
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Sulfur depletion through repetitive redox cycling unmasks the role of the cryptic sulfur cycle for (methyl)thioarsenate formation in paddy soils†
José M.
León Ninin
 *a,
Carolin Lisbeth
Dreher
b,
Andreas
Kappler
b and
Britta
Planer-Friedrich‡
a
*a,
Carolin Lisbeth
Dreher
b,
Andreas
Kappler
b and
Britta
Planer-Friedrich‡
a
aEnvironmental Geochemistry, Bayreuth Center for Ecology and Environmental Research (BayCEER), University of Bayreuth, 95440 Bayreuth, Germany. E-mail: jose.leon-ninin@uni-bayreuth.de; Tel: +49-921-553995
bGeomicrobiology, Department of Geosciences, University of Tübingen, 72076 Tübingen, Germany
First published on 24th March 2025
Abstract
Inorganic and oxymethylated thioarsenates form through the reaction of arsenite and oxymethylated arsenates with reduced sulfur, mainly as sulfide (SII−) but also as zerovalent sulfur (S0). In paddy soils, considered low-S systems, microbial reduction of the soil's “primary” sulfate pool is the principal SII− source for As thiolation. Under anoxic conditions, this primary pool is readily consumed, and the precipitation of iron (Fe) sulfides lowers SII− availability. Nonetheless, sulfate can be constantly replenished by the reoxidation of SII− coupled with the reduction of FeIII phases in the so-called cryptic S cycle (CSC). The CSC supplies a small secondary sulfate pool available for reduction and, according to previous studies, As thiolation. However, sulfate concentrations commonly found in paddy soils mask the biogeochemical processes associated with the CSC. Here, we depleted a paddy soil from excess S, Fe, and As from a paddy soil through repetitive flooding and draining (e.g., redox cycling). After 10, 20, and 30 such cycles, we followed thioarsenate formation during an anoxic incubation period of 10 days. Higher S/As ratios increased As thiolation contribution to total As up to 10-fold after 30 cycles. During the anoxic incubation, the depleted soils showed a transient first phase where the reduction of the primary sulfate pool led to inorganic thioarsenate formation. In the second phase, methylthioarsenate formation correlated with partially oxidized S species (S0, thiosulfate), suggesting CSC-driven sulfate replenishment, re-reduction, and thiolation. Methylthioarsenates formed even as inorganic thioarsenates de-thiolated, indicating thermodynamic preference under S-limited conditions. This study highlights the role of the CSC in sustaining thioarsenate formation in low-S systems.
Environmental significanceCryptic sulfur cycling has been suggested to replenish sulfate pools in low-S systems like paddy soils, supporting the formation of highly mobile and toxic thioarsenates. Here we show that sulfide production through the reduction of the soil's primary sulfate pool supports a high but transient formation of inorganic thioarsenates. Once this primary sulfate pool is exhausted, lower sulfide concentrations cause de-thiolation. In a depleted system, we observed that the re-reduction of sulfate recycled through the cryptic sulfur cycle supported the formation of methylthioarsenates. These results suggest that methylthioarsenates can form even in very low-S environments, increasing mobility, toxicity, uptake by rice plants, and accumulation in rice grains. |
Introduction
Arsenic (As) is a ubiquitously distributed element in the environment that is toxic for humans.1–3 The mobility and toxicity of As are highly redox-dependent, making it a contaminant of interest in redox-active environments, such as groundwater systems or natural and managed wetlands.1,4,5 The main As intake sources for humans are As-contaminated groundwater and As-enriched rice grains.3,6 Understanding how redox dynamics in groundwater and rice cultivation systems influence As biogeochemistry could help decrease the risk of As intake for consumers.5,7–10The mobility and toxicity of As depend on its speciation.11 Under sub-oxic and anoxic conditions, As is mobilized mainly by the reductive dissolution of As-bearing FeIII (oxy)hydroxides as arsenate or arsenite.12–14 These two species (summarized as inorganic oxyarsenic) are known carcinogens.2,3 Arsenite can be biotically methylated in consecutive steps to form the oxymethylated arsenates, mono- and dimethylarsenate (MMA and DMA, respectively).15–17 Oxymethylated arsenates are currently considered as less toxic to humans.11,18 Arsenic methylation is carried out by microbes carrying the S-adenosylmethionine methyltransferase (arsM) gene.15,19,20 Different microbial groups can methylate As depending on environmental conditions. For example, in geothermal springs, methanogens have been identified as methylators.21 In paddy soils, sulfate-reducing bacteria (SRB)22,23 and fermentative bacteria20 have been shown to methylate As. Methanogens have been identified in paddy soils as de-methylators, transforming oxymethylated arsenates back to arsenite.22,24,25
Current studies on As speciation often focus on arsenate, arsenite, MMA, and DMA, and the biogeochemical processes governing their contribution to total As. However, another group of As species called thioarsenates has been shown to contribute significantly to As speciation in different environmental systems.26–29 Thioarsenates form when reduced sulfur (S) species, mainly as sulfide (SII−), but also as zerovalent S (S0), substitute one or more hydroxyl groups in an As oxyanion.26,27,30 The formation of thioarsenates requires, thus, the reaction of arsenite or methylated oxyarsenates with reduced S species, to form inorganic or methylated thioarsenates, respectively.27,31 Depending on the system, these reduced S species could be of abiotic origin or produced by microbially mediated sulfate reduction.21,28,32
The formation of either inorganic or methylated thioarsenates is determined by the distribution of As species, availability of reduced S species, and pH conditions.27,31 Inorganic thioarsenates are preferentially formed in environments with excess SII− at neutral to alkaline pH when arsenite consecutively reacts with SII− and S0 to form mono-, di-, tri-, and tetrathioarsenate (MTA, DTA, TTA, and TetraTA, respectively) depending on S/As ratios.27,30 The role of S0 in inorganic thioarsenate formation is either related to the formation of MTA, or the oxidation of the unstable intermediate thioarsenites to thioarsenates.30 Alkaline, sulfidic geothermal springs have been described as hotspots for the formation and identification of inorganic thioarsenates.21,32
The formation of methylthioarsenates is largely governed by the availability of oxymethylated arsenates, which are readily thiolated in the presence of SII−.31,33,34 Consecutive reactions of MMA with SII− produce monomethylthioarsenate (MMMTA), monomethyldithioarsenate (MMDTA), and monomethyltrithioarsenate (MMTTA). Similarly, the thiolation of DMA produces dimethylmonothioarsenate (DMMTA) and dimethyldithioarsenate (DMDTA). Field and laboratory observations suggest that the SII− excess required to form methylthioarsenates decreases with the number of methyl groups attached to As.33,35 For example, DMA requires a lower SII− excess to be thiolated than MMA.33 A lower SII− requirement to form methylthioarsenates, together with the low stability of inorganic thioarsenates at pH values < 8,32 suggests that at neutral and acidic pH, methylthioarsenate formation is thermodynamically preferred to that of inorganic thioarsenates.
Thioarsenate formation and identification in relatively low-S terrestrial environments, such as wetlands and paddy soils, has been controversial. In such systems, SRB carrying out dissimilatory sulfate reduction (DSR) have been identified to be the main producers of SII− for As thiolation.23,28,36,37 However, the anoxic conditions required for DSR also trigger reductive dissolution of Fe(III) minerals.38 Reduced Fe scavenges SII− through the precipitation of FeS phases,39–41 decreasing its availability to form thioarsenates.28,36 Moreover, both wetlands and paddy soils are considered methanogenic environments where DSR accounts for only a small fraction of anaerobic respiration since the low sulfate contents in the soil, and hence aqueous phase concentrations are readily depleted.23 Despite these assumptions, thioarsenates have been shown as important contributors to the mobility and export of As in groundwater5 and wetland systems,29,35 as well as being formed in flooded rice fields,28,36,42 and accumulated in rice grains.43 Their presence in paddy soils and rice grains has raised concern, given their high mobility, phyto-, and cytotoxicity, comparable, in the case of dimethylthioarsenates, to that of arsenite.35,44,45 Moreover, thioarsenates have been observed and suggested to contribute to As mobility in systems with high dissolved Fe concentrations, where the availability of SII− should be limited.5,36 Thus, a better understanding of the biogeochemical processes influencing As thiolation is required.
Micro- and mesocosm experiments have suggested that the cryptic S-cycle (CSC) plays an important role in the formation of thioarsenates, providing small amounts of SII− and S0 for As thiolation.28,36 Such a CSC is related to the reoxidation of SII− to S0 coupled with the reduction of FeIII (oxyhydr)oxides, releasing FeII into the aqueous phase.46–49 Zerovalent S can be consecutively further oxidized to thiosulfate and sulfate, thus being recycled and available again as a substrate for DSR.28,46,50 Thus, even when most of the original “primary” pool of sulfate content has been consumed, SII− abiotic reoxidation forms a “secondary” pool of sulfate. The re-reduction of this recycled sulfate through DSR offers a constant source of reduced S species. While the role of CSC in As thiolation has been implied,5,28,36,51 to the best of our knowledge, it has not been directly evaluated, yet.
In the environment, the CSC has been reported in marine systems,52 low sulfate lakes,53 and wetlands.54,55 Holmkvist and Ferdelman52 described how the reoxidation of SII− explains the abundance of SRB in marine sediments with low but constant sulfate availability. The authors associated the presence of stable sulfate background concentrations with the presence of FeIII phases and SII− in sediment samples. The same study suggests that the CSC might take place independent of sulfate limitations, but that S-rich environments might mask the small secondary pool of freshly produced sulfate. According to the authors, only S-depleted environments allow the identification of the CSC. In paddy soils, the CSC has been reported due to root oxygen loss, which creates redox interfaces in which sulfate is constantly reduced to SII−, and partially reoxidized to form S0, thiosulfate, and sulfate.46,56,57 However, the identification in the bulk soil and the role of a secondary sulfate pool in thioarsenate formation has been elusive due to the masking effect of the bigger primary sulfate pool, despite products of partial SII− reoxidation being found in the bulk soil.28,36 In this sense, a S-depleted paddy soil system is required to identify the role of the CSC and its associated secondary sulfate pool in As thiolation.
Here, we present the results of a long-term microcosm experiment. Through repetitive redox cycling and aqueous phase removal, we decreased a paddy soil's background concentrations (i.e., depleting it) of S, Fe, and As, aiming to investigate the role of the CSC in As thiolation. Our hypothesis is that in a S-depleted system with a smaller primary sulfate pool, CSC will provide a constant source of SII− and S0, forming thioarsenates. Following changes in S, Fe, and As speciation, we show that even in low-sulfate, high-methanogenic systems, thioarsenates form and even dominate As speciation, with CSC providing small amounts of sulfate which, through DSR, form SII− for thioarsenate formation.
Materials and methods
Long-term microcosm experiment
Soil from the paddy cultivation area in Cixi, Province of Zhejiang, China, was used for a long-term microcosm experiment. This soil is the youngest paddy soil (50 years of paddy use – P50) of a chronosequence which has been previously used to understand how long-term paddy use affects pedologic properties58–60 and redox biogeochemistry.36,37,51 With its early stage of paddy development, the soil has shown a high potential for the formation of thioarsenates, due to low amounts of SOC and reducible Fe, and high pH.36,37 For the microcosm experiments, the <2 mm fraction of the topsoil (0–7 cm) was used. Soil properties have been published elsewhere36,58 and can be found summarized in Table S1.†Microcosms were prepared inside a glovebox (COY, with 95% N2, 5% H2) by mixing 10 g of paddy soil, 0.13 g of finely milled rice straw to support microbial activity, and 20 mL of N2 purged tap water inside 120 mL septum vials. The rice straw had an As content of 2.76 μg g−1, while the tap water background concentrations of S, Fe, and As were 14.3, 0.04, and 0.0003 μmol L−1. The vials were closed with butyl rubber stoppers and aluminum caps and incubated standing in the dark in an oven at 30 °C. To deplete the soil from excess S, Fe, and As, microcosms went through 30 redox cycles for one and a half years. Each redox cycle consisted of an anoxic period of 10 days followed by an oxic period of 5 days.
Redox fluctuations were carried out in the following way. After 10 days of anoxic incubation under the above-described conditions, the vials were taken from the oven, allowed to reach room temperature (20 °C), and shaken before removing the aluminum caps and butyl rubber stoppers. After allowing the soil to settle, the overlaying water was removed using a pipette. Finally, milled rice straw (0.13 g) was mixed into the soil to replenish the available C pool for microbes. The open vials were taken into the oven and incubated oxically for 5 days. After the oxic period, the vials were refilled with 20 mL of N2 purged tap water inside the glovebox, closed with butyl rubber stoppers and aluminum caps, and anoxically incubated again at 30 °C for 10 days.
An additional, constantly anoxic (CA), setup was used as control. These vials went through 10 anoxic incubation days before being opened inside the glovebox to release the accumulated gases and add new rice straw before being closed again and returned to the oven. On the fifth day of the oxic cycle of the main setup, the rubber stoppers of the CA bottles were punctured with a needle inside the glovebox to release accumulated gases. This release was done in order to keep similar starting conditions between CA and the redox-cycled bottles throughout the experiment and at the moment of sampling.
Samplings
After 10, 20, and 30 redox cycles (C10, C20, and C30, respectively), sacrificial time-resolved samplings took place in which aqueous phase and soils were taken at days 1, 3, 5, 7, and 10 days of the anoxic cycle. Thus, in total 15 sacrificial vials were used per redox cycle sampling. The triplicates of the CA setup were sampled only on day 10 after 20 and 30 cycles (CA20 and CA30, respectively). These sampling days were selected based on previous reports from redox dynamics in similar setups with the same soil.36,37,51 On each sampling day, the corresponding triplicate vials were taken out of the glovebox, mixed, and allowed to reach room temperature. The pressure inside the vials was measured with a handheld pressure meter (Greisinger GMH, 3100 Series) before taking a 5 mL headspace sample with a needle and a gastight syringe. This sample was injected into an evacuated glass vial and stored until CH4 and CO2 determination using a gas chromatograph with a flame ionization detector (GC-FID, SRI Instruments 8610C) equipped with a methanizer.The vials were then brought inside the glovebox, mixed to form a slurry, and their contents poured into a 50 mL centrifuge tube. The tubes were closed and wrapped with Parafilm before being centrifuged outside the glovebox (4000 rpm, 10 min). Back inside the glovebox, the aqueous phase was separated from the soil using a syringe and needle and used for the different analyses described in each section below.
Aqueous phase analyses
The aqueous phase was filtered through 0.2 μm cellulose-acetate filters (Chromafil® Xtra) and split into different aliquots for the following biogeochemical analyses. A 2 mL aliquot was stabilized with 15 μL of 30% H2O2 and 20 μL of 65% HNO3 and diluted before the determination of total As and S by Inductively Coupled Plasma Mass Spectrometry (ICP-MS, 8900 Triple Quad, Agilent). The determination was carried out using reaction mode with O2, with As being measured as AsO+ (m/z = 91) and S as SO+ (m/z = 48). Rhodium was used as an internal standard to compensate for signal drift, and a certified reference material (TMDA 62.2, Environment Canada) was used for quality control.A 700 μL aliquot for As and S speciation was stabilized with a 10 mM solution of hydroxybenzylethylenediamine (HBED, pH 7) and directly frozen in dry ice.36 For analyses, the samples were thawed inside the glovebox to avoid changes in the speciation due to oxidation. Arsenic and S speciation was determined by ICP-MS after chromatographic separation by ion chromatography (IC, 940 Professional IC Vario Metrohm) using an AS16 column (Dionex AG/AS16 IonPac column) with a 2.5–100 mM NaOH gradient, a 1.2 mL min−1 flow rate, and an injection volume of 50 μL.32 Concentrations were calculated through a calibration containing arsenite, arsenate, MMA, DMA, sulfate, and thiosulfate. Thioarsenates were identified based on previously reported retention times,27,28,31 and their concentrations were determined through their oxyarsenic homologs. Throughout the text, As species are often grouped depending on the biogeochemical processes ruling their formation. The sum of oxymethylated and methylthiolated arsenates is referred to as total methylated As, while the sum of inorganic and methylated thioarsenates is referred to as total thiolated As.
Total dissolved Fe, FeII, and SII− were determined photometrically using the ferrozine61,62 and the methylene blue method.63 After stabilization according to the methods, colorimetric reactions were done outside the glovebox. Photometric determinations were carried out in triplicate using a multiplate reader (Infinite® 200 PRO, Tecan). Sulfide was not detected throughout the experiment (LOD = 1.2 μmol L−1).
The rest of the filtered aqueous phase was used for the determination of pH and redox potential using a multiparameter (HQ40d, Hach) connected to the corresponding electrodes (PHC301, Ag/AgCl electrode and MTC101, respectively).
Solid phase analyses
Subsamples from all post-incubation soils were immediately frozen after being separated from the aqueous phase, and later freeze-dried and homogenized. Zerovalent sulfur (S0) was determined via HPLC-UV-Vis after chloroform extraction.64 Soil-extracted S0 is operationally defined as chloroform-extractable S (which includes zerovalent S in polysulfides, S bound to solid phase, and S8).Total Fe content in the original soil and after redox cycling was determined after microwave-assisted digestion (MARS Xpress, CEM) in 10 mL aqua regia. The digested samples were filtered through 0.2 μm cellulose-acetate filters (Chromafil® Xtra) and diluted 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 with ultrapure deionized water (Millipore, 18.2 MΩ cm) before analysis by ICP-MS as described above. Changes in Fe mineralogy due to repetitive redox cycling were assessed by Mössbauer spectroscopy using samples from day 10 of the anoxic cycle from C10 and C30, as well as the original soil (C0). Dried mineral powders were loaded into Plexiglas holders (area 1 cm2), forming a thin disc. Holders were inserted into a closed-cycle exchange gas cryostat (Janis cryogenics) under a backflow of He to minimize exposure to air. Spectra were collected at 5 and 77 K using a constant acceleration drive system (WissEL) in transmission mode with a 57Co/Rh source. All spectra were calibrated against a 7 μm thick α-57Fe foil that was measured at room temperature. Analysis was carried out using Recoil (University of Ottawa) and the Voigt Based Fitting (VBF) routine.65 The half width at half maximum (HWHM) was constrained to 0.12 mm s−1 during fitting. Due to some uncertainties regarding the mineral phases of the FeII and the FeIII (oxyhydr)oxides, additional μXRD measurements were carried out and can be found in the ESI Discussion.† The reference minerals were partly taken from the XRD Match! Software and partly from the mineralogical database “Rruff”.
:10 with ultrapure deionized water (Millipore, 18.2 MΩ cm) before analysis by ICP-MS as described above. Changes in Fe mineralogy due to repetitive redox cycling were assessed by Mössbauer spectroscopy using samples from day 10 of the anoxic cycle from C10 and C30, as well as the original soil (C0). Dried mineral powders were loaded into Plexiglas holders (area 1 cm2), forming a thin disc. Holders were inserted into a closed-cycle exchange gas cryostat (Janis cryogenics) under a backflow of He to minimize exposure to air. Spectra were collected at 5 and 77 K using a constant acceleration drive system (WissEL) in transmission mode with a 57Co/Rh source. All spectra were calibrated against a 7 μm thick α-57Fe foil that was measured at room temperature. Analysis was carried out using Recoil (University of Ottawa) and the Voigt Based Fitting (VBF) routine.65 The half width at half maximum (HWHM) was constrained to 0.12 mm s−1 during fitting. Due to some uncertainties regarding the mineral phases of the FeII and the FeIII (oxyhydr)oxides, additional μXRD measurements were carried out and can be found in the ESI Discussion.† The reference minerals were partly taken from the XRD Match! Software and partly from the mineralogical database “Rruff”.
Statistical analyses
Unless stated otherwise, all data are presented as mean values ± standard deviation (n = 3). Pearson correlation coefficients were used to assess the significance of all variable correlations presented. Tukey post hoc analyses were used to evaluate changes in As speciation with increasing redox cycling. Pearson correlation and Tukey post hoc analyses were carried out using R (version 4.2.1) in RStudio (R Development Core Team, 2008).Results & discussion
Depletion of S, Fe, and As with repeated redox cycling
Repeated redox cycling and removal of the aqueous phase after each anoxic period effectively depleted the soil of aqueous Fe, S, and As, and in consequence, concentrations in the aqueous phase. Aqueous Fe concentrations from the original soil (C0), peaked at 0.55 ± 0.09 mmol L−1 at 7 days of incubation (Fig. 1a). At day 7 Fe concentrations in the aqueous phase from the depleted soils (C10, C20, and C30) were lowered by 49, 43, and 61%, respectively. On average, 89% of the Fe in the aqueous phase was FeII throughout the experiment (Fig. S1†). Moreover, in the aqueous phase from the depleted soils, Fe concentrations increased constantly during the 10 day anoxic incubation period. Aqueous phase S decreased strongly with repeated redox cycling. Starting at 2.53 ± 0.08 mmol L−1 at day 1 in C0, the aqueous phase of the depleted soils lost, on average, 96% of this initial dissolved S concentration, decreasing the size of the primary S pool. In the depleted soils, a stable pool of dissolved S of around 0.10 mmol L−1 was found in the system at the beginning of each cycle. This observation suggests that during the oxic incubation period, sulfide phases were re-oxidized, releasing partially or fully oxidized soluble S forms, replenishing a small fraction of the primary sulfate pool, which was available for reduction in the next anoxic cycle (Fig. S2†).56,57 In C0 and C10, dissolved S concentration decreased during the first incubation days, indicating consumption of sulfate from the primary pool, before stabilizing by day 7 around 0.14 and 0.04 mmol L−1, respectively. In C20 and C30, stable dissolved S concentrations under 0.03 mmol L−1 were reached after day 3, showing a faster consumption of a smaller primary sulfate pool. Despite being depleted of electron acceptors, microbial activity was observed throughout the experiment, as shown by the decreasing Eh values throughout the 10 day anoxic period, and higher CO2 and CH4 production with increased redox cycling (Fig. S3†). | ||
| Fig. 1 Depletion of dissolved Fe (a), S (b), and As (c) through repeated redox cycling and washout. Data points are average values and bars indicate standard deviation (n = 3). | ||
Dissolved As concentrations also decreased with increasing redox cycling and depletion. In C0, concentrations increased sharply with anoxic incubation time, peaking on day 7 at 2.11 ± 0.09 μmol L−1. In C10, C20, and C30, dissolved As concentrations at the same incubation time were only 13, 5, and 6% of those in C0, respectively. More importantly, opposite to the trend observed in C0, As concentrations in the aqueous phase of the depleted soils decreased throughout the 10 day anoxic incubation period.
In the CA controls (Fig. S4†), dissolved Fe concentrations by the end of each cycle remained constant for CA20 and CA30 (0.7 ± 0.1 and 0.7 ± 0.03 mmol L−1, respectively) showing an increase compared to C0 (0.47 ± 0.06 mmol L−1 at day 10). Dissolved S concentrations in CA20 and CA30 were 0.13 ± 0.01 and 0.16 ± 0.01 mmol L−1, respectively. These concentrations are comparable to the stable values found on C0 from day 7 of the anoxic period reported above. Dissolved As concentration also decreased with longer CA cycling, but to a lesser extent than in depleted soils. In CA20 and CA30, aqueous phase As concentrations were 0.40 ± 0.02 and 0.57 ± 0.07 μmol L−1, compared to 2.07 ± 0.03 μmol L−1 at day 10 in C0.
The depletion of S, Fe, and As in the soils and hence the lower concentrations in the aqueous phase were a consequence of redox cycling and the removal of the standing water. Under anoxic incubation conditions, reductive dissolution of Fe(III) minerals took place, releasing FeII and the associated As into the aqueous phase.7 By removing the standing water at the end of each anoxic cycle, the dissolved fraction of FeII and As was removed from the system. The repetition of this process with repetitive redox cycling caused the depletion of the bioavailable FeIII fraction in the soils, lowering how much FeIII could be reductively dissolved in the next anoxic period. The decrease of dissolved S throughout the 10 days of the anoxic periods indicates DSR and suggests the precipitation of sulfides, likely through reaction with dissolved FeII.66,67 By removing the standing water at the end of each anoxic cycle, the non-precipitated S was removed from the system. The constant increase of dissolved Fe throughout the anoxic periods in the depleted soils suggests that FeS precipitation could be limited in these soils compared to C0, due to a lower availability of sulfate and hence, sulfide. In the CA setups, the stable concentrations of Fe and S in the aqueous phase suggest that their biogeochemistry might have reached an equilibrium in these setups, with FeS precipitation no longer taking place. The precipitation of Fe or As sulfide phases could be responsible for the lower As concentrations in the aqueous phase compared to C0.41,66,68
Changes in As speciation with repeated redox cycling
Repeated redox cycling directly affected As speciation. These results are presented as the percentage contribution of group species to total As, to correct for As depletion. Summarizing the data of the 10 anoxic incubation days of each cycle, the median contribution to total methylated As showed no trends with increasing redox cycling, with median values staying around 20–30% of total As (Fig. 2a). However, total As thiolation increased sharply with increasing redox cycling (Fig. 2b). Total contribution of thioarsenates to total As was 4% in C0, significantly increasing (p < 0.05) to 10, 40, and 39% in C10, C20, and C30, respectively.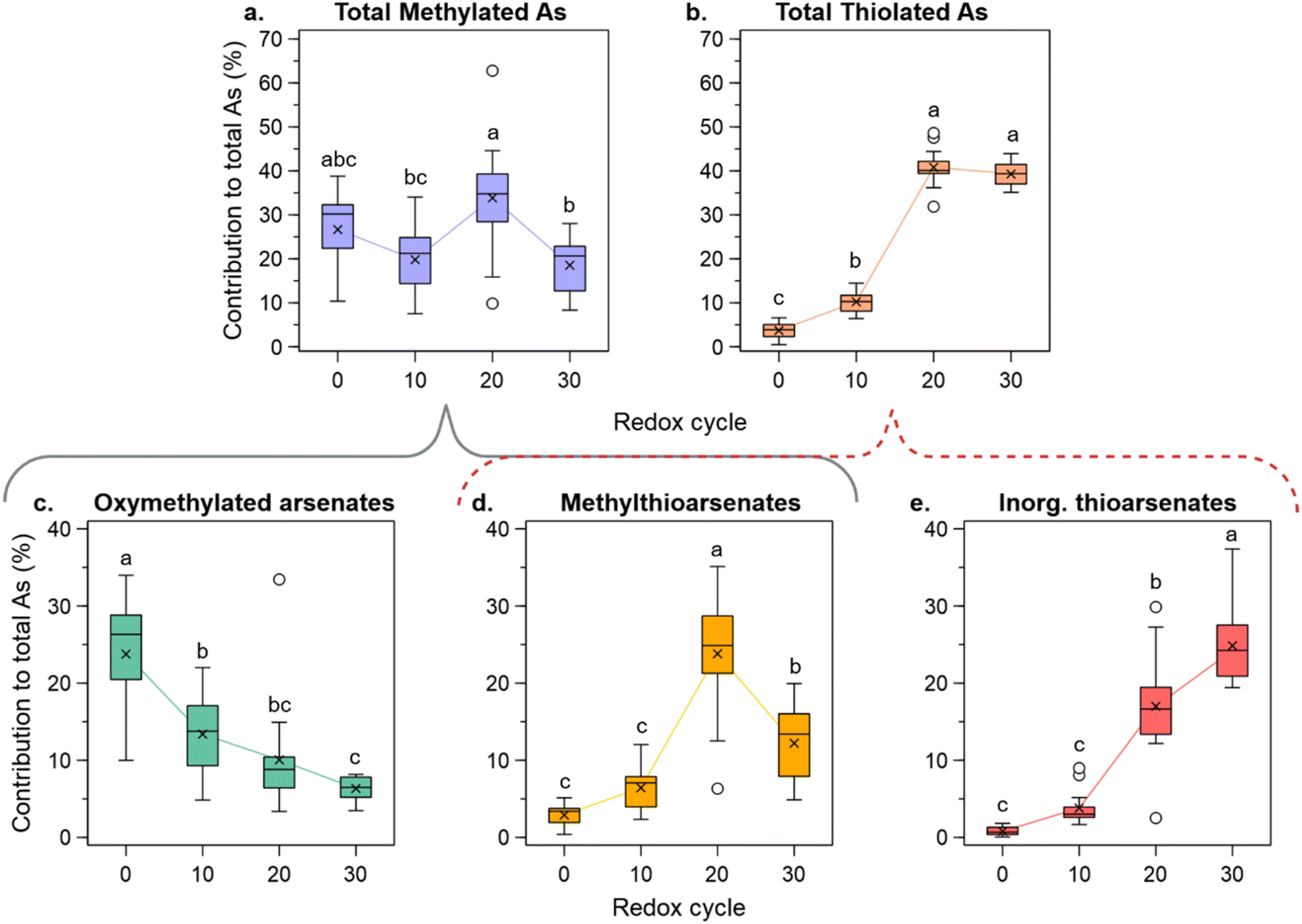 | ||
| Fig. 2 Changes in total methylated (a) and total thiolated (b) As with increasing redox cycling. Species groups are further subdivided into oxymethylated arsenates (c), methylthioarsenates (d), and inorganic thioarsenates (e). Each box in the plot summarizes the results of triplicate throughout the 10 days of anoxic incubation (n = 15), with “×” showing the mean value and white circles showing outliers (1.5 interquartile range). Different letters on top of the boxes indicate statistical significance based on Tukey post hoc analyses (p < 0.05). | ||
Breaking down these species groups into subgroups shows that while there was no net trend in total methylated As, there was a significant decrease (p < 0.05) in the contribution of oxymethylated arsenates (Fig. 2c), from a median contribution of 26% in C0 to only 7% in C30. In contrast, the contribution of methylthioarsenates (Fig. 2d) increased significantly (p < 0.05) with increasing redox cycling between C0 and C20 from 3 to 25%. Since the contribution of total methylated species remained constant throughout the repetitive redox cycling, the opposite trends observed for these two subgroups suggest that the decrease in the contribution of oxymethylarsenates is caused by their thiolation to form methylthioarsenates. Moreover, the significant increase (p < 0.05) in the median contribution of inorganic thioarsenates, from 0.6 to 24% from C0 to C30 (Fig. 2e), caused a sharp increase in the contribution of total thiolated species. Strikingly, the thioarsenate contribution to total As in these depleted systems, was much higher than values commonly reported for paddy soils, which are often <10%.28,36
An increasing S/As molar ratio in the depleted soils (C10–C30) significantly correlated (p < 0.0001) with the increased contribution of inorganic thioarsenates (Fig. 3a). In C0, despite high S/As ratios, the contribution of inorganic thioarsenates to total As was low, since S here was mostly found as sulfate (Fig. S2†). In the CA controls, S/As values were in the same magnitude as those of C20 and C30, but the contribution of inorganic thioarsenates in the CA setups was similar to that in C10. Besides higher S/As ratios, the high contribution of thioarsenates in the depleted soils compared to C0 can be linked to the lower bioavailable Fe (Fig. S5a†), since at low aqueous FeII concentrations, more reduced S is available for thiolation. Excess S over Fe correlated positively and significantly with high inorganic thioarsenate formation (Fig. S5b†). Repeated redox cycling caused a higher ratio of inorganic thioarsenates over their oxyarsenic homologs, particularly at the beginning of the anoxic incubation period in C20 and C30. Nonetheless, arsenate and arsenite were still the dominating inorganic species throughout the experiment (Fig. S6†).
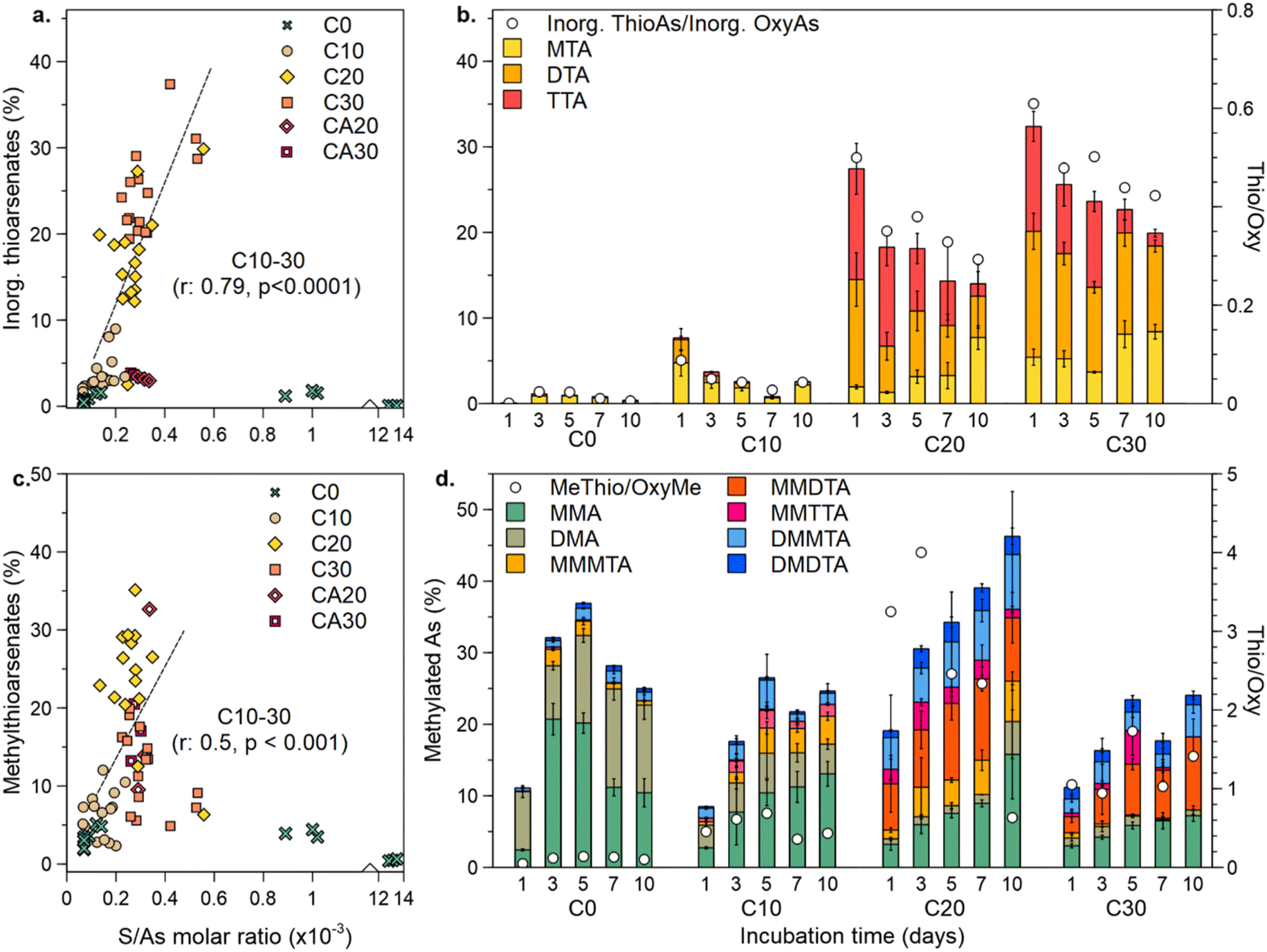 | ||
| Fig. 3 Effect of repeated redox cycling on thioarsenate formation. Influence of S/As ratios on the contribution of inorganic and methylated thioarsenates to total As (a, c, respectively) and temporal dynamics of individual inorganic and methylated As species (b, d, respectively). Regression lines in (a and b) are drawn for samples from depleted systems (C10–C30). | ||
Moreover, changes in the contribution of inorganic thioarsenates throughout the 10 days of the incubation period show further insights into how increased redox cycling influenced their formation. In the depleted soils, the contribution of inorganic thioarsenates to total As was higher at the beginning of the 10 day anoxic period (Fig. 3b). The depleted soils, especially C20 and C30, showed a higher contribution of highly thiolated inorganic thioarsenates (DTA and TTA), which decreased throughout the anoxic incubation. The constantly anoxic controls did not show a similar trend, with MTA being constantly the dominant inorganic thioarsenate and rather a decrease in inorganic thiolation with increasing cycling (Fig. S7†).
Increasing S/As ratios also had a significant influence on the contribution of methylthioarsenates (p < 0.001) (Fig. 3c). In C0, despite the high S/As ratio, the formation of methylthioarsenates was low. Moreover, the contribution of methylthioarsenates to total As changed throughout the anoxic incubation period, showing differences from the trends reported above for inorganic thioarsenates (Fig. 3d). With increasing redox cycling, the formation, contribution, and degree of thiolation of methylthioarsenates increased. In particular in C20 and C30, highly thiolated monomethylated arsenates (MMDTA and MMTTA) contributed significantly to total As. During most of the anoxic incubation period in C20 and C30, methylthioarsenates dominated over methyloxyarsenates. The contribution of methylthioarsenate to total As did not decrease with increasing incubation time, and the degree of thiolation was relatively constant throughout the anoxic period.
The above-described results for the formation and stability of inorganic and methylated thioarsenates show the contrasting behaviors of these two species groups. The formation of inorganic thioarsenates coincides kinetically with the consumption of the primary sulfate pool in the first three days of incubation (Fig. S8†), with high inorganic thioarsenate concentrations being present at high concentrations of sulfate. We suggest that this period of high DSR triggers the formation of highly thiolated inorganic thioarsenates, due to a relative excess of SII− related to the high sulfate consumption. Once SII− becomes limited, due to the consumption of most of the primary sulfate pool and likely the precipitation of FeS phases in the presence of a relative excess of dissolved Fe, inorganic thioarsenate concentrations decrease, indicating de-thiolation (Fig. S8†). Around the same time of the anoxic incubation, the contribution of methylthioarsenates increased. Methylthioarsenate formation was limited at the start of the anoxic period due to low MMA and DMA concentrations, but once these species are available, they are readily thiolated.36 Moreover, methylthioarsenates still formed when the system seemed to be SII− limited (Fig. S8†), as suggested by the dethiolation of inorganic thioarsenates and low sulfate concentrations.
Taking C20 and C30 as examples of highly depleted systems, although there is a decrease in the contribution of inorganic thioarsenates and an increase in methylthioarsenates, the contribution of total thiolated As species remains constant during the anoxic incubation period (Fig. S9†). Similar observations of inorganic thioarsenate dethiolation coupled with the formation of methylthioarsenates have been recently reported along the outflow channel of a geothermal spring.69 Under neutral pH conditions commonly found in paddy soils and in the incubation setups of this experiment (Fig. S10†), methylthioarsenates require a lower excess of SII− to form and, thus, are thought to be more thermodynamically stable and preferred thiolation products.31–33 We suggest that under SII− limited conditions in the depleted soils, the formation of MMA and DMA could trigger the de-thiolation of inorganic thioarsenates. Such competition for SII− could take place as the inorganic thioarsenates de-thiolate due to a shift in the equilibrium caused by lower SII− concentrations. The SII− released by the de-thiolation could then react with MMA or DMA to form methylthioarsenates, which are stable at lower SII− concentrations.
Changes in S speciation and influence on thioarsenate formation
Zerovalent S and thiosulfate, products of the partial reoxidation of SII−, were found in the incubations. Despite the depletion of dissolved S in the aqueous phase with increasing redox cycling, S0 accumulated in the solid phase (Fig. 4a). In C20 and C30, S0 formation did not decrease by the washout of over 95% of the aqueous phase S. More importantly, S0 was higher in depleted soils than in C0. Thus, the formation and accumulation of S0 were independent of the S washout with increased redox cycling. This newly formed S0 pool stayed relatively stable throughout the 10 day anoxic incubation, indicating comparable rates of production and consumption. In the depleted soils, S0 content correlated positively (p < 0.05) with sulfate concentration in the aqueous phase (Fig. 4b). | ||
| Fig. 4 Changes in S speciation with increasing redox cycling. Time-resolved zerovalent S during incubation (a) and soil content compared with aqueous sulfate (b). Time-resolved thiosulfate concentrations during incubation (c) and concentration compared to sulfate (d). In (a and c), data points are average values and bars indicate standard deviation (n = 3). For (b and d), only data points from the depleted systems (C10–C30) are used. | ||
Similarly, thiosulfate was found in the aqueous phase, being slightly higher at the beginning of the anoxic incubation and decreasing with time (Fig. 4c). Thiosulfate concentrations peaking at the beginning of the incubation could be related to the partial oxidation of SII− during the oxic incubation period. Nonetheless, thiosulfate is reported to be highly reactive, transforming easily back into SII− or further oxidizing into sulfate.56 Thus, thiosulfate concentrations around 0.025 μmol L−1 towards the end of the anoxic incubation period suggest its formation under anaerobic conditions. Thiosulfate concentrations also significantly correlated (p < 0.0001) with sulfate concentrations (Fig. 4d). A stable pool of S0 and thiosulfate formation under anoxic conditions, and their significant correlations with aqueous sulfate concentrations, suggest an active CSC in the depleted soils.
Reduction of FeIII is the main proposed partner for SII− reoxidation under anoxic conditions.52,70 Increasing concentrations of dissolved Fe during the anoxic incubation periods in the depleted soils indicate FeIII reduction. This reduction could be partially associated with Fe respiration. However, trends in CH4 production suggest that Fe reductive dissolution is not a central respiration pathway in the depleted soils. In C0, CH4 production had a lag phase (Fig. S3†) normally associated with the use of other thermodynamically preferred respiration pathways,71,72 followed by a phase of exponential CH4 production. Bioavailable FeIII (oxyhydr)oxides are known to delay methanogenesis.73,74 In the depleted soils, this lag was not present, showing a linear increase in CH4 production between day 1 and 10 of the anoxic incubation, suggesting the low availability of other electron acceptors for anaerobic respiration such as FeIII and sulfate. These observations suggest that abiotic FeIII reduction associated with the CSC might dominate over biotically mediated Fe reductive dissolution.
In the solid phase, Mössbauer spectroscopy showed that repeated redox cycling mainly caused changes in the distribution of FeIII (oxyhydr)oxides (Tables S2, S3 and Fig. S11†). Crystalline FeIII (oxyhydr)oxides contributed 24.6% to C0 and were decreased in half by C10. Further cycling slightly decreased their contribution. In contrast, poorly crystalline FeIII (oxyhydr)oxides increased from 2.8% in C0 to 5.4% in C10, with a slight increase with further cycling. Moreover, there were slight increases in the contribution of other identified phases, such as FeII and adsorbed FeIII with increased redox cycling (Table S3†). These Fe phases could offer a chemically reactive FeIII pool for SII− reoxidation, as proposed by Hansel et al.70 for low sulfate freshwater sediments. Similarly, Holmkvist and Ferdelman52 correlated a slowly reacting Fe pool with SII− reoxidation.
Sulfate reduction, sulfide reoxidation, and thioarsenate formation
The above-described results for As speciation suggest that the formation of inorganic and methylated thioarsenates might be ruled by different sources of reduced S species, depending mainly on thermodynamic stability and sulfate reduction kinetics. According to these results, early thiolation in paddy soils could be driven by the reduction of a big primary pool of sulfate, taking place in the first days of anoxic incubation. Later, as this primary sulfate pool is consumed, thiolation is driven by the reduction of a smaller secondary sulfate pool as a consequence of the CSC. The As speciation in the depleted soils of this experiment offers insights into this dependency.High contribution of methylthioarsenates correlated positively (p < 0.01) with excess S0 over methyloxyarsenates (as a molar ratio of the precursors (S0) and reactants (MMA + DMA) required for methylthioarsenate formation) (Fig. 5a). This correlation was not present when considering inorganic thioarsenate formation through the reaction of S0 and arsenite (Fig. 5b). In contrast, a higher contribution of inorganic thioarsenates was found at low S0/arsenite ratios. Similarly, methylthioarsenate formation correlated positively (p < 0.05) with excess thiosulfate over methyloxyarsenates (Fig. 5c), but not with inorganic thioarsenate formation when comparing thiosulfate/arsenite (Fig. 5d). These positive correlations do not imply thiolation directly through the reaction of MMA and DMA with S0 or thiosulfate, but rather S0 and thiosulfate as intermediate S species of CSC for the formation of sulfate, which will be biotically reduced to SII−, and form thioarsenates.
 | ||
| Fig. 5 Influence of excess S0, thiosulfate (ThioS), and sulfate over oxymethylated arsenates (OxyMeAs) in the formation of methylthioarsenates (a, c, and d, respectively). Influence of excess S0, thiosulfate (ThioS), and sulfate over arsenite in the formation of inorganic thioarsenates (b, d, and f, respectively). | ||
A direct evaluation of thiolation through SII− was not possible since it was not detected in any setup. Low SII− has been previously linked to the CSC since the small and constant amounts of SII− produced readily react and thus, do not accumulate.52,56 Sulfate concentrations were used to evaluate the role of the reduction of the primary sulfate pool on As thiolation, under the assumption that most of the available sulfate would be reduced by SRB to form SII−. Nonetheless, this correlation does not consider that not all produced SII− would be available for As thiolation, due to, for example, FeS precipitation. High sulfate over oxymethylated arsenate ratio correlated negatively (p < 0.05) with methylthioarsenate formation, while high inorganic thioarsenate formation correlated positively (p < 0.05) with excess sulfate over arsenite, but only when considering C20 and C30 as highly S depleted systems (Fig. 5e and f).
These correlations suggest that in a S-depleted paddy soil, the formation of inorganic and methylated thioarsenates takes place through the reduction of two different sulfate pools, summarized in Fig. 6 and contextualized for our 10 day incubation period. Since inorganic thioarsenates formed mainly at the beginning of the anoxic incubation period, and they lacked a correlation with intermediate, partially oxidized S products of the CSC, we suggest that their formation is strongly related to the reduction of the primary sulfate pool (Fig. 6, process 1), which was likely replenished through the oxidation of sulfide phases during the oxic period. The relatively high availability of SII−, low dissolved Fe, and high S/As ratios then favored the formation of highly thiolated inorganic thioarsenates (DTA and TTA).
 | ||
| Fig. 6 Conceptual model indicating the formation of inorganic thioarsenates (iThio As) and methylthioarsenates (MeThioAs) with SII− from either a primary (1) or secondary (2) sulfate pool. The top diagram in the figure displays the biogeochemical reactions involved in the production of SII− and thioarsenates. The thickness of the arrows qualitatively illustrates the importance of a reaction. Sulfur species are protonated assuming pH 7, reflecting the neutral values during the experiment (Fig. S10†). In the center, diagrams indicate changes in concentrations of key players in the reactions throughout the 10 day anoxic incubation period. Circles at the bottom of the figure show how the reduction of the primary sulfate pool decreases in importance as the soil is depleted, while SII− produced due to the reduction of a secondary sulfate pool gains importance. | ||
After 5 days, and once the primary pool of sulfate had been consumed by SRB, SII− had been likely scavenged by FeS precipitation, and methylated oxyarsenates were present, inorganic thioarsenates dethiolated. Furthermore, the positive correlation between methylthioarsenate formation and partially oxidized products of the CSC suggest that the main source of SII− for their formation in later stages of the anoxic incubation is the secondary sulfate pool formed through the CSC (Fig. 6, process 2). While this secondary sulfate pool would theoretically be available for the formation of inorganic thioarsenates, the lower excess SII− necessary for the formation of methylthioarsenates favors their formation in an S-depleted system. Moreover, due to the depletion of S, the primary sulfate pool becomes a less relevant source of SII− with increasing redox cycling.
Conclusion
The results presented here show the importance of considering thioarsenate formation when studying As speciation. It is often assumed that thioarsenates do not form in low-S systems such as wetlands or paddy soils, considered to be methanogenic environments. Here, we show that through the CSC, a supply of reduced S species is available for thiolation even in S-depleted systems and once the primary sulfate pool is consumed by SRB activity. These observations help explain the presence of thioarsenates in paddy soil porewaters even when sulfate reduction is no longer taking place. In systems with low As, where there is a high S/As ratio, thioarsenates can significantly contribute to speciation and, thus, mobility, long-range transport, and toxicity, depending on environmental conditions. In the context of paddy soils, a large share of thioarsenates can be related to a higher As uptake by rice plants, risking food safety. The results presented here do not imply that CSC only takes place in S-depleted systems. Cryptic S cycling likely takes place in S-rich systems as well, but its effects are masked by high S concentrations and changes in S speciation dominated by SRB activity during the consumption of the primary sulfate pool.More importantly, this study highlights the differences in the kinetics and thermodynamics of the formation of inorganic and methylated thioarsenates. Systems with a high and constant reduced S supply might be dominated by inorganic thioarsenates, as is the case in many geothermal systems. However, if methylated oxyarsenates are present, and reduced S becomes scarce, our results suggest the dethiolation of inorganic thioarsenates to form methylthioarsenates. These observations show the complex biogeochemistry behind As thiolation, which depends on reduced S availability and the presence of specific As species, which in turn are strongly governed by microbially mediated reactions and the biogeochemistry of C and Fe.
Data availability
The data collected during the above-described experiments and used for this study are published on Zenodo at https://doi.org/10.5281/zenodo.14336088.Author contributions
This study was conceptualized by J. M. L. N. and B. P.-F.; incubation setup and solid, aqueous, and gas phase analyses were carried out by J. M. L. N. Mössbauer spectroscopy and μXRD analyses were carried out by C. L. D. with the support of A. K. The manuscript and ESI† were written by J. M. L. N. with assistance from B. P.-F.; all authors contributed to the manuscript revisions.Conflicts of interest
The authors declare no competing interests.Acknowledgements
We thank the German Academic Scholarship Foundation for the doctoral funding support for J. M. L. N. and funding within the Federal Ministry of Education and Research project BMBF #031B0840 to B. P.-F. We are grateful to Sylvia Hafner, Jens Hamberger, Tobias Mayer, Hai Anh Nguyen, Milagros Galarza, and Alejandra Higa Mori for their experimental support during the long-term experiment. We thank Livia Urbanski and Angelika Kölbl for providing the chronosequence soil, and Ben Gilfedder for providing access to the CH4 and CO2 analyses. The Graphical Abstract and Fig. 6 were created using BioRender.References
- J. T. Trevors and B. J. Alloway, Heavy Metals in Soils: Trace Metals and Metalloids in Soils and their Bioavailability, Heavy Metals in Soils, 2013 Search PubMed.
- A. Aitio, K. Cantor, M. D. Attfield and P. A. Demers, Arsenic, metals, fibres, and dusts, IARC Monogr Eval Carcinog Risks Hum., 2012, vol. 100, Pt C, pp. 11–465 Search PubMed.
- R. Stone, Arsenic and paddy rice: A neglected cancer risk?, Science, 2008, 321(5886), 184–185 CrossRef CAS PubMed.
- I. Kögel-Knabner, W. Amelung, Z. Cao, S. Fiedler, P. Frenzel and R. Jahn, et al., Biogeochemistry of paddy soils, Geoderma, 2010, 157(1), 1–14 CrossRef.
- A. A. Nghiem, H. Prommer, M. R. H. Mozumder, A. Siade, J. Jamieson and K. M. Ahmed, et al., Sulfate reduction accelerates groundwater arsenic contamination even in aquifers with abundant iron oxides, Nat. Water, 2023, 1(2), 151–165 CrossRef CAS PubMed.
- J. Podgorski and M. Berg, Global threat of arsenic in groundwater, Science, 2020, 368(6493), 845–850 CrossRef CAS PubMed.
- P. L. Smedley and D. G. Kinniburgh, A review of the source, behaviour and distribution of arsenic in natural waters, Appl. Geochem., 2002, 17(5), 517–568 CrossRef CAS.
- J. F. Ma, N. Yamaji, N. Mitani, X.-Y. Xu, Y.-H. Su and S. P. McGrath, et al., Transporters of arsenite in rice and their role in arsenic accumulation in rice grain, Proc. Natl. Acad. Sci. U. S. A., 2008, 105(29), 9931–9935 CrossRef CAS PubMed.
- A. A. Meharg and F. J. Zhao, Arsenic & Rice, Springer Netherlands, 2012 Search PubMed.
- F.-J. Zhao, Y.-G. Zhu and A. A. Meharg, Methylated arsenic species in rice: geographical variation, origin, and uptake mechanisms, Environ. Sci. Technol., 2013, 47(9), 3957–3966 CrossRef CAS PubMed.
- B. Moe, H. Peng, X. Lu, B. Chen, L. W. L. Chen and S. Gabos, et al., Comparative cytotoxicity of fourteen trivalent and pentavalent arsenic species determined using real-time cell sensing, J. Environ. Sci., 2016, 49, 113–124 CrossRef CAS PubMed.
- S. Goldberg and C. T. Johnston, Mechanisms of arsenic adsorption on amorphous oxides evaluated using macroscopic measurements, vibrational spectroscopy, and surface complexation modeling, J. Colloid Interface Sci., 2001, 234(1), 204–216 CrossRef CAS PubMed.
- Y. Takahashi, R. Minamikawa, K. H. Hattori, K. Kurishima, N. Kihou and K. Yuita, Arsenic behavior in paddy fields during the cycle of flooded and non-flooded periods, Environ. Sci. Technol., 2004, 38(4), 1038–1044 CrossRef CAS PubMed.
- J. Zobrist, P. R. Dowdle, J. A. Davis and R. S. Oremland, Mobilization of arsenite by dissimilatory reduction of adsorbed arsenate, Environ. Sci. Technol., 2000, 34(22), 4747–4753 CrossRef CAS.
- C. Lomax, W.-J. Liu, L. Wu, K. Xue, J. Xiong and J. Zhou, et al., Methylated arsenic species in plants originate from soil microorganisms, New Phytol., 2012, 193(3), 665–672 CrossRef CAS PubMed.
- J. Qin, B. P. Rosen, Y. Zhang, G. Wang, S. Franke and C. Rensing, Arsenic detoxification and evolution of trimethylarsine gas by a microbial arsenite S-adenosylmethionine methyltransferase, Proc. Natl. Acad. Sci. U. S. A., 2006, 103(7), 2075–2080 CrossRef CAS PubMed.
- K. Viacava, K. L. Meibom, D. Ortega, S. Dyer, A. Gelb and L. Falquet, et al., Variability in arsenic methylation efficiency across aerobic and anaerobic microorganisms, Environ. Sci. Technol., 2020, 54(22), 14343–14351 CrossRef CAS PubMed.
- H. Naranmandura, M. W. Carew, S. Xu, J. Lee, E. M. Leslie and M. Weinfeld, et al., Comparative Toxicity of Arsenic Metabolites in Human Bladder Cancer EJ-1 Cells, Chem. Res. Toxicol., 2011, 24(9), 1586–1596 Search PubMed.
- F.-J. Zhao, E. Harris, J. Yan, J. Ma, L. Wu and W. Liu, et al., Arsenic methylation in soils and its relationship with microbial arsM abundance and diversity, and As speciation in rice, Environ. Sci. Technol., 2013, 47(13), 7147–7154 CrossRef CAS PubMed.
- M. C. Reid, J. Maillard, A. Bagnoud, L. Falquet, P. Le Vo and R. Bernier-Latmani, Arsenic methylation dynamics in a rice paddy soil anaerobic enrichment culture, Environ. Sci. Technol., 2017, 51(18), 10546–10554 CrossRef CAS PubMed.
- L. Wang, Q. Guo, G. Wu, Z. Yu, J. M. L. Ninin and B. Planer-Friedrich, Methanogens-Driven Arsenic Methylation Preceding Formation of Methylated Thioarsenates in Sulfide-Rich Hot Springs, Environ. Sci. Technol., 2023, 57(19), 7410–7420 CrossRef CAS PubMed.
- C. Chen, L. Li, K. Huang, J. Zhang, W.-Y. Xie and Y. Lu, et al., Sulfate-reducing bacteria and methanogens are involved in arsenic methylation and demethylation in paddy soils, ISME J., 2019, 13(10), 2523–2535 CrossRef CAS PubMed.
- C. Chen, B. Yang, Y. Shen, J. Dai, Z. Tang and P. Wang, et al., Sulfate addition and rising temperature promote arsenic methylation and the formation of methylated thioarsenates in paddy soils, Soil Biol. Biochem., 2021, 154, 108129 CrossRef CAS.
- C. Chen, B. Yang, A. Gao, L. Li, X. Dong and F.-J. Zhao, Suppression of methanogenesis in paddy soil increases dimethylarsenate accumulation and the incidence of straighthead disease in rice, Soil Biol. Biochem., 2022, 169, 108689 CrossRef CAS.
- C. Chen, L. Li, Y. Wang, X. Dong and F.-J. Zhao, Methylotrophic methanogens and bacteria synergistically demethylate dimethylarsenate in paddy soil and alleviate rice straighthead disease, ISME J., 2023, 1851–1861 CrossRef PubMed.
- S. Stauder, B. Raue and F. Sacher, Thioarsenates in sulfidic waters, Environ. Sci. Technol., 2005, 39(16), 5933–5939 CrossRef CAS PubMed.
- D. Wallschläger and C. J. Stadey, Determination of (oxy)thioarsenates in sulfidic waters, Anal. Chem., 2007, 79(10), 3873–3880 CrossRef PubMed.
- J. Wang, C. F. Kerl, P. Hu, M. Martin, T. Mu and L. Brüggenwirth, et al., Thiolated arsenic species observed in rice paddy pore waters, Nat. Geosci., 2020, 13(4), 282–287 CrossRef CAS.
- A. Eberle, J. Besold, J. M. León Ninin, C. F. Kerl, K. Kujala and B. Planer-Friedrich, Potential of high pH and reduced sulfur for arsenic mobilization – insights from a Finnish peatland treating mining waste water, Sci. Total Environ., 2021, 758, 143689 CrossRef CAS PubMed.
- B. Planer-Friedrich, C. Härtig, R. Lohmayer, E. Suess, S. H. McCann and R. Oremland, Anaerobic Chemolithotrophic Growth of the Haloalkaliphilic Bacterium Strain MLMS-1 by Disproportionation of Monothioarsenate, Environ. Sci. Technol., 2015, 49(11), 6554–6563 CrossRef CAS PubMed.
- D. Wallschläger and J. London, Determination of methylated arsenic-sulfur compounds in groundwater, Environ. Sci. Technol., 2008, 42(1), 228–234 CrossRef PubMed.
- B. Planer-Friedrich, J. London, R. B. McCleskey, D. K. Nordstrom and D. Wallschläger, Thioarsenates in geothermal waters of Yellowstone National Park: determination, preservation, and geochemical importance, Environ. Sci. Technol., 2007, 41(15), 5245–5251 CrossRef CAS PubMed.
- S. D. Conklin, M. W. Fricke, P. A. Creed and J. T. Creed, Investigation of the pH effects on the formation of methylated thio-arsenicals, and the effects of pH and temperature on their stability, J. Anal. At. Spectrom., 2008, 23(5), 711–716 RSC.
- Y.-T. Kim, H. Lee, H.-O. Yoon and N. C. Woo, Kinetics of Dimethylated Thioarsenicals and the Formation of Highly Toxic Dimethylmonothioarsinic Acid in Environment, Environ. Sci. Technol., 2016, 50(21), 11637–11645 CrossRef CAS PubMed.
- P. V. T. Knobloch, L. H. Pham, C. F. Kerl, Q. Guo and B. Planer-Friedrich, Seasonal Formation of Low-Sorbing Methylthiolated Arsenates Induces Arsenic Mobilization in a Minerotrophic Peatland, Environ. Sci. Technol., 2024, 1669–1679 CrossRef CAS PubMed.
- J. M. León Ninin, E. M. Muehe, A. Kölbl, A. Higa Mori, A. Nicol and B. Gilfedder, et al., Changes in arsenic mobility and speciation across a 2000-year-old paddy soil chronosequence, Sci. Total Environ., 2024, 908, 168351 CrossRef PubMed.
- J. M. León Ninin, N. Kryschak, S. Peiffer and B. Planer-Friedrich, Long-Term Paddy Soil Development Buffers the Increase in Arsenic Methylation and Thiolation after Sulfate Fertilization, J. Agric. Food Chem., 2024, 25045–25053 CrossRef PubMed.
- D. R. Lovley, Microbial Fe(III) reduction in subsurface environments, FEMS Microbiol. Rev., 1997, 20(3–4), 305–313 CrossRef CAS.
- E. D. Burton, S. G. Johnston and B. D. Kocar, Arsenic mobility during flooding of contaminated soil: the effect of microbial sulfate reduction, Environ. Sci. Technol., 2014, 48(23), 13660–13667 CAS.
- M. L. Farquhar, J. M. Charnock, F. R. Livens and D. J. Vaughan, Mechanisms of arsenic uptake from aqueous solution by interaction with goethite, lepidocrocite, mackinawite, and pyrite: an X-ray absorption spectroscopy study, Environ. Sci. Technol., 2002, 36(8), 1757–1762 CAS.
- B. C. Bostick and S. Fendorf, Arsenite sorption on troilite (FeS) and pyrite (FeS2), Geochim. Cosmochim. Acta, 2003, 67(5), 909–921 CAS.
- J. Wang, D. Halder, L. Wegner, L. Brüggenwirth, J. Schaller and M. Martin, et al., Redox dependence of thioarsenate occurrence in paddy soils and the rice rhizosphere, Environ. Sci. Technol., 2020, 54(7), 3940–3950 CAS.
- A. E. Colina Blanco, C. F. Kerl and B. Planer-Friedrich, Detection of thioarsenates in rice grains and rice products, J. Agric. Food Chem., 2021, 69(7), 2287–2294 CAS.
- B. Planer-Friedrich, C. F. Kerl, A. E. Colina Blanco and S. Clemens, Dimethylated thioarsenates: a potentially dangerous blind spot in current worldwide regulatory limits for arsenic in rice, J. Agric. Food Chem., 2022, 70(31), 9610–9618 CrossRef CAS PubMed.
- E. Pischke, F. Barozzi, A. E. Colina Blanco, C. F. Kerl, B. Planer-Friedrich and S. Clemens, Dimethylmonothioarsenate is highly toxic for plants and readily translocated to shoots, Environ. Sci. Technol., 2022, 56(14), 10072–10083 CAS.
- T. Wind and R. Conrad, Localization of sulfate reduction in planted and unplanted rice field soil, Biogeochemistry, 1997, 37(3), 253–278 CAS.
- S. W. Poulton, Sulfide oxidation and iron dissolution kinetics during the reaction of dissolved sulfide with ferrihydrite, Chem. Geol., 2003, 202(1), 79–94 CrossRef CAS.
- G. Jiang, K. R. Sharma, A. Guisasola, J. Keller and Z. Yuan, Sulfur transformation in rising main sewers receiving nitrate dosage, Water Res., 2009, 43(17), 4430–4440 CrossRef CAS PubMed.
- M. Mora, L. R. López, J. Lafuente, J. Pérez, R. Kleerebezem and M. C. M. van Loosdrecht, et al., Respirometric characterization of aerobic sulfide, thiosulfate and elemental sulfur oxidation by S-oxidizing biomass, Water Res., 2016, 89, 282–292 CrossRef CAS PubMed.
- S. L. Saalfield and B. C. Bostick, Changes in iron, sulfur, and arsenic speciation associated with bacterial sulfate reduction in ferrihydrite-rich systems, Environ. Sci. Technol., 2009, 43(23), 8787–8793 CrossRef CAS PubMed.
- J. M. León Ninin, A. Higa Mori, J. Pausch and B. Planer-Friedrich, Long-term paddy use influences response of methane production, arsenic mobility and speciation to future higher temperatures, Sci. Total Environ., 2024, 943, 173793 CrossRef PubMed.
- L. Holmkvist, T. G. Ferdelman and B. B. Jørgensen, A cryptic sulfur cycle driven by iron in the methane zone of marine sediment (Aarhus Bay, Denmark), Geochim. Cosmochim. Acta, 2011, 75(12), 3581–3599 CrossRef CAS.
- J. S. Berg, D. Jézéquel, A. Duverger, D. Lamy, C. Laberty-Robert and J. Miot, Microbial diversity involved in iron and cryptic sulfur cycling in the ferruginous, low-sulfate waters of Lake Pavin, PLoS One, 2019, 14(2), e0212787 CrossRef CAS PubMed.
- G.-H. C. Ng, C. E. Rosenfeld, C. M. Santelli, A. R. Yourd, J. Lange and K. Duhn, et al., Microbial and Reactive Transport Modeling Evidence for Hyporheic Flux-Driven Cryptic Sulfur Cycling and Anaerobic Methane Oxidation in a Sulfate-Impacted Wetland-Stream System, J. Geophys. Res., 2020, 125(2), e2019JG005185 CrossRef CAS.
- M. Pester, K.-H. Knorr, M. W. Friedrich, M. Wagner and A. Loy, Sulfate-reducing microorganisms in wetlands – fameless actors in carbon cycling and climate change, Front. Microbiol., 2012, 3, 72 CAS.
- T. Wind and R. Conrad, Sulfur compounds, potential turnover of sulfate and thiosulfate, and numbers of sulfate-reducing bacteria in planted and unplanted paddy soil, FEMS Microbiol. Ecol., 1995, 18(4), 257–266 CrossRef CAS.
- W. Guo, A. R. Cecchetti, Y. Wen, Q. Zhou and D. L. Sedlak, Sulfur Cycle in a Wetland Microcosm: Extended 34S-Stable Isotope Analysis and Mass Balance, Environ. Sci. Technol., 2020, 54(9), 5498–5508 CrossRef CAS PubMed.
- A. Kölbl, P. Schad, R. Jahn, W. Amelung, A. Bannert and Z. H. Cao, et al., Accelerated soil formation due to paddy management on marshlands (Zhejiang Province, China), Geoderma, 2014, 228–229, 67–89 CrossRef.
- L. Wissing, A. Kölbl, P. Schad, T. Bräuer, Z.-H. Cao and I. Kögel-Knabner, Organic carbon accumulation on soil mineral surfaces in paddy soils derived from tidal wetlands, Geoderma, 2014, 228–229, 90–103 CrossRef CAS.
- Y.-Q. Cheng, L.-Z. Yang, Z.-H. Cao, E. Ci and S. Yin, Chronosequential changes of selected pedogenic properties in paddy soils as compared with non-paddy soils, Geoderma, 2009, 151(1), 31–41 CrossRef CAS.
- L. L. Stookey, Ferrozine---a new spectrophotometric reagent for iron, Anal. Chem., 1970, 42(7), 779–781 CrossRef CAS.
- F. Hegler, N. R. Posth, J. Jiang and A. Kappler, Physiology of phototrophic iron(II)-oxidizing bacteria: implications for modern and ancient environments, FEMS Microbiol. Ecol., 2008, 66(2), 250–260 CrossRef CAS PubMed.
- J. D. Cline, Spectrophotometric determination of hydrogen sulfide in natural waters, Limnol. Oceanogr., 1969, 14(3), 454–458 CrossRef CAS.
- R. Lohmayer, A. Kappler, T. Lösekann-Behrens and B. Planer-Friedrich, Sulfur species as redox partners and electron shuttles for ferrihydrite reduction by sulfurospirillum deleyianum, Appl. Environ. Microbiol., 2014, 80(10), 3141–3149 CrossRef PubMed.
- K. Lagarec and D. G. Rancourt, Extended Voigt-based analytic lineshape method for determining N-dimensional correlated hyperfine parameter distributions in Mössbauer spectroscopy, Nucl. Instrum. Methods Phys. Res. Sect. B Beam Interact. Mater. Atoms, 1997, 129(2), 266–280 CrossRef CAS.
- X. Xu, P. Wang, J. Zhang, C. Chen, Z. Wang and P. M. Kopittke, et al., Microbial sulfate reduction decreases arsenic mobilization in flooded paddy soils with high potential for microbial Fe reduction, Environ. Pollut., 2019, 251, 952–960 CrossRef CAS PubMed.
- J. Sun, A. N. Quicksall, S. N. Chillrud, B. J. Mailloux and B. C. Bostick, Arsenic mobilization from sediments in microcosms under sulfate reduction, Chemosphere, 2016, 153, 254–261 CrossRef CAS PubMed.
- W. Wisawapipat, N. Chooaiem, S. Aramrak, N. Chittamart, S. Nookabkaew and N. Rangkadilok, et al., Sulfur amendments to soil decrease inorganic arsenic accumulation in rice grain under flooded and nonflooded conditions: Insights from temporal dynamics of porewater chemistry and solid-phase arsenic solubility, Sci. Total Environ., 2021, 779, 146352 CrossRef CAS PubMed.
- K. Yan, Q. Guo, L. Wang, Y. Liu and B. Planer-Friedrich, Potential for formation of methylated thioarsenates in geothermal environments, Geochim. Cosmochim. Acta, 2024 Search PubMed , in press.
- C. M. Hansel, C. J. Lentini, Y. Tang, D. T. Johnston, S. D. Wankel and P. M. Jardine, Dominance of sulfur-fueled iron oxide reduction in low-sulfate freshwater sediments, ISME J., 2015, 9(11), 2400–2412 CrossRef CAS PubMed.
- F. N. Ponnamperuma, The Chemistry of Submerged Soils, in Advances in Agronomy, ed. N. C. Brady, Academic Press, vol. 24, 1972, p. 29–96 Search PubMed.
- Jr. W. H. Patrick and A. Jugsujinda, Sequential reduction and oxidation of inorganic nitrogen, manganese, and iron in flooded soil, Soil Sci. Soc. Am. J., 1992, 56(4), 1071–1073 CrossRef.
- D. R. Lovley and E. J. Phillips, Competitive mechanisms for inhibition of sulfate reduction and methane production in the zone of ferric iron reduction in sediments, Appl. Environ. Microbiol., 1987, 53(11), 2636–2641 CrossRef CAS PubMed.
- P. M. van Bodegom, J. C. M. Scholten and A. J. M. Stams, Direct inhibition of methanogenesis by ferric iron, FEMS Microbiol. Ecol., 2004, 49(2), 261–268 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: ESI and figures supporting the above-described observations and discussion based on soil properties and the aqueous phase redox biogeochemistry of C, S, Fe, and As. Mössbauer spectra from original and depleted soils, and supplementary discussion on the use of μXRD for identification of mineral phases. See DOI: https://doi.org/10.1039/d4em00764f |
| ‡ Deceased. |
| This journal is © The Royal Society of Chemistry 2025 |