
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Barrier layer design reduces top electrode ion migration in perovskite solar cells†
Saivineeth
Penukula
 a,
Megh N.
Khanal
b,
Mohin
Sharma
c,
Mritunjaya
Parashar
c,
Ross A.
Kerner
d,
Min
Chen
d,
Melissa A.
Davis
d,
Rafikul Ali
Saha
e,
Eduardo
Solano
f,
Maarten B. J.
Roeffaers
e,
Joseph J.
Berry
dgh,
Joseph M.
Luther
dh,
Julian A.
Steele
ij,
Axel
Palmstrom
d,
Vincent R.
Whiteside
b,
Bibhudutta
Rout
c,
Ian R.
Sellers
b and
Nicholas
Rolston
*a
a,
Megh N.
Khanal
b,
Mohin
Sharma
c,
Mritunjaya
Parashar
c,
Ross A.
Kerner
d,
Min
Chen
d,
Melissa A.
Davis
d,
Rafikul Ali
Saha
e,
Eduardo
Solano
f,
Maarten B. J.
Roeffaers
e,
Joseph J.
Berry
dgh,
Joseph M.
Luther
dh,
Julian A.
Steele
ij,
Axel
Palmstrom
d,
Vincent R.
Whiteside
b,
Bibhudutta
Rout
c,
Ian R.
Sellers
b and
Nicholas
Rolston
*a
aRenewable Energy Materials and Devices Lab, School of Electrical, Computer and Energy Engineering (ECEE), Arizona State University, Tempe, Arizona 85281, USA. E-mail: nicholas.rolston@asu.edu
bDepartment of Electrical Engineering, University at Buffalo SUNY, Buffalo, 230 Davis Hall, New York 14260, USA
cDepartment of Physics, Ion Beam Laboratory, University of North Texas, Denton, Texas 76203, USA
dNational Renewable Energy Laboratory, Golden, Colorado 80401, USA
ecMACS, Department of Microbial and Molecular Systems, KU Leuven, Celestijnenlaan 200F, 3001 Heverlee, Belgium
fNCD-SWEET Beamline, ALBA Synchrotron Light Source, 08290 Cerdanyola del Vallès, Barcelona, Spain
gDepartment of Physics, University of Colorado Boulder, Boulder, Colorado 80309, USA
hRenewable and Sustainable Energy Institute, University of Colorado Boulder, Boulder, Colorado 80309, USA
iAustralian Institute of Bioengineering and Nanotechnology, The University of Queensland, St Lucia, QLD 4072, Australia
jSchool of Mathematics and Physics, The University of Queensland, Brisbane, QLD 4072, Australia
First published on 6th May 2025
Abstract
We report on an examination of mobile ion concentration (N0) in perovskite solar cells (PSCs) as a function of temperature and device architecture. We find that lower initial N0 is correlated to devices with higher thermal performance through in situ measurements up to 450 K. Changes in N0 are observed upon thermal aging and are impacted by the changes made at the electron collecting interface. We examine the extent to which various top electrode materials (Ag, Au, carbon) impact N0 as well as the effects of tin oxide (SnO2) or an ozone-nucleated SnO2 (O3–SnO2) barrier layer between the ETL and top electrode. Upon thermal aging, we confirm the involvement of Ag ion diffusion through the ETL dependent on the device details. We are able to quantify the degree to which Ag ions migrate or are blocked from migrating into the underlying device layers in the PSC stack. X-ray scattering shows improved suppression of the degradation products formed in the bulk of the perovskite when a blocking layer, particularly the O3–SnO2 is employed.
Broader contextIon migration is one of the important factors that affect the operational lifetime and stability of perovskite solar cells (PSCs). Even though different methodologies have been employed to show the effects of ion migration, the techniques are varied and often qualitative. Furthermore, there is no simple, quantitative method that provides a consistent correlation to the stability of PSCs. This work shows that mobile ion concentration (N0) can be correlated to PSC stability in state-of-the-art devices. N0 is a metric that can serve as a consistent and straightforward approach to quantify ion migration-related degradation modes on PSC stability. |
Introduction
Metal-halide perovskite (MHP) solar cells have achieved significant commercial interest in the renewable energy market based on rapid efficiency improvements1,2 achieving lab efficiencies of 26.7%.3 Additional advantages include the use of earth-abundant precursors, affordable manufacturing, and tunability of optoelectronic properties.4–7 However, the ion migration and chemical reactions observed under the influence of environmental stressors such as heat and light are a concern.8,9 The pace of perovskite solar cell (PSC) advances has made it difficult for field testing studies to keep pace with reports in excess of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 h limited to older devices and architectures.10 Limited field lifetimes (<1 year) for the majority of PSC modules tested by the perovskite PV accelerator for commercializing technologies (PACT)11 indicates this challenge of demonstrating sufficient reliability to bring PSCs to market. This rapid development cycle creates a need for more rapid testing methods and metrics as well as mechanistic insight related to stability and reliability issues in PSCs. Of the variety of mechanisms believed to be responsible for a change in efficiency in operation, ion migration is postulated to be a primary cause of this degradation via phase separation and reactions with charge transport layers. While these correlations have been identified, the mechanism that ultimately leads to electronic losses and irreversible corrosion of electrodes is still being revealed.12–15 Here we undertake studies to examine changes in mobile species and how these relate to device stability. Specifically, we use our previously reported measurement approach to study the change in mobile ion concentration (N0). These measurements are sensitive to mobile charges induced directly or indirectly by mobile ions and chemical reactions, providing a basis from which to see how this changes as devices are stressed, and subsequently examine the specific origins of degradation for a given device architecture.
000 h limited to older devices and architectures.10 Limited field lifetimes (<1 year) for the majority of PSC modules tested by the perovskite PV accelerator for commercializing technologies (PACT)11 indicates this challenge of demonstrating sufficient reliability to bring PSCs to market. This rapid development cycle creates a need for more rapid testing methods and metrics as well as mechanistic insight related to stability and reliability issues in PSCs. Of the variety of mechanisms believed to be responsible for a change in efficiency in operation, ion migration is postulated to be a primary cause of this degradation via phase separation and reactions with charge transport layers. While these correlations have been identified, the mechanism that ultimately leads to electronic losses and irreversible corrosion of electrodes is still being revealed.12–15 Here we undertake studies to examine changes in mobile species and how these relate to device stability. Specifically, we use our previously reported measurement approach to study the change in mobile ion concentration (N0). These measurements are sensitive to mobile charges induced directly or indirectly by mobile ions and chemical reactions, providing a basis from which to see how this changes as devices are stressed, and subsequently examine the specific origins of degradation for a given device architecture.
Recent work has shown that the top metal electrodes in PSCs spontaneously react15 or can react under electrochemical16,17 or photochemical stress.18 One strategy to prevent reactions and the formation of mobile ions is to employ a physical barrier layer.19 However, this barrier layer must be of very high quality (i.e. chemically stable and pinhole-free) to be effective. The best barrier layers are often created by atomic layer deposition (ALD) of metal oxides such as SnO2 on top of the fullerene-based electron transport layer (ETL) in the p-i-n structure of PSCs.20 The barrier properties of ALD oxides are further enhanced by ozone-nucleation (O3) of the SnO2 by exposing the C60 layer to ozone through an ultrathin (∼5 nm) non-conformally grown SnO2 which functionalizes C60 to better nucleate subsequent ALD growth and enable more robust internal barriers in PSCs that can prevent chemical reactions and block the motion of ions, water vapor, and solvents.20 The deposition of the ozone-nucleated barrier layers does not induce any new degradation modes observed under light and heat testing with T90 lifetimes of 500 h and 575 h for PSCs with SnO2 and O3–SnO2 layers, respectively, at 65 °C under approximately 1-sun illumination and quasi-maximum power point (quasi-MPP) set by a static load resistor (ISOS-L2-2I).20 Furthermore, this O3 nucleation approach was also shown to reduce the water-vapor transmission rate through the barrier layer and reduce gas, solvent, and halide migration, in turn enhancing PSC stability compared to control devices20 as well as the mechanical robustness of PSCs compared to SnO2.21
Previous work demonstrated that ion migration in PSCs can be quantified in terms of N0, which is defined as the number of mobile ions present in the MHP, whereby a significant variation in N0 (5 orders of magnitude) was observed across different samples depending on the composition and chemistry of the top electrode.22 The reasons for the variation were not well understood at the time. Here, we leverage additional characterization such as Rutherford Backscattering Spectrometry (RBS), a powerful, fast, and non-destructive technique for quantifying elemental motion throughout a PSC. Previous work employed RBS to quantify the depth profile of Pb and I in a film stack comprising TiO2/MAPbI3.23 More recently, RBS has been utilized to study the radiation hardness and elemental migration, where the RBS results clearly showed the signs of elemental migration of species such as iodine diffusing from the perovskite to the top electrode.24 In this work, RBS is used to demonstrate that the migration of Ag ions (and of iodine out of the MHP) can be largely mitigated with a thin ALD SnO2 barrier layer between the C60 layer and the top Ag electrode. This work also elaborates on several other considerations and implications of N0 that connect to material and device stability, including PSC thermal stability and bulk structural stability.
Results and discussion
Impact of top electrode chemistry on ion migration
We begin by testing our hypothesis that metal electrodes are contributing/impacting N0 in PSCs with p-i-n architectures by the diffusion of metal ions into the active layer over time or under the influence of environmental stressors such as heat, and that this diffusion can be blocked by barrier layers. N0 measurements of PSCs with and without barrier layers between the device stack and the top electrode were performed using a transient dark current measurement.22 The control PSC device stack was glass-ITO/NiOx/Cs0.2FA0.8PbI3/C60/(Ag or Au or C), as shown in Fig. 1a. A SnO2 layer was introduced for the SnO2 PSC and O3–SnO2 PSC between the C60 ETL and the top electrode (Ag or Au or C) (Fig. 1b and c) deposited on the same substrate to reduce potential variability from different MHP morphologies/microstructures.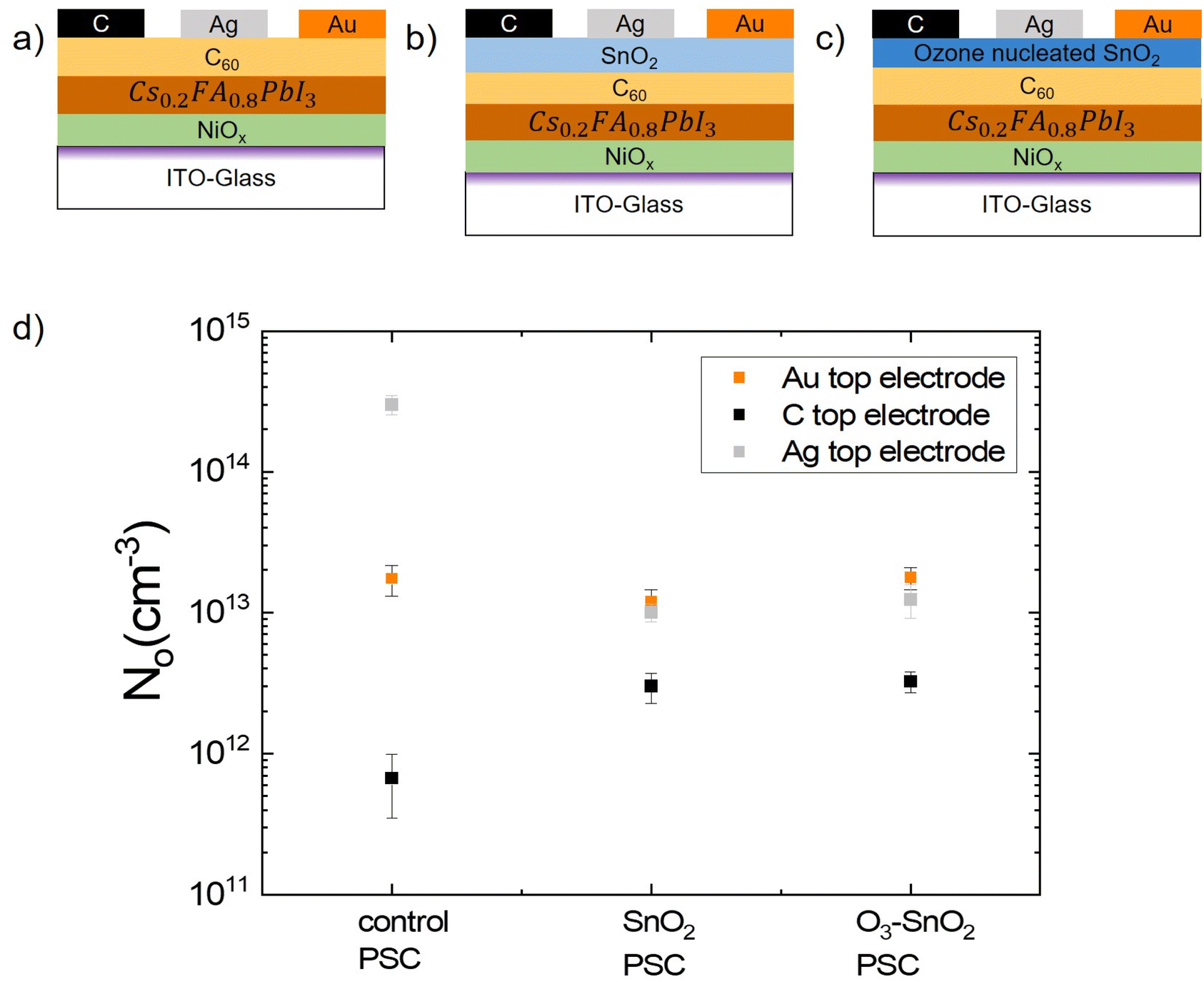 | ||
| Fig. 1 Impact of top electrode chemistry on ion migration (a) control PSC device stack structure in p-i-n configuration with Ag, Au, and C top electrode on the same substrate. (b and c) Device stack structure of PSCs with a barrier layer between the ETL and top electrode where (b) is the SnO2 PSC with SnO2 barrier layer and (c) is the O3–SnO2 PSC with ozone nucleated SnO2 barrier layer. (d) Initial N0 measurements for control PSC, SnO2 PSC, and O3–SnO2 PSC with Ag, Au, and C top electrodes. | ||
Starting with the Ag electrode case, from the square data points in Fig. 1d, the N0 of the Ag-control PSC was 3.0 × 1014 cm−3. The introduction of barrier layers results in a decreased N0 value of 1.0 × 1013 cm−3 for the Ag–SnO2 PSC and 1.2 × 1013 cm−3 for the Ag–O3–SnO2 PSC. We hypothesize that the more than 10× increase in N0 for the control PSC is due to the diffusion of mobile Ag ions into the MHP lattice through the C60 layer. By contrast, when the denser SnO2 barrier layers (both SnO2 and O3 nucleated SnO2) were used in the device stack, the diffusion of Ag ions was more effectively blocked. This phenomenon has been validated using other characterization methods where diffusion barriers were employed in the device stack to prevent the diffusion and corrosion of the top electrode.25–27 In this work, the reduction in ion migration is evident by the reduction of N0 values to a magnitude similar to that of the N0 values of the considerably more inert Au top electrode both in the case of SnO2 PSC and O3–SnO2 PSC. As such, all 3 device configurations exhibited similar N0 values for the Au electrode of ∼1.7 × 1013 cm−3, whereas the control PSC with C top electrode had a N0 of 6.7 × 1011 cm−3 and both the device configurations with barrier layers and C top electrode had a N0 of 3 × 1012 cm−3. The slightly reduced value of N0 in the control PSC when compared to the SnO2 PSC and O3–SnO2 PSC with the C electrode is potentially explained due to two factors. The first is the inertness and chemical stability of C electrodes. The second is the possibility of the C electrode improving the MHP/C60 interface through mechanical toughening an effect that we have previously demonstrated from interfacial fracture energy measurements28 which results in a more physically dense barrier that possibly suppresses the formation of halide vacancies.29
Effectiveness of SnO2 barrier layer in the prevention of Ag ion diffusion
In addition to the initial N0, we measured ion evolution under elevated temperatures to study the extent to which additional ion diffusion occurs in the MHP layer. Aging was performed on the PSCs by subjecting them to 50 °C for a period of 120 h. The percentage change in N0 for all 3 PSC device configurations with respect to the 3 top electrodes was observed after aging (Fig. S1–S3†). The Ag–SnO2 and Ag–O3–SnO2 PSCs had an increase of approximately 740 ± 335% and 90 ± 46% in N0, respectively, while the N0 of the Ag-control PSC decreased by 35 ± 9% (Fig. 2a). The N0 value is believed to be a result of the combination of opposing effects from Ag species reacting with the MHP to generate additional mobile ions and halide ions escaping the MHP lattice to reduce mobile ions after mild thermal aging. This can be seen by the reduction of N0 measured after thermal aging in the control sample without a SnO2 barrier layer an effect we attribute to halide ions escaping the MHP lattice and triggering chemical reactions with adjacent layers along with decomposition of the MHP in the bulk, an effect which is shown in Fig. 4. However, we believe that in the case of Ag–SnO2 and Ag–O3–SnO2 PSC where a barrier layer is present, the measured increase in N0 after aging is primarily a result of metal diffusion into the MHP. In absolute terms, the measured N0 for aged Ag-control PSCs was still markedly higher than the N0 of aged Ag–SnO2 and Ag–O3–SnO2 PSCs. After aging, no significant changes in N0 were observed for PSCs with Au and C top electrodes. A relatively minor decrease (<50% Δ N0) is one that we previously observed in MHPs with C electrodes. We hypothesize that the mechanism for the minor decrease is due to the possible onset of film degradation based on a slight redshift in photoluminescence after aging under these conditions,28 which could correspond with mobile ions escaping the MHP lattice. | ||
| Fig. 2 Effectiveness of SnO2 barrier layer in the prevention of Ag ion diffusion (a) percentage change in N0 for control PSC, SnO2 PSC, and O3–SnO2 PSC with Ag top electrodes after exposure of the PSCs to 50 °C for 120 h. (b) Change in RBS atomic concentration of Ag after aging in the MHP layer and ETL for control PSC, along with barrier layer for SnO2 PSC, and O3–SnO2 PSC. | ||
The above observations strongly indicate that mobile Ag species and MHP-Ag reactions are responsible for the changes in N0. To probe the redistribution of elements in the devices and confirm the observations, RBS was performed on Ag devices for unaged samples and for thermally aged samples that were subjected to the same thermal aging (50 °C for 120 h) (Fig. S4–S6 and Table S1–S6†). Subsequently, the atomic concentration of elements in all the layers of the device stack was used to understand the roles of Ag and barrier layers affecting ion diffusion in PSCs. Fig. 2b depicts the atomic concentration of Ag in the MHP layer and other PSC layers (C60 layer for Ag-control PSC, C60 and SnO2 layer for Ag–SnO2 PSC, and C60 and O3–SnO2 layer for Ag–O3–SnO2 PSC) between the MHP and the top electrode for both unaged and aged samples. Note the unaged and aged sample measurements were not performed on the exact same sample before and after aging, which could lead to minor discrepancies in the absolute numbers between samples. There was no significant change in the atomic concentration of Ag in the ETL layers before and after aging with a maximum increase in the atomic concentration of ∼0.5 × 1016 atoms per cm2 (Fig. 2b) with the amount of uncertainty as explained in ESI Note 1.† However, a significant increase in the atomic concentration of Ag in the MHP layer was found based on model fitting for all PSCs after aging with almost an eight-fold increase for Ag-control PSC from 1.0 × 1016 to 7.5 × 1016 atoms per cm2, approximately a four-fold increase for Ag–SnO2 PSC from 1.4 × 1016 to 6.0 × 1016 atoms per cm2, and approximately a two-fold increase for Ag–O3–SnO2 PSC from 1.2 × 1016 to 3.0 × 1016 atoms per cm2. Additionally, even though the atomic concentration of Ag increased in the Ag-control PSC after aging, the atomic concentration of iodine is simultaneously reduced (Fig. S7†) in the MHP layer. As previously discussed, these opposing effects on N0 likely lead to the overall reduction in N0 observed in Fig. 2a. The increase in the atomic concentration of Ag in the MHP layer for all 3 device configurations after aging confirms that the MHP is reacting with Ag without a SnO2 barrier layer, and Ag ions are diffusing into the MHP layer over time. It is also clear that O3–SnO2 PSC is most effective in reducing the diffusion of Ag into the MHP. The RBS data also shows that no iodine is evident in the layers above the MHP after aging in either the SnO2 or O3–SnO2 device configurations (Tables S4 and S6†), a further indication of the mechanism for N0 increase in both of those cases being in part due to Ag diffusion into the MHP and doping the material.
Threshold in N0 for operation and improved thermal stability of PSCs with barrier layer
Accelerated thermal stability tests in the form of in situ N0-temperature measurements were performed for Ag devices, to evaluate the correlation between ion migration and thermal stability. The in situ N0 measurements were undertaken from 300 K to 450 K with a temperature ramp rate of 10 K min−1, a tolerance of 0.5 K, and a settling time of 20 s. We note that thermal tests in the dark were selected to directly probe metal diffusion rather than other forms of instability that arise with heat + light.The point at which no electronic or ionic response was observed in the device was determined and this threshold temperature was assessed for the different architectures. We note that the apparent threshold temperatures may be either kinetic or thermodynamic effects associated with this temperature ramp experiment. The details of the kinetics of the migration are beyond the current scope of this work. Here, the HTL was either a self-assembled monolayer (SAM) (SAM-based PSC with the control architecture) or NiOx layer (control, SnO2, and O3–SnO2 PSCs) (Fig. 3a and b). A SAM-based PSC was selected as all state-of-the-art device architectures utilize SAM as the HTL and to demonstrate the versatility of the technique in measuring different device stack structures. From Fig. 3c, the SAM-based PSC and control PSC showed a threshold temperature of 370 K (∼100 °C), and the PSCs with barrier layers continued to operate with some response at a temperature of 450 K (∼180 °C), the upper limit which was tested for this study. Fig. S8† shows device failure for the control PSC with a loss in dark IV response beyond 370 K and the SAM-based PSC showing a deteriorated dark IV response at 370 K. In comparison, the SnO2 PSC showed an acceptable dark IV response at 450 K while the O3–SnO2 PSC showed an onset of degradation at 440 K, but both the PSCs still had measurable N0 values at 450 K. It is still expected that this is close to the threshold temperature for their ionic response based on the worsening of the IV curves. This effect was validated by testing a second set of samples with a similar top contact configuration in Fig. S9 and S10,† in which case the O3–SnO2 PSCs had an improved thermal stability response compared to both the control and SnO2 PSCs. In this case, N0 values for the control were measurable up to 450 K, although the J–V response exhibited similar degradation at 370 K and above. We note that a BCP layer was also included in this batch, and additional N0 measurements for control devices that contain the BCP aged at 50 °C are included in Fig. S11,† where the PSCs with BCP exhibited a trend in N0 that is very similar to control PSCs without BCP. This indicates that the introduction of barrier layers in the device stack increased the threshold operating temperature of the PSCs by at least 80 °C, allowing an unencapsulated PSC in this work to function with an Ag electrode at a temperature comparable to the state-of-the-art achieved by a metal-free top contact structure using a combination of ITO with an ALD-based nanolaminate on top of the PSC for additional extrinsic stability shown elsewhere.30
 | ||
| Fig. 3 Threshold in N0 for operation and improved thermal stability of PSCs with barrier layer (a) device stack structure in p-i-n configuration for control and SAM-based PSC with an HTL of NiOx and SAM respectively. (b) Device stack structure in p-i-n configuration for SnO2 PSC and O3–SnO2 PSC with a barrier layer of SnO2 and O3–SnO2 respectively between ETL and the top electrode. (c) N0 of PSCs versus temperature, showing threshold operating points of the devices at higher temperatures and also the threshold in N0 for operation. | ||
Interestingly, there appears to be an empirically observed upper threshold of N0 for operation at ∼3.0 × 1016 cm−3 for multiple different combinations of electron and hole-transporting layers with Ag contacts, as indicated by the purple dashed line in Fig. 3c above which there is a high possibility of device failure based on the worsening or complete loss of dark IV response for most of the samples as shown in Fig. S8.† All the PSCs showed an increase in N0 with temperature throughout the temperature range that was tested but the PSCs that had lower N0 initially (SnO2 PSC and O3–SnO2 PSC) were operational at higher temperatures (370 K to 450 K) when compared to the PSCs that had higher N0 initially (control PSC and SAM-based PSC) which failed to show a response beyond 370 K. There are multiple interpretations of this observation, all of which are in line with higher N0 values corresponding to accelerated degradation. One possible mechanism for this observation could be due to other failure modes unrelated to metal diffusion, such as reactions at the HTL/perovskite interface. This shows that having a higher N0 initially is consistent with more rapid deterioration of PSCs at higher temperatures and that having a lower initial N0 appears to be one of the factors that are associated with improved thermal stability of these p-i-n PSCs. As such, there is the possibility of implementing N0 as a screening tool or quality control for validating barrier layer efficacy in PSCs after fabrication.
Improved bulk MHP stability of PSCs with barrier layer
To study how the changes in ions correlate to microstructure changes in the films, GIWAXS was performed on control, SnO2, and O3–SnO2 PSCs before and after the PSCs were subjected to the same thermal aging (50 °C for 120 h in N2). Incident angle scans showcasing the X-ray diffraction plots in q-space at incidence angles 0.3° (representing the top surface) and 5° (representing the bulk) for all 3 device configurations before and after aging are shown in Fig. 4. Note that the unaged and aged sample measurements were not performed on the same sample. The peaks that indicate some presence of the degradation products were evident on the MHP surface for all the PSCs before and after aging was performed. All the unaged PSCs (Fig. 4a–c) showcased a clear MHP (110) peak in the bulk.31 However, after aging, the control PSC (Fig. 4d) exhibited a significant diminishing of the MHP (110) peak in the bulk. Note that a slightly higher amount of degradation was observed on the surface in the SnO2 PSC (Fig. 4e) when compared to the O3–SnO2 PSC (Fig. 4f) after aging. Both SnO2 PSC and O3–SnO2 PSC did not show a significant variation in the bulk 1D profile after aging i.e., they retained their MHP (110) peak along with no presence of degradation byproduct peaks. The integrated peak area ratios of MHP (110) and a degradation product (which we hypothesize corresponds to either 2H-FAPbI3 or a non-perovskite phase) (Table S7†) from the 1D integrated GIWAXS profiles show that this ratio reduced from unaged to aged samples in the decreasing order of control PSC, SnO2 PSC, and O3–SnO2 PSC. This reduction indicates that the control PSC has low stability both in the top surface and the bulk, whereas SnO2 PSCs and O3–SnO2 PSCs show an improvement in bulk stability after the introduction of the barrier layers in the device structure. | ||
| Fig. 4 Improved bulk MHP stability of PSCs with barrier layer X-ray diffraction plots in q-space at incidence angles 0.3° (representing the top surface) and 5° (representing the bulk) (a–c) unaged PSCs, and (d–f) PSCs subjected to 50 °C for 120 h. (a and d) Control PSC, (b and e) SnO2 PSC, and (c and f) O3–SnO2 PSC. | ||
In addition to the dark I–V measurements, a full set of measurements were performed in the light and complemented by temperature-dependent EQE for the O3–SnO2 PSC (showing a minor increase in bandgap with temperature as in line with previous reporting for PSCs with similar compositions32) as shown in Fig. S12 and S13.† Control PSCs exhibited a rapid performance degradation with an increase in temperature, whereas O3–SnO2 PSCs exhibited much better thermal stability. There was a continuous drop in power conversion efficiency (PCE) from 14.5% to 6.6% for control PSC, whereas the PCE dropped from 16% to 12% for O3–SnO2 PSCs when the devices were exposed to heat from 300 K to 450 K. The factors contributing to the drop in PCE of control PSCs are a drop in VOC and fill factor, both of which showed better stability for O3–SnO2 PSCs. Hence the improvement in N0 and the associated enhancement in the O3–SnO2 PSCs in comparison to control PSCs is complemented by better stability under extreme operational conditions (Fig. S14†) and is on par with the best reported thermal stability of PSCs, which required the use of a metal-free top contact structure comprising ITO and an ALD nanolaminate30 on top of the completed device. Our device structure shows that the reactions from a highly reactive metal (Ag) can be mitigated by preventing ion diffusion through the use of an ultra-thin, dense, and well-designed built-in barrier layer.
The activation energy (EA) of the PSCs was also determined using in situ ionic conductivity across a range of temperatures, a measurement that has been used in several other reports for ion-specific activation mechanisms.33–35 As plotted in Fig. S15 and S16,† the EA of the control PSC was 0.346 eV, the EA of the SAM-based PSC was 0.410 eV, and the EA of the O3–SnO2 PSC was 0.503 eV. This value is much higher than the EA of triple halide PSCs with a similar architecture to the control PSC in this study and without any barrier layer (0.14 eV) from previous work.22 As expected, these values support that the mobile ion activation is suppressed in the PSCs with a barrier layer when compared to the control and SAM-based PSCs. The implication of a higher EA in the O3–SnO2 PSC demonstrates that well-designed barrier layers can reduce both the formation and evolution of mobile ions under operational conditions.
The primary focus of this work was utilizing the ion blocking feature/mechanism of a dense ALD O3–SnO2 layer to clearly show the diffusion of metal into the MHP under operation and the ability to detect this diffusion using N0. A mild temperature of 50 °C was initially selected for the exposure tests to be able to observe only the temperature-dependent diffusion mechanisms on the PSCs without the influence of more rapid MHP degradation that could happen if the accelerated testing was done at higher temperatures or with light. Once an understanding regarding the diffusion of metal was achieved at 50 °C, the PSCs were exposed to much higher temperatures up to 450 K (177 °C) to observe the effects of degradation of MHP along with the diffusion of metal into the MHP. Additional experiments were performed under illumination during this high-temperature study showing the improved operational stability of O3–SnO2 devices compared to control devices (Fig. S12 and S13†) that directly correlate with the reduction in N0. Future work will include in situ PL mapping characterization of the PSCs to monitor compositional changes in the MHP caused by the metal diffusion under operation during thermal aging.
Conclusion
In this work, we quantified mobile ionic species directly or indirectly resulting from chemical reactions. We demonstrated that our N0 measurement is sensitive to Ag ions diffusing into the MHP lattice of PSCs through the changes in N0 based on top electrode chemistry and from thermal aging. We validated that O3–SnO2 is an improved barrier layer in preventing the diffusion of Ag ions along with retaining the bulk stability of the MHP while improving PSC thermal stability compared to devices without a barrier layer. This allowed us to correlate this N0 metric to current–voltage (IV) behavior and ion redistribution as measured by Rutherford Backscattering Spectrometry. It is important to note that at high enough temperatures such as 450 K, MHPs will degrade even with barrier layers due to structural degradation, an effect which was observed in the appearance of an upper threshold for N0 across device types. While many factors contribute to the real lifetime of fielded PV modules, the effectiveness, and reproducibility of barrier layers to prevent ion migration and chemical degradation are among the most critical to tackle for the stability of PSCs. Overall, our results demonstrate that N0-temperature measurements are a rapid and effective method to characterize barrier layers at perovskite/electrode interfaces and predict the chemical robustness of the full devices.To this end, there is a need for a deeper understanding of the correlation between power conversion efficiency, ion migration, and stability of PSCs. As such, we believe that the use of N0 measurements coupled with accelerated thermal and/or light aging can serve as a highly useful tool in quantifying the extent to which multiple sources of ions (whether from the top electrode or from the MHP itself) move throughout the PSC to provide a deeper understanding of ion-based degradation mechanisms.
Methods
The preparation of glass substrates before doing any of the processing on top of the substrate was performed in a step-by-step procedure as follows: indium tin oxide coated glass (ITO-glass) substrates (Xin Yan Technologies) were initially cleaned in an ultrasonic cleaner by submerging them in an industry grade soap solution of Extran (Millipore Sigma) diluted in water in the ratio of 1:10 for 10 min. Then, the ITO-glass slides were rinsed under a flow of de-ionized water with a brush to remove the residual soap on top of the substrates. This was followed by ultrasonic cleaning by submerging them in isopropyl alcohol (IPA) (Thermo Scientific) and acetone (Alfa Aesar–99.5%+) separately for 10 min. Finally, they were subjected to a UV ozone treatment for another 15 min.
Nickel–oxide (NiOx)
A NiOx sol–gel solution for depositing the hole transport layer (HTL) was prepared by mixing 1 M NiNO3. (H2O)6 (Sigma Aldrich–99.999% trace metals basis) in 94% ethylene glycol (EG) (Thermo scientific–anhydrous 99.8%) and 6% ethylenediamine (EDA) (Thermo scientific–99%); the vial was then placed in a vortex mixer, and the solution was mixed until it turned a dark blue color.Self-assembled monolayer
0.5 mg ml−1 MeO-2PACz self-assembled monolayer solution dissolved in ethanol was spin-coated on substrates at 3000 rpm for 30 s in a nitrogen glovebox, followed by annealing at 100 °C for 10 min.Cesium formamidinium lead iodide (Cs0.2FA0.8PbI3)
The MHP precursor solution for Cs0.2FA0.8PbI3 films was prepared by mixing 0.2 mol Cesium Iodide (CsI) (Sigma-Aldrich–99.999% trace metals basis), 0.8 mol Formamidinium Iodide (FAI) (Greatcell Solar Materials), and 1 mol Lead Iodide (PbI2) (TCI America–99.99% trace metals basis). A 1 M concentration solution was made by mixing 0.0519 gm of CsI, 0.1375 gm of FAI, and 0.461 gm of PbI2 in a solvent of 4:1 Dimethylformamide (DMF) (Sigma-Aldrich–Anhydrous 99.8%) and Dimethyl Sulfoxide (DMSO) (Sigma-Aldrich–Anhydrous ≥ 99.9%) with 800 μL of DMF and 200 μL of DMSO. A vortex mixer was used to mix the solution until the powders were uniformly dissolved and a yellow solution was formed.
Perovskite solar cells (PSCs)
After finishing the substrate preparation process and making the required inks, PSCs were fabricated in a step-by-step process. As the PSCs were in a p-i-n configuration, the HTL (NiOx/SAM) was first deposited on the cleaned ITO-glass by spin coating. 50 μL of NiOx solution was deposited at a speed of 5000 rpm and an acceleration of 2500 rpm s−1 for 30 s in a fume hood and then annealed at 315 °C for 1 h. The SAM layer was deposited at a speed of 3000 rpm for 30 s followed by annealing at 100 °C for 10 min. After the HTL was formed, the MHP absorber layer of Cs0.2FA0.8PbI3 was deposited using a spin coating process with anti-solvent quenching. This was done by depositing 100 μL of MHP precursor on the glass and spinning at a speed of 1000 rpm and acceleration of 500 rpm s−1 for 10 s, and then the speed was stepped up to 5000 rpm and acceleration of 1500 rpm s−1 for 10 s. In the last 3–5 s of the second step, 100 μL of chlorobenzene (anti-solvent) (Sigma-Aldrich–Anhydrous 99.8%) was deposited quickly. Then, the samples were annealed at 150 °C for 10 minutes. The ETL was deposited by evaporating 45 nm of C60 on top of the samples in an Angstrom evaporator with a shadow mask, and the top electrode was made by evaporating either 100 nm of Ag or Au on top of the device stack using a different mask. The carbon (C) top electrode was formed on top of the PSC by depositing it from the solvent-based C paste (PELCO conductive carbon glue–Ted Pella). Three different electrodes (evaporated Ag or Au and a solvent-based C) were deposited on top of the same PSC substrate to observe the variation in N0 with respect to barrier layers and the top electrode. ALD SnO2 and O3–SnO2 for the barrier layers were deposited in a Beneq TFS200 ALD reactor by 125 cycles of tetrakisdimethylamino tin(IV) and water at 90. A 15 second ozone and water treatment was applied to the O3–SnO2 samples in situ part way through the 125 cycles SnO2 deposition following the sequence: 40 cycles SnO2/15 second ozone and water/85 cycle SnO2.20Characterization
All the ionic and electronic measurements were performed with PAIOS, an all-in-one measurement equipment for photovoltaic devices and LEDs (FLUXiM AG). A hot plate was used to age the PSCs (as fabricated without encapsulation) at 50 °C in an N2 glovebox for 120 h with ex situ measurements on PAIOS. N0 was measured and calculated using the transient dark current method (Fig. S17†) as described in our previous work22 in which a voltage bias of 800 mV is applied to the PSC in a forward-bias configuration in the form of a pulse with the following characteristics: 1 ms settling time, 10 ms pulse time, and 1 ms follow-up time. The entire measurement lasts around 13 ms with the measurement cut-off around 1 ms after the bias is taken away, during which the mobile ions in the MHP drift. The measured drift current can be time-integrated and divided by the elementary charge, area, and thickness of the MHP layer respectively to determine the N0.22 For the quantification of mobile ion concentration (N0) using the transient dark current methodology, the voltage pulse is applied in forward bias for only a short time of 10 ms. This timescale was chosen to be able to only measure the intrinsic concentration of ions in the perovskite that are ready to move under a small voltage perturbation. Using such a short timescale for the measurement might result in values of N0 that are lower than what has been reported in the literature, but we believe that the values obtained represent the actual ionic concentration present in the device at the surface level or at the interfaces. The consistency of the measured N0 values has been shown in our previous works.22,28,35In situ ionic measurements were performed with the temperature control stage and module (LTS-420E) from Linkam in integration with PAIOS in increments of 10 K from room temperature (300 K) up to 450 K. EA was measured following the same methodology used in our previous work.35 The ramp rate used was 10 K min−1 and the tolerance was 0.5 K with a settling time of 20 s. The reported EA values are based on measurements of a single sample. However, the samples were measured during both ramp up and ramp down of the temperature. The reduction of temperature happened naturally and hence the samples would have significant dwell at each temperature and the calculations include averages of the measurement in both directions.
The RBS experiment was conducted in the Ion beam laboratory (IBL) at the University of North Texas (UNT) using the NEC 9SH 3 MV Pelletron accelerator.36,37 All the experiments were performed in the ion microprobe beamline using a 2 MeV He+ beam under a vacuum of 2 × 10−7 Torr. The RBS spectra were collected using a Passivated Implanted Planar Silicon (PIPS) charged particle detector from Mirion Technologies (Canberra), model No. PD25-11-300 AM, having a solid angle of 34 milli-steradian, and the operating voltage for the detector was 40 V situated at the backscattered angle of 145° (Fig. S18†). The detector arrangement in the microprobe chamber is such that the incident beam, backscattering detector, and target normal lie in the same horizontal plane.
The RBS data fitting was done using the SIMNRA software package.38 Based on the thickness values of each layer in the PSC stack, a simulated sample was generated. The concentrations of each layer were adjusted until a suitable match was achieved. Layer thickness is accepted by SIMNRA in the form of the layer's areal density (atoms cm−2). The SRIM/TRIM software program was utilized to convert the thickness into areal density39 and detailed information on the process is provided elsewhere.24 The layer information extracted from the SIMNRA was fed into the MultiSIMNRA40 software program, to further extract the contribution from the individual layers and their elemental species.
To identify the different crystalline phases in the perovskite films and devices, at different subsurface depths, synchrotron-based grazing incidence wide-angle X-ray scattering (GIWAXS) data were collected at NCD-SWEET beamline at the ALBA synchrotron (Cerdanyola del Vallès, Spain): a monochromatic (λ = 0.95741 Å) X-ray beam of 150 × 30 μm2 [H × V] was defined using a Si (111) channel cut monochromator and collimated using Be Compound Refractive Lenses (CRLs). The scattered signal was recorded using a Rayonix LX255-HS area detector placed at 251.2 mm from the sample position. Detector tilts and sample-to-detector distance were calculated using Cr2O3 as a calibrant, which was employed to calibrate the reciprocal space wavevector, q. GIWAXS frames were recorded at incident angles (αi) between 0° and 5° in a scanning fashion, shifting from the surface-sensitive evanescent regime of scattering and transitioning to a deep penetrative measurement of the film layers at relatively high angles.41 Throughout the data acquisition process, a continuous flow of N2 gas was maintained over the sample. Collected 2D images were azimuthally integrated to general 1D profiles using PyFAI42 and processed using a custom Python routine.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.Author contributions
Conceptualization, S. P. and N. R.; methodology, S. P. and N. R.; investigation, S. P., M. N. K., M. S., M. P., M. C., R. A. S., and E. S.; validation S. P. and R. A. K.; formal analysis S. P., M. N. K., M. S., M. P., and J. A. S.; data curation S. P.; visualization S. P. and N. R.; writing–original draft, S. P. and N. R.; writing–review & editing S. P., M. N. K., M. S., M. P., M. A. D., R. A. K., M. C., R. A. S., E. S., M. B. J. R, I. R. S, V. R. W., J. J. B., J. M. L., J. A. S., A. P., B. R., and N. R.; resources S. P., M. C., I. R. S., J. M. L., M. A. D., J. A. S., A. P., and B. R.; visualization S. P. and N. R.; software S. P., M. N. K., M. S., M. P., and J. A. S.; supervision M. B. J. R., I. R. S., V. R. W., J. J. B., J. M. L., J. A. S., A. P., B. R., and N. R.; project administration N. R.; funding acquisition M. B. J. R., J. J. B., J. M. J., J. A. S., A. P., B. R., and N. R.; All the authors discussed the results and commented on the manuscript.Conflicts of interest
S. P. and N. R. have filed for a patent based on this work. The other authors declare no conflict of interest.Acknowledgements
This material is based upon work supported by the National Science Foundation under Grant No. 2339233. This work was authored in part by the National Renewable Energy Laboratory for the U.S. Department of Energy (DOE) under Contract No. DE-AC36-08GO28308. A. P., J. J. B., and R. A. K acknowledges financial support from the U.S. Department of Energy's Office of Energy Efficiency and Renewable Energy (EERE) under Solar Energy Technologies Office (SETO) Agreement DE- EE0009513. The views expressed in the article do not necessarily represent the views of the DOE or the U.S. Government. J. A. S acknowledges financial support from the Australian Research Council (ARC: grant no. DE230100173. J. A. S., R. A. S., and M. B. J. R. thank the staff of the BL11 NCD-SWEET beamline for their assistance in recording the synchrotron GIWAXS data. R. A. S. and M. B. J. R. acknowledge financial support from iBOF-21-085 PERsist and Internal Funds KU Leuven (C14/23/090). B. R., M. S., and M. P. acknowledge financial support from the U.S. National Science Foundation Grant No. ECCS-2210722.References
- Z. Li, B. Li, X. Wu, S. A. Sheppard, S. Zhang and D. Gao, et al., Organometallic-functionalized interfaces for highly efficient inverted perovskite solar cells, Science, 2022, 376(6591), 416–420, DOI:10.1126/science.abm8566.
- J. J. Yoo, G. Seo, M. R. Chua, T. G. Park, Y. Lu and F. Rotermund, et al., Efficient perovskite solar cells via improved carrier management, Nature, 2021, 590(7847), 587–593, DOI:10.1038/s41586-021-03285-w.
- NREL Best Research-Cell Efficiency Chart, 2024, https://www.nrel.gov/pv/cell-efficiency.html.
- M. A. Green, A. Ho-Baillie and H. J. Snaith, The emergence of perovskite solar cells, Nat. Photonics, 2014, 8(7), 506–514, DOI:10.1038/nphoton.2014.134.
- M. Ashif and F. Mahjabeen, Promises and Challenges of Perovskite Solar Cells: A Comprehensive Review, BULLET: Jurnal Multidisiplin Ilmu, 2023, 2(5), 1147–1157 Search PubMed . Available from: https://journal.mediapublikasi.id/index.php/bullet/article/view/3685.
- P. Čulík, K. Brooks, C. Momblona, M. Adams, S. Kinge and F. Maréchal, et al., Design and Cost Analysis of 100 MW Perovskite Solar Panel Manufacturing Process in Different Locations, ACS Energy Lett., 2022, 7(9), 3039–3044, DOI:10.1021/acsenergylett.2c01728.
- R. H. Ahangharnejhad, W. Becker, J. Jones, A. Anctil, Z. Song and A. Phillips, et al., Environmental Impact per Energy Yield for Bifacial Perovskite Solar Cells Outperforms Crystalline Silicon Solar Cells, Cell Rep. Phys. Sci., 2021, 2(2), 100344 CrossRef CAS . Available from: https://www.sciencedirect.com/science/article/pii/S2666386421000291.
- K. Sakhatskyi, R. A. John, A. Guerrero, S. Tsarev, S. Sabisch and T. Das, et al., Assessing the Drawbacks and Benefits of Ion Migration in Lead Halide Perovskites, ACS Energy Lett., 2022, 7(10), 3401–3414, DOI:10.1021/acsenergylett.2c01663.
- J. Liu, M. Hu, Z. Dai, W. Que, N. P. Padture and Y. Zhou, Correlations between Electrochemical Ion Migration and Anomalous Device Behaviors in Perovskite Solar Cells, ACS Energy Lett., 2021, 6(3), 1003–1014, DOI:10.1021/acsenergylett.0c02662.
- G. Grancini, C. Roldán-Carmona, I. Zimmermann, E. Mosconi, X. Lee and D. Martineau, et al., One-Year stable perovskite solar cells by 2D/3D interface engineering, Nat. Commun., 2017, 8(1), 15684, DOI:10.1038/ncomms15684.
- Perovskite PV Accelerator for Commercializing Technologies. 2024, https://pvpact.sandia.gov/.
- E. Bi, Z. Song, C. Li, Z. Wu and Y. Yan, Mitigating ion migration in perovskite solar cells, Trends Chem., 2021, 3(7), 575–588, DOI:10.1016/j.trechm.2021.04.004.
- H. Zai, Y. Ma, Q. Chen and H. Zhou, Ion migration in halide perovskite solar cells: Mechanism, characterization, impact and suppression, J. Energy Chem., 2021, 63, 528–549 CrossRef CAS . Available from: https://www.sciencedirect.com/science/article/pii/S2095495621004290.
- Y. Zhao, W. Zhou, Z. Han, D. Yu and Q. Zhao, Effects of ion migration and improvement strategies for the operational stability of perovskite solar cells, Phys. Chem. Chem. Phys., 2021, 23(1), 94–106, 10.1039/D0CP04418K.
- L. Zhao, R. A. Kerner, Z. Xiao, Y. L. Lin, K. M. Lee and J. Schwartz, et al., Redox Chemistry Dominates the Degradation and Decomposition of Metal Halide Perovskite Optoelectronic Devices, ACS Energy Lett., 2016, 1(3), 595–602, DOI:10.1021/acsenergylett.6b00320.
- R. A. Kerner, L. Zhao, S. P. Harvey, J. J. Berry, J. Schwartz and B. P. Rand, Low Threshold Voltages Electrochemically Drive Gold Migration in Halide Perovskite Devices, ACS Energy Lett., 2020, 5(11), 3352–3356, DOI:10.1021/acsenergylett.0c01805.
- R. A. Kerner, A. V. Cohen, Z. Xu, A. R. Kirmani, S. Y. Park and S. P. Harvey, et al., Electrochemical Doping of Halide Perovskites by Noble Metal Interstitial Cations, Adv. Mater., 2023, 35(29), 2302206, DOI:10.1002/adma.202302206.
- N. N. Shlenskaya, N. A. Belich, M. Grätzel, E. A. Goodilin and A. B. Tarasov, Light-induced reactivity of gold and hybrid perovskite as a new possible degradation mechanism in perovskite solar cells, J. Mater. Chem. A, 2018, 6(4), 1780–1786, 10.1039/C7TA10217H.
- J. Zhou, Z. Liu, P. Yu, G. Tong, R. Chen and L. K. Ono, et al., Modulation of perovskite degradation with multiple-barrier for light-heat stable perovskite solar cells, Nat. Commun., 2023, 14(1), 6120, DOI:10.1038/s41467-023-41856-9.
- S. A. Johnson, K. P. White, J. Tong, S. You, A. Magomedov and B. W. Larson, et al., Improving the barrier properties of tin oxide in metal halide perovskite solar cells using ozone to enhance nucleation, Joule, 2023, 7(12), 2873–2893, DOI:10.1016/j.joule.2023.10.009.
- M. Li, S. Johnson, L. Gil-Escrig, M. Sohmer, C. A. Figueroa Morales and H. Kim, et al., Strategies to improve the mechanical robustness of metal halide perovskite solar cells, Energy Adv., 2024, 3(1), 273–280, 10.1039/D3YA00377A.
- S. Penukula, R. Estrada Torrejon and N. Rolston, Quantifying and Reducing Ion Migration in Metal Halide Perovskites through Control of Mobile Ions, Molecules, 2023, 28(13), 5026 CrossRef CAS PubMed . Available from: https://www.mdpi.com/1420-3049/28/13/5026.
- F. Lang, A. Juma, V. Somsongkul, T. Dittrich and M. Arunchaiya, Rutherford Backscattering Spectroscopy of Mass Transport by Transformation of PbI2 into CH3NH3PbI3 within np-TiO2, Hybrid. Mater., 2014, 1(1), 52–61 Search PubMed.
- M. Parashar, M. Sharma, D. K. Saini, T. A. Byers, J. M. Luther and I. R. Sellers, et al., Probing elemental diffusion and radiation tolerance of perovskite solar cells via non-destructive Rutherford backscattering spectrometry, Appl. Energy, 2024, 2(1), 016109, DOI:10.1063/5.0193601.
- H. H. Park and D. J. Fermin, Recent Developments in Atomic Layer Deposition of Functional Overlayers in Perovskite Solar Cells, Nanomaterials, 2023, 13(24), 3112 CrossRef CAS PubMed . Available from: https://www.mdpi.com/2079-4991/13/24/3112.
- E. Bi, H. Chen, F. Xie, Y. Wu, W. Chen and Y. Su, et al., Diffusion engineering of ions and charge carriers for stable efficient perovskite solar cells, Nat. Commun., 2017, 8(1), 15330, DOI:10.1038/ncomms15330.
- N. Li, Z. Shi, C. Fei, H. Jiao, M. Li and H. Gu, et al., Barrier reinforcement for enhanced perovskite solar cell stability under reverse bias, Nat. Energy, 2024, 9, 1264–1274, DOI:10.1038/s41560-024-01579-7.
- S. Penukula, F. Tippin, M. Li, K. A. Khawaja, F. Yan and N. Rolston, Use of carbon electrodes to reduce mobile ion concentration and improve reliability of metal halide perovskite photovoltaics, Energy Mater., 2024, 4(5), 400060, DOI:10.20517/energymater.2024.26 . Available from: https://www.oaepublish.com/articles/energymater.2024.26.
- Y. Zhong, J. Yang, X. Wang, Y. Liu, Q. Cai and L. Tan, et al., Inhibition of Ion Migration for Highly Efficient and Stable Perovskite Solar Cells, Adv. Mater., 2023, 35(52), 2302552, DOI:10.1002/adma.202302552.
- H. Afshari, S. Sourabh, S. A. Chacon, V. R. Whiteside, R. C. Penner and B. Rout, et al., FACsPb Triple Halide Perovskite Solar Cells with Thermal Operation over 200 °C, ACS Energy Lett., 2023, 8(5), 2408–2413, DOI:10.1021/acsenergylett.3c00551.
- Y. Yang, L. Yang and S. Feng, Interfacial engineering and film-forming mechanism of perovskite films revealed by synchrotron-based GIXRD at SSRF for high-performance solar cells, Mater. Today Adv., 2020, 6, 100068 CrossRef . Available from: https://www.sciencedirect.com/science/article/pii/S2590049820300151.
- T. Moot, J. B. Patel, G. McAndrews, E. J. Wolf, D. Morales and I. E. Gould, et al., Temperature Coefficients of Perovskite Photovoltaics for Energy Yield Calculations, ACS Energy Lett., 2021, 6(5), 2038–2047, DOI:10.1021/acsenergylett.1c00748.
- M. C. López-González, G. del Pozo, B. Arredondo, S. Delgado, D. Martín-Martín and M. García-Pardo, et al., Temperature behaviour of mixed-cation mixed-halide perovskite solar cells. Analysis of recombination mechanisms and ion migration, Org. Electron., 2023, 120, 106843 CrossRef . Available from: https://www.sciencedirect.com/science/article/pii/S156611992300099X.
- J. Xing, Q. Wang, Q. Dong, Y. Yuan, Y. Fang and J. Huang, Ultrafast ion migration in hybrid perovskite polycrystalline thin films under light and suppression in single crystals, Phys. Chem. Chem. Phys., 2016, 18(44), 30484–30490, 10.1039/C6CP06496E.
- J. Sun, S. Penukula, M. Li, M. R. Hosseinzade, Y. Tang and L. Dou, et al., Mechanical and Ionic Characterization for Organic Semiconductor-Incorporated Perovskites for Stable 2D/3D Heterostructure Perovskite Solar Cells, Small, 2024, 2406928, DOI:10.1002/smll.202406928.
- B. Rout, M. S. Dhoubhadel, P. R. Poudel, V. C. Kummari, B. Pandey and N. T. Deoli, et al., An overview of the facilities, activities, and developments at the University of North Texas Ion Beam Modification and Analysis Laboratory (IBMAL), AIP Conf. Proc., 2013, 1544(1), 11–18, DOI:10.1063/1.4813454.
- M. Sharma, M. Parashar, D. K. Saini, T. A. Byers, C. Bowen and M. N. Khanal, et al., In situ Characterization Tools for Evaluating Radiation Tolerance and Elemental Migration in Perovskites. in 2024 IEEE 52nd Photovoltaic Specialist Conference (PVSC). 2024. pp. 496–498 Search PubMed.
- M. Mayer, SIMNRA User's Guide. 1997, https://mam.home.ipp.mpg.de/ReportIPP9-113.pdf Search PubMed.
- J. F. Ziegler, M. D. Ziegler and J. P. Biersack, SRIM – The stopping and range of ions in matter (2010), Nucl. Instrum. Methods Phys. Res., Sect. B, 2010, 268(11), 1818–1823 CrossRef CAS . Available from: https://www.sciencedirect.com/science/article/pii/S0168583X10001862.
- T. F. Silva, C. L. Rodrigues, M. Mayer, M. V. Moro, G. F. Trindade and F. R. Aguirre, et al., MultiSIMNRA: A computational tool for self-consistent ion beam analysis using SIMNRA, Nucl. Instrum. Methods Phys. Res., Sect. B, 2016, 371, 86–89 CrossRef CAS . Available from: https://www.sciencedirect.com/science/article/pii/S0168583X15010459.
- J. A. Steele, E. Solano, D. Hardy, D. Dayton, D. Ladd and K. White, et al., How to GIWAXS: Grazing Incidence Wide Angle X-Ray Scattering Applied to Metal Halide Perovskite Thin Films, Adv. Energy Mater., 2023, 13(27), 2300760, DOI:10.1002/aenm.202300760.
- G. Ashiotis, A. Deschildre, Z. Nawaz, J. P. Wright, D. Karkoulis and F. E. Picca, et al., The fast azimuthal integration Python library: pyFAI, J. Appl. Crystallogr., 2015, 48(2), 510–519, DOI:10.1107/S1600576715004306.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5el00051c |
| This journal is © The Royal Society of Chemistry 2025 |