
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIncorporating a lithium-deficient layer and interfacial-confined catalysis enables the reversible redox of surface oxygen species in lithium-rich manganese-based oxides†
Junpeng
Sun
a,
Hai
Yang
 a,
Jialong
Shen
a,
Huadong
Qi
a,
Mei
Sun
b,
Yuhang
Lou
a,
Yu
Yao
a,
Xianhong
Rui
c,
Yu
Shao
d,
Xiaojun
Wu
a and
Yan
Yu
*a
a,
Jialong
Shen
a,
Huadong
Qi
a,
Mei
Sun
b,
Yuhang
Lou
a,
Yu
Yao
a,
Xianhong
Rui
c,
Yu
Shao
d,
Xiaojun
Wu
a and
Yan
Yu
*a
aHefei National Research Center for Physical Sciences at the Microscale, Department of Materials Science and Engineering, iChEM (Collaborative Innovation Center of Chemistry for Energy Materials), CAS Key Laboratory of Materials for Energy Conversion, University of Science and Technology of China, Hefei, Anhui, China. E-mail: yanyumse@ustc.edu.cn
bInstruments Center for Physical Science, University of Science and Technology of China, Hefei, Anhui, China
cSchool of Materials and Energy, Guangdong University of Technology, Guangzhou, 510006, China
dJiujiang DeFu Technology Co. Ltd, Jiujiang, Jiangxi, China
First published on 21st March 2025
Abstract
Lithium-rich manganese-based oxides (LRMOs) are promising next-generation candidate cathode materials, offering a high discharge capacity exceeding 300 mA h g−1. This exceptional capacity is attributed to the synergistic redox activity of transition metals and lattice oxygen. Nevertheless, the over-oxidation of lattice oxygen in LRMOs leads to capacity fading, severe lattice strain, and sluggish oxygen redox reaction kinetics. Herein, we introduce a lithium-deficient layer and a RuO2-promoted interface-confined catalysis network on the surface of LRMO (Ru-1). The lithium-deficient layer effectively passivates the surface lattice oxygen by reducing the Li–O–Li configurations at the atomic level. The RuO2-promoted interface-confined catalysis network successfully captures trace amounts of lost lattice oxygen and catalyzes the reversible reduction of activated O species. This configuration yields a specific discharge capacity of 307 mA h g−1 at 0.1C, with an impressive capacity retention rate of 97% after 300 cycles at 1C. The Ru-1‖graphite pouch cell exhibits a superior capacity retention rate of 85% after 450 cycles at C/3 and the Ru-1‖Li pouch cell exhibits a high energy density of 513 W h kg−1. Our strategies involving the lithium-deficient layer and interface-confined catalysis offer novel insights into protecting the surface and enhancing oxygen reusability within the LRMOs.
Broader contextIncreasing interest has been directed towards lithium-rich manganese-based oxides (LRMOs) as a leading candidate for next-generation cathode materials. However, challenges such as capacity fading, oxygen evolution, and sluggish oxygen redox kinetics due to the over-oxidation of lattice oxygen persist. To address these issues, we have innovated by integrating a lithium-deficient layer alongside a RuO2-enhanced interface-confined catalysis network on the surface of LRMOs. The lithium-deficient layer acts to passivate the lattice oxygen activity on the surface of LRMOs by minimizing Li–O–Li configurations at the atomic scale. Meanwhile, the RuO2 network effectively captures and catalyzes the reversible reduction of the trace amount of lost lattice oxygen. Our strategies of employing a lithium-deficient layer and an interface-confined catalysis mechanism provide critical insights into surface protection and oxygen reusability enhancement within LRMOs, paving the way for the development of robust, high-performance cathode materials. |
Introduction
With the growing demand for electric vehicles and portable electronic devices, developing high-energy-density Li-ion battery systems is of great significance.1 The co-redox properties of transition metals and lattice oxygen confer Li-rich and Mn-rich cathodes with a high specific discharge capacity of more than 300 mA h g−1, suggesting a promising future for the commercialization of high-energy-density batteries.2–8 Nevertheless, the anion redox processes in lithium-rich manganese-based oxides (LRMOs) that occur at voltages above 4.5 V accelerate the deterioration of the battery performance, causing severe problems like lattice oxygen loss and voltage fade.9,10 Additionally, the oxygen released during these processes contributes to capacity fading, phase transition and severe strain problems.11 These drawbacks hinder the commercial development and application of LRMO cathodes.The composition of Li-rich cathode materials can be expressed as xLi2MnO3 (1−x)LiMO2 (M = Ni, Co, and Mn). In the Li2MnO3 structure, due to the substitution of some Mn ions by Li ions in the Mn layer, a large number of axial Li–O–Li configurations are formed.12 Each oxygen atom is coordinated to six Li/M atoms. This structural arrangement decreases the hybridization of certain O 2p orbitals with the d orbitals of the transition metal (TM), leading to an increase in energy compared to fully axial Li–O–M configurations, such as those present in Ni-rich cathodes.13–16 The Li–O–Li configurations endow LRMO cathodes with the feature of lattice oxygen redox. However, the oxidized lattice oxygen is harmful to the battery system, which will cause capacity loss, voltage fade, non-uniform lattice strain, and serious safety issues.17 The O2 is initially released from the surface lattice of LRMOs, with the formation of oxygen vacancies, which are subsequently injected into the bulk lattice of LRMOs.18–20 The accumulation of these oxygen vacancies ultimately leads to the development of nanovoids, contributing to voltage fading and the formation of a nonconductive interface.21
The suppression of oxygen release from the surface lattice is crucial for the overall stability of the material. Previous studies have explored various strategies, including surface coating and the grafting of functional groups, to modify the interface.19 For instance, Zhang et al. used a heterostructured spinel Li4Mn5O12 coating strategy to protect the LRMO surface.22 The spinel coating layer not only hinders the release of oxygen but also accelerates the transformation of Li+. Similarly, Sun et al. replaced the surface lattice oxygen with sulfite, effectively shifting the oxidation state from O2− to SO32−.23 This charge transfer helps to prevent the degradation of the LRMO interface (caused by over-oxidation of surface oxygen). However, it is important to note that these coating layers do not decrease the oxygen activity of the near-surface lattice, which is intrinsically linked to the redox processes associated with the Li–O–Li configurations in the LRMO lattice. The introduced heterogeneous coating layers often fail to integrate seamlessly with the LRMO lattice, leading to uneven stress during the long cycling process and resulting in the peeling of the coating layer. Moreover, existing literature has paid insufficient attention to the oxygen species that have already escaped from the lattice, such as Li2O2, LiO2, and O2. The recycling of the released lattice oxygen can effectively reduce the capacity loss and enhance the cycling stability. Therefore, it is essential to consider strategies for reducing the content of Li–O–Li configurations at the interface and capturing the activated oxygen species.
Ruthenium dioxide (RuO2), as a gas electrode catalyst, exhibits strong affinity for activated oxygen species, effectively capturing the released lattice oxygen species (Li2O2, LiO2, and O2) and catalyzing their reversible electrochemical reduction.24,25 Herein, we propose a bifunctional method that employs an acidic Ru3+ solution to realize interfacial ion exchange between H+ and Li+ from the near-surface lattice, thereby creating a lithium-deficient interface. This Li-poor surface significantly diminishes the Li–O–Li configuration, resulting in reduced activity of the lattice oxygen and alleviating oxygen release at the surface. Meanwhile, RuO2 functions as an interfacial gas adsorption ‘island’, which captures escaped oxygen species and lowers the energy barrier for the reduction of O2, facilitating the reutilization of lost O2 from the lattice oxygen, thereby enhancing the reaction kinetics during the electrochemical reduction of released O2 on RuO2. The island-like RuO2 is firmly anchored to the surface of the LRMO, constructing an interface-confined catalysis array that effectively captures and catalyzes the reduction of trace amounts of escaped lattice oxygen. Therefore, the dynamic oxygen evolution equilibrium from the interface to the bulk in the LRMO is changed, effectively mitigating the outward diffusion of lattice oxygen and suppressing the formation of oxygen vacancies in the bulk of the LRMO. The lithium-deficient layer and interface-confined catalysis strategy gravely increase the discharge capacity of the LRMO battery from 230 mA h g−1 to 261 mA h g−1, while also improving the capacity retention rate from 75% to 97% after 300 cycles at 1C. The assembled Li metal pouch cell delivers a high energy density of 513 W h kg−1 and the Ru-1‖graphite pouch cell maintains an excellent capacity retention rate of 85% for 450 cycles. This approach provides new insight into improving the performance of LRMO batteries.
Results and discussion
The LRMO powder was synthesized using the coprecipitation method, and its morphological images are shown in Fig. S1 (ESI†). To reduce the reactivity of the surface lattice oxygen, the LRMO powder was immersed in RuCl3 solutions at varying concentrations (0.1, 0.2, and 0.6 mg mL−1). These samples are designated as Ru-0.5, Ru-1, and Ru-3, respectively. During the stirring process, Li+ ions on the surface of the LRMO are gradually exchanged with H+ ions, resulting in the formation of a gradient lithium-deficient layer and a uniform coating layer on the exterior. After filtration, washing with deionized water, and subsequent sintering, rutile RuO2 was anchored onto the surfaces of the primary LRMO particles (XRD patterns in Fig. S2a and b, ESI†). As shown in Fig. S3 and Table S1 (ESI†), the initial Coulombic efficiencies (ICEs) of the Ru-0.5, Ru-1, and Ru-3 samples at a rate of 1C are 95.8%, 97.1%, and 100.0%, respectively. Correspondingly, their discharge capacities during the first cycle are 309, 307, and 285 mA h g−1. After 300 cycles, the capacity retention rates of these samples are 96%, 98%, and 94%, respectively. As the concentration of Ru3+ and the acidity of the solution increase, more Li+ ions at the interface of the LRMO are extracted, leading to a reduction in lattice oxygen loss and consequently demonstrating a decrease in irreversible capacity and an increase in ICE. However, when the Ru3+ solution concentration is excessively high, the RuO2 deposited at the interface results in a reduction in the specific discharge capacity of the battery due to a decrease in the LRMO mass. Given its superior electrochemical performance, Ru-1 was selected for a comprehensive investigation.Fig. 1a shows the schematic diagram of the gradient lithium-deficient layer and the RuO2 interface-confined catalysis in Ru-1. The high-angle annular dark field scanning transmission electron microscopy (HAADF-STEM) images presented in Fig. 1b and c reveal that the tetragonal RuO2 is tightly anchored to the surface of the LRMO, exhibiting layered lattice striations. A 4–5 nm transition interlayer is observed between the RuO2 and the LRMO phase. As illustrated in the energy-dispersive X-ray spectroscopy (EDS) images (Fig. S4, ESI†), all elements, including island-like RuO2, are uniformly distributed in the Ru-1 sample. The powder X-ray diffraction (XRD) refinement results (Fig. S5, ESI†) demonstrate the characteristic peaks of the LRMO phase accompanied by the distinctive peak of RuO2, providing compelling evidence for the successful integration of the RuO2 coating layer in Ru-1. The detailed lattice parameters of the pristine sample and Ru-1 are shown in Table S2 (ESI†). Meanwhile, the inductively coupled plasma atomic emission spectroscopy (ICP-AES) results in Tables S3 and S4 (ESI†) detail the content of each element. The analysis reveals that in Ru-1, the chemical formula for the LRMO component is Li1.20Mn0.555Ni0.158Co0.080O2, with RuO2 constituting 0.46 wt% of the sample. During the ion exchange process, driven by the concentration gradient and the acidic RuCl3 solution, some Li+ ions from the surface of the LRMO bulk material are released into the solution. This effectively reduces the concentration of Li+ at the lithium-rich interface, thereby decreasing the number of Li–O–Li configurations and suppressing lattice oxygen oxidation. The electron energy loss spectroscopy (EELS), time-of-flight secondary ion mass spectrometry (TOF-SIMS), and X-ray photoelectron spectroscopy (XPS) results exhibit a gradient distribution of Li+ from the surface to the bulk. Fig. 1d illustrates the normalized EELS intensities for the distribution of Li and Mn at the interface boundary between RuO2 and the LRMO in Ru-1. The pale region corresponds to the RuO2 coating layer, and the blue region represents the bulk LRMO; between these two regions lies a transitional layer. The signal of Li gradually intensifies from the surface to the bulk, and the signal of Mn maintains a relatively constant level. In contrast, in the pristine sample, both the signals of Li and Mn exhibit minimal variation (Fig. 1e), indicating the presence of a gradient lithium-deficient layer in Ru-1. The raw data without fitting are shown in Fig. S6 (ESI†). Additionally, Fig. 1f and g present three-dimensional TOF-SIMS plots that further corroborate the aforementioned results. As the etching time increases, the Li signal in Ru-1 escalates progressively, whereas the Li signal in the pristine sample remains essentially unchanged. The corresponding 1D TOF-SIMS results are depicted in Fig. S7 (ESI†). To exclude the influence of the surface coating layer on the judgment of Li+ concentration, Ru-1 was first etched with argon ions for 360 seconds to eliminate the oxide coating's effect on the Li 1s signal for the XPS analysis. As shown in Fig. S8 (ESI†), after etching, Ru-1 still displays a lower Li 1s signal compared to the pristine sample, indicating semi-quantitatively that the Li+ concentration at the interface between the coating layer and the bulk material of Ru-1 is lower than that of the untreated sample.
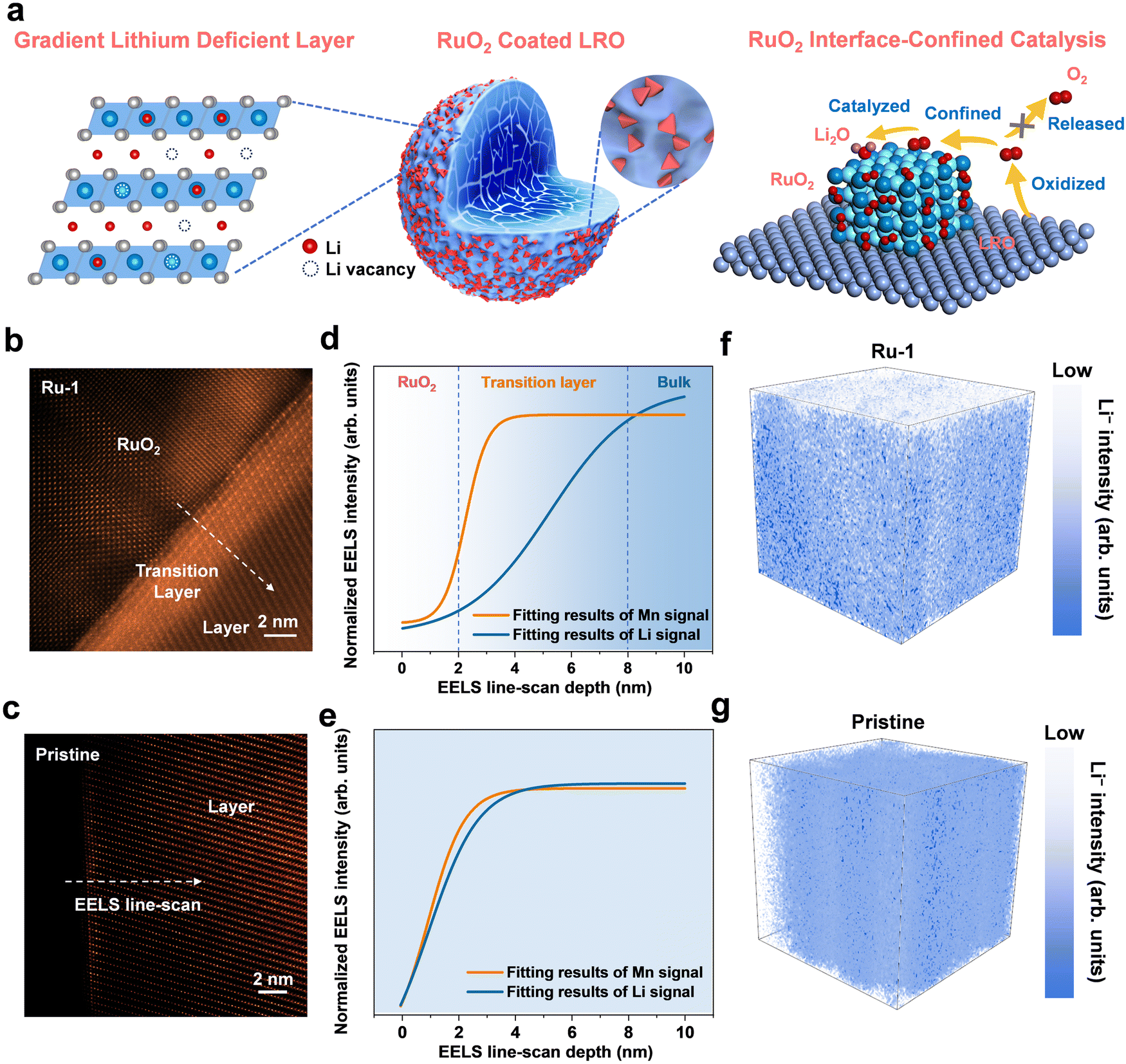 | ||
| Fig. 1 (a) Schematic diagram of the gradient lithium-deficient layer and RuO2 interface-confined catalysis in Ru-1. Characterization of a lithium-deficient layer. HADDF-STEM images of (b) Ru-1 and (c) the pristine sample. The normalized EELS intensities of the line-scan from the surface to the bulk of (d) Ru-1 and (e) pristine samples. The 3D plots of the TOF-SIMS Li− intensity of (f) Ru-1 and (g) pristine samples. The blue region corresponds to the high Li content. | ||
In situ surface-enhanced Raman scattering (SERS) spectroscopy is a powerful tool to provide insightful information on the operando surface during the electrochemical processes of batteries. According to Zhou et al., the Raman shift at 850 cm−1 can be attributed to the vibration of the lattice peroxo-like O–O bond, indicating a reversible redox process.26 As shown in Fig. 2a, during the charging process, with the voltage increasing, beyond the signal at 850 cm−1 associated with the lattice O–O bond, a Raman shift appears at 788 cm−1 in the pristine sample, corresponding to the formation of lithium peroxide (Li2O2).27 It is noteworthy that the initial formation of the Li2O2 signal is followed by a decline when the charging voltage exceeds 4.7 V, suggesting that at higher voltages, Li2O2 undergoes further oxidization to produce O2. The formation and subsequent decomposition of Li2O2 imply that lattice oxygen participates in a heterogeneous redox process within the lattice, causing the evolution and oxidation of the Li2O component. This process is irreversible and contributes to the depletion of lattice oxygen. Conversely, Fig. 2b demonstrates that the introduction of a lithium-deficient layer in Ru-1 ensures the reversible redox reaction of lattice oxygen (at 850 cm−1) without the formation of Li2O2.
 | ||
| Fig. 2 Lattice oxygen stability comparison between Ru-1 and the pristine sample. Spectra and contour plots of the in situ SRES tests and the corresponding charge and discharge curves of (a) pristine sample and (b) Ru-1. The XAS O K-edge spectra of (c) the pristine sample and (d) Ru-1. (e) DEMS spectra and the corresponding charge curves of Ru-1 and the pristine sample. | ||
The XPS results (etching depth: approximately 20 nm) of the pristine LRMO and Ru-1 are shown in Fig. S9 (ESI†). At an energy of 533 eV, the violet region corresponds to oxygen-containing species that have been deposited on the surface. The yellow area at 531.5 eV represents oxygen-containing species that are adsorbed onto the surface. The orange section at 530.9 eV is associated with peroxide species (O2)n−, and the blue region around 530 eV corresponds to lattice oxygen. When charged to voltages of 4.4 V, 4.6 V, and 4.8 V, the Ru-1 sample demonstrates peroxide ratios of 3.9%, 10.7%, and 16.0%, respectively. In contrast, the pristine LRMO sample exhibits a significantly higher proportion of peroxides, reaching 8.0%, 15.1%, and 23.1% at the same voltages. As shown in Fig. 2c and d, the O K-edge X-ray Absorption Spectroscopy (XAS) results reflect the oxidation state of lattice oxygen. The t2g peak at 532.5 eV is indicative of the electronic transition from O 1s to TM 3d t2g orbital, while the eg peak at 530.5 eV represents the electron transition to the TM 3d eg orbitals. The hump between these two peaks at 531.5 eV is regarded as the oxidation state of lattice oxygen, such as O2n−. At open circuit voltage (OCV), 4.6 V, and 4.8 V, it can be observed that the signal of O2n− at 531.5 eV in the Ru-1 is much lower than that of the pristine sample. Fig. S10 (ESI†) shows the difference of O K-edge XAS spectra between 4.6 V and OCV during the initial charge process. It is evident that there are fewer O2n−-like species at 4.6 V and 4.8 V in Ru-1, indicating that a smaller amount of lattice oxygen on the surface of Ru-1 has been oxidized owing to the introduction of a Li-poor interlayer. Furthermore, as shown in Fig. 2e, the differential electrochemical mass spectrometry (DEMS) results reveal a lower amount of O2 evolution for Ru-1 compared to the pristine sample. The XAS data of Mn, Ni, and Co L edges exhibit the dynamic ligand-to-metal charge transfer (LMCT) during the charging process.3 The LMCT process plays an important role in regulating the lattice oxygen redox activity of the LRMO. During the charge process, the electrons are initially extracted from transition metal (TM) 3d orbitals and the TM ions are oxidized to high-valence states, such as Co4+ and Ni4+. After that, due to the strong coupling of the lattice oxygen with the TM ions, the electrons of lattice oxygen are partially removed from the O 2p orbitals to the TM 3d orbitals, causing the oxidation of the lattice and the reduction of the TM ions. Initially, Ni and Co are oxidized to a higher valence state, followed by electron transfer from the oxygen non-bonding states to these high-valence metal ions, thereby triggering oxygen redox reactions.28,29 As demonstrated in Fig. S11–S13 (ESI†), the LMCT process for Ru-1 happens at 4.6 V, which is delayed compared to the pristine sample (4.4 V), meaning higher stability of the surface lattice oxygen in Ru-1. After the discharge process, the Mn in Ru-1 keeps in a higher oxidation state, consistent with its initial state, whereas a significant portion of Mn in the pristine sample is reduced to +3, suggesting substantial O2 evolution and structure rearrangement on the surface of the LRMO.
The oxidation behaviour of lattice oxygen was further elucidated through the cyclic voltammetry (CV) tests. As shown in Fig. S14a and b (ESI†), peaks A and B correspond to the oxidation processes of lattice oxygen and transition metals, respectively. The ratio of their current intensities, IA/IB, serves as a qualitative indicator of the degree of lattice oxygen oxidation. In Ru-1, this ratio is 2.67; whereas in the control sample, the ratio is as high as 3.62, indicating that the oxidation of lattice oxygen in Ru-1 is suppressed. Fig. 3a shows the cycling performance of Ru-1 and the pristine sample at 0.1C. Ru-1 exhibits higher capacity and enhanced cycling stability, maintaining a high discharge capacity exceeding 300 mA h g−1 after 120 cycles. As shown in Fig. 3b, at a current density of 1C, Ru-1 exhibits the highest reversible discharge capacity of 261 mA h g−1. After 300 cycles, it retains a high specific capacity of more than 250 mA h g−1 with a capacity retention rate of 98%. In contrast, the control material delivers a reversible specific capacity of 230 mA h g−1 and a capacity retention rate of 75% after 300 cycles. Moreover, Ru-1 shows a median voltage decay of only 509 mV after 500 cycles, corresponding to a decay of about 1.0 mV per cycle (Fig. S15, ESI†); whereas the pristine sample experiences a large voltage decay of 779 mV after 500 cycles, equating to a decay of about 1.6 mV per cycle. The suppression of voltage decay further illustrates the inhibitory effect of the coating layer on the phase transformation and ion migration within the bulk material. Ru-1 achieves an unprecedentedly high capacity and ICE. Fig. 3c shows the charge and discharge curves of Ru-1 during the first cycle at 0.1C (1C = 250 mA g−1). Ru-1 has an initial efficiency of 97%, while the pristine sample is only 86% (Fig. S16, ESI†), indicating that more lattice oxygen of the pristine sample is irreversibly oxidized and released during the charging process. As shown in Fig. S17 (ESI†), the impedance of Ru-1 is significantly decreased, indicating a higher electronic conductivity of Ru-1. As depicted in Fig. 3e, Ru-1 has superior rate performance. Even at a rate of 5C, Ru-1 still achieves a high discharge capacity of about 185 mA h g−1. In comparison to recent studies (Fig. 3f), Ru-1 demonstrates a competitively high specific discharge capacity and extended cycle stability.30–45 The detailed data can be found in Table S6 (ESI†). Furthermore, the CV curves at different scan rates for Ru-1 and pristine samples, presented in Fig. S18 (ESI†), indicate improved reaction kinetics for Ru-1. Additionally, the GITT curves (Fig. S19a, ESI†) reveal that Ru-1 exhibits a lower overpotential compared with the pristine sample, with the Li+ diffusion coefficients of Ru-1 being higher than those of the pristine sample (Fig. S19b, ESI†). Therefore, the deficiencies of Li in the LRMO structure promote the transport of Li+ and accelerate the electrochemical reaction kinetics. To evaluate the electrochemical performance of the full cell, a Ru-1‖graphite pouch cell was assembled. This configuration exhibits a high capacity retention rate of 85% after 450 cycles at C/3 (Fig. 3d) and a high energy efficiency of 91% (Fig. S20, ESI†). Furthermore, a 5.83 A h Ru-1‖Li pouch cell was constructed. As shown in Fig. 3g and h, this pouch cell exhibits a high energy density of 513 W h kg−1. The results of battery test, along with the weight and size measurements of the Ru-1‖Li pouch cell, are shown in Fig. S21 and S22 and Table S5 (ESI†). Meanwhile, a Ru-1‖Li pouch cell keeps a stable discharge capacity for 22 cycles (Fig. S23, ESI†). The electrochemical performances of the Ru-1‖Li pouch cell highlight the significant potential of Ru-1 as a cathode material for high-energy-density batteries.
 | ||
| Fig. 3 Electrochemical performances. Long cycling performances of Ru-1 and pristine sample at (a) 0.1C and (b) 1C. (c) The initial charge and discharge curves of Ru-1. (d) Long cycling performances of the Ru-1‖graphite pouch cell at C/3. (e) Rate performances of Ru-1 and the pristine sample. (f) Capacity retention rate and discharge capacity of Ru-1 compared to recent studies. (g) The initial charge and discharge curves of the 5.83 A h Ru-1‖Li pouch cell. (h) Parameters of the 5.83 A h Ru-1‖Li pouch cell. | ||
In situ XRD provides valuable insights into the phase transitions and strain changes during the charge and discharge processes. Fig. 4a and b present the in situ XRD contour plots for the pristine sample and Ru-1, along with their corresponding charge–discharge curves. The shift of the (003) peak serves as an indicator of lattice distortion within the cathode material. Apparently, a pronounced shift in the (003) peak of the pristine sample signifies substantial lattice distortion, which contributes to the irreversible oxidation of lattice oxygen. Conversely, the (003) peak of Ru-1 remains stable, suggesting minimal lattice distortion and structural change. The Rietveld refinement analysis, conducted using GSAS, indicates that Ru-1 exhibits a volume change of 2.69% during the first charge–discharge cycle, whereas the pristine sample shows a significantly higher value of 3.62%. To further investigate the volume changes and strain behaviour during the charging process, chemomechanical simulations were employed to examine the spatial distribution of Li+ concentration in the LRMO and the mechanical stress throughout the Li+ extraction process, using COMSOL software. As shown in Fig. 4c and d, driven by the electric field and concentration gradient, Li+ ions gradually deintercalate from the lattice. The pristine sample exhibits a gradient distribution of ion concentration that decreases from the interior to the exterior. With the progressive deintercalation of Li+, the ion concentration in the pristine sample manifests a gradient that diminishes from the core to the surface. Initially, the interface reaches a state of lithium depletion, experiencing a relatively smaller variation in Li+ concentration compared to its original state. However, in Ru-1, as the Li+ concentration decreases, the gradient lithium-deficient layer at the interface first attains a state of lower lithium content and then remains unchanged, resulting in a comparatively lower Li+ extraction at the interface when compared to the control group. Referring to the strain results from in situ XRD during the charging process, the stress at different delithiation states was simulated using COMSOL. The in situ XRD reveals a positive correlation between lattice strain and the absolute amount of deintercalated Li+. Consequently, the strain at the interface is comparatively smaller due to the relatively minor change in Li+ concentration at this region compared to the bulk, resulting in the formation of a buffer layer that prevents grain cracking and lattice structure damage. As a result, the peak stress in the Ru-1 amounts to 418 MPa (Table S7, ESI†), specifically within the interior of the interfacial buffer layer. Conversely, the pristine sample records a significantly higher peak stress of 567 MPa, situated at the surface, posing a risk of grain rupture. Therefore, the introduction of a lithium-deficient layer not only decreases the strain within the LRMO lattice on an atomic scale by hindering the release of lattice oxygen but also alleviates the strain at the primary particle level by controlling the lithium diffusion gradient.
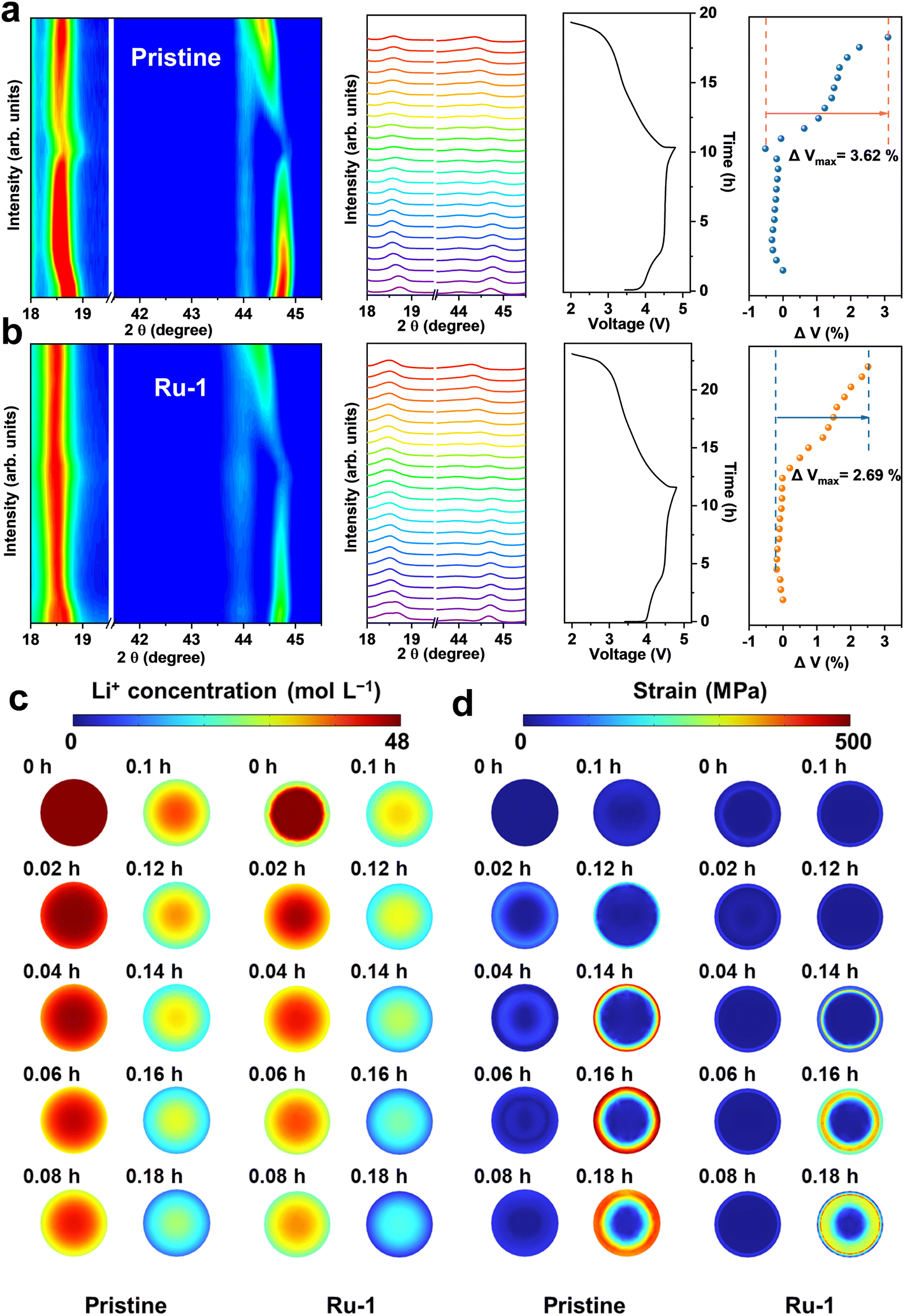 | ||
| Fig. 4 Strain changes during the electrochemical process. The contour plots and spectra of the in situ XRD results and the corresponding volume changes of (a) the pristine sample and (b) Ru-1. The volume data are derived from GSAS software. Visualization of the (c) concentration and (d) stain changes during the initial charge process. | ||
To explore the impact of the gradient lithium-deficient interlayer on interfacial stability, DFT calculations were conducted on samples with varying lithium contents. It is well established that Li2MnO3 serves as the primary oxygen-activating component in LRMO cathodes, imparting anionic redox characteristics to the lithium-rich material through its Li–O–Li configurations. As the lithium content decreases, the number of Li–O–Li configurations diminishes, significantly affecting the electronic density of states (DOS) of oxygen in Li2MnO3, thereby altering the redox properties of lattice oxygen. Using Li2MnO3 as the reference structure, a series of six models with progressively reduced lithium contents are constructed: Li30Mn18O48, Li29Mn19O48, Li28Mn20O48, Li27Mn21O48, Li26Mn22O48, and Li25Mn23O48. This is accomplished by the substitution of Li atoms in the TM layer with Mn atoms, as illustrated in Fig. S24 (ESI†). These models sequentially reduce the lithium content, mimicking the actual conditions of oxygen-activating components within a gradient lithium-deficient layer. Fig. 5a–f illustrate the distribution of partial electronic density of states for lattice O and Mn in different models. As the lithium content decreases, the proportion of O 2p electrons near the Fermi level (EF) diminishes, indicating a reduced participation of lattice oxygen in the oxidation reaction. Fig. 5g presents an integrated statistical graph of the electronic density of states for Mn and O within 1 eV below the EF. A decrease in the lithium content corresponds to a decline in the electron fraction attributed to oxygen within a 1 eV range below the EF, from 84.2% to 75.5%. Simultaneously, the electron fraction for Mn rises from 15.8% to 24.5%. This shift reflects a decreasing contribution to capacity from oxygen and a corresponding increase from manganese. Notably, the term ‘O’ refers to oxygen atoms bonded to Mn atoms; thus, the electron fraction of O consistently exceeds that of Mn. The lithium-deficient layer suppresses oxygen activity and the coupling between O 2p orbitals with the TM 3d orbitals, thereby delaying the LMCT process in the LRMO, as shown in Fig. S11–S13 (ESI†). This well-controlled oxygen activity contributes to maintaining a high reversible lattice oxygen redox process and enhancing the cycling stability of the LRMO. Subsequently, Li2MnO3, Li30Mn18O48, and Li28Mn20O48 are employed as representative models to investigate the diffusive behaviour of Li+ within the Li layer, characterized by a decremental lithium gradient. The schematic representations of the diffusion models are delineated in Fig. S25 (ESI†). Fig. 5h illustrates a progressive decline in the energy barrier for Li+ diffusion as the lithium content diminishes. This means a lower lithium content facilitates ion transport at the interface within the Li layer, thereby reducing polarization and enhancing the overall electrochemical performance, which aligns with the conclusions derived from GITT results.
 | ||
| Fig. 5 Electron density distributions and diffusion energy barriers of Li-rich components with different Li contents. PDOS plots of (a) Li30Mn18O48, (b) Li29Mn19O48, (c) Li28Mn20O48, (d) Li27Mn21O48, (e) Li26Mn22O48, and (f) Li25Mn23O48. (g) The calculated integrated area of the electron densities of Li30Mn18O48, Li29Mn19O48, Li28Mn20O48, Li27Mn21O48, Li26Mn22O48, and Li25Mn23O48. With a decrease of the Li content, the electron content of oxygen is also lowered gradually. (h) The interlayer Li+ transportation energy barriers of Li2MnO3, Li30Mn18O48, and Li28Mn20O48. | ||
In comparison to the pristine sample, Ru-1 exhibits extremely high capacity and cycling stability. To understand such performance enhancement, besides the effect of a lithium-deficient interface layer and the suppression of lattice oxygen release, a more in-depth investigation into the function of ruthenium oxide at the interface is warranted. In situ galvanostatic electrochemical impedance spectroscopy (GEIS) tests (Fig. S26, ESI†) were conducted to evaluate the reaction kinetics of the O2 formation and reduction process. By mathematically converting the GEIS data, the relaxation time distribution (DRT) results can be obtained.46 As shown in Fig. 6a, the relaxation time τ ranging from 10−2 to 10 s and from 10 to 102 s can be ascribed to charge transfer (Rct) and solid-state diffusion (Rdiff), respectively.23,46 During the charging process, the Rct of Ru-1 at high voltage surpasses that of the pristine sample, indicating a kinetically inhibited LMCT process and the limited oxidation of lattice oxygen. Nevertheless, upon discharging, the Rct effectively decreases, indicating a kinetically promoted O2 reduction process, which can be attributed to the catalytic effect of RuO2.
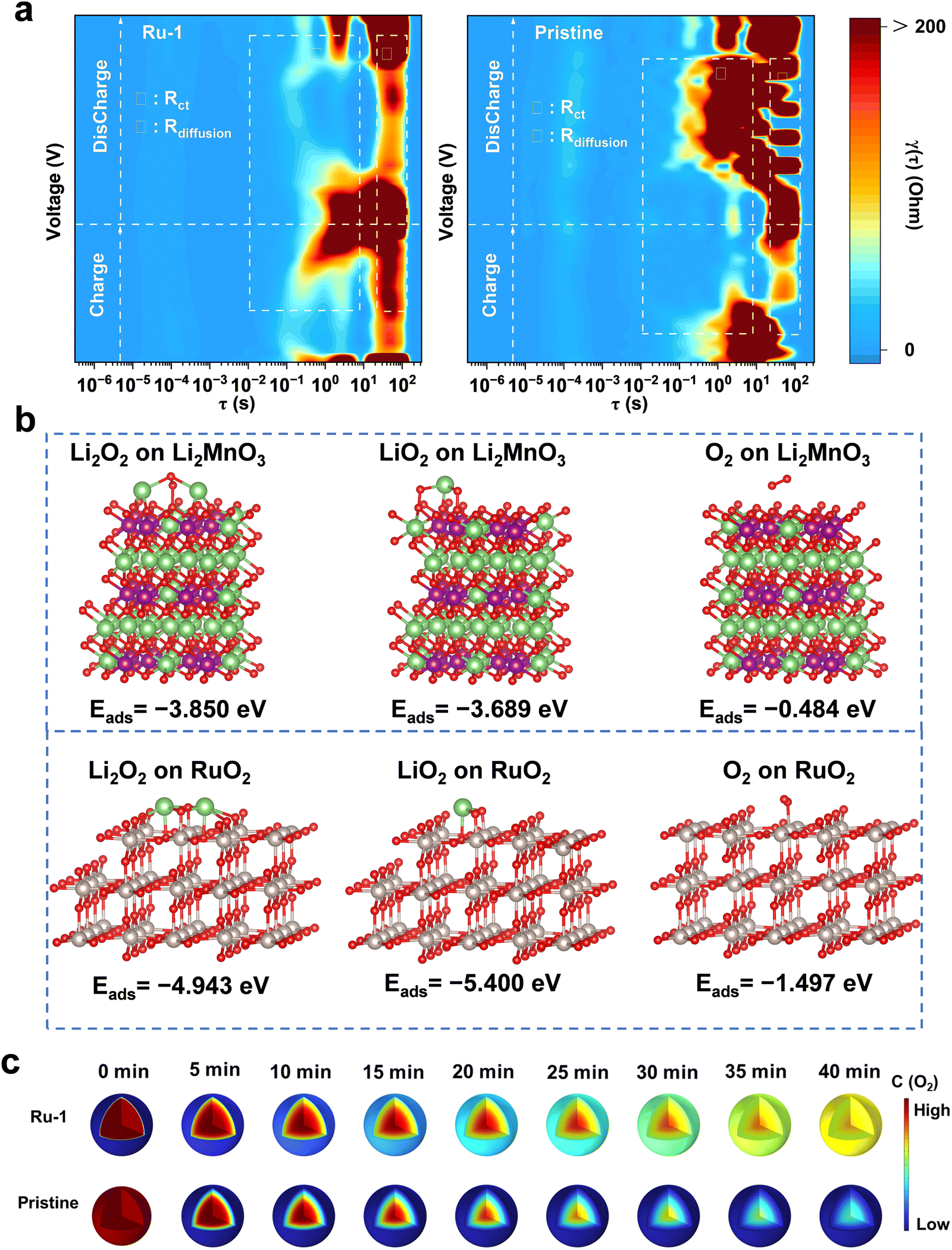 | ||
| Fig. 6 The adsorption and catalytic capability evaluation. (a) DRT profile transformation from GEIS of Ru-1 and the pristine sample. (b) The models and adsorption energies of Li2O2, LiO2, and O2 adsorbed on Li2MnO3 and RuO2. (c) O2 diffusion simulation of Ru-1 and the pristine sample with COMSOL software. | ||
Gibbs free energy pathways were calculated to reveal the catalytic capability of the RuO2 during the discharge process, specifically the oxygen electrochemical reduction process. The whole electrochemical reduction of O2 is divided into three distinct processes, each representing an electron donating. As shown in Fig. S27 (ESI†), the rate-determining processes are the electrochemical reduction of Li2O2 for both the Li2MnO3 and RuO2, which exhibit the highest free energies among the three reduction processes. Therefore, in these two materials, the reduction of Li2O2 governs the extent of O2 reduction during the discharge process, thereby characterizing the catalytic activity of O2 in the LRMO battery system, which in turn affects the discharge specific capacity. Evidently, the free energy of Li2O2 on RuO2 is 6.99 eV, which is much lower than that of Li2MnO3 (9.02 eV). This indicates that the conversion energy barrier for the whole O2 electrochemical reduction process is considerably lower on RuO2 compared to Li2MnO3, indicative of the outstanding catalyzing capability of RuO2. The remarkable adsorption and catalytic performance of RuO2 facilitate its function as an active site for O2 adsorption and electrochemical catalysis, thereby promoting the confinement and conversion of activated oxygen species.
Previous studies have demonstrated that the Li2MnO3 phase at the LRMO interface exhibits significant capabilities in gas adsorption and catalysis, leading to its selection as a reference for comparative analysis. Through DFT calculations, we evaluated the adsorptive and catalytic properties concerning the active oxygen species Li2O2, LiO2, and O2 for both Li2MnO3 and RuO2. Fig. 6b presents the adsorption energies of Li2MnO3 for Li2O2, LiO2, and O2, which are measured at −3.850 eV, −3.689 eV, and −0.484 eV, respectively. In contrast, the adsorption energies of RuO2 for these oxygen species are −4.943 eV for Li2O2, −5.400 eV for LiO2, and −1.497 eV for O2, all of which surpass those of Li2MnO3. The elevated adsorption energy implies that in the Ru-1 sample, even trace amounts of released oxygen will be effectively captured at the interface of the cathode, maintaining electrochemical contact and facilitating interfacial confinement. Conversely, in the pristine sample, the active oxygen species and emitted oxygen are dispersed throughout the battery system, which not only results in the loss of electrochemical interaction and the subsequent failure to reduce the oxidized lattice oxygen, but also introduces grave safety hazards. Meanwhile, the change in O2 concentration at the RuO2 containing interface significantly influences the overall O2 diffusion equilibrium in the primary particle. The distribution of dynamic O2 concentration is simulated using COMSOL software, as shown in Fig. 6c. For the pristine sample, O2 undergoes unhindered diffusion and rapidly escapes from the bulk. Within 40 minutes, the O2 concentration decreases from the surface to the bulk, leading to severe lattice distortion and the formation of oxygen vacancy. Nevertheless, the RuO2 oxygen sequestration array (blue shell in Fig. 6c, Ru-1, 0 min) preferentially captures oxygen molecules, thereby regulating the equilibrium of O2 diffusion and inhibiting further O2 evolution. Therefore, after 40 minutes, the oxygen concentration in Ru-1 is much higher and more evenly distributed than that of the pristine sample. The high O2 concentration within the Ru-1 lattice is thermodynamically beneficial to hinder the formation of oxygen vacancy.
XRD, Raman, HRTEM, and TOF-SIMS tests were performed on the cathode material to investigate the phase transformation and surface side reaction after 200 cycles at 1C. The phase structure of the pristine sample undergoes notable degradation, while Ru-1 maintains a well-preserved layered structure. As illustrated in Fig. S28 (ESI†), the ratio of I003/I104 is commonly employed to assess the integrity of the layered structure in the LRMO.47 After cycling, the pristine sample exhibits an I003/I104 ratio of 1.46, significantly higher than the initial value of 1.09, which suggests a substantial alteration in the material's structural anisotropy during the cycling process. Additionally, Raman spectroscopy serves as a valuable tool for monitoring the phase transformation of the cathode material. The Raman shift at 625 cm−1 indicates the presence of a spinel phase component, and the fitting results reveal that the pristine sample exhibits an increased proportion of the spinel phase structure after cycling (Fig. S29, ESI†).48 Moreover, the local phase transformation of the cathode material was analyzed. As shown in Fig. S30 (ESI†), the layered structure is well-preserved, and the lattice of the tetragonal RuO2 at the interface remains unimpaired in Ru-1. In contrast, the pristine sample displays a substantial emergence of rock-salt and spinel phases at the interface, suggesting a profound structural reorganization as a result of lattice oxygen emission. The EDS results (Fig. S31, ESI†) provide the elemental distribution of the Ru-1 sample after 200 cycles. The elements O, Mn, Ni, Co, and F (F arising from the decomposition of LiPF6) are uniformly distributed on the surface of the Ru-1 secondary particles. Ru is present in an island-like distribution on the surface, similar to the distribution observed in Fig. S4 (ESI†), indicating that RuO2 remains tightly anchored to the LRMO interface even after 200 cycles. The interfaces of the cycled Ru-1 and pristine samples were investigated through TOF-SIMS analysis. As shown in Fig. S32 (ESI†), the intensity of three-dimension depth signals of the MnF3−, NiF3−, and CoF3− in the Ru-1 sample is much lower than that of the pristine sample, indicating fewer surface side reactions and slighter transition metal dissolution in Ru-1.49 The cathode–electrolyte interface (CEI) fragments, including PO2−, PF6− and LiF2−, are significantly more concentrated at the interface in Ru-1 compared to the pristine sample, suggesting a robust and compact CEI in Ru-1.50–52 The corresponding two-dimension overlay mapping images and depth curves are shown in Fig. S33 and S34 (ESI†), respectively. The XRD, Raman, HRTEM, and TOF-SIMS analyses collectively demonstrate that the Ru-1 sample maintains a well-preserved layered structure with minimal phase transformation, while the pristine sample undergoes significant surface and bulk degradation, exhibiting increased spinel and rock-salt phases due to lattice oxygen emission, highlighting the stabilizing effect of RuO2 at the LRMO interface after prolonged cycling.
Conclusions
In summary, we have developed a bifunctional method that utilizes ion exchange between H+ ions and surface Li+ to construct a lithium-deficient layer and a RuO2 interface-confined catalysis network. The lithium-deficient layer effectively prevents the loss of surface lattice oxygen, promotes ion transportation, and alleviates strain within both the lattice and the primary particle. Concurrently, the RuO2 interface-confined catalysis network powerfully adsorbs the trace amounts of activated oxygen species and accelerates the reaction kinetics of the reduction of O2. As a result, this approach significantly improves the reversible discharge capacity of the LRMO from 230 mA h g−1 to 261 mA h g−1 at 1C and the capacity retention rate from 75% to 97%. The Ru-1‖graphite pouch cell exhibits a superior capacity retention rate of 85% after 450 cycles at C/3, and the 5.83 A h Ru-1‖Li pouch cell exhibits an energy density of 513 W h kg−1. Our work demonstrates a new feasible strategy for the reutilization of activated oxygen species within the LRMO system, effectively enhancing its battery performance and indicating a promising future for the commercialization of LRMOs.Author contributions
J. P. S., H. Y., and J. L. S. contributed equally to this work. J. P. S. designed and conducted the experiments, analyzed the data, and drafted the manuscript. H. Y. and Y. Y. supervised the research and manuscript preparation. J. L. S. and X. J. W. carried out theoretical calculations. H. D. Q. and Y. S. performed the COMSOL simulations. M. S. conducted the HADDF-STEM experiments. Y. H. L., Y. Y. and X. H. R. analyzed the XAS data. All authors approved the final version of the manuscript.Data availability
The authors will supply the relevant data in response to reasonable requests.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. 51925207, 52394170, 52394171, U24A2067, 52372239, and U23A20579), the Fundamental Research Funds for the Central Universities (WK9990000170, YD2060002042, and 20720220010), the Liaoning Binhai Laboratory (Grant No. LBLF-2023-03), and the “Transformational Technologies for Clean Energy and Demonstration” Strategic Priority Research Program of the Chinese Academy of Sciences (Grant No. XDA0400202), and the State Key Laboratory of Catalysis (2024SKL-B-003). The authors thank the USTC Supercomputing Center for providing computational resources for this project. The O K-edge, Ni, Co, and Mn L-edge XAS tests were recorded on Beamlines MCD-A and MCD-B (Soochow Beamline for Energy Materials) at the National Synchrotron Radiation Laboratory (NSRL). The SEM, TEM, XPS and ICP tests were performed in the Physical and Chemical Science Experiment Center of the University of Science and Technology of China (USTC). We thank the USTC Supercomputing Center for providing computational resources for this project.References
- M. Armand and J. M. Tarascon, Nature, 2008, 451, 652–657 CrossRef CAS PubMed.
- M. M. Thackeray, S.-H. Kang, C. S. Johnson, J. T. Vaughey, R. Benedek and S. A. Hackney, J. Mater. Chem., 2007, 17, 3112–3125 CAS.
- M. Sathiya, G. Rousse, K. Ramesha, C. P. Laisa, H. Vezin, M. T. Sougrati, M. L. Doublet, D. Foix, D. Gonbeau, W. Walker, A. S. Prakash, M. Ben Hassine, L. Dupont and J. M. Tarascon, Nat. Mater., 2013, 12, 827–835 Search PubMed.
- B. Li, K. Zhang, Y. Yang, Y. Zuo, X. Li and D. Xia, Adv. Mater., 2024, 36, 2400259 CrossRef CAS PubMed.
- J. Peng, H. Li, L. Chen and F. Wu, Precis. Chem., 2023, 1, 452–467 Search PubMed.
- C. Zhu, X. Mu, P. A. van Aken, Y. Yu and J. Maier, Angew. Chem., Int. Ed., 2014, 53, 2152–2156 CrossRef CAS PubMed.
- S. Chen, L. Shen, P. A. van Aken, J. Maier and Y. Yu, Adv. Mater., 2017, 29, 1605650 CrossRef PubMed.
- P. Puech, S. Chen, Y. Xiang, T. Hu, M. Monthioux, L. Zhang and F. Li, Carbon Future, 2024, 1, 9200026 Search PubMed.
- R. A. House, G. J. Rees, M. A. Pérez-Osorio, J.-J. Marie, E. Boivin, A. W. Robertson, A. Nag, M. Garcia-Fernandez, K.-J. Zhou and P. G. Bruce, Nat. Energy, 2020, 5, 777–785 CrossRef CAS.
- M. Sathiya, A. M. Abakumov, D. Foix, G. Rousse, K. Ramesha, M. Saubanère, M. L. Doublet, H. Vezin, C. P. Laisa, A. S. Prakash, D. Gonbeau, G. VanTendeloo and J. M. Tarascon, Nat. Mater., 2015, 14, 230–238 CrossRef CAS PubMed.
- E. Hu, X. Yu, R. Lin, X. Bi, J. Lu, S. Bak, K.-W. Nam, H. L. Xin, C. Jaye, D. A. Fischer, K. Amine and X.-Q. Yang, Nat. Energy, 2018, 3, 690–698 CrossRef CAS.
- D.-H. Seo, J. Lee, A. Urban, R. Malik, S. Kang and G. Ceder, Nat. Chem., 2016, 8, 692–697 CrossRef CAS PubMed.
- M. Saubanère, E. McCalla, J. M. Tarascon and M. L. Doublet, Energy Environ. Sci., 2016, 9, 984–991 RSC.
- Y. Xie, M. Saubanère and M. L. Doublet, Energy Environ. Sci., 2017, 10, 266–274 RSC.
- W. Guo, W. Wei, H. Zhu, Y. Hu, H. Jiang and C. Li, eScience, 2023, 3, 100082 CrossRef.
- Y. Guo, C. Guo, P. Huang, Q. Han, F. Wang, H. Zhang, H. Liu, Y.-C. Cao, Y. Yao and Y. Huang, eScience, 2023, 3, 100091 CrossRef.
- W. He, W. Guo, H. Wu, L. Lin, Q. Liu, X. Han, Q. Xie, P. Liu, H. Zheng, L. Wang, X. Yu and D.-L. Peng, Adv. Mater., 2021, 33, 2005937 Search PubMed.
- P. M. Csernica, S. S. Kalirai, W. E. Gent, K. Lim, Y.-S. Yu, Y. Liu, S.-J. Ahn, E. Kaeli, X. Xu, K. H. Stone, A. F. Marshall, R. Sinclair, D. A. Shapiro, M. F. Toney and W. C. Chueh, Nat. Energy, 2021, 6, 642–652 CrossRef CAS.
- Y. Lei, J. Ni, Z. Hu, Z. Wang, F. Gui, B. Li, P. Ming, C. Zhang, Y. Elias, D. Aurbach and Q. Xiao, Adv. Energy Mater., 2020, 10, 2002506 CrossRef CAS.
- P. Yan, J. Zheng, Z.-K. Tang, A. Devaraj, G. Chen, K. Amine, J.-G. Zhang, L.-M. Liu and C. Wang, Nat. Nanotechnol., 2019, 14, 602–608 CrossRef CAS PubMed.
- J.-J. Marie, R. A. House, G. J. Rees, A. W. Robertson, M. Jenkins, J. Chen, S. Agrestini, M. Garcia-Fernandez, K.-J. Zhou and P. G. Bruce, Nat. Mater., 2024, 23, 818–825 CrossRef CAS PubMed.
- X.-D. Zhang, J.-L. Shi, J.-Y. Liang, Y.-X. Yin, J.-N. Zhang, X.-Q. Yu and Y.-G. Guo, Adv. Mater., 2018, 30, 1801751 CrossRef PubMed.
- S. Sun, C.-Z. Zhao, H. Yuan, Z.-H. Fu, X. Chen, Y. Lu, Y.-F. Li, J.-K. Hu, J. Dong, J.-Q. Huang, M. Ouyang and Q. Zhang, Sci. Adv., 2022, 8, eadd5189 CrossRef CAS PubMed.
- E. Yilmaz, C. Yogi, K. Yamanaka, T. Ohta and H. R. Byon, Nano Lett., 2013, 13, 4679–4684 CrossRef CAS PubMed.
- Z. Fang, W. Zhao, T. Shen, D. Qiu, Y. Lv, X. Hou and Y. Hou, Precis. Chem., 2023, 1, 395–417 CrossRef CAS.
- X. Li, Y. Qiao, S. Guo, Z. Xu, H. Zhu, X. Zhang, Y. Yuan, P. He, M. Ishida and H. Zhou, Adv. Mater., 2018, 30, 1705197 CrossRef PubMed.
- Y. Qiao, S. Wu, J. Yi, Y. Sun, S. Guo, S. Yang, P. He and H. Zhou, Angew. Chem., Int. Ed., 2017, 56, 4960–4964 CrossRef CAS PubMed.
- M. D. Radin, J. Vinckeviciute, R. Seshadri and A. Van der Ven, Nat. Energy, 2019, 4, 639–646 CrossRef CAS.
- B. Li, Z. Zhuo, L. Zhang, A. Iadecola, X. Gao, J. Guo, W. Yang, A. V. Morozov, A. M. Abakumov and J.-M. Tarascon, Nat. Mater., 2023, 22, 1370–1379 CrossRef CAS PubMed.
- Y. Wei, J. Cheng, D. Li, Y. Li, Z. Zeng, H. Liu, H. Zhang, F. Ji, X. Geng, J. Lu and L. Ci, Adv. Funct. Mater., 2023, 33, 2214775 CrossRef CAS.
- W. Jiang, C. Zhang, Y. Feng, B. Wei, L. Chen, R. Zhang, D. G. Ivey, P. Wang and W. Wei, Energy Storage Mater., 2020, 32, 37–45 CrossRef.
- D. Luo, X. Ding, J. Fan, Z. Zhang, P. Liu, X. Yang, J. Guo, S. Sun and Z. Lin, Angew. Chem., Int. Ed., 2020, 59, 23061–23066 Search PubMed.
- E. Wang, Y. Zhao, D. Xiao, X. Zhang, T. Wu, B. Wang, M. Zubair, Y. Li, X. Sun and H. Yu, Adv. Mater., 2020, 32, 1906070 Search PubMed.
- V. Murugan, R. S. Arul Saravanan, K. Thangaian, T. Partheeban, V. Aravindan, M. Srinivasan, M. Sasidharan and K. K. Bharathi, ACS Appl. Energy Mater., 2021, 4, 11234–11247 CrossRef CAS.
- Z. Ye, B. Zhang, T. Chen, Z. Wu, D. Wang, W. Xiang, Y. Sun, Y. Liu, Y. Liu, J. Zhang, Y. Song and X. Guo, Angew. Chem., Int. Ed., 2021, 60, 23248–23255 CrossRef CAS PubMed.
- D. Luo, H. Xie, F. Tan, X. Ding, J. Cui, X. Xie, C. Liu and Z. Lin, Angew. Chem., Int. Ed., 2022, 61, e202203698 CrossRef CAS PubMed.
- K. Zhang, J. Qi, J. Song, Y. Zuo, Y. Yang, T. Yang, T. Chen, X. Liu, L. Chen and D. Xia, Adv. Mater., 2022, 34, 2109564 CrossRef CAS PubMed.
- Y. Fan, E. Olsson, G. Liang, Z. Wang, A. M. D’Angelo, B. Johannessen, L. Thomsen, B. Cowie, J. Li, F. Zhang, Y. Zhao, W. K. Pang, Q. Cai and Z. Guo, Angew. Chem., Int. Ed., 2023, 62, e202213806 CrossRef CAS PubMed.
- S. Li, C. Guan, W. Zhang, H. Li, X. Gao, S. Zhang, S. Li, Y. Lai and Z. Zhang, Small, 2023, 19, 2303539 CrossRef CAS PubMed.
- Y. Li, Z. Shi, B. Qiu, J. Zhao, X. Li, Y. Zhang, T. Li, Q. Gu, J. Gao and Z. Liu, Adv. Funct. Mater., 2023, 33, 2302236 CrossRef CAS.
- J. Song, F. Ning, Y. Zuo, A. Li, H. Wang, K. Zhang, T. Yang, Y. Yang, C. Gao, W. Xiao, Z. Jiang, T. Chen, G. Feng and D. Xia, Adv. Mater., 2023, 35, 2208726 CrossRef CAS PubMed.
- H. Wu, J. Dong, Y. Zhang, L. Lin, G. Gao, T. Li, X. Yi, B. Sa, J. Wang, L. Wang, J. Li, K. Amine, D.-L. Peng and Q. Xie, Adv. Funct. Mater., 2023, 33, 2303707 Search PubMed.
- Y. Yang, C. Gao, T. Luo, J. Song, T. Yang, H. Wang, K. Zhang, Y. Zuo, W. Xiao, Z. Jiang, T. Chen and D. Xia, Adv. Mater., 2023, 35, 2307138 CrossRef CAS PubMed.
- B. Zhao, C. Shen, H. Yan, J. Xie, X. Liu, Y. Dai, J. Zhang, J.-c Zheng, L. Wu, Y. Zhu and Y. Jiang, Chem. Eng. J., 2023, 465, 142928 CrossRef CAS.
- Z. Xu, X. Guo, W. Song, J. Wang, T. Qin, Y. Yuan and J. Lu, Adv. Mater., 2024, 36, 2303612 CrossRef CAS PubMed.
- Y. Lu, C.-Z. Zhao, J.-Q. Huang and Q. Zhang, Joule, 2022, 6, 1172–1198 CrossRef CAS.
- B. J. Hwang, Y. W. Tsai, D. Carlier and G. Ceder, Chem. Mater., 2003, 15, 3676–3682 Search PubMed.
- J. Hong, D.-H. Seo, S.-W. Kim, H. Gwon, S.-T. Oh and K. Kang, J. Mater. Chem., 2010, 20, 10179–10186 RSC.
- W. Li, A. Dolocan, P. Oh, H. Celio, S. Park, J. Cho and A. Manthiram, Nat. Commun., 2017, 8, 14589 CrossRef PubMed.
- Z. Zhang, L. Hu, H. Wu, W. Weng, M. Koh, P. C. Redfern, L. A. Curtiss and K. Amine, Energy Environ. Sci., 2013, 6, 1806–1810 RSC.
- X. Fan, L. Chen, O. Borodin, X. Ji, J. Chen, S. Hou, T. Deng, J. Zheng, C. Yang, S.-C. Liou, K. Amine, K. Xu and C. Wang, Nat. Nanotechnol., 2018, 13, 715–722 CrossRef CAS PubMed.
- C. Wang, Y. S. Meng and K. Xu, J. Electrochem. Soc., 2019, 166, A5184 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ee00430f |
| This journal is © The Royal Society of Chemistry 2025 |