
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSpontaneous aqueous defluorination of trifluoromethylphenols: substituent effects and revisiting the mechanism†
Zhefei
Guo‡
 a,
Geneviève W.
Tremblay‡
a,
Jingdan
Chen
b and
Shira
Joudan
*a
a,
Geneviève W.
Tremblay‡
a,
Jingdan
Chen
b and
Shira
Joudan
*a
aDepartment of Chemistry, University of Alberta, Edmonton T6G 2G2, Alberta, Canada. E-mail: joudan@ualberta.ca
bDepartment of Chemistry, University of Illinois at Urbana-Champaign, Urbana, Illinois 61801, USA
First published on 6th May 2025
Abstract
Trifluoromethylphenols (TFMPs) are environmental contaminants that exist as transformation products of aryl-CF3 pharmaceuticals and agrochemicals. Their –CF3 moiety raises concerns as it may form problematic fluorinated transformation products such as the persistent pollutant trifluoroacetic acid (TFA). This study investigates the hydrolysis and spontaneous defluorination mechanisms of 2-TFMP, 3-TFMP, 4-TFMP, and 2-Cl-4-TFMP under environmentally relevant aqueous conditions, and under alkaline pH to investigate the mechanism of defluorination. 3-TFMP did not undergo hydrolysis. The other TFMPs reacted to primarily form the corresponding hydroxybenzoic acids and fluoride. High-resolution mass spectrometry identified a benzoyl fluoride intermediate in the hydrolysis of 4-TFMP and other dimer-like transformation products of the 4- and 2-Cl-4-TFMP. Density functional theory calculations revealed that the key defluorination step likely proceeds via an E1cb mechanism, driven by β-elimination. Experimental and computational results demonstrated substituent-dependent differences in reactivity, and the importance of the deprotonation of TFMPs for the hydrolysis reaction to proceed. These findings provide mechanistic insights into the complete defluorination of TFMPs and broader implications for the environmental defluorination of other PFAS.
Environmental significanceThe environmental transformation of some aryl-CF3 pharmaceuticals and agrochemicals produces trifluoromethylphenols (TFMPs), which are environmental contaminants themselves and can be precursors to the very persistent pollutant trifluoroacetic acid (TFA). Alternatively, this study demonstrates that certain TFMPs can undergo complete hydrolytic defluorination in aqueous solutions, depending on their chemical structure. Understanding what drives this spontaneous defluorination can provide insight into why certain C–F bonds can break, despite the general stability of organofluorine compounds. Designing aryl-CF3 compounds to favor defluorination pathways without TFA formation could harness the benefits of the –CF3 moiety in pharmaceuticals and agrochemicals while reducing their environmental persistence. |
Introduction
Trifluoromethylphenols (TFMPs) are important building blocks for many pharmaceuticals and agrochemicals containing aromatic trifluoromethyl (aryl-CF3) functional groups (Fig. 1A).1,2 Fluorine (F) has a small atomic radius, the highest electronegativity, and forms strong bonds with carbon.3 By substituting all hydrogens in a methyl group (–CH3) with F atoms, the performance of aryl-CF3 products are often enhanced, such as increased metabolic stability and lipophilicity.3 However, the high stability of C–F bonds raises concerns about the persistence of these compounds and their fluorinated transformation products in the environment.4,5 Aryl-CF3 compounds are classified as per- and polyfluoroalkyl substances (PFAS) according to OECD definition,6,7 and therefore are receiving heightened interest. In addition, some have been demonstrated to be precursors to trifluoroacetic acid (TFA) in a competing mechanism to defluorination.8–10 TFA is a very persistent and very mobile pollutant that is rapidly growing in environmental concentration.11,12 4-(Trifluoromethyl)phenol (4-TFMP) is a well-recognized transformation product of fluoxetine,8,13,14 has been measured in the environment,15 and is partially converted to TFA.8–10 Another TFMP, 2-Cl-4-TFMP is a transformation product of diphenyl ether herbicides like acifluorfen, oxyfluorfen and fluoroglycofen,16–18 yet its environmental chemistry has not been studied. Understanding the environmental reactivity of TFMPs is essential for understanding the environmental fate of many aryl-CF3 pharmaceuticals and agrochemicals. | ||
| Fig. 1 (A) Representative structures of aryl-CF3 pharmaceuticals and agrochemicals containing TFMP building blocks, and reported transformation from aryl-CF3 to TFMPs; (B) TFMP structures studied in this work and their corresponding pKa values; (C) above, a scheme showing that –CF3 at the ortho or para position relative to phenol-OH can stabilize the phenolate; below, a reaction scheme showing that β-carbanion drives the defluorination reaction of –CF3. | ||
Hydrolysis is an important abiotic transformation pathway for organic pollutants. Previous research observed that 2- and 4-TFMP can undergo hydrolysis resulting in spontaneous defluorination under alkaline conditions.9,13,19–24 Reuben G. Jones first reported the base-mediated defluorination of 4-TFMP in 1947, observing that higher concentrations of 4-TFMP spontaneously defluorinated and polymerized in NaOH solutions, eventually stabilizing with no further defluorination.19 Later, in 1973, Sakai and Santi observed complete hydrolytic defluorination of 2- and 4-TFMP in buffered solutions (pH 6.5–13), yielding 2- and 4-hydroxybenzoic acid (2- and 4-HBA), respectively.20 They proposed that the defluorination mechanism is likely facilitated by β-elimination via a conjugated structure that stabilizes the phenolate anion, as illustrated in Fig. 1C. More recently, environmental chemists, including the Mabury group,13,21,22 Manfrin et al.,23 and Bhat et al.,9 reported similar defluorination reactions. 4-TFMP is the most well-studied TFMP, however, reports on its hydrolytic stability are conflicting. Some suggest 4-TFMP rapidly loses HF upon dissolution in water,13,21 while others propose that spontaneous defluorination requires additional bases that significantly deprotonate the phenol-OH group; otherwise, degradation is minimal, or the hydrolysis defluorination rate is inferred to be extremely slow.20,22
The spontaneous defluorination of certain TFMPs under environmentally relevant conditions contradicts the common perception that C–F bonds are difficult to activate and that fluoride (F−) is a poor leaving group.25 The mechanism of this spontaneous defluorination remains largely unexplored due to the lack of identified intermediates and transformation products. No plausible mechanism has been proposed to explain the reactivity differences caused by substituent effects.
Density functional theory (DFT) calculations are used in environmental chemistry to calculate the thermodynamics and kinetics of reactions, particularly for environmental chemistry reactions involving reactive transient intermediates that cannot be characterized by common analytical methods.26–28 By optimizing reactants, intermediates, products, transition states (TS) and calculating the proposed reaction pathways, it is possible to assess the plausibility of these pathways from both thermodynamic and kinetic perspectives. Combined with experimental data, DFT calculations have been successfully applied to explain the degradation of PFAS26–28 and the hydrolytic defluorination of fluorinated pharmaceuticals.29
This study systematically investigated the mechanism of TFMPs that undergo spontaneous defluorination in aqueous solutions under environmentally relevant conditions to build off previously conflicting reports of TFMP hydrolysis rates.13,21,22 To explore the substituent effects, 2-TFMP, 3-TFMP, 4-TFMP, and 2-Cl-4-TFMP were studied in aqueous solutions (Fig. 1B). pH buffer solutions from pH 6.2 to 10.8 were used to study the effect of deprotonation on hydrolysis kinetics, and Orbitrap high-resolution mass spectrometry (HRMS) was used to identify transformation products. The most plausible spontaneous defluorination mechanism was derived through DFT calculations combined with experimental data.
Materials and methods
Chemicals
All chemicals were used as received. Chemicals are described in the ESI (ESI-1).†Hydrolysis experiments and analysis
Solutions of 10 mM phosphate (pH 6.2 to 7.9) or carbonate (pH 10 to 10.8) buffers were prepared in ultrapure water. Stock solutions of 2-, 3-, 4-TFMP (5 mM) and 2-Cl-4-TFMP (1 mM) were prepared in ultrapure water from neat material. Aqueous stock solutions were stored in the dark at 4 °C, and then equilibrated to room temperature before use. Hydrolysis experiments were performed within one week of preparing the aqueous stock solutions, and were cross-calibrated against calibration standards prepared in methanol the day before the experiment. Reaction mixtures were prepared by diluting TFMPs to 10 μM in 10 mM buffers in 100 mL polypropylene volumetric flasks. Experiments were performed in triplicate in polypropylene Falcon tubes wrapped in aluminum foil to minimize exposure to ambient light. Room temperature experiments (22 °C) were run in a dark drawer with temperatures monitored using an electronic thermometer. To determine the free energy of activation, experiments at pH 10 were also performed at higher temperatures (30 °C and 40 °C) in a hot water bath, where temperatures were recorded with a mercury thermometer.At set time points for each experiment, 1 mL aliquots were mixed with a volume of 1 M HCl to quench the experiments prior to analysis: 5 μL for pH 6.2–7 experiments, 10 μL for pH 7.9 experiments, and 15 μL for pH 10–10.8 experiments. The concentrations of TFMPs and their corresponding hydrolysis products hydroxybenzoic acids (HBAs) were monitored by high performance liquid chromatography (HPLC) coupled with a UV detector. Detailed instrumental conditions are provided in the Table S1.†
Plotting ln(Ct/C0) versus time (t) provided the pseudo-first-order rate constants, k (eqn (1)). The half-lives (t1/2) of the compounds were determined from eqn (2). Eqn (3) describes the fraction of deprotonated TFMPs (in phenolate form, fphenolate) as a function of aqueous solution pH based on Henderson–Hasselbalch equation. Eqn (4) explains the degradation of TFMPs by using the fraction of TFMPs that dissociate into phenolates, where kphenolate is the rate constant when TFMPs are 100% deprotonated in their phenolate form. The Eyring equation (eqn (5)) was used to plot ln(k/T) with respect to 1/RT for each experiment (eqn (6)), where k is the pseudo-first order rate constant (s−1); T is the absolute temperature in K; R (8.314 J (mol−1 K−1)) is the gas constant; kB (1.38 × 10−23 J K−1) is the Boltzmann constant; h (6.626 × 10−34 J s) is Planck's constant; κ is the transmission coefficient (close to 1 in most cases). The slope of the eqn (6) fitting plot provided the experimental free energy of activation (ΔG‡, kcal mol−1).
 | (1) |
 | (2) |
 | (3) |
| k = kphenolate × fphenolate | (4) |
 | (5) |
 | (6) |
To determine the fluorine mass balance of pH 10 hydrolysis experiments, F− formation was tracked by the Fisherbrand Accumet AB250 pH/ISE meter and the Accumet combination fluoride ion selective electrode. Fluorine mass balance experiments were run in duplicates, and 1 mL aliquots were combined with 1 mL of the total ionic strength adjustment buffer (Fisher Scientific) prior to analysis. F− calibration standards were prepared from NaF dissolved in ultrapure water. TFMPs were also quantified by HPLC-UV in these separate experiments.
To determine how pH may affect transformation products formation, aliquots taken at different time points during pH 6.2 and pH 10.8 experiments were diluted 10 times in 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 (v/v) methanol:H2O and analyzed by ultra-high performance liquid chromatography (UHPLC) coupled with an Orbitrap HRMS. For pH 10 experiments, samples were only taken at the beginning and end. Detailed UHPLC-Orbitrap-HRMS parameters are listed in the Table S2.† Suspected transformation products are listed in Table S4.†
:50 (v/v) methanol:H2O and analyzed by ultra-high performance liquid chromatography (UHPLC) coupled with an Orbitrap HRMS. For pH 10 experiments, samples were only taken at the beginning and end. Detailed UHPLC-Orbitrap-HRMS parameters are listed in the Table S2.† Suspected transformation products are listed in Table S4.†
In situ 19F NMR was also conducted in an attempt to detect highly reactive fluorinated intermediates. Instrumental details are described in ESI-3.†
Computational methods
DFT calculations were performed by the Gaussian 16 (Revision C.01)30 at 298.15 K and 1 atm. Geometry optimization was calculated at M06-2X/6-311+g(d,p) level of theory. Integral equation formalism polarizable continuum model (IEFPCM) was used to account for solvation effects to structure optimization.31 Frequency analysis was performed to verify there were no imaginary frequencies for local minima and only one single imaginary frequency for transition states (TS). Single-point energy was calculated at M06-2X/6-311++g(2d,2p) level of theory, as the M06-2X functional is suitable for calculating main group element energies,32,33 and the same level of theory has been applied to explain the oxidative degradation of PFAS.26,27We specifically considered a hybrid solvation model for key elementary reactions to account for solvation with water. For the explicit solvation, one, two, five, or six explicit H2O molecules were added to calculations. For the implicit solvation, the solvation free energy was taken into account in the Gibbs free energy (Gf) of a molecule by using solvation model density (SMD) model34 as indicated by Chen et al.29 and Qin et al.35Gf values were also corrected for the standard state in solution as indicated by Zhang et al.26,27 For reactions with explicit H2O molecules, the Gf of H2O was corrected for its concentration (55.3 M) deviation from its standard state (1 M).29 Refer to ESI-4 and 11† for more details.
Results and discussion
Degradation kinetics
The aqueous hydrolysis of TFMPs is highly dependent on their chemical structures and the buffer pH. No degradation was observed for 2-TFMP at pH 6.2 aqueous buffer. Hydrolysis was observed for 2-TFMP from pH 7 to 10.8, and for 4-TFMP and 2-Cl-4-TFMP hydrolysis was observed across all tested pH (from pH 6.2 to 10.8) (Fig. 2). 3-TFMP exhibits strong resistance to hydrolysis even at elevated temperatures. In a solution buffered with carbonate to a pH of 10.2, and at a temperature of 40 °C for 24 hours, no obvious degradation of 3-TFMP, nor formation of its hydrolysis product 3-HBA was observed by HPLC-UV (Fig. S4†). We propose that 3-TFMP is unlikely to undergo hydrolysis in the aqueous environment. It is, however, known to undergo photolysis.9,23 | ||
| Fig. 2 Hydrolysis half-lives and rate constants of 4-TFMP, 2-TFMP, and 2-Cl-4-TFMP based on buffer pH. The solid lines for three TFMPs are fitting the curve of eqn (3) and (4). Note that the hydrolysis half-life time is plotted on the left y-axis while the hydrolysis rate constant is plotted on the right y-axis. Both y-axes are in logarithmic scale. Results are expressed as mean ± standard deviation; error bars are within the points. | ||
Fig. 2 displays the aqueous hydrolysis half-life times and rate constants of 2-TFMP, 4-TFMP and 2-Cl-4-TFMP as a function of pH. The good fitting results (R2 > 0.95) of eqn (3) and (4) to the experimental data indicates that the aqueous hydrolysis of 2-TFMP, 4-TFMP, and 2-Cl-4-TFMP is almost entirely driven by the deprotonated species, the phenolate. When fully deprotonated, each TFMP has its own fundamental hydrolysis rate constant, kphenolate, with k4-TFMP-phenolate (0.62 ± 0.05) h−1 > k2-Cl-4-TFMP-phenolate (0.115 ± 0.004) h−1 > k2-TFMP-phenolate (0.0671 ± 0.0007) h−1 (Table 1). The aqueous hydrolysis reaction rates of these three TFMPs are determined by a combination of the fraction of deprotonation and the fundamental hydrolysis rate constants of the phenolates. The fitting curves of 4-TFMP and 2-Cl-4-TFMP intersect at ∼pH 8, which implies prior to pH 8, the hydrolysis reaction rate of 2-Cl-4-TFMP is faster than that of 4-TFMP. The acid-base dissociation equilibrium of these three TFMPs dictates that a certain amount of phenolate always exists, and thus its hydrolysis can thermodynamically push the acid–base equilibrium forward. However, not all reactions are favorable kinetically. For example, in the pH 6.2 buffer over five days, only 0.5% of 4-TFMP exists in its phenolate form and slow hydrolysis was observed (2.1 × 10−3 ± 0.05 × 10−3 h−1), while 1.2% of 2-TFMP exists in its phenolate form, but no hydrolysis was observed. This is because the fundamental hydrolysis rate of the fully deprotonated 2-TFMP is too slow (nine times slower than 4-TFMP).
| Compound | pKa | Buffer/pH/temperature | Rate constant (h−1) | Half-life (h) |
|---|---|---|---|---|
| N.A. = not available; acalculated from b; bfrom ref. 22; cfrom ref. 20; values without superscripts are experimental results obtained from triplicates in this work; dcalculated from our experimental results across a range of 5 pH. | ||||

|
8.51 | 10 mM phosphate/7/22 °C | 0.0147 ± 0.0005 | 47.0 ± 1.5 |
| 10 mM carbonate/10/22 °C | 0.612 ± 0.008 | 1.13 ± 0.01 | ||
| 10 mM carbonate/10/30 °C | 1.98 ± 0.03 | 0.350 ± 0.005 | ||
| 10 mM carbonate/10.8/22 °C | 0.709 ± 0.010 | 0.976 ± 0.014 | ||
| 50 mM borate/8.5/N.A. | a 0.66 | b 1.1 | ||
| 200 mM borate/10/30 °C | c 2.3 | c 0.30 | ||
| 200 mM phosphate/7/30 °C | c 0.073 | c 9.5 | ||
| k 4-TFMP-phenolate d | 0.621 ± 0.047 | 1.11 ± 0.08 | ||

|
8.12 | 10 mM phosphate/7/22 °C | 0.0051 ± 0.0002 | 137 ± 4 |
| 10 mM carbonate/10/22 °C | 0.0661 ± 0.0008 | 10.5 ± 0.1 | ||
| 10 mM carbonate/10/30 °C | 0.217 ± 0.001 | 3.19 ± 0.01 | ||
| 10 mM carbonate/10.8/22 °C | 0.0674 ± 0.0012 | 10.3 ± 0.2 | ||
| 200 mM borate/10/30 °C | c 0.26 | c 2.7 | ||
| 200 mM phosphate/7/30 °C | c 0.0093 | c 74 | ||
| k 2-TFMP-phenolate d | 0.0671 ± 0.0007 | 10.3 ± 0.1 | ||

|
7.09 | 10 mM phosphate/7/22 °C | 0.0694 ± 0.0006 | 9.99 ± 0.09 |
| 10 mM carbonate/10/22 °C | 0.109 ± 0.004 | 6.36 ± 0.24 | ||
| 10 mM carbonate/10.8/22 °C | 0.121 ± 0.002 | 5.72 ± 0.09 | ||
| 10 mM carbonate/10/30 °C | 0.431 ± 0.004 | 1.61 ± 0.01 | ||
| k 2-Cl-4-TFMP-phenolate d | 0.115 ± 0.004 | 6.02 ± 0.21 | ||
When tested at an elevated temperature of 30 °C in pH 10 solution, we were able to compare our 4-TFMP and 2-TFMP degradation rates with those reported by Sakai and Santi more than 50 years ago.20 We found that our measured rates closely align with theirs (Table 1), despite the different buffers used: their experiments in 200 mM borate buffer, and ours in 10 mM carbonate. To the best of our knowledge, there is no previous literature to compare to our 2-Cl-4-TFMP results.
Transformation products identification
Hydrolysis of 2-, 4-, and 2-Cl-4-TFMP resulted in the formation of the corresponding HBAs (Fig. S5†). Because the hydrolysis rates of 2-Cl-4- and 2-TFMP at room temperature (22 °C) are much slower than that of 4-TFMP, raising the temperature to 30 °C and 40 °C allowed their reactions to progress sufficiently to compare to 4-TFMP. When the reactions neared completion at pH 10 and 40 °C, HBA was formed in near stoichiometric amounts (>92%) (Fig. S5†). When HBA is formed, three equivalents of F− are the other product of the reaction. The fluorine mass balance of TFMPs plus F− (normalized for the 3:1 molar formation ratio) accounted for over 92% of the initial TFMP concentration at each sampling time point (see Fig. S6†). HBA and F− formation below absolute stoichiometric yields for TFMP hydrolysis can be attributed to the formation of other small amount fluorinated transformation products as described below in Fig. 3. A detailed mechanistic diagram including electron pushing arrows can be found in Fig. S7.†
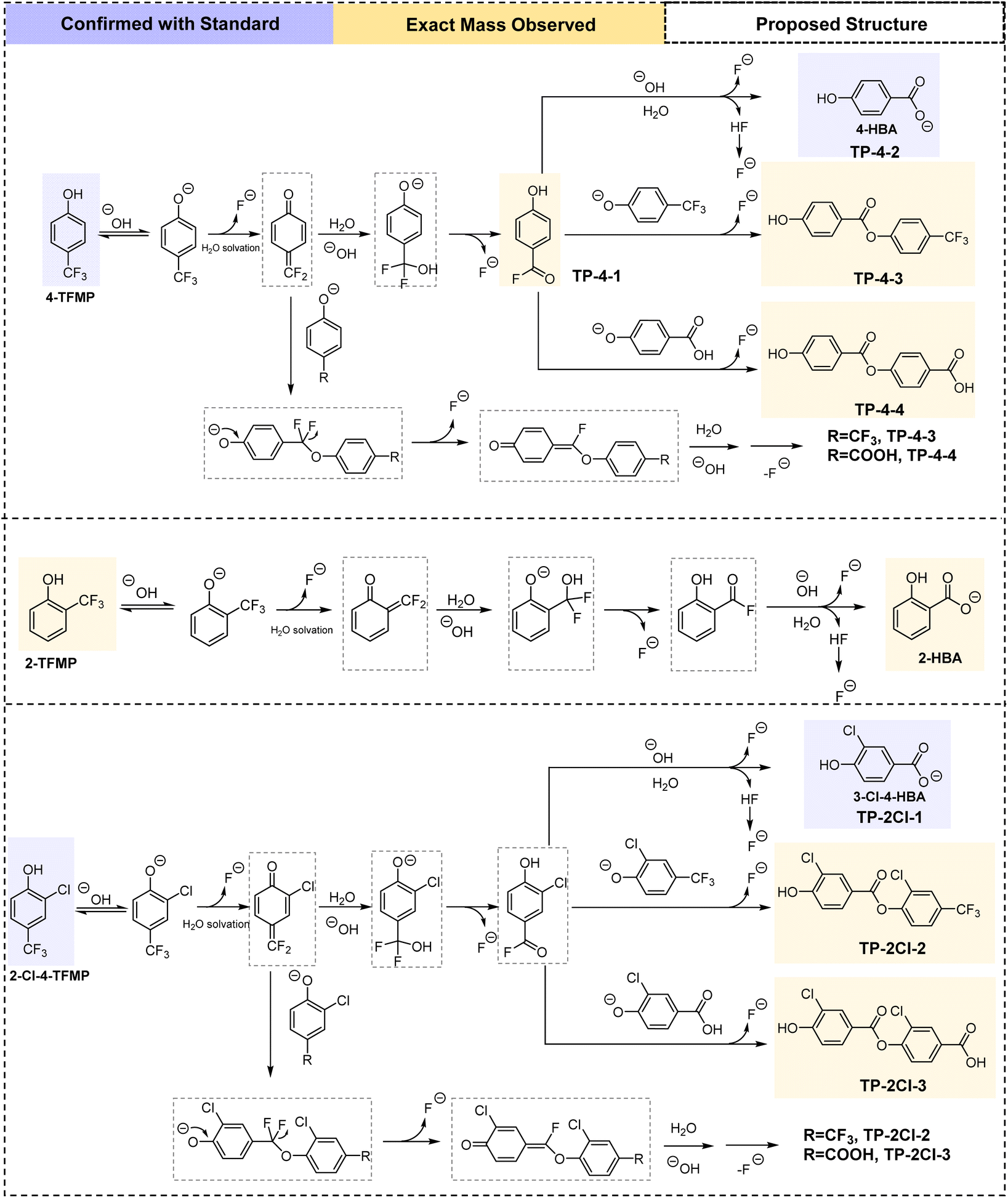 | ||
| Fig. 3 Simplified proposed reaction mechanism for 4-TFMP, 2-TFMP, and 2-Cl-4-TFMP spontaneous aqueous defluorination. A detailed arrow-pushing mechanism can be found in Fig. S7.† | ||
UHPLC-Orbitrap-HRMS and in situ19F NMR were used to search for additional transformation products, either as intermediates of the reaction from TFMPs to HBAs, or other stable products of the reaction. The benzoyl fluoride TP-4-1 was only observed in HRMS (Fig. 3) in the beginning of 4-TFMP hydrolysis experiments at pH 10. The isotope pattern of the benzoyl fluoride aligns with its molecular formula C7H4FO2− (Fig. S3†). While benzoyl fluorides have characteristic positive chemical shifts in 19F NMR, in situ19F NMR did not detect any signal in the positive region (Fig. S1†), likely because its lifetime is shorter than the acquisition time between typical 19F NMR arrays and their steady-state concentrations are very low. The observation of this benzoyl fluoride in the HRMS supports the hypothesized pathway where deprotonated 4-TFMP first eliminates F− and forms a highly reactive conjugated electrophile quinone difluoromethide. This quinone then forms an aryl difluoromethanol (–CF2OH) compound through nucleophilic addition with H2O or OH−, and unstable –CF2OH subsequently eliminates HF to form TP-4-1 (Fig. 3). Analogous benzoyl fluoride transformation products were not observed for other TFMPs, either due to lower formation, or decreased stability of the benzoyl fluorides, resulting in lower concentrations in our samples.
The HRMS of 4-TFMP and 2-Cl-4-TFMP hydrolysis experiments identified novel dimer-like transformation products in addition to HBAs, while for 2-TFMP only 2-HBA was detected (Fig. 3). TP-4-3, TP-4-4, TP-2Cl-2, and TP-2Cl-3 are ester transformation products whose exact mass was identified (Table S4†) and peak areas increased over time during hydrolysis experiments (Fig. 4 and S2†). TP-4-3 was also observed in in situ19F NMR with an increasing peak over time near the 4-TFMP chemical shift (Fig. S1†). At what is labelled t = 0, certain transformation products, such as HBAs and trifluoromethyl-containing phenolic esters (TP-4-3 and TP-2Cl-2) are present, because it is not possible to measure a true time zero for these experiments due to time required for sampling. However, these products were not detected in fresh aqueous stock solutions prepared in ultrapure water used for the experiments nor in the methanol calibration standards, thus we report them as transformation products.
 | ||
| Fig. 4 The plot of peak areas vs. time for transformation products from the aqueous hydrolysis defluorination of 4-TFMP (A) and 2-Cl-4-TFMP (B) at pH 10.8. Note that for TP-2Cl-2 and TP-2Cl-3, the peak areas are plotted on the right y-axis. | ||
We propose the esterification in TFMP hydrolysis only occurs through the deprotonated phenolate groups of either TFMP or HBA reacting via a conjugate addition with quinone difluoromethides or via a nucleophilic addition–elimination mechanism with the benzoyl fluoride intermediate (Fig. 3). Results across a range of pH support that the esterification products only form when the TFMP or HBA phenolates are present (Fig. S2 and Table S3†). 2-TFMP does not form an ester analogous to TP-4-4, because the high pKa of phenol-OH in 2-HBA (salicylic acid) makes them fully protonated (see Table S3†). The lack of the ester analogous to TP-4-3 for 2-TFMP may be due to the steric hindrance caused by the ortho-position of the fluorinated moiety relative to the phenol-OH. Our group's recent study on the photochemistry of 4-TFMP and Zhang et al.'s study on the chlorination of parabens both identified similar coupling products.10,36 The formation of these esterification products may be limited in real-world aqueous environments where concentrations are lower, but it is also possible that other aromatic pollutants or organic matter with nucleophilic group (–NH2, –OH) in the environment may react with the reactive intermediates generated during the hydrolysis.
DFT-assisted mechanistic insights into spontaneous defluorination of TFMP
Based on our experimental findings, we determined that the TFMP-phenolate is the species responsible for driving the aqueous hydrolysis and defluorination of TFMPs. Thus, for mechanistic investigations, we considered scenarios where only the TFMP-phenolate exists. Experimental data at pH 10 was used, and DFT calculations were performed for the TFMP-phenolate only.Investigation of rate-determining step
The initiating step of the spontaneous defluorination of TFMPs involves breaking a strong C–F bond. During this step, the TFMPs undergo dearomatization, leading to an energy increase. We determined that the initiating step is also the rate-determining step (Fig. 5 and S9†), and examined scenarios starting from the TFMP-phenolate and forming hydrated intermediates with varying numbers of explicit water molecules (Fig. 5 and S9†). Discussion on how solvation changes the free energy of activation (ΔG‡, kcal mol−1) is given in the ESI-11.† | ||
| Fig. 5 DFT pathways with the minimum energy barriers for 4-, 2-Cl-4-, 2-TFMP aqueous spontaneous defluorination reactions rate-determining steps under the hybrid solvation model. | ||
The DFT-calculated ΔG‡ for the hydrolytic defluorination of 4-TFMP (21.2 kcal mol−1) and 2-TFMP (22.6 kcal mol−1) closely matched the experimental values of 22.6 kcal mol−1 and 23.5 kcal mol−1, respectively (Table 2). The calculated ΔG‡ for 2-Cl-4-TFMP falls within the standard deviation range of its experimentally determined ΔG‡. 4-TFMP is the most reactive when fully deprotonated in our laboratory experiments, and has the lowest ΔG‡ of the three TFMPs determined both experimentally and theoretically. 2-Cl-4-TFMP is more reactive than 2-TFMP in hydrolysis, however, based on our ΔG‡ determination, we cannot attribute the faster reactivity to a lower energy rate-determining step. This is because of the larger relative error of our experimental ΔG‡ of 2-Cl-4-TFMP, compared to the other TFMPs. DFT calculations reported the highest ΔG‡ for 2-Cl-4-TFMP, but this does not align with our experimental observations, which suggests the current hybrid solvation model used may be insufficient to fully capture the real conditions. The rate-determining step transition state structures for three TFMPs shared similar key features: stretched C–F bonds, hydrogen-bond interactions between explicit water molecules and the phenolate, and stabilization of the transition state through interactions between the water O–H and the departing F− (Fig. 5).
| TFMP-phenolate | ΔG‡exp | R 2 | Minimum ΔG‡DFT scenario n(H2O) | ΔG‡DFT |
|---|---|---|---|---|
| 2-TFMP | 23.5 ± 1.6 | 0.995 | 6 | 22.6 |
| 4-TFMP | 22.6 ± 1.8 | 0.999 | 5 | 21.2 |
| 2-Cl-4-TFMP | 22.8 ± 3.9 | 0.971 | 5 | 25.4 |
Investigation of TFMP-phenolate electronic properties
In the chemical structure of TFMPs, the –CF3 substituents impart environmentally relevant phenol-OH pKa values due to its strong electron-withdrawing properties. Then, the deprotonated phenolates promote the cleavage of a C–F bond, releasing the first F− of ortho and para –CF3, giving dearomatized quinone difluoromethides. This process is similar to the environmental transformation of the insecticide DDT into DDE via carbanion-mediated chloride elimination.37 In contrast, the negative charge does not delocalize to the meta position, rendering the meta –CF3 group resistant to β-elimination, as seen with 3-TFMP, which resists hydrolysis even under pH 10 at 40 °C. Additional EWGs on the aromatic ring can delocalize the negative charge, thereby decreasing the potential of β-elimination and defluorination. NPA determined that the β-carbanion in 2-Cl-4-TFMP has a charge of −0.264, lower (in absolute value) than −0.288 observed in 4-TFMP (Fig. 6A), aligning well with lower observed reactivity of 2-Cl-4-TFMP compared to 4-TFMP. Bond dissociation energy (BDE) calculations further reveal that 4-TFMP and 2-Cl-4-TFMP have near-identical C–F BDEs (120.6 kcal mol−1), whereas 2-TFMP has a higher BDE of 122.3 kcal mol−1, corresponding to its consistently lowest reactivity. | ||
| Fig. 6 (A) Natural population analysis (NPA) of –CF3's β-carbons and the bond dissociation energy of C–F bonds in TFMPs (deprotonated, phenolates); CYLview was used to visualize the structures.38. (B) Downhill rearomatization of quinone difluoromethide intermediate for 4-TFMP. | ||
Quinone difluoromethide intermediate rearomatization
From the rate-determining step onward, the energy profiles of 2-, 4-, and 2-Cl-4-TFMP are generally downhill, involving barrierless reactions and reactions with low energy barriers that lead to the rearomatization of the reactive quinone difluoromethide intermediates (Fig. 6B and S10†).In Fig. 6B, the quinone difluoromethide intermediate IM-4-1 can undergo a barrierless reaction with OH− to form the same IM-4-2 (ΔG = −39.1 kcal mol−1, see potential energy surface scan results in Fig. S11†), a rearomatization process that is both thermodynamically and kinetically favorable. For 2-Cl-4-TFMP and 2-TFMP, the barrierless reaction between OH− and their corresponding quinone difluoromethides is also the more favored pathway (illustrated in Fig. S10,† with potential energy surface scan results shown in Fig. S11†). Combined with expected high OH− concentration in the basic system, these quinone difluoromethides are highly reactive electrophiles, rapidly captured by OH− or other high concentration nucleophiles such as H2O or deprotonated TFMPs substrates, making them undetectable via regular HRMS or 19F NMR.
For the intermediate IM-4-2, the unstable –CF2OH structure readily eliminates HF via a hydrogen-bonding network. Adding explicit water molecules to the TS of this HF-elimination reduced ΔG‡ from 24.8 to 13.3 kcal mol−1, consistent with prior studies.26 Alternatively, IM-4-2 can undergo intramolecular proton transfer to form IM-4-3, where the negatively charged oxygen in –CF2O− facilitates the leaving of F− with ΔG‡ of only 8.2 kcal mol−1, making it highly favorable kinetically. Similar scenarios are observed for 2-Cl-4-TFMP and 2-TFMP. After the removal of first two F−, all three TFMPs generate the corresponding (deprotonated) benzoyl fluorides, which reacts either with H2O/OH− to eliminate the last F− and to form benzoic acids or with other nucleophiles such as phenol-O− to form dimer-like transformation products.
Defluorination compared to other PFAS
In this study, para and ortho substituted TFMPs (i.e., 2-, 4-, 2-Cl-4-TFMP) achieved complete defluorination, while 3-TFMP did not. In the rate-determining step, the negative charge of the phenolate is delocalized through conjugation, driving β-elimination of a single F− to form reactive electrophilic α,β-unsaturated ketones, which is not possible for 3-TFMP. Then, the reactive α,β-unsaturated ketones immediately undergo further reactions to eliminate the second and the third F−.Although one carbon involved in the β-elimination defluorination reaction of TFMPs is an aryl-carbon (sp2 hybridized), and many PFAS molecules are composed of alkyl-carbons (sp3 hybridized), similar β-elimination defluorination mechanisms are not uncommon in PFAS chemistry and usually occur in the early stages of the overall reaction. For instance, perfluorooctanoic acid (PFOA) can undergo decarboxylation in polar aprotic solvents, forming a perfluoroalkyl carbanion that drives β-elimination of an adjacent C–F bond to give a perfluoroalkene.28 DFT calculations revealed a ΔG‡ of 19.5 kcal mol−1 for this β-elimination step,28 comparable to the ΔG‡ values observed for TFMPs in our study. For polyfluorinated compounds, it has been reported that the presence of easily removable H or Cl atoms can indirectly trigger defluorination.39,40 Multiple electron-withdrawing C–F bonds can weaken other chemical bonds, facilitating the dissociation. In this work, the phenol-O–H bonds in TFMPs are more readily dissociated than the equivalent non-fluorinated methylphenols (cresols). For example, the pKa of 4-TFMP is 8.51 compared to 10.25 for 4-methylphenol, due to the trifluoromethyl group stabilizing the carbanion. For example, 6:2, 8:2, 10:2 fluorotelomer carboxylic acid (n:2 FTCA, Cn−1F2n−1CF2CH2COOH, n = 6/8/10) can transform into n:2 fluorotelomer unsaturated carboxylic acid (n:2 FTUCA, Cn−1F2n−1CF![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHCOOH, n = 6/8/10) biotically or abiotically.41,42 This abiotic transformation is facilitated by a base,41,42 likely through the deprotonation of a C–H adjacent to the carboxyl group, leading to β-elimination and the formation of an α,β-unsaturated carbonyl compound. Similarly, 8:2 fluorotelomer aldehyde (8:2 FTAL, C7F15CF2CH2CHO) is unstable in water, and loses HF to produce the 8:2 fluorotelomer α,β-unsaturated aldehyde (8:2 FTUAL, C7F15CFCHCHO).43 A recent study suggest that an unsaturated perfluorinated compound E-perfluoro-4-methylpent-2-enoic acid [(CF3)2CFCFCFCOOH] can undergo microbial defluorination, where similar base-facilitated β-elimination defluorination contributes as part of the mechanism.39
CHCOOH, n = 6/8/10) biotically or abiotically.41,42 This abiotic transformation is facilitated by a base,41,42 likely through the deprotonation of a C–H adjacent to the carboxyl group, leading to β-elimination and the formation of an α,β-unsaturated carbonyl compound. Similarly, 8:2 fluorotelomer aldehyde (8:2 FTAL, C7F15CF2CH2CHO) is unstable in water, and loses HF to produce the 8:2 fluorotelomer α,β-unsaturated aldehyde (8:2 FTUAL, C7F15CFCHCHO).43 A recent study suggest that an unsaturated perfluorinated compound E-perfluoro-4-methylpent-2-enoic acid [(CF3)2CFCFCFCOOH] can undergo microbial defluorination, where similar base-facilitated β-elimination defluorination contributes as part of the mechanism.39
Conclusions
Here, we report a comprehensive investigation into the hydrolysis kinetics and defluorination mechanisms of TFMPs in the aqueous phase. Hydrolysis experiments were conducted in pH 6.2 to 10.8 buffer to evaluate the reactivity of 2-, 3-, 4-, 2-Cl-4-TFMP. Among these, 2-, 4-, and 2-Cl-4-TFMP showed significant spontaneous defluorination in aqueous solutions, while no hydrolysis of 3-TFMP was observed. The reactivity of TFMPs was strongly influenced by the deprotonation state of the phenol-OH. When 2-, 4-, and 2-Cl-4-TFMP TFMPs are in their fully deprotonated phenolate form, they have relative reactivities following the order 4-TFMP > 2-Cl-4-TFMP > 2-TFMP.HBAs and F− were the major products for the three TFMPs, as confirmed by their molar yields and fluorine mass balance. UHPLC-Orbitrap-HRMS and in situ19F NMR identified new transformation products, including a benzoyl fluoride intermediates for 4-TFMP and dimer-like products for 4-TFMP and 2-Cl-4-TFMP, offering additional insights into the reaction mechanisms.
The key rate-determining step for defluorination involved β-elimination, likely via an E1cb mechanism, initiated by the deprotonated phenolate. DFT calculations provided detailed insights into this process, highlighting the critical role of water molecule solvation in stabilizing the phenolate and facilitating β-elimination. The reactivity was influenced by both the C–F bond BDE and the negative charge distribution on the phenolate of the TFMPs. Additionally, DFT calculations revealed that following the rate-determining step, the rearomatization of the reactive quinone difluoromethide intermediate was highly favorable both thermodynamically and kinetically. These findings enhance our understanding of defluorination pathways for TFMPs and provide insights into the defluorination chemistry of other PFAS. This may be used to design aryl-CF3 compounds that favour defluorination in the environment, thus minimizing their environmental impact. As aryl-CF3 contaminants are more and more incorporated in the environmental analysis, our research findings also indicated that if TFMPs are to be targeted as analytes, it is important to store samples at low pH and avoid using base during sample preparation to minimize loss via hydrolysis.
Data availability
Data not included in the main text are included in the ESI.† Cartesian coordinates of deprotonated TFMPs determined by DFT are uploaded as a separate ESI File.†Author contributions
ZG: conceptualization, investigation, methodology, visualization, writing – original draft, writing – review & editing; GWT: investigation, methodology, visualization, writing – original draft, writing – review & editing; JC: investigation, writing – review & editing; SJ: conceptualization, funding acquisition supervision, writing – review & editing.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
Funding for this research was provided by the National Science and Engineering Research Council (NSERC RGPIN-2023-04369) and the Faculty of Science at the University of Alberta. Z. G. was partially supported by Future Energy Systems (FES-T10-Q05) at the University of Alberta, funded through the Canada First Research Excellence Fund. G. W. T. received support from an NSERC Undergraduate Student Research Award and the Lloyd and Margaret Cooley Memorial Studentship in Analytical Chemistry. We gratefully acknowledge access to the computing facilities provided by Digital Research Alliance of Canada (https://alliancecan.ca/en). The authors thank Joseph Utomo, Béla Reiz, and Angelina Morales-Izquierdo from the University of Alberta Department of Chemistry Mass Spectrometry Facility for their assistance with UHPLC-Orbitrap-HRMS and direct infusion-ESI-HRMS, Mark Miskolzie from the University of Alberta Department of Chemistry NMR Facility for his assistance with in situ NMR, and Christine Le (York University) and Bowen Yang (University of Alberta) for useful discussions.References
- Y. Ogawa, E. Tokunaga, O. Kobayashi, K. Hirai and N. Shibata, Current Contributions of Organofluorine Compounds to the Agrochemical Industry, iScience, 2020, 23(9), 101467 CrossRef CAS PubMed.
- M. Inoue, Y. Sumii and N. Shibata, Contribution of Organofluorine Compounds to Pharmaceuticals, ACS Omega, 2020, 5(19), 10633–10640 CrossRef CAS PubMed.
- N. A. Meanwell, Fluorine and Fluorinated Motifs in the Design and Application of Bioisosteres for Drug Design, J. Med. Chem., 2018, 61(14), 5822–5880 CrossRef CAS PubMed.
- M. Patel, R. Kumar, K. Kishor, T. Mlsna, C. U. Pittman Jr and D. Mohan, Pharmaceuticals of Emerging Concern in Aquatic Systems: Chemistry, Occurrence, Effects, and Removal Methods, Chem. Rev., 2019, 119(6), 3510–3673 CrossRef CAS PubMed.
- N. Donley, C. Cox, K. Bennett, A. M. Temkin, D. Q. Andrews and O. V. Naidenko, Forever Pesticides: A Growing Source of PFAS Contamination in the Environment, Environ. Health Perspect., 2024, 132(7), 075003 CrossRef PubMed.
- Z. Wang, A. M. Buser, I. T. Cousins, S. Demattio, W. Drost and O. Johansson, et al., A New OECD Definition for Per- and Polyfluoroalkyl Substances, Environ. Sci. Technol., 2021, 55(23), 15575–15578 CrossRef CAS PubMed.
- OECD, Reconciling Terminology of the Universe of Per- and Polyfluoroalkyl Substances: Recommendations and Practical Guidance, [Internet], Organisation for Economic Co-operation and Development, Paris, 2021, [cited 2024 Jul 17], available from: https://www.oecd-ilibrary.org/environment/reconciling-terminology-of-the-universe-of-per-and-polyfluoroalkyl-substances_e458e796-en.
- S. Tisler, F. Zindler, F. Freeling, K. Nödler, L. Toelgyesi and T. Braunbeck, et al., Transformation Products of Fluoxetine Formed by Photodegradation in Water and Biodegradation in Zebrafish Embryos (Danio rerio), Environ. Sci. Technol., 2019, 53(13), 7400–7409 CrossRef CAS PubMed.
- A. P. Bhat, T. F. Mundhenke, Q. T. Whiting, A. A. Peterson, W. C. K. Pomerantz and W. A. Arnold, Tracking Fluorine during Aqueous Photolysis and Advanced UV Treatment of Fluorinated Phenols and Pharmaceuticals Using a Combined 19F-NMR, Chromatography, and Mass Spectrometry Approach, ACS Environ. Au, 2022, 2(3), 242–252 CrossRef CAS PubMed.
- Z. Guo, A. A. Attar, Q. Qiqige, R. J. Lundgren and S. Joudan, Photochemical Formation of Trifluoroacetic Acid: Mechanistic Insights into a Fluoxetine-Related Aryl-CF3 Compound, Environ. Sci. Technol., 2025, 59(2), 1367–1377 CrossRef CAS PubMed.
- H. P. H. Arp, A. Gredelj, J. Glüge, M. Scheringer and I. T. Cousins, The Global Threat from the Irreversible Accumulation of Trifluoroacetic Acid (TFA), Environ. Sci. Technol., 2024, 58(45), 19925–19935 CrossRef CAS PubMed.
- S. Joudan, A. O. D. Silva and C. J. Young, Insufficient evidence for the existence of natural trifluoroacetic acid, Environ. Sci.: Processes Impacts, 2021, 23(11), 1641–1649 RSC.
- M. W. Lam, C. J. Young and S. A. Mabury, Aqueous Photochemical Reaction Kinetics and Transformations of Fluoxetine, Environ. Sci. Technol., 2005, 39(2), 513–522 CrossRef CAS PubMed.
- A. C. Altamura, A. R. Moro and M. Percudani, Clinical Pharmacokinetics of Fluoxetine, Clin. Pharmacokinet., 1994, 26(3), 201–214 CrossRef CAS PubMed.
- T. Chonova, S. Ruppe, I. Langlois, D. S. Griesshaber, M. Loos and M. Honti, et al., Unveiling industrial emissions in a large European river: Insights from data mining of high-frequency measurements, Water Res., 2025, 268, 122745 CrossRef CAS PubMed.
- D. Vialaton, D. Baglio, A. Paya-Perez and C. Richard, Photochemical transformation of acifluorfen under laboratory and natural conditions, Pest Manage. Sci., 2001, 57(4), 372–379 CrossRef CAS PubMed.
- L. Chen, T. Cai and Q. Wang, Characterization of Fluoroglycofen Ethyl Degradation by Strain Mycobacterium phocaicum MBWY-1, Curr. Microbiol., 2011, 62(6), 1710–1717 CrossRef CAS PubMed.
- S. K. Chakraborty, A. Bhattacharyya and A. Chowdhury, Degradation of Oxyfluorfen by Azotobacter chroococcum (Beijerink), Bull. Environ. Contam. Toxicol., 2002, 69(2), 203–209 CrossRef CAS PubMed.
- R. G. Jones, Ortho and Para Substituted Derivatives of Benzotrifluoride, J. Am. Chem. Soc., 1947, 69(10), 2346–2350 CrossRef CAS PubMed.
- T. T. Sakai and D. V. Santi, Hydrolysis of hydroxybenzotrifluorides and fluorinated uracil derivatives. General mechanism for carbon-fluorine bond labilization, J. Med. Chem., 1973, 16(10), 1079–1084 CrossRef CAS PubMed.
- D. A. Ellis and S. A. Mabury, The Aqueous Photolysis of TFM and Related Trifluoromethylphenols. An Alternate Source of Trifluoroacetic Acid in the Environment, Environ. Sci. Technol., 2000, 34(4), 632–637 CrossRef CAS.
- D. A. Jackson and S. A. Mabury, Environmental properties of pentafluorosulfanyl compounds: Physical properties and photodegradation, Environ. Toxicol. Chem., 2009, 28(9), 1866–1873 CrossRef CAS PubMed.
- A. Manfrin, A. Hänggli, J. van den Wildenberg and K. McNeill, Substituent Effects on the Direct Photolysis of Benzotrifluoride Derivatives, Environ. Sci. Technol., 2020, 54(18), 11109–11117 CrossRef CAS PubMed.
- D. C. Thompson, K. Perera and R. London, Spontaneous hydrolysis of 4-trifluoromethylphenol to a quinone methide and subsequent protein alkylation, Chem.-Biol. Interact., 2000, 126(1), 1–14 CrossRef CAS PubMed.
- H. Amii and K. Uneyama, C−F Bond Activation in Organic Synthesis, Chem. Rev., 2009, 109(5), 2119–2183 CrossRef CAS PubMed.
- Y. Zhang, A. Moores, J. Liu and S. Ghoshal, New Insights into the Degradation Mechanism of Perfluorooctanoic Acid by Persulfate from Density Functional Theory and Experimental Data, Environ. Sci. Technol., 2019, 53(15), 8672–8681 CrossRef CAS PubMed.
- Y. Zhang, J. Liu, S. Ghoshal and A. Moores, Density Functional Theory Calculations Decipher Complex Reaction Pathways of 6:2 Fluorotelomer Sulfonate to Perfluoroalkyl Carboxylates Initiated by Hydroxyl Radical, Environ. Sci. Technol., 2021, 55(24), 16655–16664 CrossRef CAS PubMed.
- B. Trang, Y. Li, X. S. Xue, M. Ateia, K. N. Houk and W. R. Dichtel, Low-temperature mineralization of perfluorocarboxylic acids, Science, 2022, 377(6608), 839–845 CrossRef CAS PubMed.
- Z. Chen, J. Chen, S. Tan, Z. Yang and Y. Zhang, Dechlorination Helps Defluorination: Insights into the Defluorination Mechanism of Florfenicol by S-nZVI and DFT Calculations on the Reaction Pathways, Environ. Sci. Technol., 2024, 58(5), 2542–2553 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, et al.Gaussian 16 Rev. C.01, Wallingford, CT, 2016 Search PubMed.
- E. Cancès, B. Mennucci and J. Tomasi, A new integral equation formalism for the polarizable continuum model: Theoretical background and applications to isotropic and anisotropic dielectrics, J. Chem. Phys., 1997, 107(8), 3032–3041 CrossRef.
- N. Mardirossian and M. Head-Gordon, How Accurate Are the Minnesota Density Functionals for Noncovalent Interactions, Isomerization Energies, Thermochemistry, and Barrier Heights Involving Molecules Composed of Main-Group Elements?, J. Chem. Theory Comput., 2016, 12(9), 4303–4325 CrossRef CAS PubMed.
- Y. Zhao and D. G. Truhlar, The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals, Theor. Chem. Acc., 2008, 120(1), 215–241 Search PubMed.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions, J. Phys. Chem. B, 2009, 113(18), 6378–6396 CrossRef CAS PubMed.
- W. Qin, K. Guo, C. Chen and J. Fang, Differences in the Reaction Mechanisms of Chlorine Atom and Hydroxyl Radical with Organic Compounds: From Thermodynamics to Kinetics, Environ. Sci. Technol., 2024, 58(40), 17886–17897 CrossRef CAS PubMed.
- Z. Zhang, K. P. Reber, N. Okwor, P. D. Gujarati, M. Vollmuth and L. Zhang, et al., Stable isotope labelling to elucidate ring cleavage mechanisms of disinfection by-product formation during chlorination of phenols, Nat. Water, 2025, 3(2), 222–230 CrossRef CAS.
- R. P. Schwarzenbach, Environmental Organic Chemistry, Wiley, 3rd edn, 2017 Search PubMed.
- Claude Legault, CYLview20, Internet, Université de Sherbrooke, 2020, Available from: http://www.cylview.org.
- Y. Yu, F. Xu, W. Zhao, C. Thoma, S. Che and J. E. Richman, et al., Electron bifurcation and fluoride efflux systems implicated in defluorination of perfluorinated unsaturated carboxylic acids by Acetobacterium spp, Sci. Adv., 2024, 10(29), eado2957 CrossRef CAS PubMed.
- S. Che, B. Jin, Z. Liu, Y. Yu, J. Liu and Y. Men, Structure-Specific Aerobic Defluorination of Short-Chain Fluorinated Carboxylic Acids by Activated Sludge Communities, Environ. Sci. Technol. Lett., 2021, 8(8), 668–674 CrossRef CAS PubMed.
- J. Liu and S. Mejia Avendaño, Microbial degradation of polyfluoroalkyl chemicals in the environment: A review, Environ. Int., 2013, 61, 98–114 CrossRef CAS PubMed.
- M. Loewen, T. Halldorson, F. Wang and G. Tomy, Fluorotelomer Carboxylic Acids and PFOS in Rainwater from an Urban Center in Canada, Environ. Sci. Technol., 2005, 39(9), 2944–2951 CrossRef CAS PubMed.
- J. W. Martin, S. A. Mabury and P. J. O'Brien, Metabolic products and pathways of fluorotelomer alcohols in isolated rat hepatocytes, Chem.-Biol. Interact., 2005, 155(3), 165–180 CrossRef CAS PubMed.
- J. J. Fifen, Z. Dhaouadi and M. Nsangou, Revision of the Thermodynamics of the Proton in Gas Phase, J. Phys. Chem. A, 2014, 118(46), 11090–11097 CrossRef CAS PubMed.
- J. R. Pliego Jr and J. M. Riveros, Gibbs energy of solvation of organic ions in aqueous and dimethyl sulfoxide solutions, Phys. Chem. Chem. Phys., 2002, 4(9), 1622–1627 RSC.
- M. D. Tissandier, K. A. Cowen, W. Y. Feng, E. Gundlach, M. H. Cohen and A. D. Earhart, et al., The Proton's Absolute Aqueous Enthalpy and Gibbs Free Energy of Solvation from Cluster-Ion Solvation Data, J. Phys. Chem. A, 1998, 102(40), 7787–7794 CrossRef CAS.
- A. E. Reed, R. B. Weinstock and F. Weinhold, Natural population analysis, J. Chem. Phys., 1985, 83(2), 735–746 CrossRef CAS.
- S. P. Ozkorucuklu, J. L. Beltrán, G. Fonrodona, D. Barrón, G. Alsancak and J. Barbosa, Determination of Dissociation Constants of Some Hydroxylated Benzoic and Cinnamic Acids in Water from Mobility and Spectroscopic Data Obtained by CE-DAD, J. Chem. Eng. Data, 2009, 54(3), 807–811 CrossRef CAS.
- CRC Handbook of Chemistry and Physics : A Ready-Reference Book of Chemical and Physical Data. 2018th–2019th, 99th edn, ed. J. R. Rumble, CRC Press, Taylor & Francis Group, 2018 Search PubMed.
- A. Arcelli and C. Concilio, Polyelectrolyte effects exerted by poly(ethyleneimine) on the ionization constant of substituted phenols, J. Chem. Soc., Perkin Trans. 2, 1989,(7), 887–892 RSC.
- L. Martínez, R. Andrade, E. G. Birgin and J. M. Martínez, PACKMOL: A package for building initial configurations for molecular dynamics simulations, J. Comput. Chem., 2009, 30(13), 2157–2164 CrossRef PubMed.
- A. K. Rappe, C. J. Casewit, K. S. Colwell, W. A. I. Goddard and W. M. Skiff, UFF, a full periodic table force field for molecular mechanics and molecular dynamics simulations, J. Am. Chem. Soc., 1992, 114(25), 10024–10035 CrossRef CAS.
- C. Bannwarth, S. Ehlert and S. Grimme, GFN2-xTB—An Accurate and Broadly Parametrized Self-Consistent Tight-Binding Quantum Chemical Method with Multipole Electrostatics and Density-Dependent Dispersion Contributions, J. Chem. Theory Comput., 2019, 15(3), 1652–1671 CrossRef CAS PubMed.
- C. Bannwarth, E. Caldeweyher, S. Ehlert, A. Hansen, P. Pracht and J. Seibert, et al., Extended tight-binding quantum chemistry methods, WIREs Comput. Mol. Sci., 2021, 11(2), e1493 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Ref. 44–54. See DOI: https://doi.org/10.1039/d4em00739e |
| ‡ Z. G. and G. W. T. contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |