
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelf-assembled infinite silver cluster with atomic precision as a scalable catalyst for CO2-electroreduction under industry-relevant reaction rates†
Leonard
Curet
a,
William
Lafargue dit-Hauret
 a,
Jordi
Benet-Buchholz
b,
Marta
Martínez-Belmonte
b,
Dominique
Foix
a,
Emilio
Palomares
*bc,
Laurent
Billon
a,
Didier
Begué
a and
Aurelien
Viterisi
*a
a,
Jordi
Benet-Buchholz
b,
Marta
Martínez-Belmonte
b,
Dominique
Foix
a,
Emilio
Palomares
*bc,
Laurent
Billon
a,
Didier
Begué
a and
Aurelien
Viterisi
*a
aUniversite de Pau et Pays de l’Adour, E2S UPPA, CNRS, IPREM UMR 5254, Technopole Hélioparc, 2 avenue du Président Pierre Angot, 64053 PAU CEDEX 09, France. E-mail: aurelien.viterisi@univ-pau.fr
bInstitute of Chemical Research of Catalonia (ICIQ), Avda. Països Catalans, 16, 43007 Tarragona, Spain
cICREA, Passeig Lluís Companys, 28, E-08010, Barcelona, Spain
First published on 18th October 2024
Abstract
The multi-gram synthesis of a phenylacetylide silver cluster catalyst and its application to the electroreduction of CO2 to carbon monoxide is described. The procedure involves one synthetic step from commercially available precursors and yields highly crystalline silver acetylide clusters in quantitative yields with no required purification. The crystal structure of the cluster was resolved from the native powder using cutting-edge electron diffraction techniques (3D-ED), and showed to consist of an infinite silver tubular core with radially disposed phenylacetylene ligands. Its catalytic properties were investigated at industry-relevant rates in a flow cell electrolyser. Faradaic efficiencies for CO above 95% were achieved at current densities reaching 350 mA cm−2. The catalyst's selectivity and stability were shown to be the result of the unique polymeric structure, a result supported by electrochemical characterisation and DFT modelling.
Broader contextThe electrochemical reduction of CO2 to feedstocks is a promising alternative for offsetting greenhouse gas emissions. While higher hydrocarbon products (so-called C2+ products) are highly revered in academic research, carbon monoxide is arguably one of the most interesting feedstocks from an industrial perspective. Indeed, phosgene, isocyanates, urea, methyl methacrylate, and acetic acid, to name only a few, are directly derived from CO, industrially. Moreover, the electroreduction of CO2 to CO is the least energetically demanding process with the highest theoretical energy efficiency. The catalyst developed in this paper combines all the attributes of industry-relevant materials, such as the scalability from widely available precursors with no required purification. The catalytic properties towards CO2 electroreduction, i.e. high faradaic efficiency for CO (>95%) at high reaction rates (350 mA cm−2), provide strong arguments in favour of potential industrial applications. A rough extrapolation of the catalysis features indicates that the CO's weight production by unit time would likely reach over 1 T hour−1 per kilogram of catalyst. The approximate cost of the latter weight of catalyst is estimated to be less than 1000 dollars, considering the price of the precursors, rendering it an insignificant parameter from an economic perspective. |
Introduction
The electrochemical reduction of CO2 to feedstocks, such as carbon monoxide (CO), methane, or higher molecular weight hydrocarbons (C2+), is a promising alternative for offsetting greenhouse gas emissions.1–3 The transformation, generally mediated by a catalyst of metallic nature under electrochemical conditions, has attracted considerable interest from the scientific community4,5 since the pioneering work of Hori.6–8 While C2+ products are highly revered in academic research, CO is arguably one of the most interesting feedstocks from an industrial perspective. Indeed, phosgene, isocyanates, urea, methyl methacrylate, and acetic acid, to name only a few, are directly derived from CO industrially. Moreover, the electroreduction of CO2 to CO is the least energetically demanding process and has the highest theoretical energy efficiency.9 Quite importantly, the adaptability to zero-gap electrolysers in conjunction with different anode reactions confers the process the highest probability of reaching the market in a reasonable timeframe.10 With these considerations in mind, a general consensus has emerged that catalysts should be assessed under industrially relevant reaction rates.11 As such, flow cells and membrane electrode assemblies are commonly accepted designs that allow overcoming the mass transport limitations resulting from the poor solubility of CO2 in aqueous media.12–16 They involve the use of gas diffusion layers (GDLs), allowing the formation of a gas/electrolyte/catalyst interface where the local concentration of CO2 rises far above its solubility limit, thus allowing the CO2RR to perform at high reaction rates.17,18 The GDLs require that the catalyst is immobilised on the surface, imposing a significant limitation on the types of catalysts that can be adapted to bulk electrolysis. Indeed, the microporous nature of carbon-based or PTFE-based GDLs commonly used in CO2 electroreduction renders this process challenging, particularly for metal catalysts.19 Their deposition onto GDLs relies on sputtering techniques that are difficult to scale,20 and reaction overpotentials are difficult to tune. Their nanoparticle counterparts, despite being more versatile from a catalytic perspective, have an intrinsic polydisperse nature.21,22 Their synthesis is consequently difficult to project on an industrial scale. Several types of molecular or hybrid catalysts have been considered as possible alternatives, such as metal–organic frameworks,23 transition-metal complexes,24–26 polyoxometalates,27 carbon-based metal-free materials28 or two-dimensional materials.29 Metal clusters have been recently described in this context since they combine attributes of both metallic and molecular catalysts, presenting themselves as discrete crystalline materials that can readily be immobilised on GDLs with controlled surface coverage.30–32 The chemical structure of metal clusters consists of a main core composed of several metal atoms linked through metal–metal bonds and organic ‘ligands’ that are orderly distributed on the outer shell.33 Discrete clusters of gold,34 copper35 and silver36–38 with different types of capping ligands have been recently shown to display remarkable catalytic properties.30–32,39 Clusters comprising alkynyl ligands have proved particularly well suited for the CO2RR, as they involve stable alkyne–silver bonds, allowing for the direct modulation of the silver cluster's geometry and electronic properties by tailoring the chemical structure of the organic moiety. However, their synthesis, similarly to most catalysts, is generally incompatible with industrial-scale production, and their performance is often limited to laboratory-scale electrolysers (Fig. 1a).24,36–38,40–45 | ||
| Fig. 1 (a) Graph depicting the faradaic efficiency for CO vs. current density of state-of-the-art silver-based clusters catalysts, in comparison with the best performing molecular catalysts; (b) general scheme of silver acetylide's synthesis with a representation of an asymmetric unit of the cluster fragment established by electron diffraction showing two Ag4 clusters subunits; (c) picture of the catalysts powder used for structure resolution by 3D-ED. | ||
To tackle the above setbacks, herein, we describe a unique silver acetylide cluster displaying an infinite tubular structure and demonstrate its ability to efficiently catalyse the CO2RR under very high reaction rates in a flow cell electrolyser. Electrolysis currents higher than 350 mA cm−2 were achieved with very high selectivity towards CO (>95%). The versatility of our catalysts is further enhanced by the fact that its synthesis involves only one extremely reproducible synthetic step carried out in aqueous media from an inexpensive commercially available alkyne precursor and silver nitrate. The reaction was scaled up to multi-gram quantities (over 20 g), and the catalyst was obtained in quantitative yields with no required purification, producing only ammonium nitrate as a by-product. The catalyst was isolated as a nanocrystalline powder from which an electron diffraction structure was fully resolved from the as-synthesised material using cutting-edge nanocrystal electron diffraction technology (3D-ED). The structure consists of infinite silver tubular clusters with perfect crystalline arrangement packed in orthorhombic Pna21 unit cell. The insoluble nature of the catalyst allowed for facile immobilisation of the as-synthesised nanocrystalline powder onto carbon paper GDLs, providing a direct structure-characteristics relationship. The resulting elevated surface area led to a turnover frequency (TOF) over 13.5 s−1. Finally, a thorough chemical and physical characterisation supported by DFT modelling provided solid evidence on the mechanistic fundaments governing the selectivity of the catalyst towards CO. Accordingly, it appears that the unique infinite tubular structure with atomically precise order is, to our understanding, an essential parameter for the stability of the catalyst over repetitive catalytic cycles, while the ligands impede catalyst poisoning by maintaining the oxidation state of the non-catalytic silver atoms.
Results and discussion
The catalytic properties of discrete all-alkynyl silver clusters towards the CO2RR have been reported very recently in the literature, with very few papers published to date.34,36–38,42 Their organic–inorganic hybrid nature infers a high degree of structural versatility due to the ease of synthesis of large libraries of silver acetylide from commercially available alkynes. However, the existing reports rely on complex synthetic procedures, generally carried out on a scale limited to tens of milligrams of starting reagents, with reported yields typically below 10%. The formed compounds generally have defined crystalline packing and unit cells with crystallite sizes in the range of a few tens of nanometres, providing a very high surface area required for higher catalytic activity while preserving a precise arrangement of the clusters in the solid state, hence, the recent coining of “atomically precise” catalysts. While the above catalysts display remarkable selectivity, their performance under high electrolysis rates is yet to be demonstrated.Scheiber and co-workers showed that silver acetylides form readily as highly insoluble species from mixing a methanolic solution of an alkyne precursor with an aqueous silver ammoniacal solution. The above procedure was employed herein and optimised to synthesise silver phenylacetylide from commercially available phenylacetylene. Upon addition of the alkyne dissolved in methanol to a solution of ammoniacal silver nitrate, the formation of a white frothy precipitate ensues almost instantly, indicative of the formation of the silver acetylide species, which was isolated as a poorly soluble solid by simple filtration, in quantitative yields (>99%) from a 20 g scale reaction (Fig. 1b).
The solubility in DMSO was, however, sufficient to characterise the cluster via1H NMR, which shows one unusually broad phenyl proton peak reminiscent of polymeric species. The 1H NMR spectrum is consistent with the quantitative formation of silver acetylide, evidenced by the absence of the acetylene peak from the starting material (see ESI†). Similarly, the absence of the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–H stretching band (3289 cm−1) in the solid-state infrared spectrum with respect to the starting alkyne is further evidence of the formation of the Ag–CC bond (Fig. S4, ESI†). Additionally, a shift of the CC stretching band from 2109 cm−1 to 2054 cm−1 further confirms the latter observation.
C–H stretching band (3289 cm−1) in the solid-state infrared spectrum with respect to the starting alkyne is further evidence of the formation of the Ag–CC bond (Fig. S4, ESI†). Additionally, a shift of the CC stretching band from 2109 cm−1 to 2054 cm−1 further confirms the latter observation.
To gain additional information on the solid-state structure of the silver acetylide, powder XRD characterisation was carried out. To our surprise, despite the acetylides’ fast formation kinetics, powder diffractograms show well-resolved peaks of archetypical polycrystalline materials. The peak width is consistent with crystallite sizes in the range of a few hundred nanometres as calculated from the Scherer equation. A comparison with calculated diffractograms from existing single-crystal structures of similar clusters did not allow for crystalline phase identification. Therefore, to obtain precise structural information on the organometallic cluster, the growth of X-ray diffraction-quality single crystals was attempted by slow diffusion of a solution of the alkyne in methanol to a solution of ammoniacal silver in similar conditions as those used for bulk synthesis. However, needle-shaped crystals unsuitable for X-ray diffraction would form regardless of the conditions employed during the crystallisation experiments. These size limitations led us to explore electron diffraction (3D-ED) as an alternative method for acquiring structural data.
Contrary to single-crystal X-ray diffraction, 3D-ED was recently demonstrated to provide diffraction data of sufficiently high quality from crystals whose size is in the range of those found in powder samples. Advanced instruments combining both electron microscopy and diffraction techniques (Synergy-ED Rigaku-JEOL) now allow for selecting and recording diffraction data from powders’ crystallites in all directions of space and resolving their structure with similar accuracy to single crystal X-ray diffraction. Consequently, 3D-ED data were collected directly from the as-synthesised catalyst powder at room temperature (Fig. 1c) and 100 K. Crystallite selection was performed directly on the electron diffractometer by TEM imaging (Fig. E1, ESI†). The samples were composed of an overwhelming majority of flat needle-shaped crystals, which were extremely difficult to index since most of the grains measured appeared to consist of several stacked plates, leaving the diffractions collected in the direction of the C-axis undefined. After collecting datasets for 63 separate grains, a suitable crystal could be localised and indexed in the space group Pna21, which allowed for structure determination (measured at 100 K) (see ESI,† for details). The later crystal turned out to be the one used for solving the structure, being the only one found that did not present deformations in the direction of the C-axis. This crystal was refined kinematically to an R1 value of 17.04% with a completeness of 90.7%. To enhance the completeness and reflections-to-parameter ratio, the dataset of the selected crystal (LC155_04_20240308, see ESI†) was merged with a second crystal (LC152LT_03strong_ 20240308, see ESI†). Although the latter crystal was of much lower quality, it allowed increasing the completeness to 97.7%. The merged dataset was refined to an R1 value of 18.61%. After the correct assignment of the unit cell and space group, a total of 44 crystal grains could be indexed and refined to a moderate quality, confirming that all the grains were of the same crystalline phase (Pna21). Simulated powder X-ray diffraction patterns from 3D-ED and comparison with experimental powder diffractograms confirmed that the crystalline phases measured by both techniques matched (Fig. E2, ESI†). A comparison with reported structures of phenylacetylene silver acetylides showed that it consisted of a polymorphic structure from a P21/c space group (see ESI†).46
Similarly to the P21/c polymorph, the silver acetylide was found to display a unique infinite tubular silver core structure composed of covalently linked silver atoms repeating along the b parameter of the unit cell, with the organic ligands radially disposed on the cluster circumference. The asymmetric unit comprises an Ag8 cluster subunit assembled with eight [Ph–CC]− ligands. The latter consists of two parallel Ag4 squares oriented at a 90° angle with respect to one another and stacked along the b-axis (Fig. 2). One of the Ag4 squares contains two μ2,η1,η2 and two μ3-η1,η1,η1 [Ph–CC]− units, while the other contains four μ3-η1,η1,η1 [Ph–CC]− units. The two Ag4 squares are linked by the bridging [Ph–CC]− ligands and Ag–Ag bonds. The extension of Ag8 along the b-axis gives a highly organised tubular silver cluster. Importantly, in addition to providing precise structural determination revealing the polymeric nature of the crystalline arrangement, the exact correspondence of the molecular packing with the nanocrystalline powder later used as the catalyst allows for the establishment of a direct structure–properties relationship. This unique feature provided by 3D-ED structure determination has no precedent in the field.
 | ||
| Fig. 2 Electron diffraction structure of silver acetylide (AgPh)n showing (a) the unit cell and crystalline packing of the cylindrical silver clusters with infinite order with radially disposed outward-pointing acetylide groups, (b) the infinite arrangement of silver atoms with the two different types of Ag4 clusters subunits depicted in blue and green, (c) the asymmetric unit viewed in the direction of the b-axis (d) a close view of a cluster fragment in space-filling style, pointing to the somewhat sterically hindered silver cluster. | ||
Electron diffraction data provided important information on the solid-state structure of the cluster, however, the inherent properties of the crystallites’ surface dictate the catalytic properties to an equally important extent. For that matter, XPS analysis was employed to characterise pristine powders and assembled GDEs. Fig. 3 shows the XPS spectra of (AgPh)n pristine powder. The C 1s spectrum is decomposed into two peaks. The first peak, centred at 284.3 eV, is attributed to the carbon bonds within the phenyl ring, while the second peak, centred at 283.5 eV, is attributed to the silver–carbon bond (C–Ag) of the acetylide (Fig. 3a). The Ag 3d spectrum shows a well-resolved spin–orbit doublet at 368.3 eV and 374.2 eV ascribed to Ag(I) species in the Ag–C environment (Fig. 3b).
 | ||
| Fig. 3 XPS measurements of the starting material (a) C 1s electrons emission. Phenyl (green) C–Ag (purple); (b) Ag 3d electrons emission. The curves were fitted with the theoretical emission patterns (red dotted lines). | ||
Importantly, quantitative analysis shows a nearly identical ratio of CC–Ag and Ag(3d) peak area, unequivocally confirming that the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 Ag/ligand ratio was determined from the 3D-ED structure. Further metal quantification from dilute solutions of (AgPh)n using microwave plasma atomic emission spectroscopy (MP-AES) provided additional evidence that the latter ratio is maintained throughout the bulk of the material (see ESI†).
:1 Ag/ligand ratio was determined from the 3D-ED structure. Further metal quantification from dilute solutions of (AgPh)n using microwave plasma atomic emission spectroscopy (MP-AES) provided additional evidence that the latter ratio is maintained throughout the bulk of the material (see ESI†).
To assess the catalytic activity of (AgPh)n, the silver cluster was deposited on carbon-based gas diffusion layers typically used for CO2 reduction. Using a standard procedure, the GDL was coated with a nanoparticles-Nafion®-(AgPh)n ink by successive deposition–evaporation sequences at 80 °C until the appropriate weight-amount of catalyst was deposited. The catalyst loading was optimised by varying the weight-amount of the catalyst with respect to the carbon nanoparticles and Nafion® to maximise the catalytic characteristics, particularly the faradaic efficiency towards the major adduct. Results show that the catalyst loading could be decreased to a great extent without a significant impact on the selectivity. However, catalyst loading values below 0.2 mg cm−2 tended to favour the undesired HER (see ESI†). A catalyst loading of 0.4 mg cm−2 was eventually found to be optimum with regard to selectivity towards the main adduct.
Further powder XRD characterisation of the pristine GDEs revealed that the catalyst retains its original nanocrystalline form throughout the deposition process. The presence of the most intense peaks of the diffraction pattern arising from the (002) and (201) diffraction planes confirms the structure's integrity. In contrast, the slight discrepancy in peak intensity with respect to the calculated diffractogram is strong evidence of the crystals orienting towards a preferred face upon deposition. This is to be expected, owing to the needle-shaped crystals (Fig. 4a). The virtually identical peaks’ FWHM with respect to the powder diffractogram is evidence that the crystallites have not undergone any significant change in size during the deposition process. This further confirms that the sonicating step prior to deposition on the GDLs and the deposition temperature are not detrimental to the catalyst from a morphological standpoint. The stability upon deposition is further confirmed by XPS, which shows unaltered Ag 3d peaks of the GDE compared with the pristine material (Fig. 4b). A slight shift of the emission peak is, however, noted and attributed to the fluorine reach environment brought about by the excess Nafion®.
 | ||
| Fig. 4 (a) Powder diffractograms of the pristine catalyst powder (AgPh)n and of the GDE after catalyst deposition; (b) XPS spectra in the Ag 3d emission region of the pristine catalyst powder and GDE after catalyst deposition. | ||
The catalytic activity of the above GDEs was first tested in a homemade (Fig. S1, ESI†) 3-electrode cell in a 1 M potassium bicarbonate electrolyte. The linear sweep voltammetry (LSV) at 10 mV s−1 was measured in both N2 and CO2 saturated conditions and showed a clear catalytic wave with a measured catalytic threshold at −0.452 V vs. RHE corresponding to a 0.383 V overpotential vs. CO2RR theoretical potential (Fig. 5a). Finally, to assess the selectivity of our catalyst, a series of chronoamperometry experiments were carried out with 0.1 V increments from −0.63 V to −1.13 V vs. RHE applied potentials (measured vs. Ag/AgCl and subsequently converted). The produced gaseous and liquid products were sampled and quantified from the cell's headspace and liquid electrolyte after 15 min (Fig. 5b).
 | ||
| Fig. 5 (a) Linear sweep voltammetry plot of (AgPh)n carried out in KHCO3 (0.5 M) under an N2 or CO2 saturated solution; (b) Faradaic efficiencies for H2, CO and formate extracted from chronoamperometry experiments carried out in incremental potential values and sampled after 30 min of reaction. | ||
Interestingly, (AgPh)n showed a high faradaic efficiency towards CO at low overpotentials, with FE reaching 90% at electrolysis potentials as low as −0.63 Volts (vs. RHE). Importantly, (AgPh)n showed an elevated propensity to suppress the competing hydrogen evolution reaction (HER). As such, the selectivity of the CO2RR remains reasonably high over a wide range of voltages, reaching electrolysis currents particularly high (15–20 mA cm−2) for laboratory-scale electrolysis cells. The quantification of the liquid products in the electrolyte by 1H NMR showed the presence of small amounts of non-volatile formate at higher overpotentials with a maximum FE of 7% at −0.93 V.
These preliminary results prompted us to apply our silver acetylide-containing GDEs to a bulk electrolyser flow cell system. The latter consisted of a home-designed and 3D-printed flow cell (Fig. S2, ESI†), and Freudenberg H24C5 was selected as the GDL support due to its demonstrated suitability for bulk electrolysers. Preliminary tests showed that their hydrophobicity remained relatively stable under electrolysis compared to other hydrophobised GDLs. Constant current chronopotentiometry experiments were carried out in KHCO3 (1 M) under currents varying from 50 mA cm−2 to 350 mA cm−2. The faradaic efficiency of the best-performing GDE is depicted in Fig. 6. The averaged FEs from four GDEs (two different GDL types) from experiments carried out on two different electrolysers with distinct designs are depicted in Fig. S20b (ESI†). An outstanding selectivity towards CO was observed over the whole range of electrolysis current density tested, whereas HER was kept to a minimum even under currents as high as 350 mA cm−2. The potentials (measured against Ag/AgCl microelectrode) of the best-performing GDE remained exceptionally low (−1.9 V vs. RHE at 350 mA cm−2, Fig. 6b). According to the quantification of the catalytically active silver (see ESI,† for details), the turnover frequency (TOF) accounted for a value over 13.5 s−1 at the highest applied current density in the best-performing GDE.
 | ||
| Fig. 6 (a) Faradaic efficiency of (AgPh)n under CO2 reduction conditions in a flow cell electrolyser (KHCO3 (1 M) as catholyte and anolyte); (b) Cathode potential of the best-performing GDE (vs. RHE with iR drop correction). | ||
Quite importantly, the catalyst showed exceptional stability over an extended electrolysis time, as shown in Fig. 7b. Indeed, continuous electrolysis under current density as high as 100 mA cm−2 led to the production of a majority of CO over the whole experiment time frame, with hydrogen composition increasing only slightly. The overall FE on CO drops from 99% to 91% after 12 h of sustained electrocatalytic conditions (Fig. 7b). However, the slight increase in H2 and decrease in CO FE was principally attributed to the gradual wetting of the GDL. Indeed, prolonged reducing conditions have been known to reduce the hydrophobicity of carbon-based GDLs, an effect which we confirmed herein using contact angle measurements (Fig. S29, ESI†). Additionally, a visual inspection of the back side of the GDEs after fixed current chronopotentiometry showed a notable amount of catholyte droplets on the carbon fibre mesh and a substantial amount of liquid in the gas compartment. The water was seen to diffuse through the pores of the GDEs in a time as short as 20 minutes, regardless of the current applied in the range of 50 to 350 mA cm−2 (Fig. S30, ESI†).
 | ||
| Fig. 7 (a) Cyclic voltammetry of Ag showing three consecutive cycles in the 0 to −1.4 V range (vs. RHE); (b) faradaic efficiency evolution over time at 100 mA cm−2. | ||
Finally, although (AgPh)n showed an overwhelming CO selectivity reminiscent of pure silver(0), the presence of liquid products in detectable amounts and the polymetallic structure of the cluster's core indicate that the organic moiety plays a mediating effect in the selectivity of the whole catalyst. The electrochemical characterisation of (AgPh)n by cyclic voltammetry in the absence of CO2 shows a clear irreversible reduction wave under negative potentials (−0.21 V vs. RHE) attributed to the reduction of Ag(I) centres from the cluster to Ag(0). The fact that this reduction wave is only present during the first cycle of the CV experiment is indicative of a stable Ag(0) intermediate (Fig. 7a).
Upon further cycling, no further reduction waves are observed other than the faradaic reduction of the surrounding water, strongly suggesting that the previously formed Ag(0) centre constitutes the active catalytic species. Importantly, the coulombic charge associated with the reduction wave can be directly linked to the number of catalytic centres formed during the first reduction event (see ESI,† for details and how it relates to the turnover frequency (TOF)). Consequently, about 7-mole% of the silver present on the GDE is electrochemically reduced to Ag(0) before catalysis, the effect of which must be accompanied by the loss of one alkynyl ligand per silver atom reduced. The polymetallic nature of the catalyst likely provides the necessary stability to accommodate the excess charge generated and maintain the cluster's structural integrity throughout the catalytic process.
To gain further insight into the structural modifications incurred to the silver cluster upon catalysis, a thorough post-catalysis analysis was carried out. For that matter, a set of fresh GDEs prepared in identical conditions were submitted to 20 min chronopotentiometry conditions under current densities spanning a 50–300 mA cm−2 range in 50 mA cm−2 increments. The GDEs were thoroughly washed, dried under vacuum after the experiments, and kept under N2 for further XPS and XRD analysis. Diffractograms of the resulting GDEs recorded in the range of 5 to 70 degrees 2θ angles show an unequivocal loss of crystallinity upon catalysis, under current as low as 50 mA cm−2. Indeed, as seen in Fig. 8a, the intensity of the characteristic (200) and (201) plane reflections from the pristine GDE are seen to decrease to such an extent that they barely protrude above the background noise level after a 50 mA cm−2 chronopotentiometry experiment. This stark decrease in crystalline volume is also observed at higher current densities, where no more diffraction peaks can be observed in the diffractograms of the chronopotentiometry carried out at 100 mA cm−2 and above. However, the loss of crystalline volume does not appear to be followed by the formation of an intermediate structure with a crystalline signature. The partial reduction of Ag(I) to Ag(0) demonstrated from the LSV experiments could have possibly induced the formation of Ag(0) in the form of metallic silver. However, a comparison of the GDEs’ diffractograms with a reference diffractogram of a silver foil shows no convincing evidence of the presence of metallic silver other than in the form of traces in the GDEs cycled at high current density. This would be intuited by the presence of a very faint peak at 2θ = 38.2° matching the (111) reflection of metallic silver, however, the missing remaining reflections do not allow for a precise assignment either. These results are, therefore, consistent with a transition of the cluster from a highly crystalline to an amorphous arrangement; however, neither fragmentation nor rearrangement to simpler forms of crystalline Ag(0) is seen to occur by XRD.
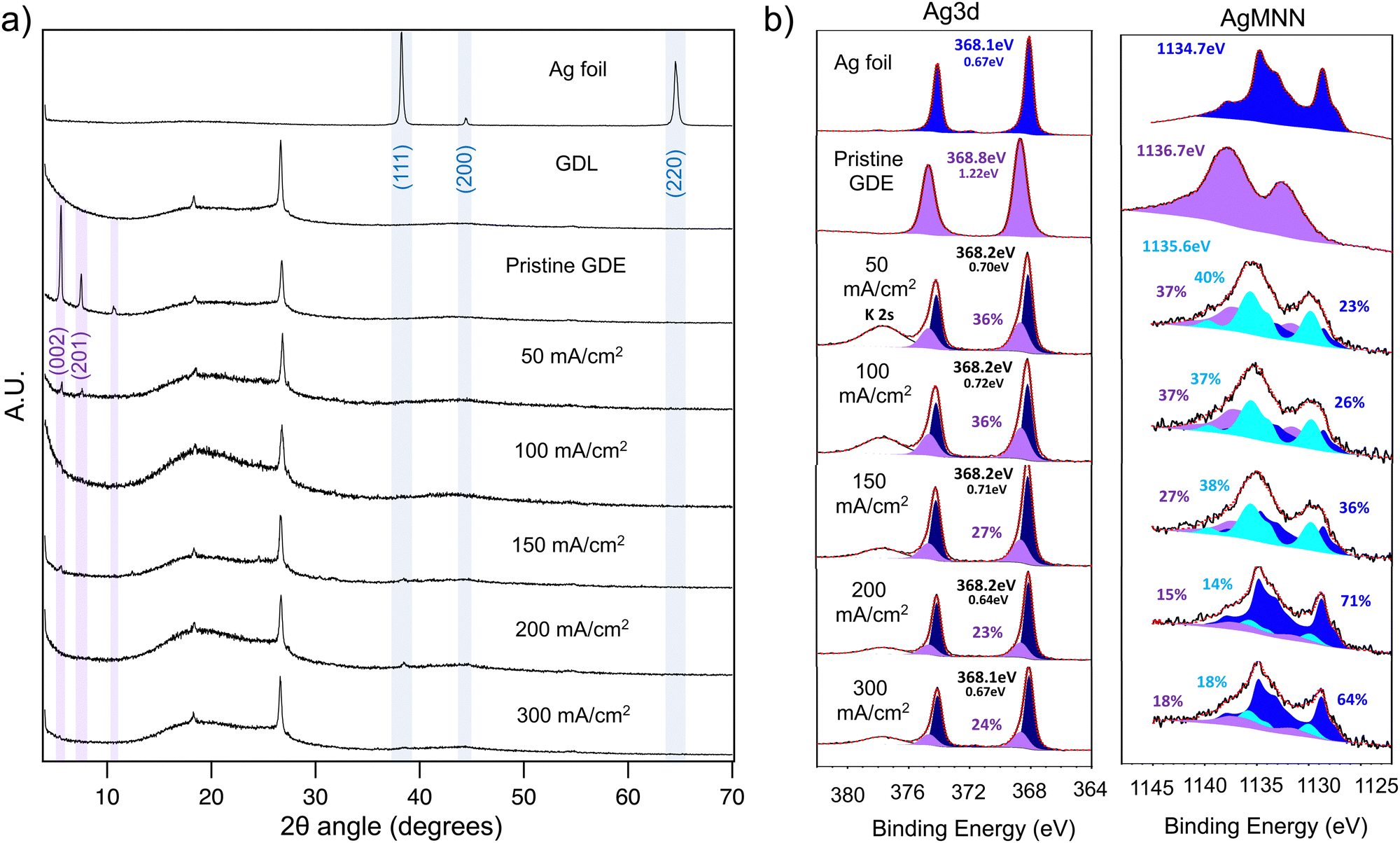 | ||
| Fig. 8 (a) Stacked powder XRD diffractograms of cycled GDEs (chronopotentiometry 20 min) together with diffractograms of reference samples; (b) stacked Ag 3d and AgMNN XPS Spectra of cycled GDEs with convoluted peaks of reference spectra. | ||
Concomitant XPS analysis of the corresponding GDEs confirms the trend observed from XRD and allows adding evidence of a structural change within the cluster structure. Indeed, post-catalysis GDEs analysis shows a significant change in the Ag 3d emission and AgMNN Auger peak shape with respect to the references’ spectrum. Indeed, the spectra of GDEs that have undergone chronopotentiometry were compared to those of metallic silver and pristine unused GDE. The characteristic emission patterns (Ag 3d and AgMNN) produced by both reference samples were fitted empirically to generate a peak signature to be used in the convolution of emission spectra of the cycled GDEs (purple and dark blue areas). Consequently, despite the fact the initial (AgPh)n is seen to evolve towards an intermediate species upon catalysis under current as low as 50 mA, the GDE still accounts for a significant amount of (AgPh)n as seen in the Ag 3d peaks convolutions (Fig. 8b). However, the amount of (AgPh)n is shown to decrease along with the increase in catalysis current. The remainder of the Ag 3d peaks’ shape is assigned to an intermediate species (Fig. 8b).
The AgMNN characteristic peaks of (AgPh)n were as well used in the convolution of the cycled GDEs’ Auger peaks, and quite importantly, the quantification of initial (AgPh)n on the latter peaks matches that from the Ag 3d with respectable accuracy (purple areas). Here again, however, a significant portion of the peak shape accounts for an intermediate species (light blue) in which the silver environment differs significantly from the starting (AgPh)n, as well as for a significant portion of Ag(0) (dark blue). The ratio between the intermediate and pure Ag(0) shifts drastically at current densities above 200 mA cm−2, with Ag(0) becoming the dominant species. Although the AgMNN peaks convoluted with the silver foil signature match that from the experimental emission spectra, the absence of characteristic diffraction peaks of metallic Ag(0) in the corresponding XRD data advocates for a different form of Ag(0) that the inorganic cubic face centred form.
The results are consistent with the fact that when submitting the catalyst to an increasing current, Ag(I) centres gradually reduce to Ag(0), providing for a number of catalytic sites proportional to the kinetics dictated by the current density. However, the Ag(0) likely remains embedded in the cluster structure, presumably inducing significant structural deformation, thus accounting for the loss of crystalline order.
These experimental findings further prompted us to perform DFT computations to further confirm the importance of the infinite structure on the stability of a catalytic mixed Ag(I)–Ag(0) intermediate. A finite model of the (AgPh)n cluster limited to eight silver (Ag8Ph8) atoms was designed, and its free energy was minimised using a 6-31+G(d) basis set. Subsequently, seven additional Intermediate structures corresponding to the sequential removal of ligands were minimised accordingly (Fig. M2, ESI†). The energy necessary to remove contiguous ligands from the finite cluster could be calculated by modelling the free energy of an isolated alkyne ligand and subtracting it from the free energy of each intermediate as per the equation in Table M6 (ESI†). Accordingly, Table M6 (ESI†) reports the free energy change (ΔE) associated with the removal of successive ligands from the cluster. The results indicate that the removal of a ligand has a high associated energetic cost regardless of the starting point (from 66 to 100 kcal mol−1), thus advocating that the removal of more than two adjacent ligands is not favoured based on thermodynamic grounds on an Ag8Ph8 hypothetical intermediate. However, these numerical results indicate that contiguous Ag(0) atoms may exist, especially under high current catalytic conditions in the experimental infinite cluster. This is further supported by the fact that once the first ligand has been removed, the energy required to remove an adjacent ligand is inferior to the amount required to remove the first one (74.4 vs. 100.8 kcal mol−1). This decreasing trend is, however, limited to two ligands as the energy required to remove a third ligand is again nearly identical to that necessary to remove the first one. It is important to note that the removal of as little as one ligand from the cluster has a strong impact on the silver atoms arrangement, showing that the Ag–Ag bond readily distorts to favour strong π–π interactions between the remaining ligands. Therefore, a 12 silver atoms cluster was further considered as a model to study the impact of the loss of ligand on the geometry of the silver core. The structure and conformation of the latter model, minimised using a 6-31+G(d) basis set, was shown to match that of an equivalent fragment from the 3D-ED structure with striking similarity (Ag12Ph12, Fig. 9a), possessing alkynyl ligands bound with η1,η1η1 or η2 hapticity in a μ2 bridging fashion. This feature is likely a confirmation that, from a synthetic perspective, the formation of the cluster's infinite structure may be a thermodynamically controlled process. Subsequent to the modelling of the cluster, a ligand was removed from a central Ag atom (Ag12Ph11), creating a hypothetical Ag(0) catalytic active species. The structure of the energy-minimised version of the latter intermediate is shown to evolve only slightly from its parent precursor at first glance, however, a very interesting feature deserves further analysis. Indeed, a comparison of the native Ag12Ph12 and the Ag12Ph11 intermediate shows that upon removal of a ligand, the remaining ligands rearrange such that they fill the gap created. The highly favourable stacking of the phenyl groups via π–π interactions is presumably the driving force governing the process. The top views of both structures, depicted in Fig. 9a, clearly show that no significant gap is perceived in the Ag12Ph11 structure.
 | ||
| Fig. 9 (a) Energy minimised structures of Ag12Ph12 and Ag12Ph11 cluster models. The ligand being removed from the neutral Ag12Ph12 is shown in light blue in both the side view (capped sticks) and top view (space filling); (b) Gibbs free energy profiles for CO2 reduction and hydrogen evolution considering both Ag4Ph4 (top diagram) and Ag4Ph3 (bottom diagram) catalytic species. C, H, atoms are represented in grey, oxygen atoms in red and silver atoms in purple. | ||
A similar approach to that employed for the Ag8Ph8 cluster reveals that the energy required to remove one ligand is of a similar magnitude in the case of the Ag12Ph12 cluster (99.6 kcal mol−1, Table M7, ESI†). However, and quite importantly, in the latter case no significant distortion of the silver atoms position is noted, contrary to the Ag8Ph8 model. This again advocates for the formation of Ag(0) centres isolated along the tubular structure with groups of two adjacent Ag(0) being as well favoured and further confirms the contribution of the infinite cluster structure to the overall structural stability of the catalytic intermediate. The removal of ligand must, however, induce sufficient structural distortion to, in turn, induce a significant loss in crystalline packing, as shown by XRD.
Following the above preliminary results, a complete mechanistic path with both species, Ag12Ph12 and Ag12Ph11, was initially envisioned. However, for accuracy reasons, a 6-311++G(d,p) basis set was deemed more suited to the task. Consequently, to bring the calculations within reach of our computation capabilities, the cluster model was further simplified to a four-Ag model. A comparative study was performed to confirm that such a simplification is meaningful to the catalytic study (see ESI,† for details). The energy of the neutral silver tetramer (Ag4Ph4) was minimised and shown to retain the original η2 hapticity in a μ2 bridging fashion of the alkynyl ligand. Similarly to the above Ag12Ph12 model, an analogue in which an alkynyl ligand was purposely removed to create an Ag(0) atom was considered, and its energy was minimised (Ag4Ph3). Thereafter, two reaction paths were modelled in which either of the above clusters was considered the catalytic species (Fig. 9b, see ESI,† for details).
Plausible electrocatalytic mechanisms and their corresponding Gibbs free energy pathways are depicted in Fig. 9b for both four-silver structures. As expected, in the case of Ag4Ph4, i.e. with all Ag(I) atoms, the formation of *COOH intermediate has a high associated ΔG (+1.7 eV), forming a rate-determining step for the formation of the CO-bound intermediate. On the other hand, the formation of the CO intermediate for Ag4Ph3, i.e. in which an alkynyl ligand has been previously removed, leading to the presence of one Ag(0) site, is very favourable from a mean thermodynamic perspective with ΔG dropping to 0.25 eV. Interestingly, the simulation of the HER reaction in identical conditions as above led to associated ΔG within a similar energy range, indicating that the HER would be even slightly favoured on thermodynamic grounds. Therefore, the experimental selectivity towards CO is attributed to kinetic factors, such as the wider availability of CO2 near the catalytic interface or the hydrophobicity of the catalyst surface shielding the Ag(0) centre from water molecules. The activation barrier of the formation of the first *COOH and *H intermediates are likely the principal parameter governing the overall selectivity of the catalyst.
Nevertheless, our theoretical models confirm the role of Ag(0) in the catalytic path and bring additional evidence of the importance of the infinite polymeric structure on the stability of the catalytic intermediate. Additionally, the shielding effect of the ligands in the catalytic species, closing the gap created by the departure of a ligand prior to the formation of the *COOH intermediate, may likely play a pivotal role in favouring the CO2RR over the HER. The ligands presumably create a kinetic barrier to the diffusion of H3O+ ions towards the silver catalytic centre, while favourable CO2–ligand interactions would favour the diffusion of CO2 to the catalytic centre.
Conclusions
In conclusion, we have described a multi-gram scale synthesis of a novel silver alkynyl cluster which self-assembles into infinite tubular silver clusters in a perfectly crystalline manner. The solid-state structure of the cluster was determined by electron diffraction (3D-ED) from the catalyst powder, confirming the molecular arrangement throughout the entire sample and allowing for establishing a direct structure–properties relationship. The insoluble nature of the material allowed for deposition from a pre-formed colloidal ink via a simple drop-casting procedure, with no significant structural change upon GDE fabrication. The catalytic properties were assessed under laboratory-scale and flow cell electrolyser conditions, showing an overwhelming selectivity for CO at low reaction overpotentials. Most importantly, GDEs fabricated from (AgPh)n were shown to perform with selectivity towards CO above 95% in bulk electrolysis under electrolysis currents as high as 350 mA cm−1 and TOF values reaching 13.5 s−1.In addition to the outstanding catalytic performance, specific electrochemistry experiments and characterisation techniques supported by molecular modelling allowed the establishment of mechanistic aspects of the CO2 electroreduction catalysed by silver clusters. CV experiments correlated with XPS measurements provided experimental evidence of the in situ formation of an Ag(0) catalytic species under reducing conditions. DFT modelling further confirmed these experimental findings, showing that the *COOH intermediate formation is only favoured if a silver atom had previously been stripped of an alkynyl ligand, essentially turning the former atom to an Ag(0) oxidation state. The CO2 molecule, therefore, is reduced from a catalytically active Ag(0) atom, part of the tubular cluster structure. Thorough characterisation of post-chronopotentiometry GDEs by XPS and XRD clearly indicates the formation of reaction intermediates involving silver atoms with three different environments and a fast loss of crystalline order upon catalysis. However, results show that the infinite nature of the cluster plays a significant role in maintaining the catalytic activity stable throughout successive cycles. Indeed, the excess charge resulting from the ligand's loss is likely accommodated by the perfectly ordered network of Ag–Ag bonded atoms, which likely favours the delocalisation of charge over the whole length of the tubular structure. In addition to supporting the Ag cluster's structure, the ligands presumably shield the Ag atoms and prevent unwanted side reactions such as the HER or others that would otherwise lead to the passivation of the catalyst surface. Therefore, the infinite nature of the Ag cluster is possibly the reason for their superior properties compared to the discrete silver acetylide clusters described thus far in this application.
All in all, the method described herein allows for the effortless synthesis of alkynyl clusters whose structure is polymeric in the crystalline sense. The latter types of clusters were shown to be better adapted to CO2 electroreduction under high reaction rates than their discrete counterparts. Importantly, the scale to which they can be synthesised with no loss of crystalline features, together with cost-effectiveness aspects, provide strong arguments in favour of potential industrial applications. An extrapolation of the catalysis features presented herein indicates that the CO's weight production rate would reach over 218 kg hour−1 per kilogram of catalyst (see ESI,† for calculation). A decrease in catalyst loading by a factor of four would, therefore, allow approaching a 1 T h−1 rate. The approximate cost of the latter weight of catalyst is estimated to be less than 1000 dollars, considering the price of the precursors, rendering it an insignificant parameter from an economic perspective.
Data availability
The data supporting this article have been included as part of the ESI.† Crystallographic data has been deposited at the Cambridge Crystallographic Data Centre (CCDC) under deposition Numbers 2375040–2375042 and can be obtained from https://www.ccdc.cam.ac.uk/.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank E2S UPPA, an ANR PIA4 project, for providing funding for the INTERMAT international Chair of E. P. and the ENSUITE Hub.Notes and references
- W. A. Smith, T. Burdyny, D. A. Vermaas and H. Geerlings, Joule, 2019, 3, 1822–1834 CrossRef CAS.
- C. Hepburn, E. Adlen, J. Beddington, E. A. Carter, S. Fuss, N. Mac Dowell, J. C. Minx, P. Smith and C. K. Williams, Nature, 2019, 575, 87–97 CrossRef CAS.
- O. S. Bushuyev, P. De Luna, C. T. Dinh, L. Tao, G. Saur, J. van de Lagemaat, S. O. Kelley and E. H. Sargent, Joule, 2018, 2, 825–832 CrossRef CAS.
- J. Qiao, Y. Liu and J. Zhang, Electrochemical Reduction of Carbon Dioxide: Fundamentals and Technologies, CRC Press, Boca Raton, 2016 Search PubMed.
- C.-H. Huang and C.-S. Tan, Aerosol Air Qual. Res., 2014, 14, 480–499 CrossRef CAS.
- Y. Hori, A. Murata, R. Takahashi and S. Suzuki, J. Chem. Soc., Chem. Commun., 1988, 17–19 RSC.
- Y. Hori, A. Murata and R. Takahashi, J. Chem. Soc., Faraday Trans. 1, 1989, 85, 2309 RSC.
- Y. Hori, A. Murata, K. Kikuchi and S. Suzuki, J. Chem. Soc., Chem. Commun., 1987, 728–729 RSC.
- J. A. Rabinowitz and M. W. Kanan, Nat. Commun., 2020, 11, 5231 CrossRef CAS.
- S. Hernandez-Aldave and E. Andreoli, Catal. Sci. Technol., 2022, 12, 3412–3420 RSC.
- T. Burdyny and W. A. Smith, Energy Environ. Sci., 2019, 12, 1442–1453 RSC.
- D. Gao, P. Wei, H. Li, L. Lin, G. Wang and X. Bao, Acta Phys. Chim. Sin., 2020, 37, 2009021 Search PubMed.
- C. Chen, J. F. Khosrowabadi Kotyk and S. W. Sheehan, Chem, 2018, 4, 2571–2586 CAS.
- T. N. Nguyen and C.-T. Dinh, Chem. Soc. Rev., 2020, 49, 7488–7504 RSC.
- D. A. Salvatore, C. M. Gabardo, A. Reyes, C. P. O’Brien, S. Holdcroft, P. Pintauro, B. Bahar, M. Hickner, C. Bae, D. Sinton, E. H. Sargent and C. P. Berlinguette, Nat. Energy, 2021, 6, 339–348 CrossRef CAS.
- J. Lin, S. Yan, C. Zhang, Q. Hu and Z. Cheng, Processes, 2022, 10, 826 CrossRef CAS.
- D. M. Weekes, D. A. Salvatore, A. Reyes, A. Huang and C. P. Berlinguette, Acc. Chem. Res., 2018, 51, 910–918 CrossRef CAS PubMed.
- E. W. Lees, B. A. W. Mowbray, D. A. Salvatore, G. L. Simpson, D. J. Dvorak, S. Ren, J. Chau, K. L. Milton and C. P. Berlinguette, J. Mater. Chem. A, 2020, 8, 19493–19501 RSC.
- C.-T. Dinh, T. Burdyny, M. G. Kibria, A. Seifitokaldani, C. M. Gabardo, F. P. García De Arquer, A. Kiani, J. P. Edwards, P. De Luna, O. S. Bushuyev, C. Zou, R. Quintero-Bermudez, Y. Pang, D. Sinton and E. H. Sargent, Science, 2018, 360, 783–787 CrossRef CAS.
- T. T. H. Hoang, S. Verma, S. Ma, T. T. Fister, J. Timoshenko, A. I. Frenkel, P. J. A. Kenis and A. A. Gewirth, J. Am. Chem. Soc., 2018, 140, 5791–5797 CrossRef CAS.
- S. Verma, X. Lu, S. Ma, R. I. Masel and P. J. A. Kenis, Phys. Chem. Chem. Phys., 2016, 18, 7075–7084 RSC.
- S. Ma, Y. Lan, G. M. J. Perez, S. Moniri and P. J. A. Kenis, ChemSusChem, 2014, 7, 866–874 CrossRef CAS.
- Z. Lei, Y. Xue, W. Chen, W. Qiu, Y. Zhang, S. Horike and L. Tang, Adv. Energy Mater., 2018, 8, 1801587 CrossRef.
- S. Ren, D. Joulié, D. Salvatore, K. Torbensen, M. Wang, M. Robert and C. P. Berlinguette, Science, 2019, 365, 367–369 CrossRef CAS.
- M. Wang, K. Torbensen, D. Salvatore, S. Ren, D. Joulié, F. Dumoulin, D. Mendoza, B. Lassalle-Kaiser, U. Işci, C. P. Berlinguette and M. Robert, Nat. Commun., 2019, 10, 3602 CrossRef.
- F. Franco, C. Rettenmaier, H. S. Jeon and B. Roldan Cuenya, Chem. Soc. Rev., 2020, 49, 6884–6946 RSC.
- Y. Cao, Q. Chen, C. Shen and L. He, Molecules, 2019, 24, 2069 CrossRef CAS.
- S. Liu, H. Yang, X. Su, J. Ding, Q. Mao, Y. Huang, T. Zhang and B. Liu, J. Energy Chem., 2019, 36, 95–105 CrossRef.
- X. Huang and Y.-B. Zhang, Coord. Chem. Rev., 2021, 427, 213564 CrossRef CAS.
- G. Ma, L. Qin, Y. Liu, H. Fan, L. Qiao, C. Yu and Z. Tang, Surf. Interfaces, 2023, 36, 102555 CrossRef CAS.
- S. Zhao, R. Jin and R. Jin, ACS Energy Lett., 2018, 3, 452–462 CrossRef CAS.
- L. Chen, L. Wang, Q. Shen, Y. Liu and Z. Tang, Mater. Chem. Front., 2023, 7, 1482–1495 RSC.
- H. Park, D. J. Shin and J. Yu, J. Chem. Educ., 2021, 98, 703–709 CrossRef CAS.
- J. Wang, F. Xu, Z. Wang, S. Zang and T. C. W. Mak, Angew. Chem., Int. Ed., 2022, 61, e202207492 CrossRef CAS PubMed.
- L. Liu, Z. Wang, Z. Wang, R. Wang, S. Zang and T. C. W. Mak, Angew. Chem., Int. Ed., 2022, 61, e202205626 CrossRef CAS PubMed.
- L. Qin, F. Sun, X. Ma, G. Ma, Y. Tang, L. Wang, Q. Tang, R. Jin and Z. Tang, Angew. Chem., Int. Ed., 2021, 60, 26136–26141 CrossRef CAS PubMed.
- L. Chen, F. Sun, Q. Shen, L. Qin, Y. Liu, L. Qiao, Q. Tang, L. Wang and Z. Tang, Nano Res., 2022, 15, 8908–8913 CrossRef CAS.
- G. Deng, J. Kim, M. S. Bootharaju, F. Sun, K. Lee, Q. Tang, Y. J. Hwang and T. Hyeon, J. Am. Chem. Soc., 2023, 145, 3401–3407 CrossRef CAS.
- T. Kawawaki, T. Okada, D. Hirayama and Y. Negishi, Green Chem., 2024, 26, 122–163 RSC.
- Q. Shen, X. Cong, L. Chen, L. Wang, Y. Liu, L. Wang and Z. Tang, Dalton Trans., 2023, 52, 16812–16818 RSC.
- X. Ma, F. Sun, L. Qin, Y. Liu, X. Kang, L. Wang, D. Jiang, Q. Tang and Z. Tang, Chem. Sci., 2022, 13, 10149–10158 RSC.
- H. Seong, K. Chang, F. Sun, S. Lee, S. M. Han, Y. Kim, C. H. Choi, Q. Tang and D. Lee, Adv. Sci., 2024, 11, 2306089 CrossRef CAS.
- K. Torbensen, C. Han, B. Boudy, N. Wolff, C. Bertail, W. Braun and M. Robert, Chem. – Eur. J., 2020, 26, 3034–3038 CrossRef CAS.
- X. Zhang, Y. Wang, M. Gu, M. Wang, Z. Zhang, W. Pan, Z. Jiang, H. Zheng, M. Lucero, H. Wang, G. E. Sterbinsky, Q. Ma, Y.-G. Wang, Z. Feng, J. Li, H. Dai and Y. Liang, Nat. Energy, 2020, 5, 684–692 CrossRef CAS.
- S. Sato, K. Sekizawa, S. Shirai, N. Sakamoto and T. Morikawa, Sci. Adv., 2023, 9, eadh9986 CrossRef CAS.
- X. Liu, Q. Yi, Y. Han, Z. Liang, C. Shen, Z. Zhou, J. Sun, Y. Li, W. Du and R. Cao, Angew. Chem., Int. Ed., 2015, 54, 1846–1850 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures, materials characterisation, 3d-ED crystal structure, electrochemical characterisation and DFT modelling data. CCDC 2375040–2375042. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ey00160e |
| This journal is © The Royal Society of Chemistry 2025 |