
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCu–ZnO nanoparticles encapsulated in ZSM-5 for selective conversion of carbon dioxide into oxygenates†
Xu
Wang
 ab,
Hwi Yeon
Woo
a,
Dongming
Shen
a,
Min Jung
Park
a,
Mansoor
Ali
a,
Faisal
Zafar
a,
Kyun Yeon
Kang
c,
Jae-Soon
Choi
c,
Eunjoo
Jang
*d and
Jong Wook
Bae
*a
ab,
Hwi Yeon
Woo
a,
Dongming
Shen
a,
Min Jung
Park
a,
Mansoor
Ali
a,
Faisal
Zafar
a,
Kyun Yeon
Kang
c,
Jae-Soon
Choi
c,
Eunjoo
Jang
*d and
Jong Wook
Bae
*a
aSchool of Chemical Engineering, Sungkyunkwan University (SKKU), 2066 Seobu-ro, Jangan-gu, Suwon, Gyeonggi-do 16419, Republic of Korea. E-mail: finejw@skku.edu; Fax: +82-31-290-7272; Tel: +82-31-290-7347
bInstitute of Advanced Study, Chengdu University, Chengdu 610106, China
cLG Chem., 188 Moonji-ro, Yuseong-gu, Daejeon, 34122, Republic of Korea
dSKKU Advanced Institute of Nanotechnology (SAINT), Sungkyunkwan University (SKKU), 2066 Seobu-ro, Jangan-gu, Suwon, Gyeonggi-do 16419, Republic of Korea. E-mail: ejjang1201@skku.edu; Fax: +82-31-299-4119; Tel: +82-31-299-4318
First published on 14th February 2025
Abstract
Engineering copper nanoparticles to achieve high dispersion and thermal stability with stable catalytic activity is crucial and challenging for the direct hydrogenation of CO2 to oxygenates via tandem catalysis over hybridized catalysts. Herein, hybridized Cu–ZnO nanoparticles were encapsulated in nano-crystalline ZSM-5 overlayers through a steam-assisted crystallization (SAC) approach by optimizing the Cu/Zn ratios of Cu–ZnO nanoparticles, the Si/Al ratio of ZSM-5, crystalline structures, and the oxidation states of active sites to achieve higher and durable direct conversion of CO2 into dimethyl ether (DME) and methanol. The spatially confined Cu–ZnO nanoparticles inside ZSM-5 frameworks facilitated suppressed nanoparticle aggregation by preserving major Cu+ phases of active copper species, which contributed to excellent catalytic performance with CO2 conversion rate of up to 20.8% and a methanol/DME selectivity of 81.6% (DME selectivity of 62.2%) with a space-time yield (STY) of 13.9 gDME (gCu−1 h−1). In situ DRIFTS, AES/XPS and XANES analyses further revealed that the spatial confinement effects in protective ZSM-5 zeolite overlayers effectively stabilized homogeneously dispersed Cu–ZnO nanoparticles with dominant distribution of Cu+ phases, which played key roles in generating formate and methoxy intermediates that are responsible for the enhanced catalytic activity and catalyst durability.
Broader contextCu–ZnO nanoparticles encapsulated in ZSM-5 revealed a higher catalytic activity and stability for CO2 hydrogenation to DME and methanol oxygenates owing to the stable preservation of the Cu+ phase via spatial confinement effects inside zeolite shells. |
1. Introduction
Significant consumption of fossil fuels, resulting in excessive carbon emissions, has led to global warming and adverse environmental effects.1,2 Considerable efforts have been devoted to developing strategies for the reduction of CO2 emissions through economically viable means.3,4 The utilization of CO2 can produce various value-added petrochemicals such as light olefins,5–8 gasoline-range hydrocarbons,9–14 aromatics,15–18 and methanol/dimethyl ether (DME).19–22 Notably, DME and methanol, which are recognized as clean and sustainable alternative fuels and crucial chemical intermediates, can be synthesized through a stepwise CO2 hydrogenation, where CO2 is initially converted into methanol on copper (Cu)-based catalysts followed by dehydration to DME on solid acid catalysts.19,20 The Cu-based catalysts and solid acid catalysts can be effectively combined together to link the reactions of CO2-to-methanol (CO2 + 3H2 ↔ CH3OH + H2O, ΔH298![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) K = −49.4 kJ mol−1) and that of methanol-to-DME (2CH3OH ↔ CH3OCH3 + H2O, ΔH298K = −23.5 kJ mol−1), while a reverse water–gas shift (RWGS) reaction (CO2 + H2 ↔ CO + H2O, ΔH298K = +41.2 kJ mol−1) as a main side reaction can produce a CO byproduct.
K = −49.4 kJ mol−1) and that of methanol-to-DME (2CH3OH ↔ CH3OCH3 + H2O, ΔH298K = −23.5 kJ mol−1), while a reverse water–gas shift (RWGS) reaction (CO2 + H2 ↔ CO + H2O, ΔH298K = +41.2 kJ mol−1) as a main side reaction can produce a CO byproduct.
For the direct synthesis of DME/methanol via CO2 hydrogenation as well as consecutive dehydration, Cu-based catalysts have been frequently modified with ZnO, ZrO2, or Al2O3 promoters to enhance thermal stability and methanol production rate.19–24 Among the promoters, ZnO has been known to be most effective for enhancing active interfacial areas24,25 for stabilizing crucial intermediates such as formates or methoxy species26–29 and dispersing active Cu metals.23,28,29 However, catalyst deactivation generally happens owing to the sintering and oxidation of Cu nanoparticles30,31 as well as agglomeration of ZnO,32 leading to the shrinkage of active Cu–ZnO interfaces and declined catalytic activity. In addition, active copper species of Cu0/Cu+ phases, where Cu0 facilitates the dissociation of adsorbed H2 and Cu+ stabilizes the methoxy or formate intermediate,33–35 are susceptible to further oxidation by H2O formed during the dehydration step. Hence, maintaining active and stable Cu0/Cu+ phases with their optimal balance is critical for CO2-to-DME tandem reactions. Although zeolites, especially ZSM-5, are commonly applied as supports, when acid catalytic sites are provided for dehydration reactions,19–22 the strong acidic sites also promote byproduct formation and coke deposition, causing catalyst deactivation.36 Introducing hierarchically structured mesoporous structures via the encapsulation of active metal oxides35 using crystalline ZSM-5 can remarkably enhance coke resistance with decreased surface carbon depositions.37,38 Therefore, controlling ZSM-5 crystallite sizes and hierarchically structured mesopores is a feasible approach to enhance the catalytic activity and thermal stability of tandem catalysts for CO2 hydrogenation and successive methanol dehydration. Although amorphous SiO2 is frequently employed as a supporting material to confine active nanoparticles with its enhanced resistance to thermal aggregation,39 SiO2 itself has a small pore volume and surface area with insignificant acidic sites that result in a lower CO2 conversion with limited intermediate diffusion. Zeolites having abundant acidic sites largely altered intermediate diffusion with its successive transformation on the acidic sites, and zeolite-encapsulated Cu nanoparticles on which CO2 hydrogenation occurs are generally reported as selective methanol synthesis catalysts.40
In this study, we synthesized ZSM-5-encapsulated Cu–ZnO nanoparticles to improve CO2-to-DME tandem reaction activity via steam-assisted crystallization (SAC), achieving an excellent DME/methanol selectivity of 81.6% with a productivity of 13.9 gDME (gCu−1 h−1) and prolonged catalyst activity for 100 h on stream. The spatial confinement effects by ZSM-5 zeolites strengthened the interactions between Cu nanoparticles and ZSM-5 zeolites, predominantly preserving Cu+ species to facilitate the generation of formate intermediates. We thoroughly investigated the effects of crystallization process with different Si/Al and Cu/Zn molar ratios, crystalline structures, and their oxidation states. The reaction mechanism over the catalyst was proposed based on the analyses using XPS, AES, XANES, and in situ DRIFTS techniques, suggesting a viable way to fabricate zeolite-encapsulated Cu–ZnO nanoparticle catalysts with their high dispersion and thermal stability, particularly for a tandem CO2-to-DME reaction.
2. Experimental section
2.1. Preparation of CZ(x)@SiO2 and CZ(x)@Z(y)-t catalysts
SiO2-encapsulated Cu–ZnO nanoparticles with different Cu/Zn molar ratios (denoted as CZ(x)@SiO2) were prepared by a previously reported reverse micelle method with copper and zinc metal precursors, where the Cu/Zn mole ratio was nominally controlled in the range of x (x = 0.3–1.5). In detail, the CZ(x)@SiO2 nanoparticles with a Cu/Zn molar ratio of x were prepared by dissolving Cu(NO3)2·3H2O and Zn(NO3)2·6H2O, which were dissolved in 5 mL deionized water (DIW), and 10.5 g of Brij-C10 (polyethylene glycol hexadecyl ether) as a non-ionic surfactant was dissolved in 50 mL cyclohexane at 50 °C. This metal precursor solution was slowly dropped into the Brij-C10 solution with stirring for 1 h. Aqueous ammonium hydroxide (3 mL) and tetraethyl orthosilicate (TEOS) (5 mL) were separately added to this solution under vigorous stirring for 2 h to obtain a blue-colored solution. The resulting fine powders were collected by centrifugation using 30 mL ethanol solvent, dried at 80 °C overnight, and calcined at 550 °C for 2 h to obtain CZ(x)@SiO2 (x = 0.3, 0.6, 0.9 and 1.5). In addition, Zn-free CuO@SiO2 (Cu@SiO2) as a reference catalyst was prepared to compare the effect of Zn.The hybridized Cu–ZnO@ZSM-5 catalysts, with solid acid ZSM-5 zeolite coating on CZ@SiO2, were prepared by controlling the crystallization duration (t) for 12, 24, or 48 h and by varying Si/Al molar ratios (y) of 40, 60, and 100, which were denoted as CZ(x)@Z(y)-t as shown in Fig. 1. For instance, CZ(0.9)@Z(60)-24 having an Si/Al ratio of 60 and a Cu/Zn ratio of 0.9 was prepared as follows: 0.5 g of as-synthesized CZ(0.9)@SiO2 was mixed with 0.0521 g aluminum nitrate precursor (Al(NO3)3·9H2O), 1.25 g of tetrapropyl ammonium hydroxide (TPAOH) (40 wt%) and 2.75 g of DIW, and stirred at room temperature for 10 h. The resulting mixture was kept in an oven at 60 °C for 24 h to obtain dry gel. The gel was transferred to a 100 mL Teflon-lined autoclave with 4 mL ammonia solution (∼28 wt%), sealed and heated up to 170 °C for successive crystallization with durations (t) of 12, 24 or 48 h. The synthesized powders were washed several times with DIW, dried at 80 °C for 24 h, and calcined at 550 °C under air for 2 h. Furthermore, CZ@SiO2 physically mixed with ZSM-5 as well as supported CZ/ZSM-5 catalysts by a wet impregnation method with similar compositions were also prepared.
2.2. Evaluations of catalytic activity
The catalytic activity was evaluated with 0.2g catalyst in a fixed-bed tubular reactor having an inner diameter of 6mm and a length of 453 mm. The catalyst was previously reduced at 300°C for 2h under a flow of 5vol% H2 balanced with N2. To carry out the direct hydrogenation of CO2 to methanol/DME, mixed gas having a molar ratio of CO2/H2/N2 = 24/72/4 (N2 as an internal standard gas) was fed into the tubular reactor at a fixed pressure of 5.0MPa, and the reaction temperature was increased up to 260°C at a space velocity (SV) of 3000 mL (gcat−1 h−1), which was the commonly applied CO2-to-DME reaction condition. The effluent gases were in situ analyzed by gas chromatography (GC, YL6100, Younglin Co.) with a thermal conductivity detector (TCD) as well as a flame ionization detector (FID), connected to a Carboxen-1000 packed column and a HP-PLOT-U capillary column, respectively. The conversion of CO2 (XCO2, mol%) and the product distributions (mol%) were calculated based on the total carbon balances at a steady state after 30 h on stream, which are described with more details in the ESI.†
2.3. Catalyst characterization
Structural properties such as specific surface area (Sg, m2 g−1), pore volume (PV, cm3 g−1) and average pore diameter (PD, nm) of the fresh CZ(x)@Z(y)-t catalysts were studied by N2 adsorption–desorption analysis at a liquid N2 temperature of −196°C using a TRISTAR-3000 instrument (Micromeritics) to calculate the specific surface area in the range of P/P0 of 0.03–0.2 based on the Brunauer–Emmett–Teller (BET) equation. The average pore diameter with pore size distribution was measured by the Barrett–Joyner–Halenda (BJH) method from the desorption branch of the isotherms. Powder X-ray diffraction (XRD) patterns in the range of 5 –90° were characterized to verify the crystalline phases of Cu–ZnO nanoparticles and ZSM-5 zeolite using an X'Pert PRO MPD diffractometer (Malvern Panalytical) equipped with a Cu Kα radiation source. Particle size distributions and elemental compositions of the CZ(x)@Z(y)-t catalysts were also determined by scanning transmission electron microscopy (STEM/TEM) and energy-dispersive X-ray spectroscopy (EDS) using a JEM-F200/JEM-2100 F instrument operating at 200 kV. The solid-state magic angle spinning nuclear magnetic resonance (MAS NMR) spectra of 27Al species were analyzed by using a 500 MHz NMR spectrometer with a Varian Unity INOVA instrument at a resonance frequency of 130.3 MHz, where the MAS probe worked at a spinning rate of 10 kHz.
The reduction behaviors of the fresh CZ(x)@Z(y)-t catalysts were verified by temperature-programmed reduction analysis (H2-TPR) under a flow of mixed reduction gas (5 vol% H2 balanced with Ar, 30mL min−1) using a BELCAT-M instrument. For the H2-TPR, 50mg fresh catalyst was purged at 300 °C for 1h at a ramping rate of 10 °C min−1 under an Ar flow (30 mL min−1), and the balanced H2 gas was switched after cooling down to 50°C. The reduction patterns of metal oxides were acquired using a TCD analyzer after passing over water trap at a temperature in the range of 100–500 °C to estimate the consumed H2 quantity (mmol gcat−1). Temperature-programmed desorption analysis of NH3 (NH3-TPD) for the fresh catalysts was performed using a BELCAT-M instrument to measure the acidic strength and the amount of surface acidic sites. Prior to the analysis, pretreatment under a flow of He gas was carried out at 500 °C for 1 h. Subsequently, it was cooled down to 100 °C, switched to NH3 probe gas for its adsorption and temperature increased to 800 °C at a ramping rate of 10 °C min−1 for monitoring desorption behaviors. In addition, temperature-programmed desorption analysis of CO2 (CO2-TPD) was performed. Before the analysis, 50 mg of sample was reduced at 300 °C for 1 h and purged under a He flow to remove surface contaminants. The adsorption of CO2 was carried out at 50 °C for 1 h, and then desorption profiles were obtained in the temperature range of 50–800 °C at a ramping rate of 10 °C min−1.
Surface properties such as metal distributions and oxidation states with their interactions on the reduced and used CZ(0.9)@Z(60)-t catalysts were separately measured using X-ray photoelectron spectroscopy (XPS) as well as Auger electron spectroscopy (AES) using a PHI Quantera II (Scanning X-Ray Microprobe) instrument equipped with Al Kα radiation at an energy of 1484.6 eV and a chamber working pressure of 5.0 × 10−7 Pa. The Cu 2p, Zn 2p and Si 2p peaks were analyzed with a resolution of 0.05 eV to elucidate the variations in binding energy (BE) and Zn/Cu surface atomic ratio after correcting their BEs using a reference BE of C 1s of 284.8 eV. In addition, X-ray absorption fine structure (XAFS) analysis was performed at Pohang Light Source-II (PLS-II) of Pohang Accelerator Laboratory (PAL) to verify the local structures and oxidation states of Cu species on the CZ(0.9)@Z(60)-24 catalyst through a transmission mode. The metallic Cu foil, Cu2O and CuO were used as reference materials. Fourier transform fittings of XAFS spectra were carried out using a k2 weighting factor, and the parameters for background removal and normalization based on the reference E0 = 8079 eV at the pre-edge range of −150.0 to −30.0, and the normalized range of 150 to 620 with a spline range of k was in 0–13. For Fourier transform parameters, the χ(k) values in the R space were obtained in the range of 3.0–12.0 Å by employing a Hanning window function, and the position of absorption edge was determined from the first maximum of the 1st derivatives of normalized μ(E). Furthermore, wavelet transform (WT) was carried out using the Hama Fortran program obtained from the European Synchrotron Radiation Facility (ESRF) official website (Rmin = 1.0 Å and Rmax = 5.0 Å via the Morlet function for the transform parameter). The EPR spectra on the CZ(0.9)@Z(60)-t catalysts were recorded using a Bruker equipment, operating at the X band with a microwave frequency of 9.421 GHz.
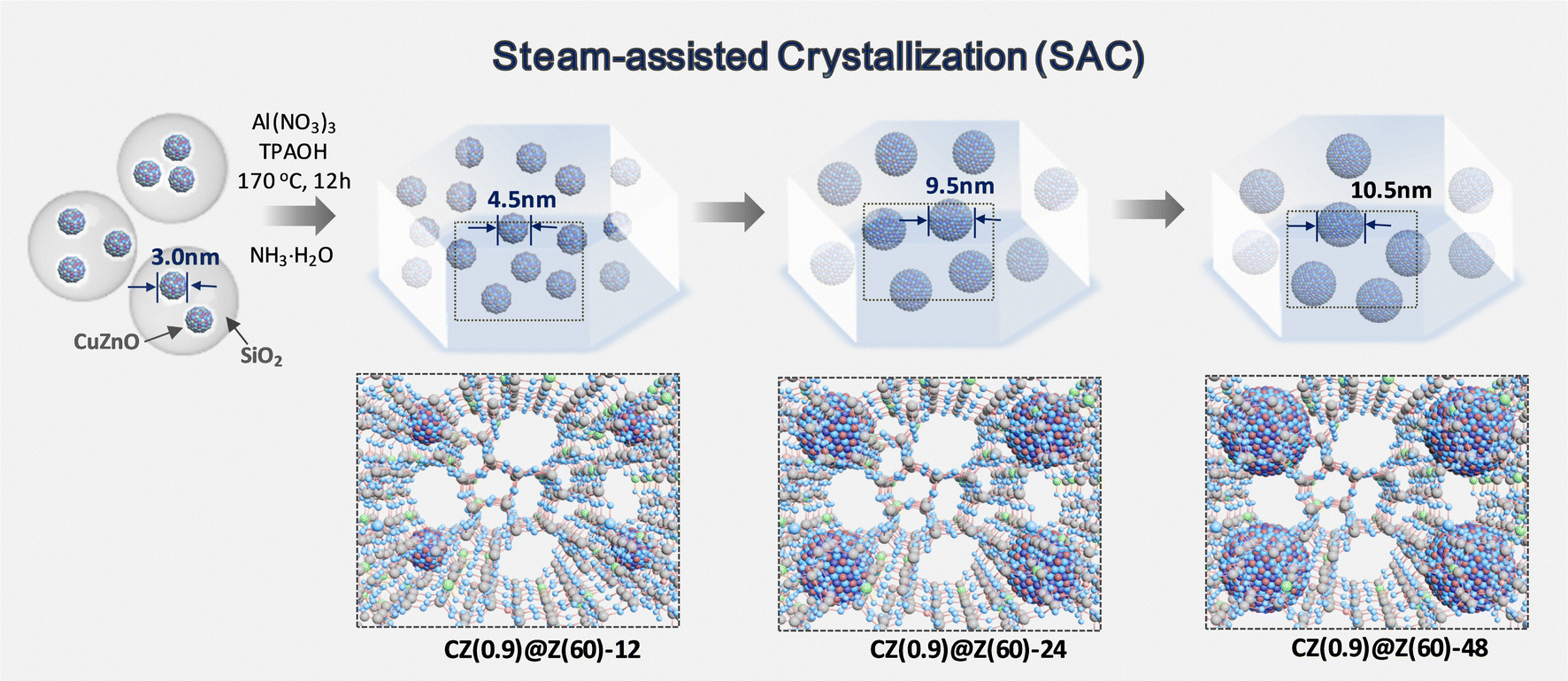 | ||
| Fig. 1 Schematic of CZ(0.9)@Z-t preparation at different crystallization times (t = 12, 24, and 48 h). | ||
In situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) analysis was performed using a FT-IR spectrometer (PerkinElmer Frontier), equipped with a diffuse reflection cell (PIKE Technologies) with a resolution of 4cm−1. Prior to the analysis, 15 mg fresh CZ(0.9)@Z(60)-24 catalyst was loaded in a DRIFT cell and reduced under a flow of 5 vol% H2/N2 at 300 °C for 1 h. After that, the sample was purged under a N2 flow for 2 h to remove surface residues and cooled down to ambient temperature to obtain the reference background spectra. Then, reactant gas (CO2/H2/N2 = 24/72/4 vol%) was injected into a reaction chamber at an ambient pressure by increasing the temperature up to 260 °C and kept for 1 h. Temperature-resolved DRIFTS spectra were recorded every 10 °C and time-resolved spectra were also recorded every five minutes at 260 °C for 1 h to identify surface intermediates. In addition, DRIFTS spectra under methanol-TPD analysis conditions at a temperature in the range of 30 to 400 °C and those of methanol dehydration to DME at 260 °C for 1 h were further verified to confirm surface intermediates during CO2-to-DME reactions. Furthermore, in situ CO-DRIFTS analysis was performed to examine the oxidation states of active metallic Cu species during the CO2-to-DME reaction. In more detail, 15 mg of catalyst was loaded in a cell and reduced at 300 °C under 5 vol% H2/N2 flow (15 mL min−1) for 2 h and cooled down to 30 °C. Subsequently, the flow was switched to N2 gas (99.99%), and purged with H2 (100 mL min−1) for 30 minutes to remove the surface residual followed by introducing CO gas (99.99%) at a flow rate of 30 mL min−1 at 30 °C for 30 minutes. Then, N2 gas was injected to purge for 30 minutes and the CO-DRIFT spectra were recorded at 30 °C.
3. Results and discussion
3.1. Structural properties of CZ(x)@Z(y)-t catalysts
The structures and surface properties of the CZ(x)@Z(y)-t catalysts were characterized to verify the reaction mechanisms. The TEM images displayed in Fig. 2(a)–(d) showed the encapsulated multiple cores in overlayer shells with different size distributions (Fig. S1, ESI†). The Cu–ZnO nanoparticles encapsulated in a thick SiO2 shell (d = 30 ± 1.6 nm) showed almost uniform particle sizes (d) of 3.0 ± 1.2 nm. As-synthesized core/shell structured CZ@SiO2, aluminum precursor, and TPAOH were subjected to a high-temperature annealing (hydrothermal) process to form CZ(0.9)@Z(60)-t catalysts with different crystallization durations (t = 12, 24 and 48 h). During the crystallization process, some portions of Cu and Zn metal elements were leached out and altered the bulk Cu/Zn ratio, as confirmed by XRF analysis (Table 1 and Table S1, ESI†), which was more accelerated in an alkaline environment. In addition, the crystallite sizes of Cu–ZnO nanoparticles inside ZSM-5 nanocrystals were gradually increased from 3.0 nm to 4.5 nm after 12 h, 9.5 nm after 24 h, and 10.5 nm after 48 h of crystallization duration. On the contrary, the Si/Al mole ratios were maintained in the range of 60.2–71.7 during the crystallization, indicating the formation of stable ZSM-5 structures even after high-temperature hydrothermal treatment. The powder XRD patterns of the fresh CZ(0.9)@Z(60)-t catalysts shown in Fig. 2(e) reveal the well-crystallized ZSM-5 structures that evolved during the crystallization step (PDF#44-0003), which are distinctively different from the initially prepared CZ@SiO2. However, the intensity of the characteristic peaks of ZSM-5 decreased after 24 h of crystallization time, attributed to the expansion of intra-particular mesopores (Fig. 2(f) and (g)). This observation corresponds well with the facts that the surface area is 339 m2 g−1 and the pore volume is 0.14 cm3 g−1 on fresh CZ(0.9)@Z(60)-24 with an average pore diameter of 7.1 nm, which showed much larger values than each respective one (326 m2 g−1, 0.08 cm3 g−1) on the CZ(0.9)@Z(60)-12 with an average pore diameter of 5.9 nm (Table 1). It was also found that the hierarchical mesopore structures were well-formed after the crystallization, which is beneficial for the fast diffusion of reaction intermediates and products, leading to enhanced catalytic activity and selectivity to oxygenates and suppressed side reactions or coke precursor formations. 27Al MAS NMR spectra displayed in Fig. 2(h) reveal that aluminum atom in the ZSM-5 structures exists in the form of framework Al (57.6 ppm) in the absence of extra-framework Al species (0 ppm). Local elemental compositions of the Cu–ZnO nanoparticles on CZ(0.9)@Z(60)-24 were further verified by TEM-EDS mapping images (Fig. S2, ESI†), which showed the uniform Cu distribution in the Cu–ZnO nanoparticles as well as revealed homogeneous distributions of Zn, Si, Al, and O elements in the entire ZSM-5 matrix with a little concentrated Zn species on the Cu–ZnO nanoparticles. The particle sizes of Cu–ZnO nanoparticles on fresh CZ(0.9)@Z(60)-24 preserved their initial average diameter (10 nm in size) and their higher particle size distributions even after 100 h of long catalytic reaction, as shown in Fig. S3 and S4 (ESI†), confirming the excellent thermal stability of the optimal CZ(0.9)@Z(60)-24 catalyst. | ||
| Fig. 2 TEM and inset HR-TEM images of the fresh catalysts: (a) CZ@SiO2, (b) CZ(0.9)@Z(60)-12, (c) CZ(0.9)@Z(60)-24, and (d) CZ(0.9)@Z(60)-48. (e) XRD patterns, (f) pore size distributions, (g) N2 adsorption–desorption isotherms, and (h) 27Al MAS NMR spectra of the fresh CZ(0.9)@Z(60)-t catalysts. | ||
| Catalyst | X CO2 (%) | Selectivitya (%) | E a (kJ mol−1)/R2 | TOFc (h−1) | N2-sorptiond | XRFe | NH3-TPDf (mmol g−1) | CO2-TPDg (mmol g−1) | XPSh | AESh |
|---|---|---|---|---|---|---|---|---|---|---|
| CO/HC/methanol/DME | S g/PV/PD (m2 g−1, cm3 g−1, nm) | R (Cu/Zn)/R(Si/Al) | R (Cu/Zn)/R(Si/Al) (reduced) | Cu0/(Cu0 + Cu+) (reduced/used) | ||||||
| a CO2 conversion (XCO2) and product distribution (selectivity to CO, hydrocarbons (HC, mainly CH4), methanol and dimethyl ether (DME)) measured under the reaction conditions of T = 260 °C, P = 5.0 MPa, space velocity (SV) = 3000 mL (gcat−1h−1) and a feed gas composition of CO2/H2/N2 = 24/72/4. The data were collected by using a steady-state value after its initial induction period. b Activation energy (Ea) estimated using the Arrhenius plot with standard deviation (linear R2) for CO2 hydrogenation at 220–280 °C under 5.0 MPa. c Turnover frequency (TOF) for CO2 conversion calculated from the active Cu amount by H2-TPR (50–400 °C). d Surface area (m2 g−1, Sg), pore volume (cm3 g−1, PV) and average pore diameter (nm, PD) measured by N2 adsorption–desorption analysis. e Cu/Zn molar ratio (R(Cu/Zn)) and Si/Al molar ratio (R(Si/Al)) measured by XRF analysis. f Total acidic sites measured by NH3-TPD analysis. g Total amount of CO2 desorption (mmol g−1) was calculated from CO2-TPD peaks. h Cu surface properties on the reduced and used catalysts as well as molar ratios were measured by AES and XPS analyses, respectively. | ||||||||||
| CZ(0.9)@Z(60)-12 | 21.7 | 21.4/1.1/18.2/59.4 | 10.1/0.97 | 50.1 | 326/0.08/5.9 | 0.79/71.7 | 1.058 | 0.654 | 1.07/27.7 | 0.16/0.12 |
| CZ(0.9)@Z(60)-24 | 20.8 | 17.9/0.5/19.4/62.2 | 15.4/0.91 | 64.7 | 339/0.14/7.1 | 0.86/60.2 | 0.819 | 0.526 | 0.40/17.7 | 0.04/0.04 |
| CZ(0.9)@Z(60)-48 | 20.5 | 18.1/1.6/21.5/58.8 | 16.8/0.92 | 97.2 | 324/0.13/7.5 | 0.50/61.9 | 0.553 | 0.503 | 0.20/16.3 | 0.06/0.06 |
| Cu@Z(60)-24 | 16.8 | 49.0/1.5/27.2/22.3 | 13.7/0.93 | 9.45 | 339/0.15/5.1 | —/61.7 | 0.382 | 0.966 | —/43.2 | 0.03/0.03 |
| CZ(0.9)@Z(40)-24 | 21.5 | 21.7/2.5/41.2/34.6 | — | 97.5 | 307/0.10/3.8 | 0.74/38.9 | 0.666 | 0.453 | — | — |
| CZ(0.9)@Z(100)-24 | 22.8 | 19.0/0.4/40.2/40.5 | — | 45.6 | 339/0.12/6.0 | 0.71/100.5 | 0.467 | 0.860 | — | — |
| CZ(0.3)@Z(60)-24 | 18.5 | 17.7/14.2/21.2/46.9 | — | 83.0 | 270/0.02/7.2 | 0.26/63.7 | 0.863 | 0.792 | — | — |
| CZ(0.6)@Z(60)-24 | 20.5 | 21.2/12.0/16.3/50.5 | — | 80.3 | 305/0.09/5.2 | 0.47/63.9 | 0.662 | 1.046 | — | — |
| CZ(1.5)@Z(60)-24 | 19.6 | 21.1/1.5/27.2/50.3 | — | 18.6 | 279/0.05/5.1 | 1.54/62.9 | 0.617 | 0.798 | — | — |
3.2. Surface properties of CZ(x)@Z(y)-t catalysts
The reduction behaviors of the CZ(0.9)@Z(60)-t catalysts were investigated by H2-TPR analysis to verify the oxidized phases of Cu–ZnO nanoparticles and their interactions with ZSM-5 zeolites. The maximum reduction peaks steadily shifted to higher temperatures as the crystallization duration increased (Fig. 3(a)). This trend appeared similarly when the Cu/Zn ratio decreased from 1.5 to 0.3 or the Si/Al ratio increased from 40 to 80 during the catalyst optimization step (Fig. 3(b) and (c)). The reduction of CZ(1.5)@Z(60)-24 occurred in the maximum temperature range of 237–423 °C, and the reduction temperature increased to the range of 317–500 °C when the Cu/Zn ratio decreased from 1.5 to 0.3. In addition, when the crystallization duration extended from 12 to 48 h, the reduction temperature regions changed from 244–400 °C to 277–487 °C. It was expected that the stronger interactions between Cu and Zn in the Cu–ZnO nanoparticles required more harsh reduction conditions, possibly due to the spinel-type Cu oxide phases.19 However, the effects of the Si/Al ratio on the reduction behaviors were not significant when the Cu/Zn ratio was fixed, indicating that the contribution of Cu/Zn ratio to the reduction was more dominant than the interactions between Cu–ZnO nanoparticles and ZSM-5 zeolites. The total quantity of consumed H2 was proportional to the Cu content in the CZ(x)@Z(y)-t catalysts, ranging from 0.50 to 2.89 mmol g−1, as summarized in Table S1 (ESI†). More specifically, the four different characteristic peaks for the reduction profiles in Fig. 3(a) could be assigned to the reduction of different copper states of CuO (Cu2+), Cu2O (Cu+) to metallic Cu0 species corresponding to the strengths of the interaction with ZSM-5. The peak that appeared at a temperature below 280 °C was attributed to the reduction of the small-sized isolated Cu–ZnO nanoparticles. The reduction peaks were involved in one-step reduction of CuO to surface metallic Cu0 or two-step successive reduction of Cu2+ species to Cu+ and surface Cu0 phases, which were separately assigned to the peaks appearing at a temperature lower than 300 °C and at a temperature higher than 400 °C for the transition from Cu+ to Cu0 species.41–44 In addition, the reduction peaks in the range of 300–360 °C originated from the reduction of the encapsulated and strongly interacted CuO nanoparticles with ZSM-5 zeolite overlayers as well as with the ZnO moiety by possibly forming spinel-type CuAl2O4 or ZnAl2O4,19 eventually affecting the thermal stability of the Cu–ZnO nanoparticles against aggregation during CO2-to-DME reactions. Therefore, the CZ(0.9)@Z(60)-12 catalyst, prepared with a relatively short crystallization duration, still contained weakly interacted Cu(–ZnO) nanoparticles compared to the catalysts experiencing a longer crystallization time. The reduction peaks above 400 °C can be assigned to the reduction of successive Cu+ phase to bulk metallic Cu0 species,43,44 suggesting that the possible ion-exchange of Cu+ on the ZSM-5 surfaces decreased the number of stronger Brønsted acid sites. The phenomena are also beneficial for suppressing potential hydrocarbon formation via the methanol-to-hydrocarbon pathway. While the Cu/Zn ratio was controlled from 0.3 to 1.5 or to even Zn-free catalysts, the reduction behaviors were considerably changed, where the reduction of the Cu@Z(60)-24 occurred in the lowest temperature region due to the weak interaction between Cu and ZSM-5 layers.25 | ||
| Fig. 3 H2-TPR patterns of the fresh CZ(x)@Z(y)-t catalysts with different (a) crystallization conditions (t), (b) Cu/Zn ratios (x), and (c) Si/Al ratios (y) with variations in the Cu/Zn ratio. | ||
NH3-TPD analyses of the CZ(x)@Z(y)-t catalysts were performed to investigate the surface acidic properties, and the results are summarized in Fig. 4(a)−(c), Table 1 and Table S1 (ESI†). In general, the crystallization process for zeolite synthesis affects the surface acidity significantly by changing the crystallinity and defect sites. Three characteristic desorption peaks of NH3 probe molecules could be assigned to weak (W), medium (M) and strong (S) acidic sites at their respective desorption temperatures of ∼180, 250 and 400 °C with the possible water desorption or NH3 desorption on Lewis acid sites at ∼580 °C.42,45 The weak acidic sites originated from the weakly adsorbed NH3 molecules on the surface Si–OH groups,43 and medium and strong acidic sites correspond to the strongly absorbed NH3 molecules on Lewis and Brønsted acidic sites, respectively.46,47 The Lewis acid sites, assigned to medium acidic sites, originated from metal ion-exchanged ZSM-5 surfaces with the characteristic peaks above 500 °C in the NH3-TPD analysis. In addition, the peaks can be only reduced above 400 °C based on the H2-TPR patterns, where the appearance of the reduction peaks above 400 °C can be possibly attributed to the reduction of Cu+-exchanged ZSM-5 sites.48 As the crystallization duration increased from 12 to 48 h (Table 1 and Table S1, ESI†), the total amounts of weak, medium, and total acidic sites decreased from 1.058 to 0.553 mmol g−1. However, the strong acidic sites revealed similar trends, showing minimum amounts of strong acidic sites (0.078 mmol g−1) on fresh CZ(0.9)@Z(60)-48. While the Si/Al ratio or the Cu/Zn ratio increased, both total and strong acidic sites decreased due to lower Al and Cu contents, respectively. Based on the XRF results, as summarized in Table 1 and Table S1 (ESI†), it was noticed that the Cu species leached out more easily than Zn species during the hydrothermal synthesis step. Furthermore, the Py-IR analysis revealed that the Lewis acid sites were found to be more abundant than Brønsted acid sites on the CZ(0.9)@Z(60)-t catalysts (Fig. S5, ESI†), and both Lewis and Brønsted sites were also found to be active for methanol dehydration. In summary, the smallest acidic sites (0.382 mmol g−1) in Cu@Z(60)-24 were responsible for the lowest DME selectivity (22.3%), while medium acidic sites (0.819 mmol g−1) in CZ(0.9)@Z(60)-24 showed the highest DME selectivity (62.2%), suggesting that the appropriate number of acidic sites significantly adjusted the DME formation rate from the methanol intermediate by a dehydration reaction over the ZSM-5 surface.
 | ||
| Fig. 4 NH3-TPD patterns of the fresh CZ(x)@Z(y)-t catalysts with different (a) crystallization conditions (t = 12, 24, and 48 h), (b) Cu/Zn ratios (x = 0.3, 0.5, and 1.5), and (c) Si/Al ratios (y = 40, 60, and 100). CO2-TPD patterns on fresh (d) CZ(0.9)@Z(60)-t catalysts (t = 12, 24, and 48 h), (e) different Cu/Zn molar ratios (x = 0.3, 0.5, and 1.5), and (f) different Si/Al molar ratios (y = 40, 60, and 100). | ||
The CO2-TPD profiles on the CZ(x)@Z(y)-t catalysts according to different crystallization durations and other preparation conditions were used to estimate the surface basicity and oxygen vacant sites (Fig. 4(d)–(f)). Most catalysts showed three distinct desorption peaks of CO2, assigned to weak (W), medium (M) and strong (S) basic sites.49 The weak basic sites at the lowest CO2 desorption temperature below 200 °C originated from the surface OH groups, and the medium basic sites resulted from adsorbed CO2 on metal–oxygen pair-like Zn–O as linearly adsorbed O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CO−M species.50 The strong basic sites appearing at a higher desorption temperature above 500 °C were attributed to the bridge-bonded M–O–C–O–M over the surface O2− anion sites.50–52 It was noticed that the amounts of the desorbed CO2 molecules decreased as the crystallization time increased (0.654 mmol g−1 on the CZ(0.9)@Z(60)-12 and 0.503 mmol g−1 on the CZ(0.9)@Z(60)-48) and the Si/Al ratio decreased (0.860 mmol g−1 on the CZ(0.9)@Z(100)-24 and 0.453 mmol g−1 on the CZ(0.9)@Z(40)-24) (Table 1 and Table S1, ESI†). These observations were attributed to the strong interactions of oxygen vacant sites, as confirmed by EPR analysis with the characteristic g factor of 2.07,53 as shown in ESI,† Fig. S6 as well as the adjusted basic sites from the Cu–ZnO nanoparticles inside the ZSM-5 layers. The decreased number of active sites for CO2 adsorption after longer crystallization durations can further suppress CO2 conversion, which were possibly attributed to the loss of oxygen vacant sites by the leaching phenomena of CuO species during the hydrothermal synthesis process. The CO2 adsorption capacity showed high values in the range of 0.792–1.046 mmol g−1 when the Cu/Zn ratios were lower in the range of 0.3–0.6 than the optimum Cu/Zn ratio of 0.9, which clearly explained the enhanced byproduct selectivity on the CZ(x)@Z(y)-t catalysts.
CO−M species.50 The strong basic sites appearing at a higher desorption temperature above 500 °C were attributed to the bridge-bonded M–O–C–O–M over the surface O2− anion sites.50–52 It was noticed that the amounts of the desorbed CO2 molecules decreased as the crystallization time increased (0.654 mmol g−1 on the CZ(0.9)@Z(60)-12 and 0.503 mmol g−1 on the CZ(0.9)@Z(60)-48) and the Si/Al ratio decreased (0.860 mmol g−1 on the CZ(0.9)@Z(100)-24 and 0.453 mmol g−1 on the CZ(0.9)@Z(40)-24) (Table 1 and Table S1, ESI†). These observations were attributed to the strong interactions of oxygen vacant sites, as confirmed by EPR analysis with the characteristic g factor of 2.07,53 as shown in ESI,† Fig. S6 as well as the adjusted basic sites from the Cu–ZnO nanoparticles inside the ZSM-5 layers. The decreased number of active sites for CO2 adsorption after longer crystallization durations can further suppress CO2 conversion, which were possibly attributed to the loss of oxygen vacant sites by the leaching phenomena of CuO species during the hydrothermal synthesis process. The CO2 adsorption capacity showed high values in the range of 0.792–1.046 mmol g−1 when the Cu/Zn ratios were lower in the range of 0.3–0.6 than the optimum Cu/Zn ratio of 0.9, which clearly explained the enhanced byproduct selectivity on the CZ(x)@Z(y)-t catalysts.
The surface environments of Cu–ZnO nanoparticles of the representative catalysts under fresh, reduced and used conditions were studied with XPS and AES analyses (Fig. 5). The binding energy (BE) of the Cu 2p peak of fresh CZ(0.9)@Z(60)-24 and Cu@Z(60)-24 was determined to be ∼933 eV with compound peaks at higher BEs of ∼935 eV, which are separately assigned to Cu+ and Cu2+ oxidation states. The Cu2+ phase can be coordinated to the framework oxygen atoms or the surface CuO phase,47,54,55 and the Cu+ species originated from the partially reduced Cu2O phase under reductive conditions.56–58 It is noteworthy that the Cu2+ peak intensity on Cu@Z(60)-24 was found to be much larger than that of CZ(0.9)@Z(60)-24, as shown in Fig. 5(a). The ZnO promoter on CZ(0.9)@Z(60)-24 slightly decreased the BE of Cu 2p at 933.0 eV, suggesting that the ZnO moiety abated the interactions between CuO and ZSM-5.59 The Al 2p peaks are shown in Fig. 5(a), in which three characteristic peaks were observed at the BEs of 73.6–73.8, 75.2–75.9 and 77.4–78.6 eV. The peaks below 74 eV can be assigned to the framework Al species in–Si–O–Al– of the ZSM-5 structures, and the peaks above 75 eV are attributed to the closely interacting Al species with Cu–ZnO nanoparticles through transferring electrons from Al to Cu atoms.56 The peak above ∼75 eV on Cu@Z(60)-24 was found to be considerably larger than that of CZ(0.9)@Z(60)-24, which strongly indicated the much stronger interactions of the CuO phase with Al sites on the ZSM-5 overlayers. The Si 2p spectra at ∼102.3 and 103.4 eV shown in Fig. 5(a) are assigned to the framework Si atoms such as –Si–O–Al– and –Si–O–Si– on the ZSM-5 overlayers having lower oxidation states,60,61 which were found to be similar on Cu@Z(60)-24 and CZ(0.9)@Z(60)-24. However, the O 1s spectra displayed in Fig. 5(a) show two distinctive peaks at 531.1 and 532.6 eV, where the peak at higher BE can be assigned to oxygen species on the ZSM-5 frameworks and the peak at lower BE can be assigned to the oxygen species from the surface metal oxides,57 and the lower BE peak appeared larger on Cu@Z(60)-24 due to the stronger interactions of CuO nanoparticles with ZSM-5 overlayers. After the reduction treatment, the Cu2+ peaks at ∼935 eV on CZ(0.9)@Z(60)-t significantly declined by maintaining the peak intensities at ∼933 eV while Zn-free Cu@Z(60)-24 had an intense Cu2+ peak with a Cu+/Cu0 shoulder peak at 933 eV (Fig. 5(b)). This explains that the Cu species in Cu@Z(60)-24 is more difficult to be reduced due to their stronger interactions with ZSM-5 overlayers. On the contrary, the Cu nanoparticles on used Cu@Z(60)-24 were oxidized more easily during the reaction, which showed the much larger peak intensity at ∼935 eV compared to the ZnO-promoted CZ(0.9)@Z(60)-t catalyst. This also suggests that the ZnO moiety can prevent the reoxidation of active Cu0/Cu+, preserving the original ratio of active sites (Cu0/(Cu+ + Cu0)) (based on the AES results presented in Table 1) by inhibiting thermal aggregations of Cu–ZnO nanoparticles. Furthermore, a slight shift to a lower BE of Cu 2p and Zn 2p peaks was observed due to the increased interactions between Cu–ZnO nanoparticles and zeolite overlayers according to increased crystallization times, as shown in Fig. 5(b), which was found to be consistent with H2-TPR results as well.
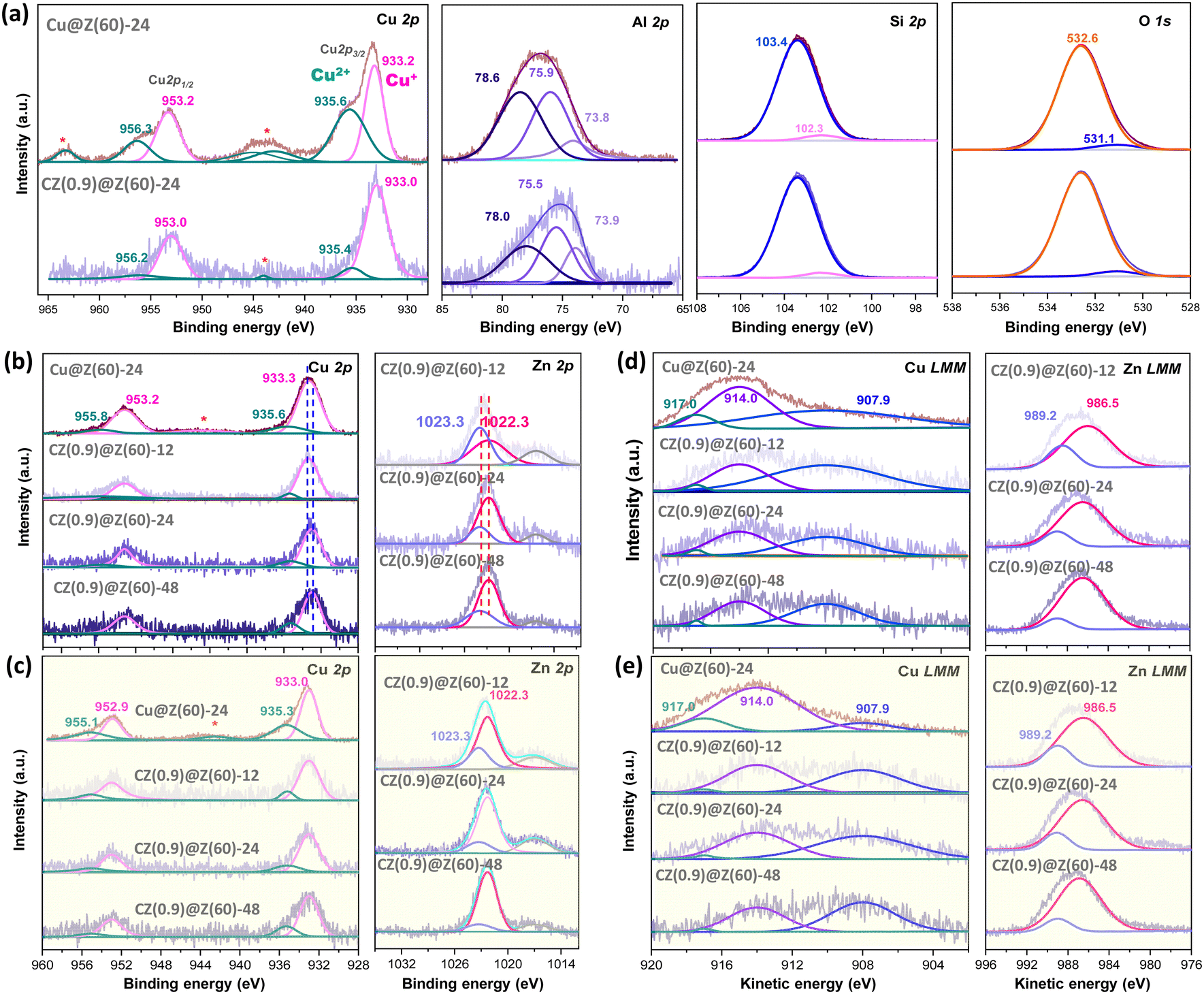 | ||
| Fig. 5 XPS spectra of Cu 2p, Al 2p, Si 2p, and O 1s for (a) fresh Cu@Z(60)-24 and CZ(0.9)@Z(60)-24. XPS spectra of Cu 2p and Zn 2p of (b) reduced and (c) used catalysts. Cu LMM and Zn LMM AES spectra of (d) reduced and (e) used catalysts (*denotes satellite). | ||
The atomic Cu/Zn ratios decreased from 1.07 to 0.20 as the crystallization duration increased from 12 to 48 h, and the surface ratios of the Cu/Zn and Si/Al on the reduced CZ(x)@Z(y)-t catalysts measured by the XPS analysis were found to be much smaller than the corresponding ratios measured by the XRF analysis (Table 1). This observation strongly suggests the preferential formation of the strong Cu–Al2O3 interactions inside ZSM-5 overlayer matrices (less exposed on the surfaces, confirmed by XPS analysis), which resulted in more stabilized Cu–ZnO nanoparticles under the crystallization or reaction condition. The Zn 2p XPS spectra of the reduced and used samples displayed in Fig. 5(b) and (c) appeared at 1022.3 and 1023.3 eV separately assigned to ZnO and Zn(OH)+ species, indicating there were strong interactions between the ZnO and ZSM-5 overlayers or partially reduced ZnO species.62 Furthermore, the oxidation states of the outermost Cu surfaces were measured by AES analysis (Fig. 5(d) and (e)) on the reduced and used catalysts, representing the distinguished oxidation states of Cu species (Cu0 and Cu+) with the kinetic energy in the range of 904–918 eV. The lower Cu0/(Cu0 + Cu+) ratio on the CZ(0.9)@Z(60)-t catalysts in the range of 0.03–0.16 also revealed that a little amount of metallic Cu0 and dominant Cu+ phases were stably preserved by maintaining their initial oxidation states due to the presence of protective crystalline ZSM-5 zeolite overlayers by encapsulating Cu–ZnO nanoparticles during the hydrothermal synthesis process. However, the lower ratio of Cu0/(Cu0 + Cu+) was also attributed to the stronger interaction between Cu–ZnO nanoparticles and ZSM-5 overlayers as well as the large-sized CuO nanoparticles in the absence of ZnO promoter. Additionally, Zn LMM AES was analysed to further understand the reduced Zn states, as displayed in Fig. 5(d) and (e), and the peaks at 989.2 and 986.5 eV could be assigned to the partially reduced Znδ+ species (0 < δ < 2) and Zn2+ in ZnO phase.63 The oxidation states of Cu species on the reduced CZ(x)@Z(y)-t catalysts were further confirmed by XANES and CO-probe in situ DRIFTS analysis (Fig. 6). It was disclosed that the reduced CZ(0.9)@Z(60)-24 catalyst predominantly presented Cu+ instead of the Cu0 phase (Fig. 6(a)–(c)). The Cu local coordination environments were observed by wavelet transform (WT) in Fig. 6(d), and the Cu oxidation state was determined by CO-DRIFTS analysis, as observed in Fig. 6(e), showing the bands around 2162, 2148, and 2131 cm−1 for the CO adsorption on Cu2+ in SiO2 matrices,64 symmetric and asymmetric stretching modes of Cu+(CO)265 and Cu+(CO) species,64,65 respectively. The dominant Cu+ and little portion of Cu0 phase were detected based on the Cu LMM peak with partially reduced Znδ+ species (0 < δ < 2) according to the Zn LMM in the AES analysis, which stably preserved even after the reduction and reaction without any significant change in the Cu+/Cu0 distribution.
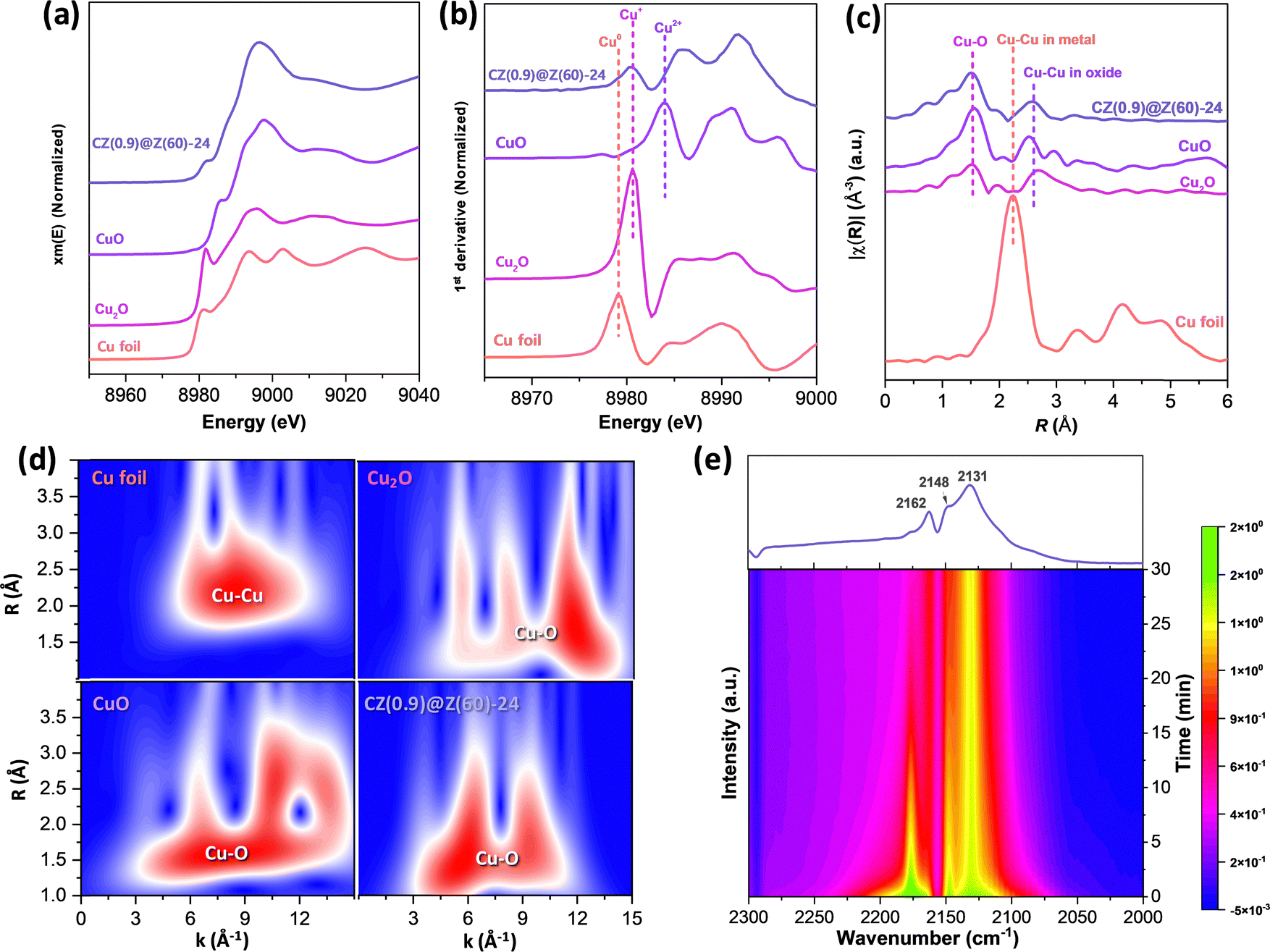 | ||
| Fig. 6 Characterization results of local structures and Cu oxidation states of reduced CZ(0.9)@Z(60)-24: (a) normalized Cu K-edge XANES spectra. (b) Normalized 1st derivative of the Cu K-edge XANES spectra. (c) Fourier transformations in R-space. (d) Wavelet transform (WT) of the Cu foil, Cu2O, CuO and reduced CZ(0.9)@Z(60)-24 and (e) in situ CO-DRIFTS analysis at 30 °C. | ||
3.3. CO2 hydrogenation activity to DME and methanol over CZ(x)@Z(y)-t catalysts
The hybridized bifunctional catalysts of Cu–ZnO nanoparticles encapsulated in ZSM-5 zeolite were successively prepared via the SAC approach and showed excellent catalytic performances with time on stream, as displayed in Fig. 7 and Fig. S7 (ESI†). In addition, the CZ(x)@SiO2 catalysts were synthesized with different Cu/Zn metal ratios of (x) as reference catalysts. The amorphous SiO2 overlayer shells with inner Cu–ZnO nanoparticles were transformed into the crystalline ZSM-5 zeolite with their different sizes of Cu–ZnO nanoparticles of 4.5, 9.5 to 10.5 nm during their crystallization durations of 12, 24, and 48 h, respectively. The increase in the crystallite sizes of mixed metal (oxides) of the Cu–ZnO nanoparticles under the hydrothermal condition was responsible for the decreased active surface area, enhancing the activation energy barriers with lower CO2 conversion to oxygenates (Table 1). The catalytic activity for CO2 hydrogenation to DME and methanol oxygenates on CZ(x)@Z(y)-t catalysts with different Cu/Zn ratios (x), Si/Al ratios (y), and crystallization time durations (t) was measured under the reaction conditions of T = 260 °C and P = 5.0 MPa (Fig. 7 and Fig. S7, ESI†), which showed stable activity after the induction period of ∼10 h on stream. All the CZ(0.9)@Z(60)-t catalysts with a Cu/Zn ratio of 0.9 and an Si/Al ratio of 60, revealing similar CO2 conversion in the range of 20.5–21.4% regardless of the crystallization duration. The highest DME selectivity of 62.2% and the lowest CO selectivity of 17.9% were observed on optimal CZ(0.9)@Z(60)-24, as shown in Fig. 7(a), and summarized in Table 1 and Table S3 (ESI†). The slight decrease in CO2 conversion on CZ(0.9)@Z(60)-24 compared to that of CZ(0.9)@Z(60)-12 was possibly due to the decreased Cu–ZnO content (less than 5 wt% as shown in Table S1 (ESI†) during the SAC preparation step, resulting in a less amount of active metallic Cu0 sites. The acidic and basic sites of the CZ(x)@Z(y)-t catalysts were also measured by NH3-TPD and CO2-TPD analyses (Table 1), and more abundant acidic and basic sites on CZ(0.9)@Z(60)-12 with respective values of 1.058 and 0.654 mmol g−1 than the other CZ(0.9)@Z(60)-t catalysts revealed a lower methanol selectivity (18.2%) due to an increased successive reaction to form CO, facilitated by surface Brønsted acidic sites. The effects of the Cu/Zn molar ratios on the CZ(x)@Z(60)-24 catalysts were examined for CO2 hydrogenation, and are displayed in Fig. 7(b). As the Cu/Zn ratio increased from 0.3 to 0.9, CO2 conversion rate and DME selectivity were enhanced from 18.5 to 20.8% and from 46.9 to 62.2%, respectively. However, the CO2 conversion and DME selectivity decreased (19.6 and 50.3%, respectively) when the Cu/Zn ratio further increased up to 1.5. It is worth noting that the CZ(x)@Z(60)-24 catalysts with a Cu/Zn ratio lower than 0.6 revealed higher selectivity toward hydrocarbons. By decreasing the Cu/Zn ratios in the CZ(x)@Z(60)-24 catalysts, the amount of reducible and active Cu phase was significantly declined, as confirmed by H2-TPR analysis shown in Fig. 4, which caused the concentrations of the formed methanol intermediate over the Cu–ZnO nanoparticles to be significantly decreased for their further selective conversion to DME, which seems to possibly cause the selective formation of methane from the kinetic prospective.66 | ||
| Fig. 7 Catalytic activity of the CZ(x)@Z(y)-t catalysts: (a) CZ(0.9)@Z(60)-t at different crystallization times (t = 12, 24 and 48 h). (b) CZ(x)@Z(60)-24 for different Cu-to-Zn molar ratios (x = 0.3, 0.6, 0.9, 1.5 and only Cu metal). (c) CZ(0.9)@Z(y)-24 for different Si/Al molar ratios (y = 40, 60 and 100). (d) Catalytic activity of CZ(0.9)@Z(60)-24 with time on stream at 260 °C and 5.0 MPa. | ||
The Cu@Z(60)-24 catalyst without any Zn promoter showed the lowest CO2 conversion of 16.8% with a higher CO selectivity of 49.0%. This was mainly attributed to the poorly dispersed, large-sized Cu nanoparticles, explaining the contribution of the Zn promoter to isolate the Cu phases and create more Cu–Zn interfaces,24,25 which were responsible for a higher methanol synthesis activity and Zn promotion had significant effects on the catalytic activity and selectivity. The effects of Si/Al ratios, controlled from 40 to 100 in the CZ(0.9)@Z(y)-24 catalysts for the catalytic performances, are shown in Fig. 7(c), which showed similar CO2 conversions with different product distributions. In contrast, the observed lower catalytic activity on the reference physically mixed and wet-impregnated CZ(0.9)/ZSM-5(60) catalyst is briefly shown in Table S3 (ESI†), which clearly indicated that the ZSM-5 encapsulation strategy of Cu–ZnO nanoparticles significantly enhanced catalytic performances. The highest DME selectivity of 62.2% was observed for an Si/Al ratio of 60 due to its highly crystallized ZSM-5 structures with abundant strong acidic sites (0.118 mmol g−1 obtained from NH3-TPD analysis, as shown in Table 1).19 CH4 byproduct formation was also enhanced for CZ(0.9)@Z(40)-24 having a lower Si/Al ratio of 40 with 2.5% selectivity. Therefore, the catalyst compositions and structures were optimized at a Cu/Zn ratio of 0.9, an Si/Al ratio of 60, and a crystallization time of 24 h, resulting in the optimal CZ(0.9)@Z(60)-24 catalyst, where a medium turnover frequency (TOF) of 64.7 h−1 and an activation energy (Ea) of 15.4 kJ mol−1 were obtained. The medium particle size of Cu–ZnO (diameter, d = 9.5 nm) showed the medium amount of acidic and basic sites (0.819 and 0.526 mmol g−1, respectively), leading to an increased CO2 conversion and DME selectivity due to sufficient acidic sites and active CO2 reactants on the surface metallic Cu0 sites. CZ(0.9)@Z(60)-24 was further tested for 100 h on stream to verify the catalytic stability (Fig. 7(d)), revealing selective production of DME (66%) at high CO2 conversion (∼22%) with an excellent stability even after 100 h of reaction. The observed induction period was probably involved in the surface reconstructions by the adsorption of surface intermediates formed during CO2 hydrogenation,67 which was also supported by the particle size changes from ∼9.5 nm before the reaction to ∼10 nm after 100 h reaction over the CZ(0.9)@Z(60)-24 catalyst, as displayed in Fig. S1 and S3 (ESI†).
3.4. Proposed reaction mechanisms for CO2 hydrogenation over CZ(x)@Z(y)-t catalysts
In situ DRIFTS analysis of the CO2 hydrogenation reaction, temperature-programed desorption of methanol (methanol-TPD) and methanol dehydration on CZ(0.9)@Z(60)-24 were further carried out to verify surface intermediates, and altered analyses from 30 to 400 °C for methanol-TPD analysis were also performed (Fig. 8). The characteristic peaks appearing at 2953, 2892, 2842 and 1585 cm−1 originated from C–H and C–O vibrations of the surface formate intermediate.68–71 Those peaks slightly increased as the reaction temperature increased up to 260 °C, and the duration extended up to 60 minutes because of the enhanced formation of intermediates by CO2 activation. The newly formed peak appearing at 3015 cm−1 was attributed to the formation of surface methane,72 and the absorption bands at 2978, 2960 and 2855 cm−1, introduced at a higher temperature above 200 °C, were attributed to the C–H vibration modes of surface methoxy intermediates or methanol formed.72–75 It was expected that the peak at 2899 cm−1 originated from the DME product, strongly bonded to the surface acidic sites of the ZSM-5 overlayers (Fig. 8(a)–(d)).73 The formation of surface formate and methoxy intermediates explains that methanol was possibly produced by the well-known formate-pathway and could be dehydrated to form DME on the acidic ZSM-5 surfaces. This pathway was facilitated on the optimal CZ(0.9)@Z(60)-24 catalyst with a large amount of strong acidic sites and Cu+/Cu0 sites. Furthermore, the formation of C–H bonds was accelerated by facilitated CO2 dissociation to generate surface CO* intermediates selectively, confirmed by the observed peaks in the range of 2250–2400 cm−1 for adsorbed CO2 and at 2154 and 1868 cm−1 for surface CO* species,74–78 formed by the RWGS reaction on the oxygen vacant basic sites. To clarify the adsorption properties of methanol intermediates over CZ(0.9)@Z(60)-24, methanol-TPD and methanol dehydration reactions were carried out by in situ DRIFTS analysis. The C–H vibration peaks at ∼2949, 2854, and ∼2956 cm−1 were assigned to methanol or surface methoxy species and the C–H vibration peak (2990 cm−1) for the DME product is shown in Fig. 8(d). It was noted that the C–H vibration peak of formate intermediates (2843 cm−1) appeared with the formation of C–O vibration (2156 cm−1) and that the desorption occurred at the temperature range from 200 to 300 °C (Fig. 8(e)). This observation suggests that the CO* adsorbate can also be generated on the active Cu+ sites in CZ(0.9)@Z(60)-24 for further hydrogenation or decomposition.79 In addition, the formation of surface methoxy intermediates (C–H vibration at 2950, 2844 cm−1), formate (C–H vibration at 2844 cm−1) and DME product (C–H vibration at 2899 cm−1) through the reaction of methanol with the surface methoxy species formed by dehydration were also observed, as shown in Fig. 8(f). Based on the in situ DRIFTS analyses, the mechanisms for CO2-to-DME tandem reactions are summarized as follows, and the schematic descriptions are displayed in Fig. 9. First, CO2 and H2 reactants were simultaneously activated on the active Cu–ZnO nanoparticles to from methanol/methoxy species through formate intermediates with their successive hydrogenation to methoxy species, and the dehydration of methoxy and methanol on the acidic sties of the ZSM-5 overlayers produced DME products. Accordingly, the catalytic activity and stability for direct CO2 hydrogenation to DME on the core–shell structured CZ(0.9)@Z(60)-24 catalyst were correlated well with the formation rate of surface methoxy intermediates via the preferential reaction pathway from the formate intermediate to methanol. The optimized CZ(0.9)@Z(60)-24 catalyst showed much higher DME/methanol selectivity above 80% at a comparable CO2 conversion above 20% compared to the previously reported results, as summarized in Table S2 (ESI†). In addition, CZ(0.9)@Z(60)-24 revealed a higher DME selectivity with a lower CO selectivity than those of the physically mixed reference CZ@Si/ZSM-5 catalysts, as summarized in Table S3 (ESI†). High crystallinity, abundant strong acidic and basic sites, and large amounts of active Cu+ on the thermally stable Cu–ZnO nanoparticles, which were encapsulated with proper ZSM-5 overlayers, were found to be most crucial for an enhanced catalytic activity and stability during direct CO2 hydrogenation to oxygenates. | ||
| Fig. 8 In situ DRIFTS spectra over CZ(0.9)@Z(60)-24 with (a)–(c): mixed gas feeding (H2/CO2 = 3/1) at different reaction temperatures ranging from 50 to 260 °C, and kept at a constant reaction temperature of T = 260 °C and P = 0.5 MPa for 60 minutes every 5 minutes, (d) and (e) methanol-TPD at different reaction temperatures ranging from 30 to 400 °C, and (f) methanol dehydration to DME at 260 °C under ∼10 mol% methanol vapor/N2 flow (30 mL min−1). | ||
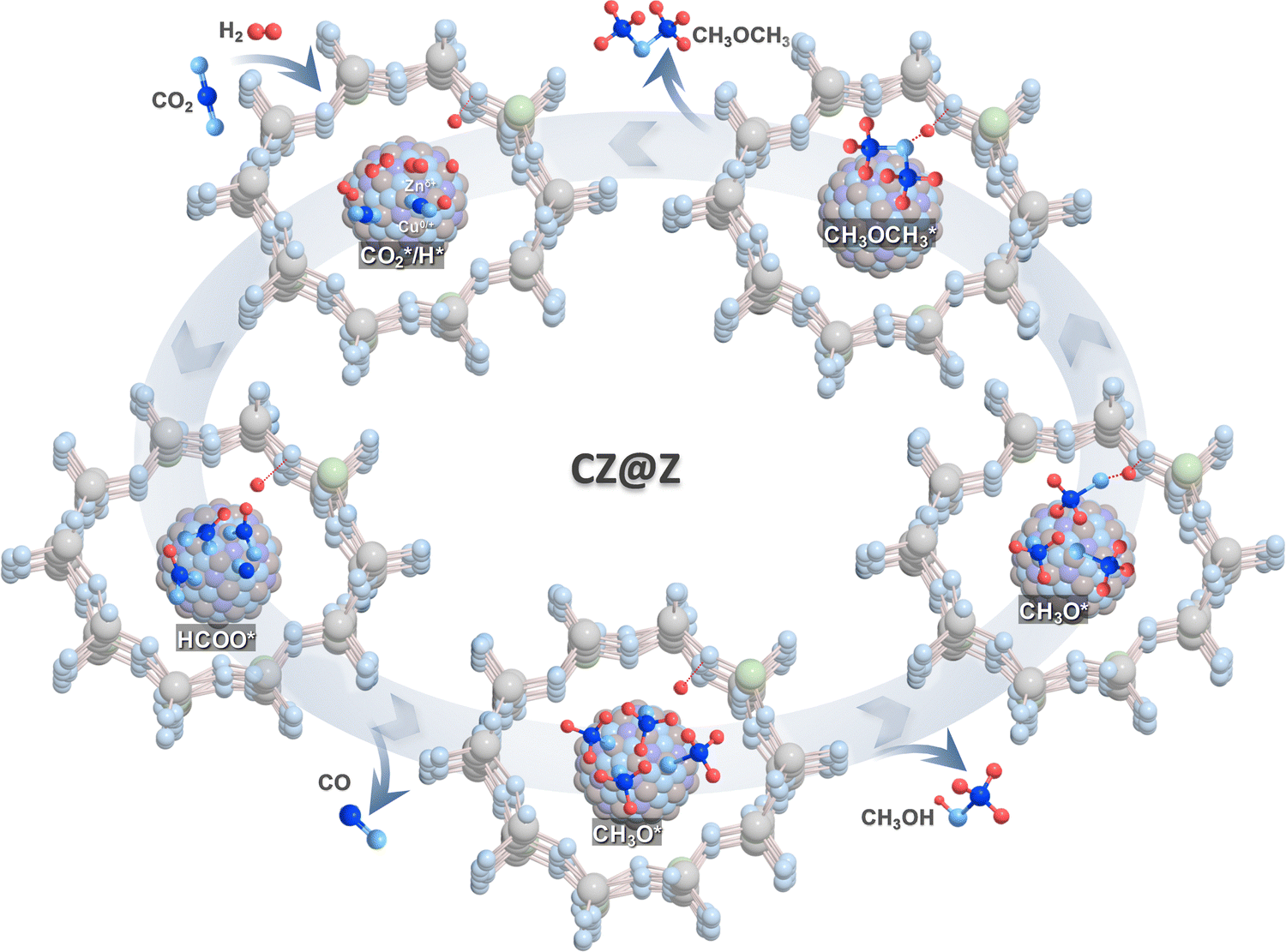 | ||
| Fig. 9 Proposed reaction mechanisms for direct CO2 hydrogenation to DME and methanol over ZSM-5-encapsulated Cu–ZnO nanoparticles. | ||
4. Conclusions
Cu–ZnO nanoparticles encapsulated in nanocrystalline ZSM-5 zeolite overlayers were prepared via a SAC preparation approach. Highly dispersed Cu–ZnO nanoparticles maintained active Cu+ species via stronger interactions between Cu/Zn and ZSM-5 overlayers, which revealed their thermal stability against sintering under the optimized reduction and reaction conditions of Cu/Zn ratio of 0.9, an Si/Al ratio of 60.2, and 24 h of crystallization process. The catalyst revealed an excellent performance of CO2 hydrogenation to methanol and its successive dehydration compared to the previous reports and exhibited a CO2 conversion rate of 20.8%, a STY of DME products of 13.9 gDME (gCu−1 h−1), and a methanol/DME oxygenate selectivity of 81.6% (62.2% DME). The spatial confinements of Cu–ZnO nanoparticles inside ZSM-5 overlayers strengthened the interactions between Cu and ZSM-5, which preserved dominant Cu+ species and facilitated the formation of key intermediates such as formate and methoxy species for enhanced CO2 conversion, high DME selectivity, and prolonged catalyst durability for CO2-to-DME tandem reactions. These findings for the surface structures of catalytic active sites and reaction mechanisms were well supported by detailed characterization results including in situ DRIFTS, AES/XPS, and XANES analyses. Our reports also paved the possible pathway for an efficient carbon removal method by simply providing effective and stable Cu–ZnO nanoparticles stabilized with ZSM-5 zeolite overlayers for CO2-to-DME tandem reactions.Author contributions
J. W. B. and E. J. conceived and designed the idea, revised and perfected the manuscript. X. W. performed the catalytic reactions, conducted the characterizations and related experiments as well as wrote the manuscript. H. Y. W., M. J. P., D. S., M. A., F. Z., K. Y. K., J. S. C. analyzed and interpreted the data from characterizations and catalytic evaluation experiments.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
We all declare no financial/commercial conflicts of interest.Acknowledgements
This research was supported by the Korea Electric Power Corporation of the Republic of Korea (R21XA01-29). The authors would sincerely like to acknowledge the financial support from the National Research Foundation of Korea (NRF) grant funded by the South Korea government (RS-2024-00405818).References
- P. Erickson, M. Lazarus and G. Piggot, Nat. Clim. Change, 2018, 8, 1037–1043 CrossRef CAS.
- J. S. Sawyer, Nature, 1972, 239, 23–26 CrossRef CAS.
- R. M. Cuéllar-Franca and A. Azapagic, J. CO2 Util., 2015, 9, 82–102 CrossRef.
- N. Von Der Assen, P. Voll, M. Peters, A. Bardow, N. Von Der Assen, P. Voll, M. Peters and A. Bardow, Chem. Soc. Rev., 2014, 43, 7982–7994 RSC.
- R. W. Dorner, D. R. Hardy, F. W. Williams and H. D. Willauer, Energy Environ. Sci., 2010, 3, 884–890 RSC.
- S. Wang, L. Zhang, P. Wang, X. Liu, Y. Chen, Z. Qin, M. Dong, J. Wang, L. He, U. Olsbye and W. Fan, Chem, 2022, 8, 1376–1394 CAS.
- X. Wang, D. Wu, J. Zhang, X. Gao, Q. Ma, S. Fan and T. S. Zhao, Appl. Catal., A, 2019, 573, 32–40 CrossRef CAS.
- A. J. Barrios, D. V. Peron, A. Chakkingal, A. I. Dugulan, S. Moldovan, K. Nakouri, J. Thuriot-Roukos, R. Wojcieszak, J. W. Thybaut, M. Virginie and A. Y. Khodakov, ACS Catal., 2022, 12, 3211–3225 CrossRef CAS.
- J. Wei, Q. Ge, R. Yao, Z. Wen, C. Fang, L. Guo, H. Xu and J. Sun, Nat. Commun., 2017, 8, 15174 CrossRef PubMed.
- P. Gao, S. Li, X. Bu, S. Dang, Z. Liu, H. Wang, L. Zhong, M. Qiu, C. Yang, J. Cai, W. Wei and Y. Sun, Nat. Chem., 2017, 9, 1019–1024 CrossRef CAS PubMed.
- X. Shang, G. Liu, X. Su, Y. Huang and T. Zhang, EES Catal., 2023, 1, 353–368 RSC.
- X. Wang, C. Zeng, N. Gong, T. Zhang, Y. Wu, J. Zhang, F. Song, G. Yang and Y. Tan, ACS Catal., 2021, 11, 1528–1547 CrossRef CAS.
- R. McGinnis, Joule, 2020, 4, 509–511 CrossRef.
- Z. He, M. Cui, Q. Qian, J. Zhang, H. Liu and B. Han, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 12654–12659 CrossRef CAS PubMed.
- Y. Ni, Z. Chen, Y. Fu, Y. Liu, W. Zhu and Z. Liu, Nat. Commun., 2018, 9, 3457 CrossRef PubMed.
- Y. Wang, L. Tan, M. Tan, P. Zhang, Y. Fang, Y. Yoneyama, G. Yang and N. Tsubaki, ACS Catal., 2018, 9, 895–901 CrossRef.
- G. Tian, X. Liang, H. Xiong, C. Zhang and F. Wei, EES Catal., 2023, 1, 677–686 RSC.
- C. Zhou, J. Shi, W. Zhou, K. Cheng, Q. Zhang, J. Kang and Y. Wang, ACS Catal., 2019, 10, 302–310 CrossRef.
- (a) X. Wang, S. Y. Jeong, H. S. Jung, D. Shen, M. Ali, F. Zafar, C. H. Chung and J. W. Bae, Appl. Catal., B, 2023, 327, 122456 CrossRef CAS; (b) H. Ham, S. W. Baek, C. H. Shin and J. W. Bae, ACS Catal., 2019, 9, 679–690 CrossRef CAS.
- M. D. Porosoff, B. Yan and J. G. Chen, Energy Environ. Sci., 2016, 9, 62–73 RSC.
- F. Frusteri, G. Bonura, C. Cannilla, G. D. Ferrante, A. Aloise, E. Catizzone, M. Migliori and G. Giordano, Appl. Catal., B, 2015, 176, 522–531 CrossRef.
- L. Yao, X. Shen, Y. Pan and Z. Peng, Energy Fuels, 2020, 34, 8635–8643 CrossRef CAS.
- (a) S. Kattel, P. J. Ramírez, J. G. Chen, J. A. Rodriguez and P. Liu, Science, 2017, 355, 1296–1299 CrossRef CAS PubMed; (b) Y. F. Shi, S. Ma and Z. P. Liu, EES Catal., 2023, 1, 921–933 RSC.
- F. Jiang, Y. Yang, L. Wang, Y. Li, Z. Fang, Y. Xu, B. Liu and X. Liu, Catal. Sci. Technol., 2022, 125, 551–564 RSC.
- X. Cui, W. Yan, H. Yang, Y. Shi, Y. Xue, H. Zhang, Y. Niu, W. Fan and T. Deng, ACS Sustainable Chem. Eng., 2021, 9, 2661–2672 CrossRef CAS.
- T. Fujitani, I. Nakamura, S. Ueno, T. Uchijima and J. Nakamura, Appl. Surf. Sci., 1997, 121, 583–586 CrossRef.
- D. Gómez, C. Candia, R. Jiménez and A. Karelovic, J. Catal., 2022, 406, 96–106 CrossRef.
- F. Liao, Y. Huang, J. Ge, W. Zheng, K. Tedsree, P. Collier, X. Hong and S. C. Tsang, Angew. Chem., Int. Ed., 2011, 123, 2210–2213 CrossRef.
- M. Behrens, F. Studt, I. Kasatkin, S. Kühl, M. Hävecker, F. Abild-Pedersen, S. Zander, F. Girgsdies, P. Kurr, B. L. Kniep, M. Tovar, R. W. Fischer, J. K. Nørskov and R. Schlögl, Science, 2012, 336, 893–897 CrossRef CAS PubMed.
- J. T. Sun, I. S. Metcalfe and M. Sahibzada, Ind. Eng. Chem. Res., 1999, 383, 868–3872 Search PubMed.
- Y. Choi and H. G. Stenger, Appl. Catal., B, 2002, 38, 259–269 CrossRef CAS.
- B. Liang, J. Ma, X. Su, C. Yang, H. Duan, H. Zhou, S. Deng, L. Li and Y. Huang, Ind. Eng. Chem. Res., 2019, 58, 9030–9037 CrossRef CAS.
- (a) J. Yu, M. Yang, J. Zhang, Q. Ge, A. Zimina, T. Pruessmann, L. Zheng, J. D. Grunwaldt and J. Sun, ACS Catal., 2020, 10, 14694–14706 CrossRef CAS; (b) Z. Q. Wang, Z. N. Xu, S. Y. Peng, M. J. Zhang, G. Lu, Q. S. Chen, Y. Chen and G. C. Guo, ACS Catal., 2015, 5, 4255–4259 CrossRef CAS.
- (a) E. Fernández-Villanueva, P. G. Lustemberg, M. Zhao, J. Soriano Rodriguez, P. Concepción and M. V. Ganduglia-Pirovano, J. Am. Chem. Soc., 2024, 146, 2024–2032 CrossRef PubMed; (b) A. Jangam, P. Hongmanorom, M. Hui Wai, A. Jeffry Poerjoto, S. Xi, A. Borgna and S. Kawi, ACS Appl. Energy Mater., 2021, 4, 12149–12162 CrossRef CAS.
- (a) L. Wang, L. Wang, X. Meng and F. S. Xiao, Adv. Mater., 2019, 31, 1901905 CrossRef CAS PubMed; (b) S. Xing, S. Turner, D. Fu, S. van Vreeswijk, Y. Liu, J. Xiao, R. Oord, J. Sann and B. M. Weckhuysen, JACS Au, 2013, 3, 1029–1038 CrossRef PubMed; (c) E. Eom, M. Song, J. C. Kim, D. Kwon, D. N. Rainer, K. Gołąbek, S. C. Nam, R. Ryoo, M. Mazur and C. Jo, JACS Au, 2022, 2, 2327–2338 CrossRef CAS PubMed.
- G. Laugel, X. Nitsch, F. Ocampo and B. Louis, Appl. Catal., A, 2011, 402, 139–145 CrossRef CAS.
- M. Rutkowska, D. Macina, N. Mirocha-Kubień, Z. Piwowarska and L. Chmielarz, Appl. Catal., B, 2015, 174, 336–343 CrossRef.
- (a) A. A. Rownaghi, F. Rezaei, M. Stante and J. Hedlund, Appl. Catal., B, 2012, 119, 56–61 CrossRef; (b) M. Lyu, J. Zheng, C. Coulthard, J. Ren, Y. Zhao, S. C. E. Tsang, C. Chen and D. O'Hare, Chem. Sci., 2023, 14, 9814–9819 RSC.
- C. Gao, F. Lyu and Y. Yin, Chem. Rev., 2020, 121, 834–881 CrossRef PubMed.
- (a) U. J. Etim, Y. Chen and Z. Zhong, Chem. Eng. J., 2024, 485, 155783 CrossRef; (b) Y. Chai, B. Qin, B. Li, W. Dai, G. Wu, N. Guan and L. Li, Natl. Sci. Rev., 2023, 10, nwad043 CrossRef CAS PubMed; (c) W. G. Cui, Y. T. Li, L. Yu, H. Zhang and T. L. Hu, ACS Appl. Mater. Interfaces, 2021, 13, 18693–18703 CrossRef CAS PubMed; (d) R. Kanomata, K. Awano, H. Fujitsuka, K. Kimura, S. Yasuda, R. Simancas, S. Bekhti, T. Wakihara, T. Yokoi and T. Tago, Chem. Eng. J., 2024, 485, 149896 CrossRef CAS.
- S. Wang, Z. Huang, Y. Luo, J. Wang, Y. Fang, W. Hua, Y. Yue, H. Xu and W. Shen, Catal. Sci. Technol., 2020, 10, 6562–6572 RSC.
- H. Wang, J. Jia, S. Liu, H. Chen, Y. Wei, Z. Wang, L. Zheng, Z. Wang and R. Zhang, Environ. Sci. Technol., 2021, 55, 5422–5434 CrossRef CAS PubMed.
- X. Liu, X. Wu, D. Weng and L. Shi, J. Rare Earths, 2016, 34, 1004–1009 CrossRef CAS.
- J. Cheng, D. Zheng, C. Dai, R. Xu, N. Liu, G. Yu, N. Wang and B. Chen, Environ. Sci.: Nano, 2022, 9, 2372–2387 RSC.
- G. Bonura, M. Migliori, L. Frusteri, C. Cannilla, E. Catizzone, G. Giordano and F. Frusteri, J. CO2 Util., 2018, 24, 398–406 CAS.
- P. Nakhostin Panahi, Environ. Prog. Sustainable Energy, 2017, 36, 1049–1055 CAS.
- H. Xue, X. Guo, S. Wang, C. Sun, J. Yu and D. Mao, Catal. Commun., 2018, 112, 53–57 CrossRef CAS.
- K. Kubo, H. Iida, S. Namba and A. Igarashi, Catal. Commun., 2012, 29, 162–165 CAS.
- Y. Zhang, D. Li, Y. Zhang, Y. Cao, S. Zhang, K. Wang, F. Ding and J. Wu, Catal. Commun., 2014, 55, 49–52 CAS.
- R. Singh, K. Tripathi, K. K. Pant and J. K. Parikh, Fuel, 2022, 318, 123641 CAS.
- X. Dong, F. Li, N. Zhao, F. Xiao, J. Wang and Y. Tan, Appl. Catal., B, 2016, 191, 8–17 CAS.
- C. Huang, J. Wen, Y. Sun, M. Zhang, Y. Bao, Y. Zhang, L. Liang, M. Fu, J. Wu, D. Ye and L. Chen, Chem. Eng. J., 2019, 374, 221–230 CrossRef CAS.
- W. Chen, J. Xu, F. Huang, C. Zhao, Y. Guan, Y. Fang, J. Hu, W. Yang, Z. Luo and Y. Guo, Appl. Surf. Sci., 2023, 618, 156539 CrossRef CAS.
- T. Zhang, J. Liu, D. Wang, Z. Zhao, Y. Wei, K. Cheng, G. Jiang and A. Duan, Appl. Catal., B, 2014, 148, 520–531 CrossRef.
- R. Moreno-Tost, J. Santamaría-González, E. Rodríguez-Castellón, A. Jiménez-López, M. A. Autié, E. González, M. C. Glacial and C. D. L. Pozas, Appl. Catal., B, 2004, 50, 279–288 CrossRef CAS.
- V. Boosa, S. Varimalla, M. Dumpalapally, N. Gutta, V. K. Velisoju, N. Nama and V. Akula, Appl. Catal., B, 2021, 292, 120177 CrossRef CAS.
- B. Dou, G. Lv, C. Wang, Q. Hao and K. Hui, Chem. Eng. J., 2015, 270, 549–556 CrossRef CAS.
- O. B. Ayodele, Appl. Catal., B, 2022, 312, 121381 CrossRef CAS.
- A. M. Rabie, M. A. Betiha and S. E. Park, Appl. Catal., B, 2017, 215, 50–59 CrossRef CAS.
- C. M. A. Parlett, L. J. Durndell, A. Machado, C. Giannantonio, W. B. Duncan, S. H. Nicole, W. Karen and F. L. Adam, Catal. Today, 2014, 229, 46–55 CrossRef CAS.
- K. Narasimharao and H. S. Kamaluddin, Mater. Chem. Phys., 2023, 32, 101675 CAS.
- K. Wang, H. Ge and Y. Qin, ChemCatChem, 2022, 14, e202200022 CrossRef CAS.
- (a) Y. Shao, M. Kosari, S. Xi and H. C. Zeng, ACS Catal., 2022, 12, 5750–5765 CrossRef CAS; (b) M. Tian, X. Tian, E. Ma, J. Hao, Z. Zuo and W. Huang, ACS Sustainable Chem. Eng., 2023, 11, 13616–13627 CrossRef CAS.
- A. Dandekar and M. A. Vannice, J. Catal., 1998, 178, 621–639 CrossRef CAS.
- J. Woo, K. Leistner, D. Bernin, H. Ahari, M. Shost, M. Zammit and L. Olsson, Catal. Sci. Technol., 2018, 8, 3090–3106 RSC.
- H. Jiang, Z. Hou and Y. Luo, ACS Catal., 2020, 10, 13518–13523 CrossRef CAS.
- (a) J. E. N. Swallow, E. S. Jones, A. R. Head, J. S. Gibson, R. B. David, M. W. Fraser, M. A. van Spronsen, S. Xu, G. Held, B. Eren and R. S. Weatherup, J. Am. Chem. Soc., 2023, 145, 6730–6740 CrossRef CAS PubMed; (b) F. Tao and M. Salmeron, Science, 2024, 386, eadq0102 CrossRef CAS PubMed.
- R. Khobragade, M. Roškarič, G. Žerjav, M. Košiček, J. Zavašnik, N. Van de Velde, I. Jerman, N. N. Tušar and A. Pintar, Appl. Catal., A, 2021, 627, 118394 CrossRef CAS.
- K. Chen, H. Fang, S. Wu, X. Liu, J. Zheng, S. Zhou, X. Duan, Y. Zhuang, S. C. Edman Tsang and Y. Yuan, Appl. Catal., B, 2019, 251, 119–129 CrossRef CAS.
- Y. Yan, R. J. Wong, Z. Ma, F. Donat, S. Xi, S. Saqline, Q. Fan, Y. Du, A. Borgna, Q. He, C. R. Müller, W. Chen, A. A. Lapkin and W. Liu, Appl. Catal., B, 2022, 306, 121098 CrossRef CAS.
- (a) Y. Wang, Y. Liu, L. Tan, X. Lin, Y. Fang, X. F. Lu, Y. Hou, G. Zhang and S. Wang, J. Mater. Chem. A, 2023, 11, 26804–26811 RSC; (b) S. Liu, Q. Zhao, X. Han, C. Wei, H. Liang, Y. Wang, S. Huang and X. Ma, Trans. Tianjin. Univ., 2023, 29, 293–303 CrossRef CAS; (c) A. N. Matveyeva and S. O. Omarov, Trans. Tianjin. Univ., 2024, 30, 337–358 CrossRef CAS.
- A. Atakan, E. Erdtman, P. Mäkie, L. Ojamäe and M. Odén, J. Catal., 2018, 362, 55–64 CrossRef CAS.
- H. Zhou, W. Zhu, L. Shi, H. Liu, S. Liu, Y. Ni, Y. Liu, Y. He, S. Xu, L. Li and Z. Liu, J. Mol. Catal. A: Chem., 2016, 417, 1–9 CrossRef CAS.
- A. Święs, A. Kowalczyk, B. Gil and L. Chmielarz, RSC Adv., 2022, 12, 9395–9403 RSC.
- Q. Sheng, R. P. Ye, W. Gong, X. Shi, B. Xu, M. Argyle, H. Adidharma, M. Fan, H. Adidharma and M. Fan, J. Environ. Sci., 2020, 92, 106–117 CrossRef CAS PubMed.
- A. S. Malik, S. F. Zaman, A. A. Al-Zahrani, M. A. Daous, H. Driss and L. A. Petrov, Appl. Catal., A, 2018, 560, 42–53 CrossRef CAS.
- J. W. Bae, S. H. Kang, Y. J. Lee and K. W. Jun, Appl. Catal., B, 2009, 90, 426–435 CrossRef CAS.
- Y. Chen, H. Hong, J. Cai and Z. Li, ChemCatChem, 2021, 13, 656–663 CrossRef CAS.
- X. Wang, A. A. Arvidsson, M. O. Cichocka, X. Zou, N. M. Martin, J. Nilsson, S. Carlson, J. Gustafson, M. Skoglundh, A. Hellman and P. A. Carlsson, J. Phys. Chem. C, 2017, 121, 27389–27398 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Particle size distribution of the fresh Cu–ZnO nanoparticles encapsulated in SiO2 (Fig. S1), HADDF images and EDS elemental mapping results of fresh CZ(0.9)@Z(60)-24 (Fig. S2), TEM image and size distribution of used CZ(0.9)@Z(60)-24 (Fig. S3), TEM and EDS elemental mapping images of used CZ(0.9)@Z(60)-24 after 100 h of reaction (Fig. S4), Py-IR spectra of the fresh CZ(0.9)@Z(60)-t catalyst (Fig. S5), EPR spectra of the fresh CZ(0.9)@Z(60)-t catalysts (Fig. S6), catalytic activity with time-on-stream (TOS) on all the CZ(x)@Z(y)-t catalysts (Fig. S7), summarized results of bulk composition and surface properties (Table S1), comparisons of the catalytic activity of CZ(0.9)@Z(60)-24 with previous reports on CO2 hydrogenation (Table S2), and catalytic performances of the hybridized reference CZ@Si and CZ@ZSM-5, supported CZ/ZSM-5 catalysts (Table S3). See DOI: https://doi.org/10.1039/d4ey00273c |
| This journal is © The Royal Society of Chemistry 2025 |