high C2H4 selectivity, increasing their industrial viability. Adopting CO(2)RR technologies at an industrial scale would allow for minimal changes to existing industrial infrastructure and chemical processes whilst reducing the carbon footprint of the molecules and materials in use today, provided that the required CO(2) and (electrical) energy are procured from non-fossil sources. |
1. Introduction
Electrochemical CO2 reduction is a theoretically viable technology to produce various industrially desirable molecules. Products include chemical feedstocks such as CH4,1–3 C2H4,4–6 C2H6,7,8 C3H8,9 EtOH,10–13 PrOH14–17 and various other oxygenates.18–22 Importantly, it is possible to generate these products in a single electrochemical reactor, directly from CO2 and H2O as carbon- and proton sources, respectively. When supplying such a device with renewable power and CO2 procured from non-fossil-based sources, one can produce green23 commodity chemicals with a low carbon footprint. Multiple techno-economic assessments have reported on the industrial viability of carbon-based electrolysis for particular products.24–27 These studies find that ‘simple’, i.e., less reduced, 2-electron (2e−) products have the strongest business case, typically comprising CO, HCOOH and syngas.27 In part, this is because energy efficiency and catalyst selectivity – denoted by the faradaic efficiency (FE) in an electrochemical context – are critical in deciding whether the techno-economics are favorable.24 Nonetheless, even though the electrochemical synthesis of these 2e− products appears promising, industrialization of the technology has been slow with only few (mostly start-up) companies involved in upscaling the technology, including Twelve, CERT Systems, Toshiba, Siemens, Dioxycle, Avantium and GAFT. Overall, CO2 reduction reaction (CO2RR) technologies are still at a low technology readiness level (TRL), especially when considering further reduced (≥4e−) products like C2H4.Industrially, carbon–carbon coupled products appear compelling considering their prevalence in existing processes. However, electrocatalysts that generate such C2+ products typically perform poorly from either an energy efficiency perspective or from a selectivity perspective, making them less economically attractive. Furthermore, additional costs are incurred when a catalyst requires acidic CO2 to react at an alkaline interface to yield high selectivities,28,29 considering that bicarbonate and carbonate ions will form and potentially migrate to the anode where they are converted back into CO2 and mix with the produced O2 in the absence of a separator or when diaphragms or anion-exchange membranes are used as separators.30 Although various strategies exist to mitigate the crossover of these ions, such as e.g., preventing their migration via using cation-exchange membranes (CEMs)31 and/or bipolar membranes (BPMs);32,33 suppressing their concentration through using either humidified CO2 or pure water-based/acidic electrolytes at the cathode;34 or to circumvent their formation entirely by reacting CO instead of CO2, each of these strategies have their own drawbacks. For example, BPM systems incur voltage penalties related to the splitting of water, CEM systems (when using non-acidic anolytes) will result in cation buildup at the cathode, acidic systems require operation at high current densities to increase the local pH so-as to increase the catalyst's C2H4 selectivity in addition to needing acidic anolyte in order to prevent cation buildup, and using CO as a substitute reactant results in additional costs being incurred to convert CO2 to CO via an additional process such as e.g., the reverse water–gas shift (rWGS) process.
Although the electrochemical production of C2+ products is not straightforward and (currently) significantly more costly than 2e− products, C2+ products have the advantage that there are a wide range of mature chemical processes and infrastructure available for their transportation and interconversion on large scale. For instance, C2H435 and EtOH36 are ubiquitous platform chemicals with large markets, where C2H4 is favorable over EtOH from a system perspective on account of it being easier to separate as it is a gas under typical reaction conditions. In addition, replacing conventional, complex lineups for C2H4 production from CO2 with a single-step conversion would in principle allow for significant process simplification. Additionally, alcohols tend to chemically degrade the membranes that are typically used in electrochemical cells,37,38 whereas C2H4 will not.
Based upon these considerations, we investigate herein the potential industrial applicability of the electrochemical production of C2H4 by studying existing literature on the catalyst formulation of electrochemical systems with high C2H4 selectivity to identify trends for improving C2H4 selectivity. We focus our investigation on specifically the cathode electrocatalyst as this component directly impacts the product distribution and cathodic activation losses when operating at a given rate of production. Hence, cathode performance dictates the system's selectivity and a significant proportion of the total energy losses. In addition, maximum current density, which is also largely determined by the electrocatalyst's properties, scales linearly with system size (and thus capital expenditure, CapEx) when normalized to production capacity. Furthermore, catalyst durability plays an integral role with respect to industrial viability with a minimum of 5000 h of operability having been proposed as a figure of merit for commercial deployment of carbon-based electrolysis technology.39 Beyond a minimum threshold (5000 h only equating to ca. 7 months), catalyst durability also influences maintenance frequency and the rate at which consumables (e.g., gas diffusion electrodes or catalyst coated membranes) must be replaced when the catalyst is the limiting component, driving up operational expenditure (OpEx). Therefore, the properties of the cathode catalyst are one of the decisive factors in determining the economic viability of an electrochemical C2H4 production plant. To this end, we reviewed high-performance C2H4 catalyst systems from the literature to determine (a) what is experimentally achievable and (b) which factors determine why certain catalyst systems exhibit superior performance.
In general, determining standardized catalyst performance for industrial application requires a comprehensive study under (i) industrial conditions in (ii) an optimized device. Namely, determining e.g., maximum achievable current densities and energy efficiencies of an industrial system depends not only on the catalyst material itself but also on e.g., its distribution and loading, and the reactant/product mass transport properties of the electrode. Determining lifetime similarly necessitates industrial testing conditions and an optimized system for obtaining representative results. Importantly, accurately estimating standardized catalyst performance through extrapolation of results obtained under non-industrial conditions with unoptimized systems is a difficult task to achieve. This realization is an important one, given that optimizing every single component of an electrochemical cell (or stack) and measuring performance under industrial conditions is typically beyond the scope of most scientific works, which are the source of information for this perspective. As such, values for the maximum current density, cell voltage and lifetime as reported in scientific publications are of limited use in assessing the industrial applicability of the corresponding catalyst systems.
The remaining commonly reported metric is product selectivity, which is mostly determined by intrinsic catalyst activity and less by engineering constraints. Although not without its flaws (such as the effect of feedstock conversion level on selectivity40), such limitations can generally be remedied via a thorough experimental design. Because of this, we consider product selectivity to be a valuable indicator of industrial performance, and we use it in this study to investigate catalysts’ ‘industrial potential’. We achieved this through compiling a list of highly selective C2H4 electrocatalysts, which we subsequently analyzed to identify global trends with respect to favorable catalyst properties and/or characteristics for achieving exceptional C2H4 selectivity. We place an emphasis on bi- and multi-elemental catalyst systems on account of their increased degrees of freedom, which is beneficial for optimization.
Overall, our study shows that 75–80% C2H4 can be obtained reliably under industrially relevant conditions using either CO2 or CO as reactants. Here, we use the word ‘reliably’ to denote ‘reported by multiple authors for different catalyst systems’, rather than ‘having high system stability’. Tandem-type and supported-type catalysts were found to perform relatively more poorly than other types of systems, though their activity can be improved under specific circumstances. We hypothesize that substrates with inherent C2H4 activity make for good supporting materials and provide ample non-copper based materials that could be used to test this. We repeatedly observe the importance of the microenvironment, with nanoconfining morphologies and polymeric additives bringing about the largest C2H4 selectivity enhancement. However, we also identified that reproducibility of catalyst performance and independent reproduction of (promising) results are lacking, for which we propose several strategies for improvement. Finally, we discuss how additional industrial relevance can be achieved with relatively little extra experimental burden.
2. Data discussion
Summary tables of the high selectivity C2H4 electrocatalysts identified in this study, making a minimum of 40% C2H4 for metallic and oxide-derived Cu systems and a minimum of 25% C2H4 for multi-elemental systems, are provided in the ESI.† They have been grouped (categorized) primarily based on their elemental composition, with the exception of polymer-based systems which were grouped separately. We chose to categorize the catalyst systems in this way to maintain some degree of chemical comparability within each group. This has resulted in the following tables:• Pure metallic copper catalysts, summarized in Table S1 (ESI†).
• Oxide-derived copper catalysts, summarized in Table S2 (ESI†).
• Bi-elemental Cu/M catalysts with M = Al, B, Mg, Zn, Sn, Pd, Pb, Ni, Co, Ga, Fe, Au, Ag, [Zr & Hf], Ti, Si, Lanthanides (Ce, [La, Pr, Nd, Eu, Sm & Gd]) and carbon/C are summarized in Tables S3–S22 (ESI†) whilst a final group consisting of bi-elemental catalysts with insufficient sources (≤3) consisting of [Pt, Sb, Bi, Sr, Se, Mo, Mn, Ru, Rh, Sc, Ge, In & W] is summarized in Table S23 (ESI†).
• Multi-elemental Cu/∑M catalysts (≥3 metallic elements), summarized in Table S24 (ESI†).
• Polymeric core/shell-type Cu catalysts consisting of a metallic or oxide-derived copper core with either an organic or inorganic layer/shell, summarized in Table S25 (ESI†).
• Metallic or oxide-derived Cu catalysts post-modified with an organic and/or inorganic coating/overlayer, summarized in Table S26 (ESI†).
The first Section 2.1 will focus on metallic and oxide-derived copper catalysts, to define a baseline of what pure copper is capable of. However, many works have investigated if the inherent selectivity of copper can be improved through the addition of various other components (‘copper-based’ systems). Many such copper-based systems can be categorized as bi-elemental systems, denoted herein as Cu/M (e.g., Cu + co-element M). Bi-elemental systems with M = Al, B, Mg, Zn, Sn, Pd, Pb, Ni, Co, Ga, Fe, Au, Ag, [Zr & Hf], Ti, Si, Lanthanides (Ce, [La, Pr, Nd, Eu, Sm & Gd]) and carbon/C have been summarized in Tables S3–S22 (ESI†), whilst bi-elemental systems having ≤3 sources have been grouped together in a miscellaneous category, comprising [Pt, Sb, Bi, Sr, Se, Mo, Mn, Ru, Rh, Sc, Ge, In & W] and are summarized in Table S23 (ESI†). Considering that the bi-elemental systems were found to be less common than metallic and oxide-derived copper systems, we opted to decrease our selection criteria to systems making ≥25% C2H4 instead of ≥40% C2H4 to increase the likelihood of achieving statistically relevant numbers of sources on a per-element basis for our analysis. A small number of publications even report on multi-elemental systems (total metallic elements of ≥3) with ≥25% C2H4 selectivity, denoted herein as Cu/∑M, as summarized in Table S24 (ESI†). Finally, we identified two additional categories for systems that are differentiated by the presence of an organic and/or inorganic component – either in the form of a polymeric core/shell-type morphology (comprising a metallic or oxide-derived copper core and an organic or inorganic shell, Table S25, ESI†) or metallic or oxide-derived Cu catalysts that have been post-modified with an organic and/or inorganic overlayer, summarized in Table S26 (ESI†).
Compared to metallic and oxide-derived copper, the copper-based systems are significantly more heterogeneous, e.g., having more (initial) chemical states accessible, with many interfaces existing between chemically distinct particles and individual atoms, having large differences in electrical and thermal conductivities, having large variations in surface hydrophobicity, etc. Importantly, even with our reduced selection criterium of ≥25% C2H4, the number of publications reporting on bi- and multi-elemental catalysts of a given composition that meet this criterium typically does not exceed 10. As such, we analyze the entirety of the dataset (including metallic and oxide-derived Cu) from a holistic perspective rather than on a per-elemental basis to identify global trends regarding what makes for a highly selective C2H4 electrocatalyst from Section 2.2 and beyond. With this we mean to say that we look at the overall dataset for trends that are shared across systems rather than elemental composition-specific trends, which we will interchangeably refer to as holistic or global observations/trends.
Although we will look at the dataset from a holistic perspective on account of the small sample size on a per-element basis, brief descriptions of all the tabulated systems have been provided in the ESI.† We opt for not including this information in the main text to allow us to focus on the main goal of this study: to identify and discuss global trends with respect to favorable catalyst properties and/or characteristics for achieving high C2H4 selectivity from electrochemical CO(2) reduction on copper-based catalysts. However, it is the dataset on a whole that gives us the confidence to make the claims we do herein, which is why all investigated systems are included in the ESI† as opposed to only those systems that are directly relevant. Importantly, alkaline CO2 systems are omitted from our analyses (unless stated otherwise) on account of the unsustainable costs associated with maintaining a local alkaline pH in the presence of CO2 which necessitates continual HCO3−/CO32− removal.30 Further details regarding how catalysts were identified and selected are provided in the ESI.† A high-level overview of the catalyst systems studied in this work is provided in Fig. 1, depicting the spread of the C2H4 FEs for various catalyst systems with differing elemental compositions together with the number of identified publications for the various categories.
 | ||
| Fig. 1 High-level overview of the catalyst systems compiled in this work, depicting the maximum C2H4 FE distribution for the catalyst systems as grouped based on their elemental composition, together with the number of sources identified for each of these categories. | ||
2.1. Feedstock agnosticism
For the purposes of this manuscript, metallic copper-based electrocatalysts are used as a reference point of what level of performance is achievable in the absence of convoluting effects, with a summary of their selectivities and characteristics being presented in Table S1 (ESI†). We consider the dataset sufficiently large to omit alkaline CO2RR systems (15 out of 56 entries) from our analysis. Overall, we reliably observe maximum C2H4 FEs between 50–60%, with outliers yielding 77%41 and 93%.42 Notably, for systems that yield ≥55% C2H4, the feedstock distribution is relatively equal with 6 entries using CO2 as a reactant and 4 entries using CO (i.e., a CO2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CO ratio of 1.5×). In addition, many of these systems (7 out of 10 entries) were operated at >|−100| mA cm−2. However, we observe that the majority of the metallic Cu systems consist of the same catalyst, namely sputter-deposited copper. As such, the number of unique metallic Cu catalyst systems is considerably less than the number of entries in the table implies, because each publication is added as a new entry even when the reported catalyst is equal to a different publication.
:CO ratio of 1.5×). In addition, many of these systems (7 out of 10 entries) were operated at >|−100| mA cm−2. However, we observe that the majority of the metallic Cu systems consist of the same catalyst, namely sputter-deposited copper. As such, the number of unique metallic Cu catalyst systems is considerably less than the number of entries in the table implies, because each publication is added as a new entry even when the reported catalyst is equal to a different publication.
As a variation on- and follow-up to metallic copper, we compiled in Table S2 (ESI†) the performance of oxide-derived copper. Oxide-derived was interpreted freely to mean any catalyst with the generic formula CuX, where X constitutes either (i) a highly electronegative element or (ii) an alkali metal. In practice, this translates to X = Li, Na, K, Rb, Cs, N, O, P, S, F, Cl, Br and/or I, and combinations thereof. In alignment with the metallic copper dataset, the oxide-derived dataset was sufficiently large to allow us to omit alkaline CO2RR systems (60 out of 157 entries) from our analysis. We find that the maximum C2H4 FEs reported for oxide-derived Cu catalysts outperform metallic Cu catalysts by ca. 10%, with C2H4 FEs reliably observed between 60–70% (vs. 50–60% for metallic Cu) and outliers yielding 76–85%.5,43–45 For systems that yield ≥55% C2H4, we find a relatively asymmetrical feedstock distribution with 38 entries using CO2 and 8 entries using CO as a reactant (i.e., a CO2:CO ratio of 4.75). Furthermore, it can be observed that relatively few systems (14 out of 46 entries) were operated at >|−100| mA cm−2. Considering the ca. 10% outperformance in maximum C2H4 FEs of oxide-derived Cu vs. metallic Cu, it is also interesting to look at the ≥65% C2H4 range as a comparison. By doing so, we find that the feedstock imbalance (CO2:CO ratio) for oxide-derived systems is significantly reduced, dropping from 4.75 to 2.3 (vs. 1.5 for metallic Cu) whilst high-current density systems become somewhat more common, increasing from 14 out of 46 entries to 9 out of 23 entries (vs. 7 out of 10 entries for metallic Cu).
Metallic and oxide-derived Cu systems exhibit similarities as well. For instance, both CO2 and CO can serve as the feedstock for high selectivity (≥55% C2H4) systems. Additionally, top-tier catalysts (on a selectivity basis) also exhibit similar performance, yielding 77–93%41,42 for metallic Cu systems vs. 76–85%5,43–45 for oxide-derived Cu systems. The main difference seems to be that oxide-derived Cu catalysts yield maximum C2H4 FEs that are ca. 10% higher than metallic Cu catalysts when looking at the ≥55% range. We hypothesize this performance gap it is not unreasonable when considering the propensity of oxide-derived copper to reconstruct into smaller (higher active area) particles which are rich in undercoordinated sites.46 Although not necessarily more active, Kim et al. show that such sites can act as CO reservoirs, resulting in higher overall turnover frequencies (TOFs) towards C2H4 of the ‘real’ catalytically active sites.47
This overview of metallic and oxide-derived Cu systems serves as a benchmark of what can be reasonably expected of primarily copper-based electrocatalysts in terms of C2H4 performance. It also yields our first global observation, namely that both CO2 and CO can be used as feedstock for high selectivity C2H4 electrocatalysts with little difference in maximum achievable performance provided that the alkaline CO2RR effect is accounted for (achieved herein through omitting such systems from the analysis). Although the current discussion focused on metallic and oxide-derived copper systems, the feedstock agnosticism is found to be present also when we consider the entirety of the dataset. This can be seen from Table 1, wherein catalyst systems with the highest C2H4 selectivities as identified in the current work (≥70% C2H4) are summarized. Although this table is discussed in further detail in Section 2.6, it can be observed that both CO2 and CO are present as feedstocks for these top-performing systems, including for various copper-based systems.44,48,49

2.2. Support effect
In this section, we investigate a series of observations we colloquially refer to as the ‘support effect’, pertaining to systems wherein the active catalyst is supported on (or physically mixed with) nm μm−1-sized secondary particles with a different chemical composition than the primary (C2H4-forming) catalyst. We have identified several trends regarding such supported-type catalysts that occur repeatedly across a variety of different systems, allowing us to garner insights for improving C2H4 selectivity. The majority of supported-type systems identified in this work consist of carbon-supported catalyst systems, which are included in Table S22 (ESI†) as part of the Cu/C summary. We find that these carbon-supported catalytic systems exhibit relatively poor C2H4 selectivity overall (i.e., ≤60% C2H4), though outliers yielding between 60–70%50–53 and even up to 81%54 C2H4 exist. Importantly, Cu supported on commercially available, pristine carbon nanoparticles (CNPs, e.g., Ketjen Black46 and Vulcan XC-7255) reliably yield ≤55% C2H4. Increased C2H4 selectivity is primarily observed for systems containing chemically modified carbon supports as prepared via e.g., hetero atom-functionalization,54,56,57 organic functionalization with hetero atom-containing ligands,58 or by in situ decomposition of copper-based complexes50–53 such as metal–organic frameworks (MOFs), covalent organic frameworks (COFs) or transition metal complexes (TMCs). The general poor C2H4 selectivity of supported-type systems is not limited to (unmodified) carbon-based support materials either, with copper catalysts supported on oxidic secondary particles such as e.g., ZrOx,59 ZnOx,60 TiOx,61 SiOx62–64 and CeOx65–67 similarly yielding 50–60% C2H4 at best. Importantly, the industrial viability of oxidic supports is debatable, considering such materials could potentially dissolve in situ, as reported for e.g., SiOx.64,68 A notable exception to the poor overall selectivity of (oxide-) supported copper systems exists in the form of Cu supported on AlOx-based secondary particles, with several such systems having been reported to exhibit high C2H4 selectivity (≥70% C2H4) even under non-alkaline CO2RR conditions.69,70Likely, the poor selectivity of supported-type catalyst systems (i.e., ≤60% C2H4) can be attributed to electrochemical competition between the supporting particles and the primary (C2H4-forming) catalyst through the support's inherent (parasitic) electrocatalytic activity. Namely, most particles used as support materials favor 2e− products (e.g., H2, CO, HCOOH). Crucially, we have identified that supported-type copper systems can outperform unsupported copper systems when ‘ideal’ support materials are used. Ideal in this context refers to supports that are (i) stable in situ, (ii) electrochemically inert and (iii) electrically conductive. This has been demonstrated by Yeo et al.,71 who used exfoliated Mg/Al-based layered double hydroxide (LDH) nanosheets to support commercial Cu nanoparticles (NPs). In their study, they observe similar CORR activity for unsupported vs. supported Cu NPs under ‘standard’ conditions, whilst addition of the supporting material allowed for increased catalyst loadings and operation at increased current densities without negatively impacting C2H4 selectivity. In fact, operation at elevated current densities improved C2H4 selectivity and was possible because it concerned a supported-type catalyst.
Lastly, we observe that it is beneficial to employ a support material which itself has some (≤5% C2H4) intrinsic activity for C2H4 formation. For example, Haihong et al.72 show that Cu single atom catalyst (SAC) sites supported on two-dimensional (2D) Ti3C2Tx MXene nanosheets (Tx denoting surface functional groups) can yield up to 71% C2H4 for the CORR. Importantly, the bare substrate sans Cu sites also exhibits C2H4 activity. Furthermore, the support plays an active role in the catalytic process considering that the supported Cu NPs they also test in their study perform considerably poorer than the Cu-SAC sites deposited on the same secondary particles, whilst normally SACs preferentially generate C1 products rather than carbon-coupled products.73,74 Although supported copper systems with non-copper substrates that have inherent C2H4 activity are rare, we managed to identify four additional publications reporting on such systems, namely Cu supported on: Ti nanotubes61 (55% C2H4), Mg/Al LDHs71 (46% C2H4), carbon-based quantum dots75 (46% C2H4) and Cu supported on a [Ni8(BDP)6] MOF76 (53% C2H4). From the limited data available, we hypothesize that (non-Cu) substrates with inherent C2H4 activity might be key in designing novel, high selectivity C2H4 supported electrocatalysts. To this end, we have identified various non-copper based materials having intrinsic capacity for making (small amounts of) C2H4 that could be investigated as catalyst supports, including Ti-,49,61 Pt-,77,78 Ag,79,80 Ni,81–87 Au-, Cd-,88,89 Ru-,90 Zn-,91 Ce-,92 Pd- and Sn-based93 materials and/or intermetallics thereof,71,85,94–105 various TMCs,106,107 COFs,88,89 MOFs76,108 and enzymes,109–111 and finally a number of metal-free75,112–116 and metal-doped117–120 types of carbons.
We conclude that supported-type catalyst systems have the potential to yield improved C2H4 selectivity under highly specific conditions as described in this section. However, the balance between the support and the copper catalyst is fragile and creating a supported system that outperforms conventional, unsupported (arguably ‘self-supported’) copper-based catalyst systems is an exacting task, with relatively few supported-type systems achieving high C2H4 selectivity.
2.3. Morphological benefits
The next global observation is regarding the beneficial effect of having a 2D nanosheet structure with respect to improving C2H4 selectivity. The importance of the nanosheet morphology was already partially evident when we discussed the support effect, where we noted the superior performance of Cu NP supported on 2D Mg/Al-based LDH nanosheets and Cu-SAC sites supported on Ti3C2Tx nanosheets, with both substrates also exhibiting intrinsic C2H4 activity. However, more supported-type systems containing nanosheets that exhibit C2H4 FEs between 50–80% can be identified, albeit without the intrinsic capability of the secondary particles for making C2H4. These systems include e.g., CuO NPs supported on CuSiO3 lamella63 (52% C2H4), CuOx NCs on Al2O3 nanosheets70 (71% C2H4), CuO NPs on Al2CuO4 nanosheets69 (79% C2H4), Cu/Pd mixed NPs supported on Bi2S3 nanosheets121 (57% C2H4) and CuOx NPs supported on Sm2O3 nanosheets122 (49% C2H4). Importantly, the effect is not limited to only nanosheet-based supported-type systems, with Sn-doped CuO nanosheets,123 Al-doped CuOx nanosheets,124 B-doped CuO ‘nanobundles’ (composed of nanowires and nanosheets),125 graphene oxide nanodots on CuO nanosheets126 and Zn–Cu nanosheet arrays127 all having been reported to exhibit maximum C2H4 FEs in the range of 50–60%. Finally, several truly excellent C2H4 electrocatalysts also consist of nanosheets, with defective Cu nanosheets,5 alloyed hexagonal CuNi128 nanosheets and alloyed hexagonal CuCo129 nanosheets all exhibiting maximum C2H4 FEs of ca. 80%. Together, these results demonstrate the importance of the nanosheet morphology as a driver for high C2H4 selectivity.Although the original publications do not report on this nanosheet effect nor its origin, we hypothesize that the trend might be related to the way that nanosheets stack – with the long sheets preventing intermediates from diffusing away rapidly. Hence, we are arguing for a nanoconfinement effect rather than specifically a nanosheet effect. To corroborate this hypothesis, we enumerate various non-nanosheet catalyst systems where we found the morphology to be especially conducive to trapping intermediates: CuOx NPs trapped in a porous carbon matrix containing Ni-SAC sites,130 yielding a maximum C2H4 FE of 72%; porous Cu2O microparticles impregnated with a Co-based TMC,43 generating 76% C2H4 both in the presence and absence of the cobalt complex; Cu-SAC sites dispersed within the nanopores of a cathodically stable Zr-based MOF,131 making 62% C2H4; ordered CuO particles with a zeolite-like structure, coated with a CuSiO3 layer,132 giving a maximum C2H4 FE of 82%; a Cu/Ce bi-elemental catalyst with a hollow nanofiber morphology reported to make 78% C2H4;133 and finally, a catalyst system consisting of copper sites decorating defective, hollow Au/Ag nanoframes134 that can make up to 77% C2H4. These catalysts’ morphologies are depicted in Fig. 2 and compared to the typical morphology for nanosheet-based catalysts as identified in this work.
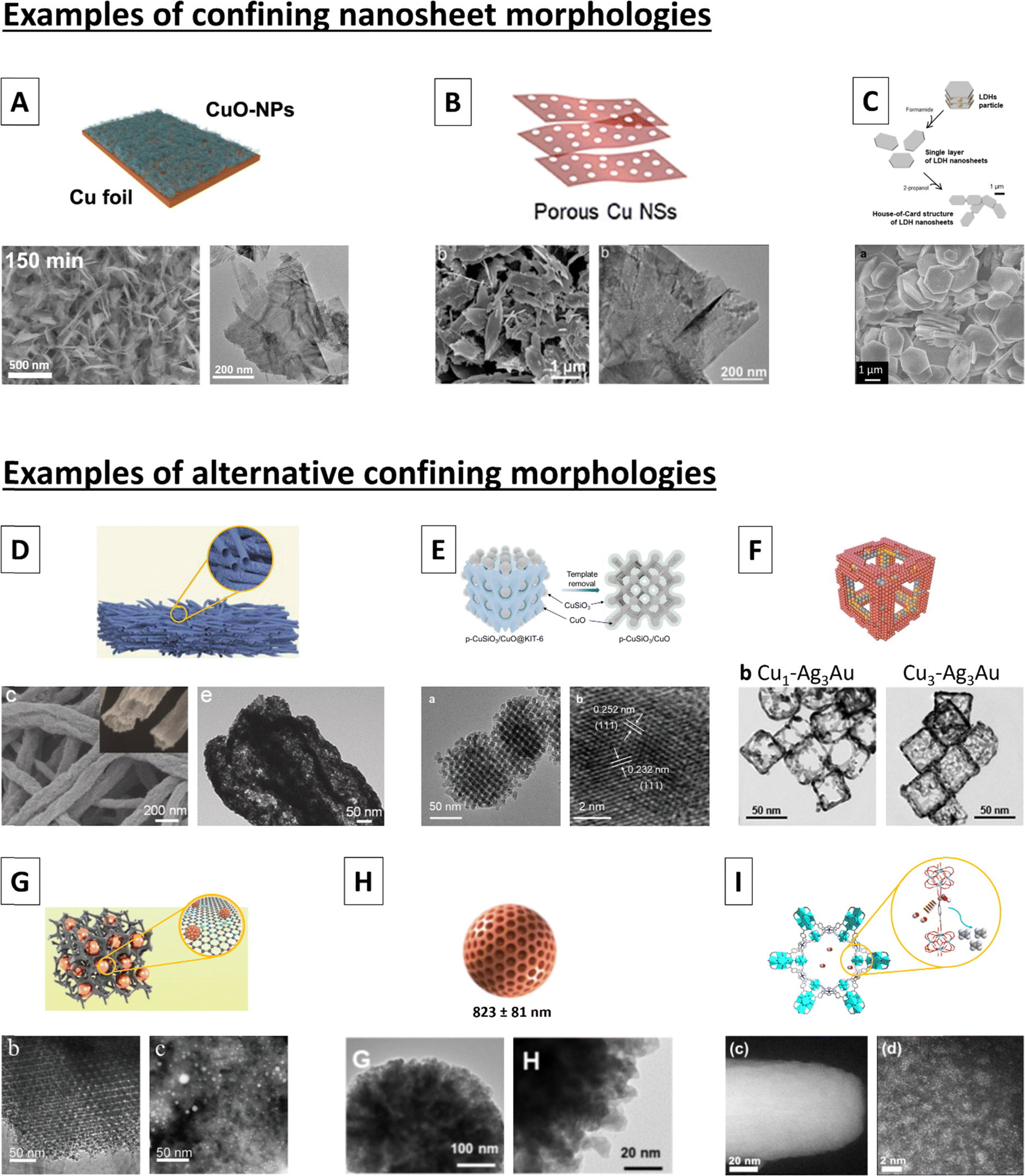 | ||
| Fig. 2 (A)–(C) Morphologies of 2D nanosheet systems compared to (D)–(I) catalyst systems exhibiting nanoconfining morphologies as identified in this perspective. Individual sub-figures are adaptations from the following sources: A,6 B,135 C,71 D,133 E,132 F,134 G,130 H,43 I.131 | ||
Besides sharing a morphology that can effectively trap intermediates, the catalysts previously described and depicted in Fig. 2 all provide exceptionally high C2H4 selectivities. Because of this, we hypothesize that the morphological nanosheet effect stems from increased residence time of the intermediates through nanoconfinement effects.
2.4. Suppressed selectivity of tandem-type systems
In this section we discuss global observations relating to tandem-type catalyst systems, which we define herein as dual-component catalyst systems that contain a dedicated CO2-to-CO conversion catalyst and a separate CO-to-C2H4 conversion catalyst. Considering the focus of this manuscript, our definition only pertains to systems where such catalysts pairs are present in a single device. Hence, the catalysts are generally (though not necessarily136,137) part of the same cathode, which means that the applied bias is equal for both the CO and C2H4 forming constituents. Because of this, aligning the catalysts’ potential such that their respective optima for CO and C2H4 activity match is important for achieving good overall CO2-to-C2H4 selectivity138 in addition to matching the formation and depletion rates of CO through optimizing the relative loadings of the individual components. Many different materials are found to catalyze the electroconversion of CO2 to CO,139 including e.g., Au,140 Ag,141 Zn142 and Ni-SACs.143,144 This high catalyst variability has resulted in the body of literature that we have identified and analyzed being quite extensive, benefitting the statistical significance of our findings.From a holistic perspective, we find that most of these tandem-type catalyst systems yield maximum C2H4 FEs in the range of 50–60%. A small number of outliers exhibiting higher C2H4 selectivities were identified, but exactly those tandem-type systems were observed to have additional differentiating features that strongly correlated with their improved performance. To specify, various tandem-type electrocatalysts yielding ≥60% C2H4 also had nanoconfining morphological features such as e.g., copper sites distributed on the ribs of CO-forming hollow Au/Ag nanoframes134 (77% C2H4); CuOx NPs distributed in the micropores of a carbon matrix containing CO-forming Ni-SAC sites130 (72% C2H4); porous Cu2O microparticles impregnated with a CO-forming Co-based TMC43 (76% C2H4) and a Cu-SAC catalyst dispersed within the nanopores of a morphologically stable CO-forming Zr/Ir-based MOF131 (71% C2H4). For those systems, we hypothesize it is the nanoconfinement effect that allows for their exceptional C2H4 selectivity as opposed to addition of the CO-forming component yielding extraordinary results. In part, this is substantiated by the results of the Cu2O/Co-TMC system43 and the Cu-SAC/Zr,Ir-MOF system,131 for which very similar performance was obtained when CO was used as the reagent in absence of the CO-forming component.
Besides confounding nanoconfinement morphologies, several other high-selectivity tandem-type catalyst systems were identified to contain organic and/or inorganic constituents. Examples of such organic/inorganic additives include e.g., polymeric coatings, addition of ionic liquids to the catalyst layer, thiol-bound surface modifying agents, PTFE coatings, cross-linked ionomer overlayers and carbon-derived overlayers. The presence of such (typically polymeric) components is often found to be beneficial for C2H4 selectivity such as reported by e.g., Zhiji et al. for Cu paired with substituted pyridinium additives145 and by Chen et al. for polyamine-incorporated Cu electrodes.4 This effect is discussed in detail in a perspective by Nam et al.146 wherein they label it the ‘molecular enhancement effect’, showing that various different products can be promoted depending on the specific nature of the molecular constituent. Importantly, all of the remaining tandem-type electrocatalysts that yield ≥60% C2H4 were found to contain such polymeric constituents, consisting of a Cu/Ag system where a Cu NP/Nafion layer was deposited on top of an Ag foil147,148 (76% C2H4) and a Cu/Zn system modified with a Nafion/PVDF coating149 (74% C2H4). In line with our nanoconfinement argument, we hypothesize that the exceptional C2H4 selectivity of these specific tandem-type systems is driven by the molecular enhancement effect rather than originating from the tandem action.
Our last observation regarding tandem-type systems is that they are absent from the best-performing C2H4 electrocatalysts (≥70% C2H4 FE, Table 1) when we omit catalyst systems that are confounded with either nanoconfinement and/or polymeric/molecular enhancement effects. This absence of top-tier, purely tandem-type catalyst systems is noteworthy considering the large quantity of such types of systems present in the dataset. It seems that requiring the presence of two largely independent, though electrically connected, catalysts being located closely together in a single electrochemical cell is not necessarily beneficial to achieving higher C2H4 selectivities. However, we also find that the suppressed activity of tandem-type systems can be improved to a considerable degree by incorporating additional selectivity-enhancing features into the catalyst system, like the previously described confining morphologies or tuning the microenvironment through the addition of e.g., polymeric components.
2.5. Polymeric (molecular) enhancement effect
Although we discussed enhanced C2H4 selectivity for specifically tandem-type systems having been modified with organic and/or inorganic constituents, we find that this effect is generalizable to other types of catalyst systems as well. For instance, we identified a Cu/Pb catalyst supported on a polyaniline-modified carbon substrate150 that substantially out-performs other Cu/Pb systems. However, we observe that the effect is most common for metallic copper and/or oxide-derived copper catalyst systems that contain organic and/or inorganic additives. This is illustrated in Tables S25 and S26 (ESI†), wherein we have summarized catalyst systems consisting of a copper (oxide) core having an organic or inorganic coating/shell, and (oxide-derived) Cu catalysts that have been post-modified with an organic and/or inorganic overlayer, respectively. Although morphologically distinct, we believe the trends we observe for these two types of systems have the same origin. As such, we discuss them in unison.Given the extensive dataset available to us, the effect is best illustrated quantitatively. In the case of copper and copper oxide systems modified with (in-)organic additives, we find that 31 out of 70 table entries (ca. 44%) exhibit C2H4 FEs of ≥55%. This is comparable to what we observe for metallic copper and oxide-derived copper catalysts, for which we find that a combined 104 out of 213 entries (ca. 49%) yield C2H4 FEs of ≥55%. However, when we focus on the proportion of top-performing catalysts (≥70% C2H4) within the ≥55% segment, we observe that 13 out of 31 (ca. 42%) of Cu systems modified with organic and/or inorganic components exhibit excellent C2H4 selectivity whilst this ratio is substantially lower for ‘generic’ metallic copper and oxide-derived copper catalysts, at 24 out of 104 entries (ca. 23%). This statistical outperformance is retained when we instead compare to the >55% segment of the entirety of the dataset sans polymerically enhanced systems, for which we find that 55 out of 224 entries yield exceptional C2H4 selectivity (ca. 25%). Importantly, when we correct for the alkaline CO2RR enhancement effect through omitting such systems from our analysis, the relative outperformance of organic/inorganic-assisted catalyst systems remains, though the differences become smaller. Specifically, 6 out of 17 (ca. 35%) vs. 13 out of 56 (ca. 23%) vs. 35 out of 122 (ca. 29%) of the systems have outstanding C2H4 selectivity in the case of catalyst systems with organic/inorganic additives, ‘generic’ copper and oxide-derived copper systems, and the entirety of the dataset excluding organic/inorganic-assisted systems, respectively. These results exemplify the beneficial, selectivity-enhancing effect that organic and/or inorganic additives can have with respect to C2H4 formation.
Many others have also reported on this effect,4,145,146,151–156 out of which we would like to highlight a recent perspective by Song et al.151 that we found to be highly informative. Due to the large number of chemically distinct possible additives, numerous explanations have been proposed to underly the molecular enhancement effect, including, but not limited to, (i) increased CO2 adsorption capacity, (ii) activation of the CO2 molecule, (iii) tuning of the local environment through e.g., changing hydrophobicity, changing hydrogen bonding or changing the charge distribution near the surface or (iv) regulating mass transport properties through changing the (local) morphology, including creating a confining environment.151 Though the exact mechanism might differ from system to system, the overall trend is an increase in C2H4 selectivity.
2.6. Importance of heterogeneity
Our last global observation is regarding the effect of catalyst heterogeneity for achieving selective C2H4 production. Compared to the previous topics, this concept is more difficult to substantiate. However, having investigated the body of literature reported in this study, we consider this concept to have sufficient scientific merit. The hypothesis that heterogeneity plays a role in enhancing C2H4 activity is based on a diverse, seemingly disjointed set of themes that were found to be relatively consistent across multiple catalyst systems, which we will discuss in this section. The themes are also graphically represented in Fig. 3, depicting the levers which were found to play a role in increasing system heterogeneity and, by association, C2H4 selectivity. | ||
| Fig. 3 Themes centered around catalyst heterogeneity that were identified to be beneficial for C2H4 selectivity. | ||
The first theme looks at heterogeneity from an atomic perspective, by observing that systems containing thermodynamically unstable oxidation states have increased heterogeneity on account of such unstable sites typically being (i) randomly distributed, (ii) having highly variable (local) electron densities and (iii) having an ill-defined lifetime considering they exist in a state from which they could quickly transition into a different, chemically more stable state. Holistically, we observe that these heterogeneous systems comprising thermodynamically unstable oxidation states tend to outperform fully reduced catalyst systems. This topic has been debated in literature extensively and is included as a theme predominantly on account of its prominence in literature on copper-based catalyst systems, though it is also evident from oxide-derived copper studies. Many of those copper-based catalyst studies provide in situ results that show copper to exist in a partially oxidized state under (cathodic) operating conditions, which is typically found to persist for longer upon the addition of the co-element studied.48,66,157–161 Often, this effect is hypothesized to be related to the (temporary) existence of a Cu+ oxidation state,157,158,162,163 similar to what is reported for oxide-derived Cu systems.164,165 Importantly, these same studies also typically report that the *CO coverage is increased concomitantly.
The second theme looks at heterogeneity more from a morphological perspective, observing that catalyst systems with large quantities of (different types of) defects, especially when in the form of 2D surfaces, oftentimes exhibit excellent C2H4 selectivities. This superior catalytic performance is hypothesized to be grounded in the need for a variety of catalytic sites. For example, Scholten et al. have shown that a shortage of defect sites severely inhibits the C–C coupling reaction166 whilst Kim et al. show that diversified surface sites aid in catalysis even if not catalytically active themselves through acting as a reservoir of readily accessible CO that is converted by the actual active sites into C2H4.47 In addition, if large numbers of defects are introduced through leaching away material (as is often done), holes5 and pores43 are formed in 2D and 3D structures, respectively. Besides introducing numerous defect sites, such methods also yield complicated geometries that effectively trap intermediate species, which, as we discussed previously, can also enhance C2H4 selectivity.
A third reoccurring theme in favor of heterogeneity pertains to the importance of the local nano-environment, where we observe that close contact between chemically distinct sites yields enhanced C2H4 activity vs. simple physical mixing of components.91,134,167–173 Heterogeneity of such systems is increased as additional, intermediate chemical states become viable when chemically distinct species are in direct contact through (partial) charge transfer. The benefit of an intimate interface was demonstrated in a study by Sichao et al. who showed that C–C coupling was significantly enhanced for phase-separated Cu/Pd NPs compared to both ordered and disordered alloyed Cu/Pd NPs.174 The fourth theme is similar to the third, and concerns the observation that a non-uniform distribution of non-equal elements yields improved catalytic activity vs. a homogeneous elemental distribution, an effect that is commonly reported for e.g., core/shell-type systems (multi-phase catalyst systems consisting of a core comprised of material A wrapped with a chemically distinct shell comprised of material B),175 and systems in which small amounts of (nonuniformly distributed) dopants are present – typically at the surface.123,176–179
The last theme in favor of heterogeneity is based on the observation that, out of the bi-elemental catalysts, Cu/Ni, Cu/Pd and Cu/Co systems seem to make for some of the best C2H4 electrocatalysts reported to date. When considered independently of copper, Ni, Pd and Co share a notable property, which is that they exhibit a wide range of possible CO2RR chemistries besides high C2H4 selectivity; a characteristic we denote ‘chemical promiscuity’. For example, Ni-based SACs make for superb CO forming catalysts,143 whereas metallic Ni has been reported to make minor quantities of CH4 and C2H4 in addition to H2.81,87,180 By contrast, nickel phosphides have been reported to make complex C3-4 oxygenates181 and acetone18 as well as various other products.182 Pd exhibits a similarly broad product spectrum,183 being capable of selective HCOOH184,185 and CO186 production when coupled with non-Cu metals, and yielding e.g., methanol,187 acetate174 and propanol188 when coupled with Cu. In turn, Co189 has been shown to be able to selectively produce CO190 and HCOOH191 in addition to C–C coupled products such as e.g., ethanal19,192 and EtOH193 depending on the exact catalyst composition. This capacity of making a range of carbon-based and carbon-coupled electroreduction products is relatively uncommon, as most electrocatalysts (with the exception of Cu) are traditionally categorized as being either H2, CO or HCOOH-forming.194 We posit that these elements’ varied chemistry denotes their adaptability, being able to vary their chemical properties to a sufficiently large degree to facilitate various different chemistries, e.g., heterogeneity in the form of their chemical promiscuity.
Although the concept of heterogeneity might seem disproportionally correlated with the superior C2H4 selectivity of oxide-derived Cu catalysts, it is more broadly applicable. This is best illustrated by looking at the most selective catalyst systems identified in this study (C2H4 FEs ≥70%), which are listed in Table 1. We find that metallic and oxide-derived copper systems make up roughly a third (24/68) of the entries in the table, with this ratio largely unchanged if we omit alkaline CO2RR systems from the comparison, at 13/41 entries. The other two-thirds comprise systems wherein additional components are present, either in the form of organic and/or inorganic modifying agents, or in the form of co-element(s). We use this observation as our last argument in favor of system heterogeneity as a driver for C2H4 performance. We posit that the heterogeneity effect predominantly improves selectivity through modulating the local environment, optimizing both the adsorption strengths of intermediates at the catalyst sites as well as the availability of intermediates near, and transportation of intermediates to, these active sites. Clearly, more direct and unambiguous evidence for this hypothesis would be highly desirable, as it impacts on the way we “rationally design” optimal catalysts.
Looking at the catalyst systems described in Table 1, we find that it seems feasible to achieve FEs for C2H4 of at least 75–80% in an industrial setting. Indeed, various authors have reported independently on (diverse) catalyst systems with FEs in the low 80 s at applied current densities of ≥|−150| mA cm−2 under reaction conditions where parasitic carbonate losses are not a concern.128,129,195 Although publications reporting even higher FEs (≥90%) have been reported by a single group for various catalytic systems,42,127,196 these studies were conducted at low current densities (i.e., ≤|−10| mA cm−2). Because of this, we are hesitant to declare such high FEs to be industrially feasible, though the results look promising. Albeit exhibiting slightly lower C2H4 selectivities, the rest of the catalyst systems described in Table 1197–223 are noteworthy in their own right but describing them in more detail is not feasible in the current work.
2.7. Absence of alignment between studies
Although a large amount of knowledge was obtained from the publications analyzed herein, the overall learnings have been limited. We observe that heterogeneity, this time in the form of differences across publications, plays a substantial part in this. Because of this, we posit that improved standardization would benefit the field. Various factors contribute to the inhomogeneity of the results, including:• Reactant type (CO2vs. CO).
• Mass transport properties (dissolved gas vs. gaseous, electrolyte flow vs. stirring vs. static).
• Membrane type (absent, diaphragm, cation-exchange, anion-exchange, bipolar).
• Temperature and temperature control (uncontrolled, electrolyte heating, cell heating).
• Pressure and pressure control (uncontrolled, back pressure regulated, with internal standard(s) present).
• Substrate type (e.g., plates, foams, gas diffusion layers (GDLs)) and substrate elemental composition.
• The choice of electrolyte and its compatibility with the reactant.
• The way in which the data are reported (e.g., in the form of graphs without exact numbers, in individual tables and/or summation tables, or as current efficiencies and/or partial current densities).
As described previously, many factors are important in determining the industrial applicability of a particular catalyst system for the electroformation of C2H4. However, most of these metrics (e.g., maximum achievable current densities, overall energetic costs, system lifetime) necessitate investigating an optimized system, which is generally beyond the scope of scientific literature. As such, we do not advocate for scientific studies to provide industry these metrics (although this would certainly be beneficial). Instead, what we have identified to be missing from existing literature regarding CO(2)RR electrocatalyst development is data comparability, data reproducibility and independent party result replication.
A strong contributor to the difficulties related with inter-publication comparability and result reproducibility is the diversity of electrolyzer systems used across studies, or rather, the effect that cell design has on mass transport properties. Two broad categories of electrolyzers exist: H-cell systems and gas diffusion electrode (GDE) systems. Depending on which type is used, the reactant is supplied either in the dissolved state (H-cell systems) or in the gaseous state (GDE systems). Although various sub-configurations exist, such as e.g., which type of support employed, how gases flow through the system and how water is supplied to the interfaces, the main differentiating factor is mass transport characteristics. Previous works by Chae et al.224 and Tan et al.,225 detail the extent to which the resulting differences in mass transport properties influence the measurement outcome for an otherwise identical catalyst. One way to prove the absence of mass transport limitations in a study could be by reporting the CORR activity alongside the CO2RR activity, considering that the much poorer solubility of CO vs. CO2 (1 mM vs. 33 mM) would significantly worsen mass transportation issues. E.g., Bernasconi et al. use this concept to show that their GDE system does not actually form a 3-phase reaction interface.226 Guaranteeing the reliability of results and ensuring reproducibility across institutes necessitates proving that the measurements were conducted in the absence of mass transportation limitations. From an industrial perspective, we are strongly in favor of using GDE systems as they not only significantly reduce the chances of running into mass transportation limited regimes, but also enable operation at industrially relevant current densities.
A second factor that limits inter-study comparability and reproducibility is the difference in (and absence of) control of process conditions. These process conditions include e.g., (partial) pressure control, temperature control and means of electrolyte delivery and agitation. These process conditions are often not reported, and thus (presumably) not actively measured nor controlled. However, they can fluctuate considerably depending on the local laboratory conditions, exact operating conditions, and the type of electrolyzer employed. Importantly, variations in these process conditions will significantly influence the product distribution, as has been reported for e.g., temperature40,227,228 and feedstock conversion level40 (by way of the reactant's (partial229–231) pressure220,232,233). As such, we consider it a necessity to at least measure and report these process conditions. However, we firmly believe a good measurement involves actively controlling them, such that fluctuations in the operating conditions (e.g., current density, cell voltage) do not result in significant changes in the process conditions.
The third consideration is regarding the lack of a standardized catalyst that can be used as a benchmark to prove the validity of the electrolyzer setup. Although a generic copper catalyst is often included, there is too much variation in its performance on account of differences in e.g. morphology, crystallographic orientation, particle size and size distribution. In addition, GDE manufacturing methodologies and employed substrates can vary significantly, resulting in large variation in catalytic performance. Therefore, we strongly argue that a well-documented, easily obtainable, and affordable catalyst with thoroughly verified performance is required. Likely, the best candidate would be a catalyst that can be made in every lab without the need of extreme conditions or expensive equipment. Although various such catalysts are reported in the papers that make up the dataset described in this perspective, we are of the opinion that a full-fledged study regarding the identification of a robust benchmark catalyst would be of great benefit to the field. Such a study might involve e.g., identifying affordable and reproducible means to synthesize copper nanoparticles, which are subsequently turned into GDEs (and possibly other types of electrodes) via specific, well-documented procedures employing off-the-shelf components, and then have their performance and performance reproducibility investigated under standardized (and actively controlled) conditions. Preferably a brief durability test would also be included, such that authors who adopt this standardized testing methodology could identify possible time-dependent problems their local setup might have.
Besides significant inter-study electrolyzer variability, a lack of control and quantification of important process conditions and the unavailability of a stable, well-defined, and reproducible baseline catalyst system, we also identified the absence of standardized measurement protocols as an additional factor in the poor reproducibility of results across research groups. To draw a parallel with the more mature field of solar cells, a large amount of effort was dedicated to establishing such so-called consensus performance testing protocols.234–236 In part, the absence of these types of protocols in the field of CO(2) electrolysis can be explained by the relative immaturity of the field in concert with the increased complexity of CO(2)RR measurements in general. Although it is evident from other fields that such protocols are an important component in facilitating reproducibility and measurement reliability, it is difficult to adapt field-specific protocols (such as available for solar cells) to a different field, instead necessitating a bottom-up approach in developing such protocols although inspiration can be taken from other fields.
For the field of CO2RR to be able to accelerate, we posit that a minimum of standardization must be implemented so-as to enable objective comparison of research outputs between different groups. Taking inspiration from the field of solar cells, establishing such consensus protocols requires the inputs of numerous independent researchers to be refined into a set of concrete steps regarding which sets of experiments need to be conducted at a minimum, and how to conduct them. Organizing focus group discussions with prominent experts in the field such as during e.g., international field-relevant conferences seems to be a viable route to establishing such protocols.236 Importantly, such a minimum protocol should not exclude research groups based on their financial capability whilst still allowing for measurement results to be compared like-for-like through systemic instructions on which parameters to assess, how to determine them, and how to prove and/or guarantee the validity of the results.
A final issue pertains to the absence of result verification by independent parties. At this moment, <5 of the studies we discuss in this perspective (out of many hundreds) have had their results tested by a third party. Periodically validating the top-performing newly identified catalysts in a systemic manner at an identical workstation would be of significant value to industry, but also to the field at large. An invited publication could be employed to assure that such a practice becomes implemented. We are of the opinion that such a practice would be highly beneficial to the development of CO2 to ethylene electrocatalysts.
2.8. Facilitating industrial relevancy
Although we do not advocate for scientific catalyst studies to directly provide all parameters that are relevant for industry, minor changes can be implemented to improve a work's industrial relevance. Foremost, we advocate for reporting tabulated Faraday efficiency data on a per-product basis for all measurements discussed in the manuscript, including total FEs, current densities and applied overpotentials together with measured cathode-reference electrolyte resistance values, if available. The difficulty associated with accurately comparing results across research groups with respect to non-tabular formats is illustrated in Fig. 4, wherein common graphical representations are illustrated for which extracting accurate FEs is non-trivial/time consuming. Secondly, we advocate for including a brief durability measurement. Although we previously argued that estimating industrially relevant catalyst lifetime necessitates investigating a fully optimized system, an initial indication of lifetime can be obtained with much less effort without the need for a fully optimized system. Arguably, measuring short-term performance benefits from being conducted in a standardized system instead of an optimized system, making it more scientifically feasible. We advocate for measuring performance stability over a period of minimally 12 h, but preferably ≥100 h as certain effects are only observable on longer timescales.38,237,238 In addition, the time-dependent performance should be compared to the behavior of a standardized catalyst measured under otherwise identical conditions to prove that intrinsic catalyst stability is measured rather than auxiliary degradation processes. Although such a durability measurement does not provide much insight into industrial applicability nor catalyst behavior under industrial conditions for prolonged periods of time, it will provide valuable information about the system and the catalyst itself.239 For further information, we direct the reader to a dedicated review on the topic of stability during CO2 electrolysis.240 | ||
| Fig. 4 Various types of graphical reporting styles that increase the difficulty of inter-study result comparison. Individual sub-figures were adapted with permission from the following sources: A & B,241 C & D,242 E & F,243 G, H & I,244 J,165 K & L.245 | ||
A final set of industrially relevant measurements that can be implemented with reasonable ease are regarding catalyst optimization. Catalyst layer thickness (i.e., loading), catalyst-to-ionomer ratio, partial reactant pressure, temperature and electrolyte composition have all been shown to have a significant impact on electrocatalytic performance. Ideally, one would compare catalyst performance trends as a function of all these parameters to be able to rigorously assess which catalyst is objectively better than another. Reporting performance under a range of different (standardized) conditions as determined by e.g. a design of experiment (DoE) approach would yield a more complete picture of catalyst performance and allow for mathematically sound data interpolation to ascertain the conditions where maximum performance is achieved. Although experimentally intensive, investigating the effect of many different variables simultaneously allows for experiment optimization through partial factorial design, significantly reducing the experimental burden. If this would be implemented by default (e.g., for the best-performing catalyst in a study), comparing different publications becomes both easier and more meaningful.
3. Conclusions/outlook
In this study, we sought to identify global trends with respect to favorable catalyst properties and/or characteristics for achieving high C2H4 selectivity from electrochemical CO(2) reduction on copper-based catalysts. High C2H4 selectivity is what we consider to be the best indicative descriptor of potential industrial applicability. We investigated approximately 630 publications reporting on >850 copper-based catalyst systems exceeding a pre-defined C2H4 FE threshold and extracted 6 global trends that reoccur across many catalyst systems with highly varied chemical and morphological characteristics.Specifically, we found that (trend 1) tandem-type and (trend 2) supported-type systems yield suppressed C2H4 selectivity compared to other catalyst systems: ca. 50–60% C2H4vs. ≥70%. However, the C2H4 selectivity of such tandem- and supported-type systems can be improved by (i) implementing confining morphologies, (ii) adding polymeric constituents and/or (iii) employing chemically modified forms of carbon (for supported-type systems). Overall, we conclude that supported-type systems have greater potential than tandem-type systems for industrial application on account of higher demonstrated C2H4 selectivities with an arguably simpler system considering there is no need to match potentials between two distinct catalyst species nor to match catalyst loadings such that reaction rates are aligned.
To continue, we found that catalysts with a nanosheet morphology exhibit above-average C2H4 selectivities (trend 3), which we attribute to the nanoconfining morphology originating from the stacking of these nanosheets. We expand this reasoning to other systems, showing that morphologies conducive to intermediate trapping can yield excellent C2H4 selectivities. We also identified that organic and/or inorganic modifying agents can substantially increase C2H4 selectivity (trend 4), in line with established literature.4,145,146,151–156 The next trend (trend 5) concerns the observation that heterogeneity is important for obtaining selective C2H4 formation, as evidenced by the large differences between top-performing (≥70% C2H4, Table 1) catalyst systems. Overall, we conclude that optimizing the microenvironment is vital for achieving high C2H4 selectivity.
Lastly (trend 6) we observe that both CO2 and CO can be used as a feedstock with little effect on maximum achievable C2H4 performance, applying to metallic and oxide-derived Cu catalysts in general, and to copper-based catalyst systems exhibiting ≥70% FEs for C2H4. This is beneficial from an industrial perspective on account of increased freedom in designing process lineups involving CO2- and CO-reducing electrolyzers.
Although the discussed dataset is comprehensive, we found that a lack of standardization and control of process conditions hindered the learnings that could be extracted. For example, although several independent groups have achieved 75–80% C2H4 for chemically distinct catalyst systems under industrially relevant current densities, most of these catalyst systems’ performances have not been replicated to date. We conclude that reproducibility and a lack of independent result verification is currently a setback for this field. Therefore, we advocate for a thorough study that focuses on identifying, characterizing and rigorously assessing the performance of a simple catalyst system that can be used as a benchmark material across research groups. Also, standardized measurement protocols in line with what is available for more mature electrochemical subdomains such as solar cells need to be developed, ideally including periodic replication of top-performing systems by independent parties. Finally, we discuss various strategies for increasing the industrial relevancy of electrocatalyst-focused research projects.
We propose that next steps should involve investigating the energy efficiencies, lifetime, and behavior under reactant-constrained conditions (i.e., high single-pass conversion rates) of the catalyst systems with the highest C2H4 selectivities described in this work. A follow-up should also include assessing whether the global C2H4 performance-enhancing trends discussed within this article can be combined to further optimize the selectivity of electrocatalysts with high intrinsic catalytic activity. Our findings provide a framework for designing robust and reproducible electrocatalysts with increased C2H4 selectivity, propelling the realization of CO(2)RR technology at industrial scales. The implementation of this technology at an industrial level will allow for reducing the carbon footprint of the molecules and materials in use today with minimal changes to existing industrial infrastructure and chemical processes, provided that the required CO(2) and (electrical) energy are procured from non-fossil sources.
Author contributions
Stefan J. Raaijman: conceptualization, data curation, formal analysis, investigation, visualization, writing – original draft, writing – review & editing. Maarten P. Schellekens: conceptualization, data curation, formal analysis, investigation, visualization, writing – original draft. Yoon Jun Son: visualization. Marc T. M. Koper: conceptualization, validation, writing – review & editing, supervision. Paul J. Corbett: conceptualization, formal analysis, funding acquisition, project administration, resources, supervision, validation, visualization, writing – review & editing.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank our colleagues Ben Rowley and Kai Han of Shell Global Solutions International B.V. for sending relevant publications our way.Notes and references
- Y. Cui, C. Kong, C. Yang, Y. Su, Y. Cheng, D. Yao, G. Chen, K. Song, Z. Zhong, Y. Song, G. Wang, Z. Li and L. Zhuang, ACS Catal., 2023, 13, 11625–11633 CAS.
- X. Zhou, J. Shan, L. Chen, B. Y. Xia, T. Ling, J. Duan, Y. Jiao, Y. Zheng and S.-Z. Qiao, J. Am. Chem. Soc., 2022, 144, 2079–2084 CAS.
- Y. Zheng, Y. Wang, Y. Yuan and H. Huang, ChemNanoMat, 2021, 7, 502–514 CAS.
- X. Chen, J. Chen, N. M. Alghoraibi, D. A. Henckel, R. Zhang, U. O. Nwabara, K. E. Madsen, P. J. A. Kenis, S. C. Zimmerman and A. A. Gewirth, Nat. Catal., 2021, 4, 20–27 CAS.
- B. Zhang, J. Zhang, M. Hua, Q. Wan, Z. Su, X. Tan, L. Liu, F. Zhang, G. Chen, D. Tan, X. Cheng, B. Han, L. Zheng and G. Mo, J. Am. Chem. Soc., 2020, 142, 13606–13613 CrossRef CAS PubMed.
- W. Liu, P. Zhai, A. Li, B. Wei, K. Si, Y. Wei, X. Wang, G. Zhu, Q. Chen, X. Gu, R. Zhang, W. Zhou and Y. Gong, Nat. Commun., 2022, 13, 1877 Search PubMed.
- N. Zhu, X. Zhang, P. Wang, N. Chen, J. Zhu, X. Zheng, Z. Chen, T. Sheng and Z. Wu, ACS Sustainable Chem. Eng., 2024, 12, 2969–2977 CrossRef CAS.
- K. D. Yang, W. R. Ko, J. H. Lee, S. J. Kim, H. Lee, M. H. Lee and K. T. Nam, Angew. Chem., Int. Ed., 2017, 129, 814–818 CrossRef.
- M. Esmaeilirad, Z. Jiang, A. M. Harzandi, A. Kondori, M. Tamadoni Saray, C. U. Segre, R. Shahbazian-Yassar, A. M. Rappe and M. Asadi, Nat. Energy, 2023, 8, 891–900 CrossRef CAS.
- G. Wu, C. Zhu, J. Mao, G. Li, S. Li, X. Dong, A. Chen, Y. Song, G. Feng, X. Liu, Y. Wei, J. Wang, W. Wei and W. Chen, ACS Energy Lett., 2023, 8, 4867–4874 CrossRef CAS.
- B.-B. Zhang, Y.-H. Wang, S.-M. Xu, K. Chen, Y.-G. Yang and Q.-H. Kong, RSC Adv., 2020, 10, 19192–19198 RSC.
- K. A. Adegoke, S. G. Radhakrishnan, C. L. Gray, B. Sowa, C. Morais, P. Rayess, E. R. Rohwer, C. Comminges, K. B. Kokoh and E. Roduner, Sustainable Energy Fuels, 2020, 4, 4030–4038 RSC.
- J. Ding, H. Bin Yang, X.-L. Ma, S. Liu, W. Liu, Q. Mao, Y. Huang, J. Li, T. Zhang and B. Liu, Nat. Energy, 2023, 8, 1386–1394 CrossRef CAS.
- W. Niu, Z. Chen, W. Guo, W. Mao, Y. Liu, Y. Guo, J. Chen, R. Huang, L. Kang, Y. Ma, Q. Yan, J. Ye, C. Cui, L. Zhang, P. Wang, X. Xu and B. Zhang, Nat. Commun., 2023, 14, 4882 CrossRef CAS PubMed.
- X. Wang, P. Ou, A. Ozden, S.-F. Hung, J. Tam, C. M. Gabardo, J. Y. Howe, J. Sisler, K. Bertens, F. P. García de Arquer, R. K. Miao, C. P. O’Brien, Z. Wang, J. Abed, A. S. Rasouli, M. Sun, A. H. Ip, D. Sinton and E. H. Sargent, Nat. Energy, 2022, 7, 170–176 CrossRef CAS.
- H. Phong Duong, J. G. Rivera de la Cruz, N.-H. Tran, J. Louis, S. Zanna, D. Portehault, A. Zitolo, M. Walls, D. V. Peron, M. W. Schreiber, N. Menguy and M. Fontecave, Angew. Chem., Int. Ed., 2023, e202310788 CAS.
- C. Long, K. Wan, Y. Chen, L. Li, Y. Jiang, C. Yang, Q. Wu, G. Wu, P. Xu, J. Li, X. Shi, Z. Tang and C. Cui, J. Am. Chem. Soc., 2024, 146, 4632–4641 CrossRef CAS PubMed.
- M.-G. Kim, Y. Choi, E. Park, C.-H. Cheon, N.-K. Kim, B. K. Min and W. Kim, ACS Appl. Energy Mater., 2020, 3, 11516–11522 CrossRef CAS.
- J. Yin, Z. Yin, J. Jin, M. Sun, B. Huang, H. Lin, Z. Ma, M. Muzzio, M. Shen, C. Yu, H. Zhang, Y. Peng, P. Xi, C.-H. Yan and S. Sun, J. Am. Chem. Soc., 2021, 143, 15335–15343 CrossRef CAS PubMed.
- K. Zhao, X. Nie, H. Wang, S. Chen, X. Quan, H. Yu, W. Choi, G. Zhang, B. Kim and J. G. Chen, Nat. Commun., 2020, 11, 2455 CrossRef CAS PubMed.
- L. Wang, D. C. Higgins, Y. Ji, C. G. Morales-Guio, K. Chan, C. Hahn and T. F. Jaramillo, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 12572–12575 CrossRef CAS PubMed.
- A. Ostovari Moghaddam, S. Mehrabi-Kalajahi, A. Abdollahzadeh, R. Salari, X. Qi, R. Fereidonnejad, S. A. Akaahimbe, M. Nangir, D. A. Uchaev, M. A. Varfolomeev, A. Cabot, A. S. Vasenko and E. A. Trofimov, J. Phys. Chem. Lett., 2024, 15, 5535–5542 CrossRef CAS.
- P. T. Anastas and J. C. Warner, Green chemistry: theory and practice, Oxford University Press, 2000 Search PubMed.
- M. Jouny, W. Luc and F. Jiao, Ind. Eng. Chem. Res., 2018, 57, 2165–2177 CrossRef CAS.
- J. M. Spurgeon and B. Kumar, Energy Environ. Sci., 2018, 11, 1536–1551 RSC.
- H. Shin, K. U. Hansen and F. Jiao, Nat. Sustain., 2021, 4, 911–919 CrossRef.
- G. Leonzio, A. Hankin and N. Shah, Chem. Eng. Res. Des., 2024, 208, 934–955 CrossRef CAS.
- C.-T. Dinh, T. Burdyny, M. G. Kibria, A. Seifitokaldani, C. M. Gabardo, F. P. García de Arquer, A. Kiani, J. P. Edwards, P. De Luna, O. S. Bushuyev, C. Zou, R. Quintero-Bermudez, Y. Pang, D. Sinton and E. H. Sargent, Science, 2018, 360, 783–787 CrossRef CAS PubMed.
- R. L. Cook, R. C. MacDuff and A. F. Sammells, J. Electrochem. Soc., 1990, 137, 607–608 CrossRef CAS.
- T. Alerte, J. P. Edwards, C. M. Gabardo, C. P. O’Brien, A. Gaona, J. Wicks, A. Obradović, A. Sarkar, S. A. Jaffer, H. L. MacLean, D. Sinton and E. H. Sargent, ACS Energy Lett., 2021, 6, 4405–4412 CrossRef CAS.
- W. Fang, W. Guo, R. Lu, Y. Yan, X. Liu, D. Wu, F. M. Li, Y. Zhou, C. He, C. Xia, H. Niu, S. Wang, Y. Liu, Y. Mao, C. Zhang, B. You, Y. Pang, L. Duan, X. Yang, F. Song, T. Zhai, G. Wang, X. Guo, B. Tan, T. Yao, Z. Wang and B. Y. Xia, Nature, 2024, 626, 86–91 CrossRef CAS PubMed.
- A. Pătru, T. Binninger, B. Pribyl and T. J. Schmidt, J. Electrochem. Soc., 2019, 166, F34 CrossRef.
- X. She, L. Zhai, Y. Wang, P. Xiong, M. M.-J. Li, T.-S. Wu, M. C. Wong, X. Guo, Z. Xu, H. Li, H. Xu, Y. Zhu, S. C. E. Tsang and S. P. Lau, Nat. Energy, 2024, 9, 81–91 CrossRef CAS.
- T. Zhang, J. Zhou, T. Luo, J.-Q. Lu, Z. Li, X. Weng and F. Yang, Chem. – Eur. J., 2023, 29, e202301455 CrossRef CAS PubMed.
- H. Zimmermann and R. Walzl, Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, 7th edn, 2011, vol. 13, pp. 465–529 Search PubMed.
- N. Kosaric, Z. Duvnjak, A. Farkas, H. Sahm, S. Bringer-Meyer, O. Goebel and D. Mayer, Ullmann's Encyclopedia of Industrial Chemistry, 2011, pp. 1–72 DOI:10.1002/14356007.a09_587.pub2.
- Y. Sun, F. Bai, J. Liu, S. Sun, Y. Mao, X. Liu, Y. Huang and Y. Chen, J. Phys. Chem. Lett., 2024, 15, 9122–9128 CrossRef CAS PubMed.
- B. Sahin, S. Kimberly Raymond, F. Ntourmas, R. Pastusiak, K. Wiesner-Fleischer, M. Fleischer, E. Simon and O. Hinrichsen, ACS Appl. Mater. Interfaces, 2023, 15, 45844–45854 CrossRef CAS PubMed.
- A. J. Martín, G. O. Larrazábal and J. Pérez-Ramírez, Green Chem., 2015, 17, 5114–5130 RSC.
- M. P. Schellekens, S. J. Raaijman, M. T. M. Koper and P. J. Corbett, Chem. Eng. J., 2024, 483, 149105 CrossRef CAS.
- C. Choi, S. Kwon, T. Cheng, M. Xu, P. Tieu, C. Lee, J. Cai, H. M. Lee, X. Pan, X. Duan, W. A. Goddard and Y. Huang, Nat. Catal., 2020, 3, 804–812 CrossRef CAS.
- I. Merino-Garcia, J. Albo and A. Irabien, Nanotechnology, 2018, 29, 014001 CrossRef CAS PubMed.
- Y. Wang, Q. Cheng, H. Zhang, L. Ma and H. Yang, Chem. Eng. J., 2024, 492, 152254 CrossRef CAS.
- H. P. Duong, N.-H. Tran, G. Rousse, S. Zanna, M. W. Schreiber and M. Fontecave, ACS Catal., 2022, 12, 10285–10293 CrossRef CAS.
- R. Wang, J. Liu, L.-Z. Dong, J. Zhou, Q. Huang, Y.-R. Wang, J.-W. Shi and Y.-Q. Lan, CCS Chem., 2023, 5, 2237–2250 CrossRef CAS.
- Y. Jiang, X. Wang, D. Duan, C. He, J. Ma, W. Zhang, H. Liu, R. Long, Z. Li, T. Kong, X. J. Loh, L. Song, E. Ye and Y. Xiong, Adv. Sci., 2022, 9, 2105292 CrossRef CAS PubMed.
- C. Kim, N. Govindarajan, S. Hemenway, J. Park, A. Zoraster, C. J. Kong, R. R. Prabhakar, J. B. Varley, H.-T. Jung, C. Hahn and J. W. Ager, ACS Catal., 2024, 14, 3128–3138 CAS.
- M. Xie, Y. Shen, W. Ma, D. Wei, B. Zhang, Z. Wang, Y. Wang, Q. Zhang, S. Xie, C. Wang and Y. Wang, Angew. Chem., Int. Ed., 2022, 61, e202213423 CAS.
- H. Bao, Y. Qiu, X. Peng, J.-A. Wang, Y. Mi, S. Zhao, X. Liu, Y. Liu, R. Cao, L. Zhuo, J. Ren, J. Sun, J. Luo and X. Sun, Nat. Commun., 2021, 12, 238 CAS.
- H. Sun, L. Chen, L. Xiong, K. Feng, Y. Chen, X. Zhang, X. Yuan, B. Yang, Z. Deng, Y. Liu, M. H. Rümmeli, J. Zhong, Y. Jiao and Y. Peng, Nat. Commun., 2021, 12, 6823 CAS.
- C. Liu, X.-D. Zhang, J.-M. Huang, M.-X. Guan, M. Xu and Z.-Y. Gu, ACS Catal., 2022, 12, 15230–15240 CAS.
- J. Du, B. Cheng, L. Jiang and Z. Han, Chem. Commun., 2023, 59, 4778–4781 CAS.
- C. F. Wen, M. Zhou, X. Wu, Y. Liu, F. Mao, H. Q. Fu, Y. Shi, S. Dai, M. Zhu, S. Yang, H. F. Wang, P. F. Liu and H. G. Yang, J. Mater. Chem. A, 2023, 11, 12121–12129 CAS.
- H. Zhang, C. Zhu, R. Liu, Q. Fang, S. Song and Y. Shen, Appl. Catal., B, 2024, 343, 123566 CAS.
- L. Fan, Q. Geng, L. Ma, C. Wang, J.-X. Li, W. Zhu, R. Shao, W. Li, X. Feng, Y. Yamauchi, C. Li and L. Jiang, Chem. Sci., 2023, 14, 13851–13859 CAS.
- Y. Huo, X. Peng, X. Liu, H. Li and J. Luo, ACS Appl. Mater. Interfaces, 2018, 10, 12618–12625 CAS.
- Y. Shen, Y. Pan, H. Xiao, H. Zhang, C. Zhu, Q. Fang, Y. Li, L. Lu, L. Ye and S. Song, J. Mater. Chem. A, 2024, 12, 9075–9087 CAS.
- C. F. Wen, M. Zhou, P. F. Liu, Y. Liu, X. Wu, F. Mao, S. Dai, B. Xu, X. L. Wang, Z. Jiang, P. Hu, S. Yang, H. F. Wang and H. G. Yang, Angew. Chem., Int. Ed., 2022, 61, e202111700 CrossRef CAS PubMed.
- P.-P. Guo, Z.-H. He, S.-Y. Yang, W. Wang, K. Wang, C.-C. Li, Y.-Y. Wei, Z.-T. Liu and B. Han, Green Chem., 2022, 24, 1527–1533 RSC.
- Z. Yang, X. Wen, X. Guo, Y. Chen, L. Gao, R. Wei, X. Pan, J. Zhang and G. Xiao, Sep. Purif. Technol., 2024, 332, 125870 CrossRef CAS.
- A. Stalinraja, K. Gopalram, S. Venkatesan, M. J. Swamynathan, S. Ghosh and T. Selvaraj, Electrochim. Acta, 2022, 431, 141078 CrossRef CAS.
- Z. Yang, X. Wen, X. Guo, Y. Chen, R. Wei, L. Gao, X. Pan, J. Zhang and G. Xiao, J. Colloid Interface Sci., 2023, 650, 1446–1456 CrossRef CAS PubMed.
- X. Yuan, S. Chen, D. Cheng, L. Li, W. Zhu, D. Zhong, Z. J. Zhao, J. Li, T. Wang and J. Gong, Angew. Chem., Int. Ed., 2021, 133, 15472–15475 CrossRef.
- T. Zhao, J. Li, J. Liu, F. Liu, K. Xu, M. Yu, W. Xu and F. Cheng, ACS Catal., 2023, 13, 4444–4453 CrossRef CAS.
- S. Hong, H. G. Abbas, K. Jang, K. K. Patra, B. Kim, B.-U. Choi, H. Song, K.-S. Lee, P.-P. Choi, S. Ringe and J. Oh, Adv. Mater., 2023, 35, 2208996 CrossRef CAS PubMed.
- Y. Zhang, H. Zhang, S. Xie, Z. Hou, T. Xu, Y. Shang and Z. Yan, New J. Chem., 2022, 46, 17244–17250 RSC.
- S. Chu, X. Yan, C. Choi, S. Hong, A. W. Robertson, J. Masa, B. Han, Y. Jung and Z. Sun, Green Chem., 2020, 22, 6540–6546 RSC.
- Z.-Y. Zhang, H. Tian, H. Jiao, X. Wang, L. Bian, Y. Liu, N. Khaorapapong, Y. Yamauchi and Z.-L. Wang, J. Mater. Chem. A, 2024, 12, 1218–1232 RSC.
- S. Sultan, H. Lee, S. Park, M. M. Kim, A. Yoon, H. Choi, T.-H. Kong, Y.-J. Ko, H.-S. Oh, Z. Lee, H. Kim, W. Kim and Y. Kwon, Energy Environ. Sci., 2022, 15, 2397–2409 RSC.
- X. Wang, Y. Jiang, K. Mao, W. Gong, D. Duan, J. Ma, Y. Zhong, J. Li, H. Liu, R. Long and Y. Xiong, J. Am. Chem. Soc., 2022, 144, 22759–22766 CrossRef CAS PubMed.
- S. Kwon, J. Zhang, R. Ganganahalli, S. Verma and B. S. Yeo, Angew. Chem., Int. Ed., 2023, 62, e202217252 CrossRef CAS PubMed.
- Y. Zhou and B. S. Yeo, J. Mater. Chem. A, 2020, 8, 23162–23186 RSC.
- M. Li, F. Zhang, M. Kuang, Y. Ma, T. Liao, Z. Sun, W. Luo, W. Jiang and J. Yang, Nano-Micro Lett., 2023, 15, 238 CrossRef CAS PubMed.
- S. Yi, Z. Wang, Z. Wu, X. Yang, H. Wang, J. Yang, X. She and H. Xu, Appl. Surf. Sci., 2025, 681, 161492 CAS.
- C. Chen, X. Yan, S. Liu, Y. Wu, Q. Wan, X. Sun, Q. Zhu, H. Liu, J. Ma, L. Zheng, H. Wu and B. Han, Angew. Chem., Int. Ed., 2020, 132, 16601–16606 Search PubMed.
- L. Huang, Z. Liu, G. Gao, C. Chen, Y. Xue, J. Zhao, Q. Lei, M. Jin, C. Zhu, Y. Han, J. S. Francisco and X. Lu, J. Am. Chem. Soc., 2023, 145, 26444–26451 CrossRef CAS PubMed.
- K. Hara, N. Sonoyama and T. Sakata, Bull. Chem. Soc. Jpn., 2006, 70, 745–754 Search PubMed.
- G. Centi, S. Perathoner, G. Winè and M. Gangeri, Green Chem., 2007, 9, 671–678 RSC.
- R. Shiratsuchi and G. Nogami, J. Electrochem. Soc., 1996, 143, 582 CrossRef CAS.
- W. Zhang, J. Ai, T. Ouyang, L. Yu, A. Liu, L. Han, Y. Duan, C. Tian, C. Chu, Y. Ma, S. Che and Y. Fang, J. Am. Chem. Soc., 2024, 146, 28214–28221 CAS.
- H. Yoshio, M. Akira, T. Ryutaro and S. Shin, Chem. Lett., 1987, 1665–1668 Search PubMed.
- R. L. Cook, R. C. MacDuff and A. F. Sammells, J. Electrochem. Soc., 1990, 137, 187 CrossRef CAS.
- Y. Hori, H. Wakebe, T. Tsukamoto and O. Koga, Electrochim. Acta, 1994, 39, 1833–1839 CrossRef CAS.
- T. Saeki, K. Hashimoto, N. Kimura, K. Omata and A. Fujishima, J. Electroanal. Chem., 1996, 404, 299–302 CrossRef.
- S. Nellaiappan and S. Sharma, ACS Appl. Energy Mater., 2019, 2, 2998–3003 CrossRef CAS.
- Y. Zhou, A. J. Martín, F. Dattila, S. Xi, N. López, J. Pérez-Ramírez and B. S. Yeo, Nat. Catal., 2022, 5, 545–554 CrossRef CAS.
- R. E. Vos and M. T. M. Koper, ACS Catal., 2024, 14, 4432–4440 CrossRef CAS PubMed.
- Y. Xiao, J. Lu, K. Chen, Y. Cao, C. Gong and F.-S. Ke, Angew. Chem., Int. Ed., 2024, 63, e202404738 CrossRef CAS PubMed.
- X. Suo, F. Zhang, Z. Yang, L. Wang, M. Lei, J. A. Gaugler, M. Li, J. Fan, B. P. Thapaliya, I. Popovs, A. S. Ivanov, L. C. Gallington, D.-E. Jiang, Z. Liu and S. Dai, Chem Catal., 2023, 3, 100506 CrossRef CAS.
- B. Cao, F.-Z. Li, S. Han, Q. Xu and J. Gu, ChemElectroChem, 2025, 12, e202400595 CAS.
- Y. Zhang, Q. Zhou, X.-Y. Lu, X.-Y. Zhang, F. Gong and W.-Y. Sun, CCS Chem., 2024, 6, 2950–2960 CrossRef CAS.
- Z. Yang, X. Guo, Y. Chen, L. Gao, R. Wei, X. Pan and G. Xiao, Chem. Eng. J., 2024, 495, 153506 CrossRef CAS.
- C. Liang, B. Kim, S. Yang, L. Yang, C. Francisco Woellner, Z. Li, R. Vajtai, W. Yang, J. Wu, P. J. A. Kenis and P. M. Ajayan, J. Mater. Chem. A, 2018, 6, 10313–10319 CAS.
- S. Singh, R. K. Gautam, K. Malik and A. Verma, J. CO2 Util., 2017, 18, 139–146 CrossRef CAS.
- X. Wang, Z. Jiang, P. Wang, Z. Chen, T. Sheng, Z. Wu and Y. Xiong, Angew. Chem., Int. Ed., 2023, 62, e202313646 CrossRef CAS PubMed.
- R. Kortlever, I. Peters, C. Balemans, R. Kas, Y. Kwon, G. Mul and M. T. M. Koper, Chem. Commun., 2016, 52, 10229–10232 RSC.
- S. Y. Hwang, J. Y. Maeng, G. E. Park, S. Y. Yang, S. Y. Kim, C. K. Rhee and Y. Sohn, Chemosphere, 2023, 338, 139616 CrossRef CAS PubMed.
- Y. J. Kim, J. Y. Maeng, S. Y. Hwang, C. K. Rhee and Y. Sohn, Appl. Catal., B, 2023, 338, 123017 CrossRef CAS.
- Y. J. Kim, J. Y. Maeng, S. Y. Hwang, J. H. Yang, I. Yoon, C. W. Myung, C. K. Rhee and Y. Sohn, Nano Energy, 2023, 118, 108995 CrossRef CAS.
- S. Y. Kim, S. Y. Hwang, J. Y. Maeng, C. K. Rhee and Y. Sohn, Int. J. Hydrogen Energy, 2024, 51, 571–587 CrossRef CAS.
- D. van den Berg, B. Izelaar, S. Fu and R. Kortlever, Catal. Sci. Technol., 2024, 14, 555–561 RSC.
- D. A. Torelli, S. A. Francis, J. C. Crompton, A. Javier, J. R. Thompson, B. S. Brunschwig, M. P. Soriaga and N. S. Lewis, ACS Catal., 2016, 6, 2100–2104 CrossRef CAS.
- A. R. Paris and A. B. Bocarsly, ACS Catal., 2017, 7, 6815–6820 CrossRef CAS.
- J. Y. Maeng, S. Y. Hwang, C. K. Rhee and Y. Sohn, Appl. Surf. Sci., 2023, 631, 157576 CrossRef CAS.
- Y. Ji, X. Lv, R. Wei, A. Guan, C. Yang, Y. Yan, M. Kuang and G. Zheng, Angew. Chem., Int. Ed., 2024, 63, e202411194 CAS.
- T. V. Magdesieva, T. Yamamoto, D. A. Tryk and A. Fujishima, J. Electrochem. Soc., 2002, 149, D89 CrossRef CAS.
- J. Du, B. Cheng, H. Yuan, Y. Tao, Y. Chen, M. Ming, Z. Han and R. Eisenberg, Angew. Chem., Int. Ed., 2023, 62, e202211804 CrossRef CAS PubMed.
- F. Guo, R.-X. Li, S. Yang, X.-Y. Zhang, H. Yu, J. J. Urban and W.-Y. Sun, Angew. Chem., Int. Ed., 2023, 62, e202216232 CrossRef CAS PubMed.
- C. C. Lee, Y. Hu and M. W. Ribbe, Angew. Chem., Int. Ed., 2015, 127, 1235–1238 CrossRef.
- K. Tanifuji, N. Sickerman, C. C. Lee, T. Nagasawa, K. Miyazaki, Y. Ohki, K. Tatsumi, Y. Hu and M. W. Ribbe, Angew. Chem., Int. Ed., 2016, 128, 15862–15865 CrossRef.
- N. S. Sickerman, K. Tanifuji, C. C. Lee, Y. Ohki, K. Tatsumi, M. W. Ribbe and Y. Hu, J. Am. Chem. Soc., 2017, 139, 603–606 CrossRef CAS PubMed.
- J. Wu, S. Ma, J. Sun, J. I. Gold, C. Tiwary, B. Kim, L. Zhu, N. Chopra, I. N. Odeh, R. Vajtai, A. Z. Yu, R. Luo, J. Lou, G. Ding, P. J. A. Kenis and P. M. Ajayan, Nat. Commun., 2016, 7, 13869 CrossRef CAS PubMed.
- X. Zou, M. Liu, J. Wu, P. M. Ajayan, J. Li, B. Liu and B. I. Yakobson, ACS Catal., 2017, 7, 6245–6250 CrossRef CAS.
- R. Feng, Q. Zhu, M. Chu, S. Jia, J. Zhai, H. Wu, P. Wu and B. Han, Green Chem., 2020, 22, 7560–7565 RSC.
- T. Zhang, W. Li, K. Huang, H. Guo, Z. Li, Y. Fang, R. M. Yadav, V. Shanov, P. M. Ajayan, L. Wang, C. Lian and J. Wu, Nat. Commun., 2021, 12, 5265 CrossRef CAS PubMed.
- R. M. Yadav, Z. Li, T. Zhang, O. Sahin, S. Roy, G. Gao, H. Guo, R. Vajtai, L. Wang, P. M. Ajayan and J. Wu, Adv. Mater., 2022, 34, 2105690 CrossRef CAS PubMed.
- A. F. Pérez-Cadenas, C. H. Ros, S. Morales-Torres, M. Pérez-Cadenas, P. J. Kooyman, C. Moreno-Castilla and F. Kapteijn, Carbon, 2013, 56, 324–331 CrossRef.
- A. Abdelwahab, J. Castelo-Quibén, M. Pérez-Cadenas, A. Elmouwahidi, F. J. Maldonado-Hódar, F. Carrasco-Marín and A. F. Pérez-Cadenas, Catalysts, 2017, 7, 25 CrossRef.
- W. Cai, J. Yin, C. Hu, H. Han, J. Ma, Y. Cao and Y. Zhao, Catal. Lett., 2022, 153, 2718–2727 CrossRef.
- Y. Cui, C. Yang, H. Lin, S. Rui, D. Yao, Y. Liao, C. Zhang, Y. Fang, X. Wang, Z. Zhong, Y. Song, G. Wang, L. Zhuang and Z. Li, ACS Catal., 2023, 13, 169–178 CrossRef CAS.
- B. M. Pirzada, F. AlMarzooqi and A. Qurashi, Ultrason. Sonochem., 2025, 112, 107189 CrossRef CAS PubMed.
- J. Liu, P. Li, J. Bi, S. Jia, Y. Wang, X. Kang, X. Sun, Q. Zhu and B. Han, J. Am. Chem. Soc., 2023, 145, 23037–23047 CrossRef CAS PubMed.
- Y. Jiang, C. Choi, S. Hong, S. Chu, T.-S. Wu, Y.-L. Soo, L. Hao, Y. Jung and Z. Sun, Cell Rep. Phys. Sci., 2021, 2, 100356 CrossRef CAS.
- J. Wan, L. Lin, T. Yang and Y. Li, Electrochim. Acta, 2022, 421, 140488 CrossRef CAS.
- Q. Wan, J. Zhang, B. Zhang, D. Tan, L. Yao, L. Zheng, F. Zhang, L. Liu, X. Cheng and B. Han, Green Chem., 2020, 22, 2750–2754 RSC.
- R. Du, Q. Wu, S. Zhang, P. Wang, Z. Li, Y. Qiu, K. Yan, G. I. N. Waterhouse, P. Wang, J. Li, Y. Zhao, W.-W. Zhao, X. Wang and G. Chen, Small, 2023, 19, 2301289 CrossRef CAS PubMed.
- I. Merino-Garcia, J. Albo, J. Solla-Gullón, V. Montiel and A. Irabien, J. CO2 Util., 2019, 31, 135–142 CrossRef CAS.
- Y. Yan, Z. Zhao, J. Zhao, Y. Xu, Y. Xu, Y. Zhao, W. Tang and J.-M. Lee, ACS Mater. Lett., 2021, 3, 1143–1150 CrossRef CAS.
- Y. Yan, Z. Zhao, J. Zhao, W. Tang, W. Huang and J.-M. Lee, J. Mater. Chem. A, 2021, 9, 7496–7502 RSC.
- B. Chen, L. Gong, N. Li, H. Pan, Y. Liu, K. Wang and J. Jiang, Adv. Funct. Mater., 2024, 34, 2310029 CrossRef CAS.
- Q. Mo, S. Li, C. Chen, H. Song, Q. Gao and L. Zhang, ACS Sustainable Chem. Eng., 2024, 12, 6093–6101 CrossRef CAS.
- Q. Li, J. Wu, L. Lv, L. Zheng, Q. Zheng, S. Li, C. Yang, C. Long, S. Chen and Z. Tang, Adv. Mater., 2024, 36, 2305508 CrossRef CAS PubMed.
- D. Tan, B. Wulan, X. Cao and J. Zhang, Nano Energy, 2021, 89, 106460 CrossRef CAS.
- L. Xiong, X. Zhang, H. Yuan, J. Wang, X. Yuan, Y. Lian, H. Jin, H. Sun, Z. Deng, D. Wang, J. Hu, H. Hu, J. Choi, J. Li, Y. Chen, J. Zhong, J. Guo, M. H. Rümmerli, L. Xu and Y. Peng, Angew. Chem., Int. Ed., 2021, 60, 2508–2518 CrossRef CAS PubMed.
- D. Wang, Q. Song, K. Li, Y. Zhou, J. Mao, C. Zhang, Y. Lou, C. Pan, J. Zhang, Y. Zhu and Y. Zhang, J. Mater. Chem. A, 2024, 12, 11968–11974 RSC.
- J. Y. Kim, Y. Kim, C. H. Ryu and H. S. Ahn, Green Chem., 2023, 25, 5290–5295 RSC.
- D. Shu, M. Wang, F. Tian, H. Zhang and C. Peng, J. CO2 Util., 2021, 45, 101444 CrossRef CAS.
- M. Liu, Q. Wang, T. Luo, M. Herran, X. Cao, W. Liao, L. Zhu, H. Li, A. Stefancu, Y.-R. Lu, T.-S. Chan, E. Pensa, C. Ma, S. Zhang, R. Xiao and E. Cortés, J. Am. Chem. Soc., 2024, 146, 468–475 CrossRef CAS PubMed.
- F.-Y. Gao, R.-C. Bao, M.-R. Gao and S.-H. Yu, J. Mater. Chem. A, 2020, 8, 15458–15478 RSC.
- Y. Cui, Y. Cheng, C. Yang, Y. Su, D. Yao, B. Liufu, J. Li, Y. Fang, S. Liu, Z. Zhong, X. Wang, Y. Song and Z. Li, ACS Sustainable Chem. Eng., 2023, 11, 11229–11238 CrossRef CAS.
- M. Z. Iqbal, S. Imteyaz, C. Ghanty and S. Sarkar, J. Ind. Eng. Chem., 2022, 113, 15–31 CrossRef CAS.
- J. Rosen, G. S. Hutchings, Q. Lu, R. V. Forest, A. Moore and F. Jiao, ACS Catal., 2015, 5, 4586–4591 CrossRef CAS.
- J. Chen, D. Wang, X. Yang, W. Cui, X. Sang, Z. Zhao, L. Wang, Z. Li, B. Yang, L. Lei, J. Zheng, L. Dai and Y. Hou, Angew. Chem., Int. Ed., 2023, 62, e202215406 CrossRef CAS PubMed.
- J. C. Weiss, Y. He, D. A. Cullen, A. Benavidez, J. D. Jernigen, H. Zhang, L. Osmieri and P. Zelenay, Small, 2025, 21, 2412162 CrossRef CAS PubMed.
- Z. Han, R. Kortlever, H.-Y. Chen, J. C. Peters and T. Agapie, ACS Cent. Sci., 2017, 3, 853–859 CrossRef CAS PubMed.
- D.-H. Nam, P. De Luna, A. Rosas-Hernández, A. Thevenon, F. Li, T. Agapie, J. C. Peters, O. Shekhah, M. Eddaoudi and E. H. Sargent, Nat. Mater., 2020, 19, 266–276 CrossRef CAS PubMed.
- T. Akter, H. Pan and C. J. Barile, J. Phys. Chem. C, 2022, 126, 10045–10052 CrossRef CAS.
- T. Akter and C. J. Barile, J. Mater. Chem. A, 2023, 11, 11354–11363 RSC.
- H. Pan, T. Akter and C. J. Barile, ACS Appl. Energy Mater., 2022, 5, 12860–12868 CAS.
- S. Han, W. Xia, S. Jia, T. Yao, J. Jiao, M. Wang, X. Dong, J. Yang, D. Zhou, M. He, H. Wu and B. Han, ChemCatChem, 2024, 16, e202300918 CAS.
- D. Song, Y. Lian, M. Wang, Y. Su, F. Lyu, Z. Deng and Y. Peng, eScience, 2023, 3, 100097 CrossRef.
- M. Li, S. Kuang, H. Liu, Q. Fan, S. Zhang and X. Ma, J. Phys. Chem. C, 2023, 127, 3952–3959 CAS.
- L. Xue, Z. Gao, T. Ning, W. Li, J. Li, J. Yin, L. Xiao, G. Wang and L. Zhuang, Angew. Chem., Int. Ed., 2023, 62, e202309519 CAS.
- J. C. Bui, C. Kim, A. J. King, O. Romiluyi, A. Kusoglu, A. Z. Weber and A. T. Bell, Acc. Chem. Res., 2022, 55, 484–494 CAS.
- O. Christensen, S. Zhao, Z. Sun, A. Bagger, J. V. Lauritsen, S. U. Pedersen, K. Daasbjerg and J. Rossmeisl, ACS Catal., 2022, 12, 15737–15749 CrossRef CAS.
- M. Jun, D. Kim, M. Kim, M. Kim, T. Kwon and K. Lee, ACS Omega, 2022, 7, 42655–42663 CrossRef CAS PubMed.
- D. Xiao, X. Bao, M. Zhang, Z. Li, Z. Wang, Y. Gao, Z. Zheng, P. Wang, H. Cheng, Y. Liu, Y. Dai and B. Huang, Chem. Eng. J., 2023, 452, 139358 CrossRef CAS.
- S. Min, Z. Wang, X. Xu, J. He, M. Sun, W. Lin and L. Kang, Appl. Surf. Sci., 2024, 663, 160150 CrossRef CAS.
- M. Ma, L. Xiong, Y. Dong, Q. Bai, W. Hua, Z. Zheng, F. Lyu, Y. Lian, Z. Wei, H. Yuan, Z. Jiao, J. Cheng, D. Song, M. Wang, Z. Xing, J. Zhong, S. Han, Z. Deng and Y. Peng, Adv. Funct. Mater., 2024, 34, 2315667 CrossRef CAS.
- W. Li, L. Li, Q. Xia, S. Hong, L. Wang, Z. Yao, T.-S. Wu, Y.-L. Soo, H. Zhang, T. W. B. Lo, A. W. Robertson, Q. Liu, L. Hao and Z. Sun, Appl. Catal., B, 2022, 318, 121823 CrossRef CAS.
- J. Feng, L. Wu, S. Liu, L. Xu, X. Song, L. Zhang, Q. Zhu, X. Kang, X. Sun and B. Han, J. Am. Chem. Soc., 2023, 145, 9857–9866 CrossRef CAS PubMed.
- X. Yan, C. Chen, Y. Wu, S. Liu, Y. Chen, R. Feng, J. Zhang and B. Han, Chem. Sci., 2021, 12, 6638–6645 RSC.
- R. P. Singh, P. Arora, S. Nellaiappan, C. Shivakumara, S. Irusta, M. Paliwal and S. Sharma, Electrochim. Acta, 2019, 326, 134952 CrossRef CAS.
- R. M. Arán-Ais, F. Scholten, S. Kunze, R. Rizo and B. Roldan Cuenya, Nat. Energy, 2020, 5, 317–325 CrossRef.
- X. He, L. Lin, X. Li, M. Zhu, Q. Zhang, S. Xie, B. Mei, F. Sun, Z. Jiang, J. Cheng and Y. Wang, Nat. Commun., 2024, 15, 9923 CrossRef CAS PubMed.
- B. Roldán Cuenya, F. Scholten, K.-L. C. Nguyen, J. P. Bruce and M. Heyde, Angew. Chem., Int. Ed., 2021, 60, 19169–19175 CrossRef PubMed.
- Y. Zhang, P. Li, C. Zhao, G. Zhou, F. Zhou, Q. Zhang, C. Su and Y. Wu, Sci. Bull., 2022, 67, 1679–1687 Search PubMed.
- Y. Zheng, J. Zhang, Z. Ma, G. Zhang, H. Zhang, X. Fu, Y. Ma, F. Liu, M. Liu and H. Huang, Small, 2022, 18, 2201695 CrossRef CAS PubMed.
- X. Mao, C.-W. Chang, Z. Li, Z. Han, J. Gao, M. Lyons, G. Sterbinsky, Y. Guo, B. Zhang, Y. Wang, X. Wang, D. Han, Q.-H. Yang, Z. Feng and Z. Weng, Adv. Energy Mater., 2024, 14, 2400827 CrossRef CAS.
- S. Wang, H. Chen, W. Lin, W. Zhou, X. Lv, J. Wang and J. Fu, Ind. Eng. Chem. Res., 2022, 61, 16445–16452 CrossRef CAS.
- J. Feng, W. Zhang, D. Shi, Y. Jia, Y. Tang, Y. Meng and Q. Gao, Chem. Sci., 2024, 15, 9173–9182 RSC.
- X. Wu, Z. Tong, Y. Liu, Y. Li, Y. Cheng, J. Yu, P. Cao, C. Zhuang, Q. Shi, N. Liu, X. Liu, H. Liang and H. Li, Nano Res., 2024, 17, 7194–7202 CrossRef CAS.
- D. Kim, H. Yun, J. Kim, C. W. Lee and Y. J. Hwang, J. Mater. Chem. A, 2024, 12, 23780–23788 RSC.
- Y. Ji, Z. Chen, R. Wei, C. Yang, Y. Wang, J. Xu, H. Zhang, A. Guan, J. Chen, T.-K. Sham, J. Luo, Y. Yang, X. Xu and G. Zheng, Nat. Catal., 2022, 5, 251–258 CrossRef CAS.
- J. Huang, J. Dai, J. Zhu, R. Chen, X. Fu, H. Liu and G. Li, J. Catal., 2022, 415, 134–141 CrossRef CAS.
- J. Zhang, C. Guo, S. Fang, X. Zhao, L. Li, H. Jiang, Z. Liu, Z. Fan, W. Xu, J. Xiao and M. Zhong, Nat. Commun., 2023, 14, 1298 CrossRef CAS PubMed.
- X. Chen, D. A. Henckel, U. O. Nwabara, Y. Li, A. I. Frenkel, T. T. Fister, P. J. A. Kenis and A. A. Gewirth, ACS Catal., 2020, 10, 672–682 CrossRef CAS.
- D. Zhou, C. Chen, Y. Zhang, M. Wang, S. Han, X. Dong, T. Yao, S. Jia, M. He, H. Wu and B. Han, Angew. Chem., Int. Ed., 2024, 63, e202400439 CrossRef CAS PubMed.
- Y. Zhou, F. Che, M. Liu, C. Zou, Z. Liang, P. De Luna, H. Yuan, J. Li, Z. Wang, H. Xie, H. Li, P. Chen, E. Bladt, R. Quintero-Bermudez, T.-K. Sham, S. Bals, J. Hofkens, D. Sinton, G. Chen and E. H. Sargent, Nat. Chem., 2018, 10, 974–980 CrossRef CAS PubMed.
- M. Azuma, K. Hashimoto, M. Hiramoto, M. Watanabe and T. Sakata, J. Electroanal. Chem. Interfacial Electrochem., 1989, 260, 441–445 CrossRef CAS.
- K. U. D. Calvinho, A. B. Laursen, K. M. K. Yap, T. A. Goetjen, S. Hwang, N. Murali, B. Mejia-Sosa, A. Lubarski, K. M. Teeluck, E. S. Hall, E. Garfunkel, M. Greenblatt and G. C. Dismukes, Energy Environ. Sci., 2018, 11, 2550–2559 CAS.
- X.-H. Liu, X.-L. Jia, Y.-L. Zhao, R.-X. Zheng, Q.-L. Meng, C.-P. Liu, W. Xing and M.-L. Xiao, Adv. Sen. Energy Mater., 2023, 2, 100073 CrossRef.
- A. Mustafa, Y. Shuai, B. G. Lougou, Z. Wang, S. Razzaq, J. Zhao and J. Shan, Chem. Eng. Sci., 2021, 245, 116869 CAS.
- R. Kortlever, C. Balemans, Y. Kwon and M. T. M. Koper, Catal. Today, 2015, 244, 58–62 CrossRef CAS.
- X. Bai, W. Chen, C. Zhao, S. Li, Y. Song, R. Ge, W. Wei and Y. Sun, Angew. Chem., Int. Ed., 2017, 129, 12387–12391 CrossRef.
- H. Huang, H. Jia, Z. Liu, P. Gao, J. Zhao, Z. Luo, J. Yang and J. Zeng, Angew. Chem., Int. Ed., 2017, 56, 3594–3598 CAS.
- L. Lu, X. Sun, J. Ma, D. Yang, H. Wu, B. Zhang, J. Zhang and B. Han, Angew. Chem., Int. Ed., 2018, 57, 14149–14153 CAS.
- M. Rahaman, K. Kiran, I. Z. Montiel, V. Grozovski, A. Dutta and P. Broekmann, Green Chem., 2020, 22, 6497–6509 CAS.
- M. Usman, M. Humayun, M. D. Garba, L. Ullah, Z. Zeb, A. Helal, M. H. Suliman, B. Y. Alfaifi, N. Iqbal, M. Abdinejad, A. A. Tahir and H. Ullah, Nanomaterials, 2021, 11, 2029 CAS.
- M. Wang, K. Torbensen, D. Salvatore, S. Ren, D. Joulié, F. Dumoulin, D. Mendoza, B. Lassalle-Kaiser, U. Işci, C. P. Berlinguette and M. Robert, Nat. Commun., 2019, 10, 3602 Search PubMed.
- S. Gao, Y. Lin, X. Jiao, Y. Sun, Q. Luo, W. Zhang, D. Li, J. Yang and Y. Xie, Nature, 2016, 529, 68–71 CAS.
- Q. Li, Y. Hou, J. Yin and P. Xi, Catalysts, 2023, 13, 1384 CAS.
- J. Du, S. Li, S. Liu, Y. Xin, B. Chen, H. Liu and B. Han, Chem. Sci., 2020, 11, 5098–5104 CAS.
- Y. Hori, Modern Aspects of Electrochemistry, Springer, New York, 2008, vol. 42, pp. 89–189 Search PubMed.
- H. Wu, L. Huang, J. Timoshenko, K. Qi, W. Wang, J. Liu, Y. Zhang, S. Yang, E. Petit, V. Flaud, J. Li, C. Salameh, P. Miele, L. Lajaunie, B. Roldán Cuenya, D. Rao and D. Voiry, Nat. Energy, 2024, 9, 422–433 CrossRef CAS.
- A. Marcos-Madrazo, C. Casado-Coterillo, J. Iniesta and A. Irabien, Membranes, 2022, 12, 783 CrossRef CAS PubMed.
- J. Han, B. Tu, P. An, J. Zhang, Z. Yan, X. Zhang, C. Long, Y. Zhu, Y. Yuan, X. Qiu, Z. Yang, X. Huang, S. Yan and Z. Tang, Adv. Mater., 2024, 36, 2313926 CrossRef CAS PubMed.
- S. S. Lavate and R. Srivastava, Energy Adv., 2024, 3, 2801–2811 RSC.
- Y. Wang, J. Wang, R. Cai, J. Zhang, S. Xia, Z. Li, C. Yu, J. Wu, P. Wang and Y. Wu, Adv. Funct. Mater., 2024, 2417764 Search PubMed.
- M. Zhong, K. Tran, Y. Min, C. Wang, Z. Wang, C.-T. Dinh, P. De Luna, Z. Yu, A. S. Rasouli, P. Brodersen, S. Sun, O. Voznyy, C.-S. Tan, M. Askerka, F. Che, M. Liu, A. Seifitokaldani, Y. Pang, S.-C. Lo, A. Ip, Z. Ulissi and E. H. Sargent, Nature, 2020, 581, 178–183 CrossRef CAS PubMed.
- Y. Yao, T. Shi, W. Chen, J. Wu, Y. Fan, Y. Liu, L. Cao and Z. Chen, Nat. Commun., 2024, 15, 1257 CrossRef CAS PubMed.
- Y. Sha, J. Zhang, X. Cheng, M. Xu, Z. Su, Y. Wang, J. Hu, B. Han and L. Zheng, Angew. Chem., Int. Ed., 2022, 61, e202200039 CrossRef CAS PubMed.
- Z.-Y. Zhang, H.-B. Wang, F.-F. Zhang, J.-W. Li, X.-Z. Hu, S.-W. Yan, Y.-M. Bai, X. Zhang, G.-R. Shen, P.-F. Yin, J. Yang, C.-K. Dong, J. Mao, H. Liu and X.-W. Du, Rare Met., 2024, 43, 1513–1523 CrossRef CAS.
- W. Zhou, H. Zheng, X. Li, N. Meng, C. Feng, H. Yang, Q. Hu and C. He, Adv. Funct. Mater., 2024, 34, 2311226 CAS.
- H. Luo, B. Li, J.-G. Ma and P. Cheng, Angew. Chem., Int. Ed., 2022, 61, e202116736 CrossRef CAS PubMed.
- H. Tao, F. Wang, Z. Zhang and S. Min, Sustainable Energy Fuels, 2023, 7, 2991–2996 RSC.
- B. Cheng, J. Du, H. Yuan, Y. Tao, Y. Chen, J. Lei and Z. Han, ACS Appl. Mater. Interfaces, 2022, 14, 27823–27832 CrossRef CAS PubMed.
- W. Xue, H. Liu, X. Chen, X. Yang, R. Yang, Y. Liu, M. Li, X. Yang, B. Y. Xia and B. You, Sci. China Chem., 2023, 66, 1834–1843 CrossRef CAS.
- G. Dong, G. Wang, J. Cheng, M. Li, Z. Liang, D. Geng and W. Tang, Appl. Catal., B, 2024, 342, 123444 CrossRef CAS.
- Y. Wang, X. Wei, Y. Li, J. Luo, L. Chen and J. Shi, Chem. Eng. J., 2024, 485, 149800 CrossRef CAS.
- F. Li, A. Thevenon, A. Rosas-Hernández, Z. Wang, Y. Li, C. M. Gabardo, A. Ozden, C. T. Dinh, J. Li, Y. Wang, J. P. Edwards, Y. Xu, C. McCallum, L. Tao, Z.-Q. Liang, M. Luo, X. Wang, H. Li, C. P. O’Brien, C.-S. Tan, D.-H. Nam, R. Quintero-Bermudez, T.-T. Zhuang, Y. C. Li, Z. Han, R. D. Britt, D. Sinton, T. Agapie, J. C. Peters and E. H. Sargent, Nature, 2020, 577, 509–513 CrossRef CAS PubMed.
- L. Bian, Z.-Y. Zhang, H. Tian, N.-N. Tian, Z. Ma and Z.-L. Wang, Chin. J. Catal., 2023, 54, 199–211 CrossRef CAS.
- C. Zhang, Y. Gu, Q. Jiang, Z. Sheng, R. Feng, S. Wang, H. Zhang, Q. Xu, Z. Yuan and F. Song, Nano-Micro Lett., 2024, 17, 66 CrossRef PubMed.
- J. Liu, K. Yu, Z. Qiao, Q. Zhu, H. Zhang and J. Jiang, ChemSusChem, 2023, 16, e202300601 CrossRef CAS PubMed.
- Q. He, H. Li, Z. Hu, L. Lei, D. Wang and T.-T. Li, Angew. Chem., Int. Ed., 2024, 63, e202407090 CrossRef CAS PubMed.
- J.-Y. Kim, D. Hong, J.-C. Lee, H. G. Kim, S. Lee, S. Shin, B. Kim, H. Lee, M. Kim, J. Oh, G.-D. Lee, D.-H. Nam and Y.-C. Joo, Nat. Commun., 2021, 12, 3765 CrossRef CAS PubMed.
- A. Thevenon, A. Rosas-Hernández, J. C. Peters and T. Agapie, Angew. Chem., Int. Ed., 2019, 131, 17108–17114 CrossRef.
- X. Y. Zhang, Z. X. Lou, J. Chen, Y. Liu, X. Wu, J. Y. Zhao, H. Y. Yuan, M. Zhu, S. Dai, H. F. Wang, C. Sun, P. F. Liu and H. G. Yang, Nat. Commun., 2023, 14, 7681 CrossRef CAS PubMed.
- H. Bai, T. Cheng, S. Li, Z. Zhou, H. Yang, J. Li, M. Xie, J. Ye, Y. Ji, Y. Li, Z. Zhou, S. Sun, B. Zhang and H. Peng, Sci. Bull., 2021, 66, 62–68 CrossRef CAS PubMed.
- W. Yang, H. Liu, Y. Qi, Y. Li, Y. Cui, L. Yu, X. Cui and D. Deng, J. Energy Chem., 2023, 85, 102–107 CrossRef CAS.
- J. Wang, W. Li, J. Peng, S. Shang, X. Fu, G. He and Q. Zhang, Vacuum, 2024, 222, 113017 CrossRef CAS.
- Y. Wang, Z. Wang, C.-T. Dinh, J. Li, A. Ozden, M. Golam Kibria, A. Seifitokaldani, C.-S. Tan, C. M. Gabardo, M. Luo, H. Zhou, F. Li, Y. Lum, C. McCallum, Y. Xu, M. Liu, A. Proppe, A. Johnston, P. Todorovic, T.-T. Zhuang, D. Sinton, S. O. Kelley and E. H. Sargent, Nat. Catal., 2019, 3, 98–106 Search PubMed.
- Z. Wang, Y. Li, X. Zhao, S. Chen, Q. Nian, X. Luo, J. Fan, D. Ruan, B.-Q. Xiong and X. Ren, J. Am. Chem. Soc., 2023, 145, 6339–6348 CAS.
- Y. Chae, K. Kim, H. Yun, D. Kim, W. Jung, Y. J. Hwang, U. Lee, D. K. Lee, B. K. Min, W. Choi and D. H. Won, J. Mater. Chem. A, 2023, 11, 7025–7033 RSC.
- Y. C. Tan, W. K. Quek, B. Kim, S. Sugiarto, J. Oh and D. Kai, ACS Energy Lett., 2022, 7, 2012–2023 CAS.
- F. Bernasconi, A. Senocrate, P. Kraus and C. Battaglia, EES Catal., 2023, 1, 1009–1016 CAS.
- S. T. Ahn, I. Abu-Baker and G. T. R. Palmore, Catal. Today, 2017, 288, 24–29 CAS.
- H. Yoshio, K. Katsuhei, M. Akira and S. Shin, Chem. Lett., 1986, 897–898 Search PubMed.
- H. Song, J. T. Song, B. Kim, Y. C. Tan and J. Oh, Appl. Catal., B, 2020, 272, 119049 CAS.
- M. Moradzaman, C. S. Martínez and G. Mul, Sustainable Energy Fuels, 2020, 4, 5195–5202 CAS.
- M. Ma, W. Deng, A. Xu, D. Hochfilzer, Y. Qiao, K. Chan, I. Chorkendorff and B. Seger, Energy Environ. Sci., 2022, 15, 2470–2478 CAS.
- B. Sahin, J. J. Leung, E. Magori, S. Laumen, A. Tawil, E. Simon and O. Hinrichsen, Energy Technol., 2022, 10, 2200972 CAS.
- R. Kas, R. Kortlever, H. Yılmaz, M. T. M. Koper and G. Mul, ChemElectroChem, 2015, 2, 354–358 CAS.
- M. V. Khenkin, E. A. Katz, A. Abate, G. Bardizza, J. J. Berry, C. Brabec, F. Brunetti, V. Bulović, Q. Burlingame, A. Di Carlo, R. Cheacharoen, Y.-B. Cheng, A. Colsmann, S. Cros, K. Domanski, M. Dusza, C. J. Fell, S. R. Forrest, Y. Galagan, D. Di Girolamo, M. Grätzel, A. Hagfeldt, E. von Hauff, H. Hoppe, J. Kettle, H. Köbler, M. S. Leite, S. Liu, Y.-L. Loo, J. M. Luther, C.-Q. Ma, M. Madsen, M. Manceau, M. Matheron, M. McGehee, R. Meitzner, M. K. Nazeeruddin, A. F. Nogueira, Ç. Odabaşı, A. Osherov, N.-G. Park, M. O. Reese, F. De Rossi, M. Saliba, U. S. Schubert, H. J. Snaith, S. D. Stranks, W. Tress, P. A. Troshin, V. Turkovic, S. Veenstra, I. Visoly-Fisher, A. Walsh, T. Watson, H. Xie, R. Yıldırım, S. M. Zakeeruddin, K. Zhu and M. Lira-Cantu, Nat. Energy, 2020, 5, 35–49 CrossRef.
- E. Zimmermann, K. K. Wong, M. Müller, H. Hu, P. Ehrenreich, M. Kohlstädt, U. Würfel, S. Mastroianni, G. Mathiazhagan, A. Hinsch, T. P. Gujar, M. Thelakkat, T. Pfadler and L. Schmidt-Mende, APL Mater., 2016, 4, 091901–091907 Search PubMed.
- M. O. Reese, S. A. Gevorgyan, M. Jørgensen, E. Bundgaard, S. R. Kurtz, D. S. Ginley, D. C. Olson, M. T. Lloyd, P. Morvillo, E. A. Katz, A. Elschner, O. Haillant, T. R. Currier, V. Shrotriya, M. Hermenau, M. Riede, K. R. Kirov, G. Trimmel, T. Rath, O. Inganäs, F. Zhang, M. Andersson, K. Tvingstedt, M. Lira-Cantu, D. Laird, C. McGuiness, S. Gowrisanker, M. Pannone, M. Xiao, J. Hauch, R. Steim, D. M. DeLongchamp, R. Rösch, H. Hoppe, N. Espinosa, A. Urbina, G. Yaman-Uzunoglu, J.-B. Bonekamp, A. J. J. M. van Breemen, C. Girotto, E. Voroshazi and F. C. Krebs, Sol. Energy Mater. Sol. Cells, 2011, 95, 1253–1267 CrossRef CAS.
- B. Sahin, M. Kraehling, V. Facci Allegrini, J. Leung, K. Wiesner-Fleischer, E. Magori, R. Pastusiak, A. Tawil, T. Hodges, E. Brooke, E. C. Corbos, M. Fleischer, E. Simon and O. Hinrichsen, J. CO2 Util., 2024, 82, 102766 CAS.
- Q. Xu, S. Garg, A. B. Moss, M. Mirolo, I. Chorkendorff, J. Drnec and B. Seger, Nat. Catal., 2023, 6, 1042–1051 CAS.
- T. Zhang, Z. Li, X. Lyu, J. Raj, G. Zhang, H. Kim, X. Wang, S. Chae, L. Lemen, V. N. Shanov and J. Wu, J. Electrochem. Soc., 2022, 169, 104506 CAS.
- J. M. Álvarez-Gómez and A. S. Varela, Energy Fuels, 2023, 37, 15283–15308 Search PubMed.
- K. P. Kuhl, E. R. Cave, D. N. Abram and T. F. Jaramillo, Energy Environ. Sci., 2012, 5, 7050–7059 CAS.
- X. Wang, C. Tomon, T. Bobrowski, P. Wilde, J. R. C. Junqueira, T. Quast, W. He, N. Sikdar, J. Weidner and W. Schuhmann, ChemElectroChem, 2022, 9, e202200675 CAS.
- Q. Chen, X. Luo, G. Mu, X. Mao, H. Yin, E. H. Lester and T. Wu, ACS Sustainable Chem. Eng., 2024, 12, 15134–15146 CAS.
- Z. Wei, W. Wang, T. Shao, S. Yang, C. Liu, D. Si, R. Cao and M. Cao, Angew. Chem., Int. Ed., 2025, 64, e202417066 CrossRef CAS PubMed.
- Y. Zhou, R. Ganganahalli, S. Verma, H. R. Tan and B. S. Yeo, Angew. Chem., Int. Ed., 2022, 61, e202202859 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ey00287c |
| This journal is © The Royal Society of Chemistry 2025 |