Dual-anion ionic liquid electrolytes: a strategy for achieving high stability and conductivity in lithium metal batteries†
Received
8th January 2025
, Accepted 10th March 2025
First published on 13th March 2025
Abstract
Ionic liquid electrolytes (ILEs) provide promising thermal and electrochemical stability characteristics for safer lithium metal batteries (LMBs). However, their development faces challenges due to their low ionic conductivity and poor wettability on separators. In this study, we introduce a dual-anion locally concentrated ionic-liquid electrolyte (D-LCILE), designed with a diluent and two distinct anions to significantly improve the ionic conductivity and wettability. These improvements were confirmed through electrochemical impedance spectroscopy (EIS) measurements on stainless steel symmetric cells, contact angle tests, and rate capability assessments on a 300 μm thick lithium metal half-cell. Notably, the dual-anion design enhances the interfacial stability, as density functional theory (DFT) calculations revealed a more stable solvation shell structure, further supported by molecular dynamics (MD) simulations. Additionally, scanning electron microscopy (SEM) experiments confirmed the deposition of a thin and, dense lithium layer, while X-ray photoelectron spectroscopy (XPS) depth profile analysis showed a stable solid electrolyte interphase (SEI) with increased LiF content. Performance tests on a 20 μm-thick Li‖LiFePO4 full cell revealed an average Coulombic efficiency exceeding 99.90% and capacity retention >99.93% after 200 cycles at 1C, making D-LCILE a highly promising candidate for next-generation, high-performance LMBs.
Broader context
The pursuit of high-performance and safe energy storage solutions is essential for advancing sustainable energy systems, particularly with the increasing global demand for renewable energy integration and electrification. Among next-generation battery technologies, lithium metal batteries (LMBs) have gained significant attention due to their high theoretical capacity and low electrochemical potential. However, dendrite growth, unstable solid electrolyte interphase (SEI), and safety concerns remain major obstacles to commercialization. This study introduces a dual-anion locally concentrated ionic liquid electrolyte (D-LCILE), incorporating bis(trifluoromethanesulfonyl)imide (TFSI−) and bis(fluorosulfonyl)imide (FSI−) anions with a diluent. This formulation overcomes trade-offs in conventional ionic liquid electrolytes (ILEs) and locally concentrated ionic liquid electrolytes (LCILEs) by improving ionic conductivity, interfacial stability, and wettability. The synergistic effect of TFSI− and FSI− anions enables a LiF-rich SEI, enhanced lithium-ion mobility, and mitigated viscosity. These findings are supported by time-of-flight secondary ion mass spectrometry (ToF-SIMS), electrochemical impedance spectroscopy (EIS), molecular dynamics (MD) simulations, and X-ray photoelectron spectroscopy (XPS) depth profiling, revealing a fluorine content increase from 11.56% to 27.62%. Full-cell and symmetric cell cycling tests confirm superior Coulombic efficiency and capacity retention with D-LCILE, establishing it as a promising electrolyte for safe, high-energy-density LMBs in renewable energy storage and electrified transportation.
|
1 Introduction
The development of safe, high-performance energy storage technologies is essential for building a sustainable society. Among various battery types, lithium metal batteries (LMBs) stand out as a promising candidate in this field, owing to the high theoretical capacity and low electrochemical potential of lithium metal, which result in exceptionally high energy densities.1–4 These characteristics make LMBs especially suitable for applications requiring long-term, high-capacity energy storage. However, although lithium metal provides an outstanding energy density, it poses significant challenges that must be addressed to realize its commercial potential. Key issues include the growth of lithium dendrites and the instability of the solid electrolyte interphase (SEI), both of which cause safety risks and reduced cycle life. Ensuring stable and safe LMB performances requires advanced electrolyte solutions that can stabilize lithium deposition and promote the formation of an effective SEI.5,6 Ionic liquid electrolytes (ILEs) have gained significant attention for their intrinsic non-flammability and high thermal stability, which provide promising solutions to address the safety concerns associated with LMBs. Unlike conventional organic electrolytes, ILEs have no vapor pressure, thus reducing the risk of fire and fitting well with the requirement of safe, sustainable energy storage. However, conventional ILEs suffer from low ionic conductivity and poor wettability on lithium metal and separator surfaces, hindering ion transport and increasing the internal cell resistance. These limitations can reduce the performance of LMBs, especially under high-rate conditions, emphasizing the need for electrolyte designs that balance safety with improved ionic mobility and interfacial stability.7–11 Locally concentrated ionic liquid electrolytes (LCILEs) have emerged as an innovative system to improve the ionic mobility and SEI stability, specifically addressing the limitations in conductivity and wettability affecting traditional ILEs.11–16 By creating high local concentrations of active ions, LCILEs enhance the SEI formation, promote the formation of LiF-rich interphases, and improve the lithium-ion transport, which are all essential features for stable cycling in LMBs.17–19 A unique advantage of LCILEs is that introducing a diluent at the same salt concentration can reduce the electrolyte viscosity, resulting in an improved fluidity and, ionic mobility, and reducing the internal resistance without sacrificing the locally concentrated environment. However, despite these improvements, LCILEs continue to face significant challenges, such as residual viscosity issues under high-rate conditions and inadequate wettability on lithium surfaces and separators, highlighting the need for further optimization.20–22 To address these specific limitations, we introduce a novel dual-anion locally concentrated ionic liquid electrolyte (D-LCILE), which uniquely combines bis(trifluoromethanesulfonyl)imide anion (TFSI−) and bis(fluorosulfonyl)imide anion (FSI−) with a co-solvent diluent to simultaneously enhance the ionic conductivity, reduce the viscosity, and improve the interfacial stability. This dual-anion design reduces the viscosity, enhances the ion mobility, and improves the interfacial stability through several mechanisms. First, the use of two structurally distinct anions minimizes strong ionic interactions, preventing uniform ion structures, and favoring diverse ion distributions.15,23–26 This design control ion pairing and aggregate formation, both of which typically affect the viscosity. In particular, the larger and chemically more stable TFSI− ion interacts more dynamically with the solvent, further reducing the viscosity. This is also related to the improvement in Li-ion mobility and viscosity, which arises from the bimodal effect of the two anions with different sizes. Moreover, the presence of anions of different sizes promotes the formation of distinct solvation shells around lithium ions, optimizing the flexibility and fluidity of these structures, thus increasing ionic mobility.27,28 The dual-anion structure also contributes to the SEI stability by forming dense solvation shells induced by ions of different sizes around lithium metal, promoting the deposition of a thin, homogeneous lithium layer and the formation of a stable SEI. Specifically, FSI− facilitates the formation of LiF, a highly stable SEI component that resists dendritic growth, while TFSI−, in combination with the diluent, reduces the viscosity, further improving the lithium-ion mobility and surface wettability.29 This improved design makes D-LCILE an advanced electrolyte solution combining high ionic conductivity and structural stability, effectively addressing the limitations of conventional ILEs and LCILEs for safe and efficient lithium metal batteries. To evaluate the performance of the D-LCILEs, we employed a range of electrochemical and structural characterization techniques. Raman spectroscopy and molecular dynamics (MD) simulations confirmed the improved solvation structure and ionic mobility, while X-ray photoelectron spectroscopy (XPS) and scanning electron microscopy (SEM) measurements demonstrated the formation of a uniform SEI with high LiF content. Electrochemical impedance spectroscopy (EIS) and cyclic voltammetry (CV) measurements further confirmed the enhanced ionic conductivity and electrochemical stability of D-LCILE. A full cell comprising an LFP cathode and a thin lithium metal anode, with bis(2,2,2-trifluoroethyl) ether (BTFE) as the diluent in the D-LCILE electrolyte and incorporating LiTFSI and LiFSI salts, exhibited a capacity retention of 99.93% over 200 cycles at a 1.0C rate. Thus, D-LCILE provides a balanced solution that mitigates both viscosity and wettability limitations, which makes it an advanced electrolyte for safe and efficient lithium metal batteries, fitting well with sustainable energy goals.
2 Results and discussion
2.1 Electrolyte properties and ionic transport
Four distinct electrolytes were synthesized using EMIFSI as solvent, incorporating LiFSI and LiTFSI salts with BTFE as diluent: molar fraction compositions are detailed in Table S1 (ESI†). Comprehensive structural and solvation analyses were performed using Raman spectroscopy to examine the solvation dynamics within the ILEs and LCILEs. The analysis specifically differentiated free anions, contact ion pairs (CIPs), and aggregates (AGGs) by focusing on the S–N–S stretching peaks between 700 and 750 cm−1, characteristic of both FSI− and TFSI− anions.30,31 Owing to the larger, more complex structure of TFSI−, the Raman peak shifted toward higher S–N–S stretching energies, shows an expanded solvation shell, as illustrated in Fig. 1(a). Fig. 1(a) also shows that the dual-anion electrolyte (D-CILE) exhibited significant peak shifts to higher energy levels compared to the single-anion electrolyte (CILE), a trend observed in the LCILE and D-LCILE systems with BTFE.32–34 As shown in Fig. 1(d), the use of dual anions or a diluent such as BTFE enlarges the primary solvation shell, resulting in a higher proportion of AGGs. Fig. 1(b) and (c) reveal that the AGG proportion increased by approximately 74%, while the CIP proportion decreased by 15.1%. The addition of BTFE and the dual-anion configuration further reduces the proportion of CIPs while increasing that of AGGs [Fig. 1(b) and (e)], resulting in a ∼128% AGG increase and a 6.8% CIP decrease in LCILE. The D-LCILE electrolyte enhanced the AGG formation by 54% and decreased the CIP proportion by 30% compared to LCILE when both dual-anion configuration and diluent were used, as shown in Fig. 1(e) and (f). Compared to conventional electrolytes such as 1 M LiPF6 in EC/DEC (v/v = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), which typically exhibit solvent-separated ion pairs (SSIPs), CIPs, AGGs, and free anions, the EMIFSI-based systems did not contain SSIP structures [Fig. 1(d)]. This absence results from the ionic nature of EMIFSI, where direct solvation by a molecular solvent is minimal. The specific reason why the use of BTFE diluent increases the proportion of AGGs is that BTFE, being a non-polar solvent, lowers the solvation energy of the solvent, facilitating the formation of clusters rather than individual ion bonds.35 This enhances the likelihood of the solvent anion, FSI−, interacting with Li+ ions and creates a locally concentrated solution by reducing the overall concentration of the solution. Locally concentrated ion clusters primarily take the form of AGGs. Furthermore, since BTFE does not act as a solvent, it does not interfere with or engage in interactions between [EMIm+] and [FSI−], which allows direct bonding between Li+ and FSI−/TFSI− ions, thereby promoting AGG formation. When TFSI− is used as a dual anion, its larger size compared to FSI− results in a lower interaction energy with Li+ ions, enabling weaker interactions. This slightly reduces the viscosity of the solution, increases the flexibility of the solvation shell, and allows relatively freer ion placement within the solvation shell, influencing Li+ desolvation energy.16–19 Additionally, the large size of TFSI− improves the separation of electrons and ions at the electrode surface, enhancing the stability of the EDL.36–38 The presence of the BTFE diluent indirectly influences the ionic configurations by occupying the volume without dissolving FSI− or TFSI−, which expands the Li+ primary solvation shell without increasing the inter-ionic distances. Fig. 1(e) and (f) show that the inclusion of BTFE altered the ionic distributions, enhancing the aggregation between FSI− and TFSI− ions without increasing the separation. The dual-anion combination of FSI− and TFSI− creates further differences in the solvation behavior: FSI− forms compact ion pairs due to its smaller size and higher polarity, while TFSI− establishes looser, extended interactions because of its larger size and lower polarity. This configuration leads to a heterogeneous primary solvation shell around Li+, where BTFE further promotes aggregation by modifying the spatial arrangement of ions. This expanded yet densely packed solvation environment affects the ionic mobility and stability, highlighting how dual-anion and diluent configurations control the solvation shells and ion interactions to enhance the electrolyte performance.
:1), which typically exhibit solvent-separated ion pairs (SSIPs), CIPs, AGGs, and free anions, the EMIFSI-based systems did not contain SSIP structures [Fig. 1(d)]. This absence results from the ionic nature of EMIFSI, where direct solvation by a molecular solvent is minimal. The specific reason why the use of BTFE diluent increases the proportion of AGGs is that BTFE, being a non-polar solvent, lowers the solvation energy of the solvent, facilitating the formation of clusters rather than individual ion bonds.35 This enhances the likelihood of the solvent anion, FSI−, interacting with Li+ ions and creates a locally concentrated solution by reducing the overall concentration of the solution. Locally concentrated ion clusters primarily take the form of AGGs. Furthermore, since BTFE does not act as a solvent, it does not interfere with or engage in interactions between [EMIm+] and [FSI−], which allows direct bonding between Li+ and FSI−/TFSI− ions, thereby promoting AGG formation. When TFSI− is used as a dual anion, its larger size compared to FSI− results in a lower interaction energy with Li+ ions, enabling weaker interactions. This slightly reduces the viscosity of the solution, increases the flexibility of the solvation shell, and allows relatively freer ion placement within the solvation shell, influencing Li+ desolvation energy.16–19 Additionally, the large size of TFSI− improves the separation of electrons and ions at the electrode surface, enhancing the stability of the EDL.36–38 The presence of the BTFE diluent indirectly influences the ionic configurations by occupying the volume without dissolving FSI− or TFSI−, which expands the Li+ primary solvation shell without increasing the inter-ionic distances. Fig. 1(e) and (f) show that the inclusion of BTFE altered the ionic distributions, enhancing the aggregation between FSI− and TFSI− ions without increasing the separation. The dual-anion combination of FSI− and TFSI− creates further differences in the solvation behavior: FSI− forms compact ion pairs due to its smaller size and higher polarity, while TFSI− establishes looser, extended interactions because of its larger size and lower polarity. This configuration leads to a heterogeneous primary solvation shell around Li+, where BTFE further promotes aggregation by modifying the spatial arrangement of ions. This expanded yet densely packed solvation environment affects the ionic mobility and stability, highlighting how dual-anion and diluent configurations control the solvation shells and ion interactions to enhance the electrolyte performance.
 |
| | Fig. 1 Raman spectra in the 650–800 cm−1 region: (a) CILE, D-CILE, LCILE, and D-LCILE; (d) free anion, CIP, and AGGs fractions; (b), (c), (e) and (f) peak deconvolutions of (b) CILE, (c) D-CILE, (e) LCILE, and (f) D-LCILE. | |
The findings in Fig. 1 are further supported by the radial distribution function (RDF) analysis conducted through classical molecular dynamics simulations, as shown in Fig. 2. In this analysis, only the anions and Li ions of each electrolyte system were isolated, enabling the RDF evaluation of the distance between Li ions and nitrogen atoms. The results show a ∼48% increase in the g(r) value (from 23.77 to 35.22) from CILE to D-CILE, providing additional evidence of an enlarged solvation shell. In the case of LCILE, the g(r) value reached 46.72, representing an increase of around 97%, in agreement with the Raman analysis results discussed above. Although D-LCILE displayed a g(r) value of 48.86, showing a relatively small increase of ∼4.6% over LCILE, this minimal discrepancy with the Raman analysis results is likely due to limitations of the simulation approach.39,40
 |
| | Fig. 2 (a) MD simulation snapshots of CILE, D-CILE, LCILE, and D-LCILE; (b) radial distribution functions and coordination number of Li–N interactions in corresponding to the snapshots. | |
As shown in Fig. 3(a), the wettability of each electrolyte was assessed by measuring the contact angle on the PE separator, in order to determine the suitability of the electrolytes. The ionic liquid electrolyte, CILE, exhibited a contact angle of 75.39° on the PE separator, while D-CILE showed a reduced contact angle of 59.99°. In comparison, LCILE displayed an even lower contact angle of 49.40°, while D-LCILE exhibited the lowest contact angle of 35.91°. These results suggest that the dual-anion configuration enhances the wettability on the separator compared to the single-anion structure, with the addition of a diluent further improving the compatibility with the PE separator.41,42 Fig. S2 (ESI†) confirms that this trend was also observed for 20 μm thin lithium metal, indicating that dual anions and diluents can indirectly improve the Li-ion mobility within the electrolyte. As shown in Fig. 3(b), symmetrical SS‖SS cells were fabricated using each electrolyte and subjected to EIS measurements. The Nyquist plots were analyzed to calculate the ionic conductivity using the following equation:43,44
| |  | (1) |
Where
σ is the ionic conductivity,
l is the thickness of the separator,
Rb denotes the bulk resistance, and
A represents the area of the electrode.
 |
| | Fig. 3 Characteristics of ionic liquid-based electrolytes (CILE, D-CILE, LCILE, and D-LCILE): (a) wettability on PE separator, (b) ionic conductivity, (c) Li+ transference number measured by Bruce–Vincent method, and (d) diffusion coefficient measured from MSD plot. | |
The results revealed that CILE, D-CILE, and LCILE had ionic conductivities of 0.165, 0.180, and 0.476 mS cm−1, respectively, while D-LCILE showed the highest conductivity of 1.053 mS cm−1, indicating that the presence of a diluent significantly influenced the ionic conductivity. As shown in Fig. 3(c), symmetrical Li‖Li cells were fabricated, and subjected to EIS measurements, similar to the above. The Nyquist plots were analyzed, and the Li-ion transference numbers were estimated using the Bruce–Vincent method, according to the following equation:12,16,44,45
| |  | (2) |
Where
I0 and
Iss are the initial and steady-state currents, Δ
V is the voltage difference,
R0 denotes the initial resistance, and
Rss represents the steady-state resistance.
The results showed Li-ion transference numbers of 0.465 for CILE and 0.819 for D-CILE. In contrast, LCILE and D-LCILE exhibited transference numbers of 0.875 and 0.879 respectively, demonstrating that the use of dual anions and diluents led to significant increases in the Li-ion transference number, with the diluent having a particularly strong effect. To further evaluate the ionic mobility, classical molecular dynamics (CMD) simulations were conducted to calculate the mean squared displacement (MSD) of Li ions, using the following equation:
| | 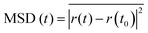 | (3) |
The MSD analysis provides insights into the diffusion behavior of Li ions over time. A steeper slope of the MSD plot indicates a higher diffusion coefficient (D), which reflects a greater ionic mobility. The diffusion coefficient value can be determined from the MSD using the following relationship:
| |  | (4) |
The diffusion coefficient for each electrolyte can be estimated, by calculating the slope of the MSD plot, enabling a comparative analysis along with the ionic conductivity. As shown in Fig. 3(d), the variation in slope became more pronounced in the presence of a diluent, indicating that the lithium-ion diffusion was significantly enhanced in this case.46,47 The slope of the MSD plot for D-CILE was on average larger than that of CILE, further suggesting enhanced ionic mobility in D-CILE. Compared to CILE the slopes of the LCILE or D-LCILE, plots increased by nearly four times. While the increased slope for D-LCILE is in good agreement with the observed improvements in ionic conductivity, the slope observed for LCILE appears to be overestimated in comparison, revealing some discrepancies that require further investigation. Furthermore, despite the addition of BTFE to the concentrated ionic liquid, the electrolyte exhibited flame-retardant property es when ignited with a torch, as shown in Fig. S5 (ESI†).
Li‖SS cells were fabricated to assess the electrochemical stability using LSV at a scan rate of 0.1 mV s−1, and the results are shown in Fig. 4. The oxidation stability window for the CILE and D-CILE ionic liquids was observed to be around 5.4 V, while the LCILEs exhibited a stability window of approximately 4.2 V owing to the influence of the diluent.13–15,17 These results indicate that a sufficient operating voltage could be achieved when using LFP cathodes and that the electrolytes are also suitable for commercial applications with NCM cathodes.48
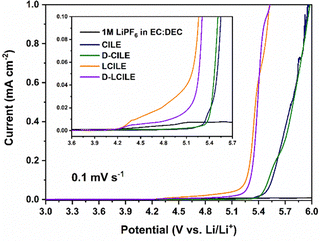 |
| | Fig. 4 Linear sweep voltammograms of Li‖SS cells in 1 M LiPF6 in EC:DEC (1:1, v/v), CILE, D-CILE, LCILE, and D-LCILE at a scan rate of 0.1 mV s−1. | |
2.2 Interfacial stability and SEI formation
To analyze the interfacial properties of the SEI and anode according to the type of electrolyte, the advanced Aurbach method was applied to Li‖Cu cells to examine the lithium-ion consumption in ionic liquid electrolytes.49,50 The average Coulombic efficiency over 10 cycles was used to estimate the lithium-ion consumption. The results indicated that LCILE, a single-anion electrolyte with an added diluent, exhibited an average Coulombic efficiency of 98.89%, whereas D-LCILE, with a dual-anion configuration, showed an efficiency of 99.18%. This suggests that D-LCILE had a higher average lithium recovery rate, indicating that the dual-anion structure resulted in more reversible reactions and suppressed the electrolyte decomposition due to lithium-ion consumption.51 Additionally, as shown in Fig. 5(b), the overpotential during initial lithium nucleation on the Cu substrate was higher in D-LCILE (65.1 mV) than LCILE (52.4 mV). The increased overpotential in D-LCILE can be attributed to the formation of a denser electric double layer (EDL) at the interface, which hinders the lithium-ion nucleation and thus requires more energy.52 The compact ionic layer in D-LCILE, observed in the MD simulation (Fig. 2) reveals a closer ion proximity in the dual-anion system, and the Raman spectroscopy results in Fig. 1 further confirm a denser ionic environment owing to the dual-anion configuration. The dense ionic layer created by the simultaneous presence and decomposition of FSI and TFSI ions increases the energy barrier for nucleation, resulting in the observed higher overpotential.36–38 However, it should be noted that a higher overpotential during plating/stripping does not necessarily indicate a greater interfacial stability. Further investigations are required to fully understand this relationship.
 |
| | Fig. 5 Electrochemical performance of LCILE and D-LCILE in a Li‖Cu cell configuration: (a) average coulombic efficiency for LCILE and D-LCILE measured by the advanced Aurbach method at 0.4 mA cm−2, and (b) voltage overpotential at 0.4 mA cm−2. | |
To investigate the characteristics of the SEI layer, Li‖Cu cells were fabricated and subjected to charge–discharge cycling to observe the lithium plating behavior. Following cycling at a capacity of 1 mA h cm−2 and a current density of 1 mA cm−2, we obtained both top and cross-sectional SEM images, as shown in Fig. 6. The ionic liquid electrolytes CILE and D-CILE, with high viscosity and low wettability, exhibited poor ionic conductivity, resulting in unsuccessful lithium plating. Fig. S2 (ESI†) confirmed the wettability of each electrolyte on lithium metal, indicating that conventional ionic liquid electrolytes may not effectively facilitate the lithium transfer. The top-view SEM images of the Cu side in Fig. 6(a)–(d) illustrate the lithium ion plating behavior. As shown in Fig. 6(a), the lithium plating in CILE had an irregular and sparse appearance. Although D-CILE showed improvements over CILE [Fig. 6(b)], it still exhibited an irregular and thick lithium plating. These irregular Li metal structures were visible in both the top-view images in Fig. 6(a) and (b) and the cross-sectional views in Fig. 6(e) and (f). In contrast, Fig. 6(c) shows that a more regular lithium plating pattern was achieved with LCILE (incorporating a diluent), with a thickness of 18.44 μm as confirmed in Fig. 6(g). When both a dual-anion configuration and a diluent were used in D-LCILE [Fig. 6(d)], the lithium plating was dense and highly uniform, with a significantly lower thickness of 13.49 μm, as shown in Fig. 6(h). These findings suggest that the dual-anion configuration of the electrolyte, particularly when combined with a diluent, promotes the formation of a more stable and efficient SEI layer, a critical improvement for enhancing the overall performance and longevity of lithium metal batteries.16–19 In the LCILE systems, the increased proportion of AGGs contributes to the formation of a denser SEI.35,38,53 Specifically, during the charging process, while Li+ ions migrate from the cathode to the anode, anions typically exhibit the opposite movement. However, due to the interaction between Li+ ions and anions forming solvation shells, there is a higher likelihood of anions migrating alongside Li+ ions to the anode side during charging. This migration results in a higher local concentration of anions on the surface of the Li metal electrode, promoting the formation of an SEI enriched with F-rich SEI.54,55 For the dual-anion systems utilizing TFSI−, although TFSI− itself may not significantly contribute to the F-rich SEI formation due to its structural stability and resistance to decomposition, its inherent stability and larger size enhance the formation of a robust EDL at the electrode surface. The increased overpotential in D-LCILE is likely due to a denser EDL at the interface, which hinders the lithium-ion nucleation and thus requires more energy.36–38 The compact ionic layer in D-LCILE, as observed in the MD simulation in Fig. 2, shows closer ion proximity in the dual-anion system, and the Raman spectroscopy results in Fig. 2 further confirm the presence of a denser ionic environment resulting from the dual-anion configuration. The dense ionic layer formed by the simultaneous presence and decomposition of FSI and TFSI ions results in an increased energy barrier for nucleation, leading to the observed higher overpotential.52 However, it should be noted that a higher overpotential during plating/stripping does not necessarily indicate an improved interfacial stability.
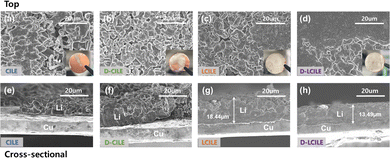 |
| | Fig. 6 SEM images of Li plated on a Cu substrate in different electrolytes at a current density of 1 mA cm−2 and an areal capacity of 1 mA h cm−2: (a)–(d) top-view images and (e)–(h) cross-sectional images. | |
The SEI layer forms naturally at the interface between electrolyte and lithium metal. To identify the components contributing to SEI formation, a 20 μm thin Li metal symmetric cell was fabricated and subjected to charge–discharge cycling at 0.5 mA cm−2 and 1 mA h cm−2. After 20 cycles, the cell was disassembled, and the composition of the Li metal electrode surface was analyzed using XPS. Depth profiling by XPS allowed us to analyze elemental changes with SEI depth. Fig. 7(a)–(c) show that the outer layer of the SEI was composed of carbon and oxygen-based organic components, as evidenced by a rapid decrease in the carbon content as etching proceeded. At the same time, a significant increase was observed in the fluorine content, indicating that the SEI layer consisted of an organic-dominated outer plane and an inner plane rich in fluorine-based inorganic components. As shown in Fig. 7(d)–(g), further analysis of the O 1s and F 1s spectra confirmed that the concentration of LiF increased with the SEI depth, while the outer layer contained organic compounds such as Li2CO3 or Li2O. This trend was observed for both LCILE and D-LCILE, although D-LCILE displayed a significantly higher fluorine content, with an increase from 11.56% at 0 s of etching to 27.62% at 250 s, compared to the increase from 6.15% to 19.43% observed for LCILE over the same interval. Conversely, the carbon content in LCILE decreased from 45.52% at 0 s to 9.00% at 250 s, whereas in D-LCILE, it dropped from 39.85% to 6.71% during the same etching period. According to previous studies, SEI layers formed primarily from carbon-based organic compounds tend to dissolve into the electrolyte over time, owing to ongoing interfacial reactions.56–58 This results in weak thermal and mechanical stabilities, causing frequent damage and breakdown of the SEI layer, which in turn leads to an unstable interface and repeated SEI regeneration. In contrast, the inorganic component LiF, found in higher concentrations in the SEI of D-LCILE, possesses high mechanical strength and electrochemical stability, preventing dissolution and damage; this leads to a more stable interface that effectively suppresses dendrite formation, enhancing the durability of the SEI.59
 |
| | Fig. 7 XPS depth analysis of Li metal surface in LCILE and D-LCILE after 50 cycles: atomic fraction evolutions from depth profiles in (a) LCILE (b) D-LCILE; (c) relative elemental compositions at 0 and 250 s; O 1s and F 1s depth profiles in (d) and (e) LCILE and (f) and (g) D-LCILE. | |
To investigate the interfacial stability and long-term plating/stripping behavior of lithium, Li symmetric cells were fabricated using 20 μm thin lithium metal. During lifespan tests using these cells, which have a limited lithium supply, short-circuiting was observed. As shown in Fig. 8(a) and (c), CILE and D-CILE exhibited unstable behavior in the initial stages of charge–discharge cycling. This instability is attributed to internal short circuits caused by voltage hysteresis, low wettability, and poor conductivity on Li metal. Conventional ionic liquid electrolytes are known to exhibit low wettability on Li metal, preventing stable charge–discharge cycling. In contrast, the locally concentrated ionic liquid electrolytes, LCILE and D-LCILE, demonstrated excellent wettability on Li metal. Consequently, Fig. 8(a) shows that these two electrolytes operated stably for 20 cycles at a current density of 0.5 mA cm−2 and a capacity of 1 mA h cm−2. However, as shown in Fig. 8(d), LCILE experienced a sudden voltage drop at the 23rd cycle, indicating a short circuit. This phenomenon is not considered a complete short circuit; previous studies suggest that this behavior results from continuous electron flow through micro short circuits formed within the cell, along with charge transfer through ionic carriers. In this experiment, the behavior observed at the 23rd cycle with LCILE was identified as a short circuit caused by growth of micro-dendrites within the separator.60,61 In contrast, D-LCILE operated stably without voltage drops for 60 cycles, indicating a longer lifespan. A similar trend was observed even at a higher current density of 3 mA cm−2. As shown in Fig. 8(b) and (e), while LCILE experienced a voltage drop and short circuit at the 58th cycle, D-LCILE remained stable for over 200 cycles, demonstrating a significantly higher stability. This suggests that the dual-anion structure is more effective at suppressing short circuits compared to the single-anion counterpart. To further evaluate the electrolyte stability at high current densities, we performed critical current density (CCD) measurements, as shown in Fig. 8(f). LCILE maintained a stable performance up to 3 mA cm−2, with partial instability beginning at 5 mA cm−2. In contrast, D-LCILE remained stable up to 5 mA cm−2, with instability observed only at 10 mA cm−2. These results demonstrate the higher stability of D-LCILE compared to LCILE, particularly at elevated current densities.
 |
| | Fig. 8 Symmetric cell test with 20 μm-thick Li: (a) voltage profiles in CILE, D-CILE, LCILE, and D-LCILE at 0.5 mA cm−2 and 1 mA h cm−2, (b) voltage profiles in LCILE and D-LCILE at 3 mA cm−2 and 1 mA h cm−2, (c) first five cycles, (d) five cycles after 80 hours at 0.5 mA cm−2 and 1 mA h cm−2, and (e) eight cycles after 36 h at 3 mA cm−2 and 1 mA h cm−2, (f) critical current density measurements of LCILE and D-LCILE. | |
As shown in Fig. 9, AIMD simulations were conducted at 298 K using lithium metal along with EMIFSI and EMITFSI ion pairs to examine the anion dissociation. The results revealed that, for the FSI anion, dissociation occurred as early as 0.13 ps, with F detaching from the anion, followed by a second fluorine dissociation event observed at 1.05 ps. In contrast, the TFSI anion exhibited strong bonding within the CF3 group, preventing F dissociation over a 5 ps period. Instead, the decomposition of TFSI led to the formation of species such as CF3 and SO2. These findings suggest that the FSI anion contributes significantly to the formation of a LiF-rich SEI, while the TFSI anion, enhances the Li-ion transfer, owing to its higher stability.15 These results are consistent with the data presented in Fig. 4.
 |
| | Fig. 9 Dissociation mechanism of FSI and TFSI anions analyzed using DFT. | |
2.3 Full cell performance and cycling
Full cells were fabricated with each electrolyte using LFP and 20 μm thin lithium metal electrodes to evaluate the cycling performance. Post-cycling EIS analysis was also conducted. Fig. 10(a) and (b) show the Nyquist plots for LCILE and D-LCILE after 3, 10, 30, and 50 cycles, respectively. After three cycles, the Warburg impedance indicated a smaller slope in LCILE compared to D-LCILE, suggesting a more favorable mass transfer in the latter.62 Additionally, D-LCILE maintained a lower internal resistance throughout the cycling process, with consistently reduced charge transfer resistance (Rct), consistent with the SEI stability observed in Fig. 6 and 7. The SEI layer formed in D-LCILE, with its dual-anion structure comprising FSI and TFSI, exhibited a reduced charge transfer resistance owing to a higher LiF proportion and an enhanced stability, resulting in a higher interfacial stability over repeated charge–discharge cycles and an extended cell lifespan.59,63Fig. 10(c) presents the cycle performance of 20 μm thick Li‖LFP cells with LCILE and D-LCILE. Consistent with the experimental results shown in Fig. 8, D-LCILE with dual-anion configuration maintained stable capacity and high Coulombic efficiency over 200 cycles. In contrast, LCILE, with single-anion structure, failed to reach 100 cycles, showing significant declines in both capacity and Coulombic efficiency. The superior cycling performance of D-LCILE is likely due to its stable SEI formation, which minimizes the continuous electrolyte consumption and reduces the capacity fading over time, ultimately extending the cell lifespan. As shown in Fig. 10(d), CILE and D-CILE displayed irregular charge responses and eventual cell failure, indicating that they are not suitable for full-cell applications, owing to their very low wettability on the PE separator and insufficient ionic conductivity. These observations are consistent with the results presented in Fig. 8(a) and (c). An increase in Coulombic efficiency was observed with LCILE, as evidenced by the voltage profile in Fig. 10(e), where the constant voltage (CV) charging phase extended progressively with cycling. This led to increased charge capacity, decreased discharge capacity, and a subsequent decline in Coulombic efficiency, a pattern commonly seen in cells affected by soft shorts.60,64 In contrast, the voltage profile of D-LCILE in Fig. 10(f) reveals a stable performance over 200 cycles, showing a reversible and stable voltage profile without an extended CV charging phase. These different electrochemical properties of the electrolytes are consistent with the test results for the 20 μm thick Li symmetric cell. The GCD performance of cells with D-LCILE showed high-capacity retention and stable cycling, with a retention rate of 99.93% over 200 cycles at a 1.0C rate, highlighting the potential of D-LCILE for high-energy density and long-lifespan applications. Similar trends were observed in Li‖LFP half-cells using a thicker (300 μm) lithium metal electrode, where D-LCILE retained 101.97% of its initial capacity over 300 cycles at a 1.0C rate, as shown in Fig. S8 and S9 (ESI†).
 |
| | Fig. 10 Evaluation of thin Li‖LFP full cell in CILE, D-CILE, LCILE, and D-LCILE: (a) Nyquist plots of LCILE and, (b) D-LCILE during cycling; (c) cycling performance of full cell in LCILE and D-LCILE; voltage profiles of cell in (d) CILE and D-CILE, (e) LCILE, and (f) D-LCILE. | |
3 Conclusions
This study demonstrates the potential of D-LCILE as an advanced electrolyte for LMBs. By combining TFSI− and FSI− anions with a diluent, D-LCILE achieved significant improvements in key areas crucial to the LMB performance. Compared to traditional single-anion systems, D-LCILE exhibited a higher ionic conductivity of 1.053 mS cm−1, larger Li+ transference numbers, and enhanced wettability on PE separators; the combination of these properties resulted in an enhanced ion mobility and a reduced internal cell resistance. Additionally, the flame-retardant properties of D-LCILE further contribute to its safety, making it a viable candidate for high-energy-density storage applications. The stability and mechanical integrity of the SEI in D-LCILE were significantly enhanced, as evidenced by the high LiF content and reduced carbon content observed in the XPS depth profile. The dual-anion structure promoted the formation of a dense and uniform SEI, which contributed to improve the Coulombic efficiency, achieving an average value of 99.18% in Li‖Cu cells and effectively suppressing dendritic growth. The SEM analysis of the lithium plating morphology confirmed the formation of a more stable and thinner SEI layer (13.49 μm) in D-LCILE compared to LCILE, which mitigated dendrite formation and extended the battery life. Cycling tests further demonstrated the performance of D-LCILE in practical applications. In full Li‖LFP cells, D-LCILE exhibited a capacity retention of 99.93% over 200 cycles at a 1.0C rate, highlighting its ability to maintain long-term cycling stability. In Li symmetric cells, D-LCILE exhibited stable cycling without voltage drops for up to 60 cycles, along with critical current density stability up to 5 mA cm−2, outperforming LCILE and demonstrating superior stability at high current densities. These findings demonstrate the suitability of D-LCILE for high-demand applications requiring long cycle life and high-power capabilities. Owing to these combined advantages, D-LCILE offers a balanced solution for safe and efficient lithium metal batteries, addressing both stability and conductivity challenges in energy storage applications.
4 Experimental
4.1 Cell configuration
A Li iron phosphate (LFP) cathode was prepared by dispersing LFP powder (MTI Korea), conductive carbon (Super P, TIMCAL), and poly(vinylidene difluoride) (PVdF, HSV 900, Kynar) in N-methyl-2-pyrrolidone (NMP, Sigma Aldrich, anhydrous, 99.5%) solvent. The slurry was mixed at 2000 rpm for 5 minutes using a planetary centrifugal mixer (AR100, Thinky). The resulting slurry was cast onto a 13-μm-thick aluminum foil (Korea JCC) and dried in a convection oven at 80 °C for 3 h. The cathode active material's loading mass was about 6 mg cm−2. The dried electrodes were punched into 14 mm diameter discs, then further dried in a vacuum oven at 120 °C for 12 h. The prepared electrodes were stored in an argon-filled glovebox (O2 < 0.1 ppm, H2O < 0.1 ppm, Korea Kiyon). For comparative purposes, we used a conventional electrolyte consisting of, 1 M LiPF6 in ethylene carbonate (EC) and diethyl carbonate (DEC) in 1:1 (v/v) ratio (Enchem). A polyethylene (PE) membrane (9 μm, Enerever Battery Solution) was used as the separator. Negative electrodes included etched copper foil (9 μm, MTI Korea), 300-μm-thick Li metal (Honjo Metal Co.), and a 20-μm-thick Li electrode laminated on a 9-μm-thick Cu substrate (Honjo Metal Co.), all punched into 14 mm diameter discs in a glovebox. To eliminate moisture, lithium bis(fluorosulfonyl)imide (LiFSI, ultra dry, 99.9% trace metals basis, Sigma Aldrich), lithium bis(trifluoromethanesulfonyl)imide (LiTFSI, anhydrous, 99.99% trace metals basis, Sigma Aldrich), and 1-ethyl-3-methylimidazolium bis(fluorosulfonyl)imide (EMIFSI, TCI Chemicals, >98%, HPLC) were vacuum-dried at 60 °C for 2 days. The diluent, bis(2,2,2-trifluoroethyl) ether (BTFE, 98%, Sigma Aldrich), was dried with molecular sieves in a glovebox.
4.2 Electrolyte preparation
Concentrated ionic liquid electrolytes (CILEs) were prepared by dissolving the dried LiFSI and LiTFSI in EMIFSI in molar ratios of 0.6:0:1 and 0.4:0.2:1, to produce CILE and D-CILE, respectively. The locally concentrated ILEs (LCILE and D-LCILE) were prepared by adding BTFE to CILE and D-CILE in a 1:1 molar ratio to EMIFSI. The molar ratios of the electrolytes are detailed in Table S1 (ESI†). Each electrolyte mixture was stirred magnetically for over 12 h at room temperature to ensure homogeneity.
4.3 Electrochemical measurements
Electrochemical tests were conducted on CR2032 coin cells (Sinopro MRX Co.) assembled with a PE separator and 70 μL of electrolyte. For Li‖Cu asymmetric cells and Li‖LFP half-cells, 300-μm-thick Li metal discs served as negative electrode, while thin Li electrodes were used in Li symmetric cells and thin Li‖LFP cells. Stainless-steel symmetric cells (SS‖SS) were assembled with the separator soaked in electrolyte. All cells were tested at 30 °C. EIS tests were conducted using a BioLogic SP-300 potentiostat in a frequency range of 0.1 Hz to 1 MHz to obtain Nyquist plots. Linear sweep voltammetry (LSV) measurements were performed from the open-circuit voltage to 7 V at a scan rate of 0.1 mV s−1 to evaluate the electrochemical stability. Galvanostatic charge–discharge (GCD) tests were conducted using a WonATech WBC 3000 L battery cycler to assess the performance of Li‖Cu, Li‖Li, and LFP cells.
4.4 Physical and chemical characterization
Raman spectroscopy (Thermo Fisher DXR2xi) was employed to investigate the coordination structures within the electrolytes. Contact angle measurements with the PE separator were conducted using a GSX instrument (Surface Tech Co.) to assess the electrolyte wettability. Fire retardancy tests were performed by igniting 70 μL of each electrolyte. SEM (Hitachi S-4800, 5 keV) and XPS (Thermo Fisher NEXSA) measurements were conducted on cycled samples, which were retrieved from the disassembled cells, washed three times with diethyl carbonate (DEC), and stored under vacuum. Cross-sections of Li-plated Cu substrates were prepared using a razor blade for SEM analysis. XPS depth profiling was performed by etching samples to a depth of 390 nm using an Ar monatomic source. The XPS samples were transferred in vacuum using an evacuated vacuum transfer module (VTM).
4.5 Initio and molecular dynamics calculations
Density functional theory (DFT) calculations were conducted using the Vienna ab initio simulation package (VASP) version 6.4.2, applying the projected augmented wave (PAW) method and the Perdew–Burke–Ernzerhof (PBE) generalized gradient approximation (GGA) for exchange–correlation functionals.65–68 van der Waals corrections were implemented using the DFT-D3 method with Becke–Johnson damping. All geometries were fully relaxed, with an energy cutoff of 550 eV, and the convergence thresholds for the total energy and forces set at 10−5 eV and 0.01 eV Å−1, respectively. Γ-centered 1 × 1 × 1 k-point sampling was employed in the calculations. Ab initio molecular dynamics (AIMD) simulations were conducted in the NVT ensemble with a Nose–Hoover thermostat (Nose-mass parameter = 1) to maintain a temperature of 298 K for 5 ps, using a time step of 1 fs.69
4.6 Classical molecular dynamics calculations
MD simulations were performed with the LAMMPS package, using the polymer consistent force field plus (PCFF+) for describing interatomic interactions under 3D periodic boundary conditions.70,71 Nonbonded interactions were treated with the particle–particle particle-mesh (PPPM) method, applying a cutoff distance of 9.5 Å and a buffer width of 2.0 Å. Electrostatic interactions were calculated to a precision of 0.00001. The system was equilibrated in the NPT ensemble for 100 ps at 298 K and 1 atm with a Nose–Hoover thermostat and barostat, followed by a 100 ps equilibration in the NVT ensemble. Production simulations were conducted in the NVE ensemble for 10 ns with constant energy. Unit cells containing 1500–2000 ions were generated using Monte Carlo simulations. The cubic unit cell lattice parameters were 25.8 Å for CILE, 26.2 Å for D-CILE, 29.4 Å for LCILE, and 29.6 Å for D-LCILE.
Author contributions
J. L. and W. C. formulated the project goals and research plan. W. C. and J. Y. supervised the work. J. L. performed the primary data curation and analysis and contributed to visualization and methodology optimization. J. L. and H. K. worked on data validation and visualization. H. K. assisted in refining the experimental methodology. J. Y. directed project administration, conceptualized the framework, and secured funding. All authors discussed the results and contributed to the preparation of the final draft of the manuscript.
Data availability
The data supporting the findings of this study are available within the article and its ESI.† Additional raw data related to the characterization and computational simulations, including electrochemical measurements, molecular dynamics simulation files, and spectroscopic data, are available from the corresponding author upon reasonable request. Requests for data should be directed to Jeeyoung Yoo (https://jyoo@knu.ac.kr).
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This research was supported by the National Research Foundation (NRF) funded by the Korean government (MSIT) (No. RS-2024-00452853 and RS-2024-00431341).
References
- X. B. Cheng, R. Zhang, C. Z. Zhao and Q. Zhang, Chem. Rev., 2017, 117(15), 10403–10473 CrossRef CAS PubMed.
- D. Lin, Y. Liu and Y. Cui, Nat. Nanotechnol., 2017, 12, 194–206 CrossRef CAS PubMed.
- J.-M. Tarascon and M. Armand, Nature, 2001, 414, 359–367 CrossRef CAS PubMed.
- W. Xu, J. Wang, F. Ding, X. Chen, E. Nasybulin, Y. Zhang and J. G. Zhang, Energy Environ. Sci., 2014, 7, 513–537 RSC.
- N. W. Li, Y. X. Yin, C. P. Yang and Y. G. Guo, Adv. Mater., 2016, 28, 1853–1858 CrossRef CAS PubMed.
- K. Huang, S. Bi, B. Kurt, C. Xu, L. Wu, Z. Li, G. Feng and X. Zhang, Angew. Chem., Int. Ed., 2021, 60, 19232–19240 CrossRef CAS PubMed.
- M. Galiński, A. Lewandowski and I. Stepniak, Electrochim. Acta, 2006, 51(26), 5567–5580 CrossRef.
- M. Armand, F. Endres, D. R. MacFarlane, H. Ohno and B. Scrosati, Nat. Mater., 2009, 8, 621–629 CrossRef CAS PubMed.
- A. Basile, A. I. Bhatt and A. P. O’Mullane, Nat. Commun., 2016, 7, 11794 CrossRef PubMed.
- H. Sun, G. Zhu, Y. Zhu, M. C. Lin, H. Chen, Y. Y. Li, W. H. Hung, B. Zhou, X. Wang, Y. Bai, M. Gu, C. L. Huang, H. C. Tai, X. Xu, M. Angell, J. J. Shyue and H. Dai, Adv. Mater., 2020, 32(26), 2001741 CrossRef CAS PubMed.
- H. Yoon, P. C. Howlett, A. S. Best, M. Forsyth and D. R. MacFarlane, J. Electrochem. Soc., 2013, 160, A1629–A1637 CrossRef CAS.
- H. Zhang, W. Qu, N. Chen, Y. Huang, L. Li, F. Wu and R. Chen, Electrochim. Acta, 2018, 285, 78–85 CrossRef CAS.
- U. Pal, D. Rakov, B. Lu, B. Sayahpour, F. Chen, B. Roy, D. R. MacFarlane, M. Armand, P. C. Howlett, Y. S. Meng and M. Forsyth, Energy Environ. Sci., 2022, 15, 1907–1919 RSC.
- A. Heist and S.-H. Lee, J. Electrochem. Soc., 2019, 166, A1860–A1866 CrossRef CAS.
- J. Alvarado, M. A. Schroeder, T. P. Pollard, X. Wang, J. Z. Lee, M. Zhang, T. Wynn, M. Ding, O. Borodin, Y. S. Meng and K. Xu, Energy Environ. Sci., 2019, 12, 780–794 RSC.
- Z. Wang, F. Zhang, Y. Sun, L. Zheng, Y. Shen, D. Fu, W. Li, A. Pan, L. Wang, J. Xu, J. Hu and X. Wu, Adv. Energy Mater., 2021, 11(17), 2003752 CrossRef CAS.
- X. Liu, A. Mariani, M. Zarrabeitia, M. E. Di Pietro, X. Dong, G. A. Elia, A. Mele and S. Passerini, Energy Storage Mater., 2022, 44, 370–378 CrossRef.
- X. Liu, T. Diemant, A. Mariani, X. Dong, M. E. Di Pietro, A. Mele and S. Passerini, Adv. Mater., 2022, 34(49), 2207155 CrossRef CAS PubMed.
- X. Liu, A. Mariani, H. Adenusi and S. Passerini, Angew. Chem., Int. Ed., 2023, 62(17), e202219318 CrossRef CAS PubMed.
- A. M. Wright, Z. Wu, G. Zhang, J. L. Mancuso, R. J. Comito, R. W. Day, C. H. Hendon, J. T. Miller and M. Dincă, Chem, 2018, 4, 2894–2901 CAS.
- L. Yang, N. M. Hagh, J. Roy, E. Macciomei, J. R. Klein, U. Janakiraman and M. E. Fortier, J. Electrochem. Soc., 2024, 171, 060504 CrossRef CAS.
- Z. Liu, W. Guo, J. Tan, H. Yan, C. Bao, Y. Tian, Q. Liu and B. Li, Chem. Eng. J., 2024, 490, 151812 CrossRef CAS.
- R. Miao, J. Yang, X. Feng, H. Jia, J. Wang and Y. Nuli, J. Power Sources, 2014, 271, 291–297 CrossRef CAS.
- V. A. Afrifah, J. M. Kim, Y. M. Lee, I. Phiri, Y. G. Lee and S. Y. Ryou, J. Power Sources, 2022, 548, 232017 CrossRef CAS.
- V. Sharova, A. Moretti, T. Diemant, A. Varzi, R. J. Behm and S. Passerini, J. Power Sources, 2018, 375, 43–52 CrossRef CAS.
- J. Zheng, M. H. Engelhard, D. Mei, S. Jiao, B. J. Polzin, J. G. Zhang and W. Xu, Nat. Energy, 2017, 2, 17012 CrossRef CAS.
- Y. Zhou, S. Wu, X. Ren, H. Li, S. Li and T. Yan, Chin. Chem. Lett., 2024, 110048 CrossRef CAS.
- Y. Maeda, Y. Sonta, Y. Sasaki and H. Mori, Polymer, 2023, 269, 125704 CrossRef CAS.
- H. Wan, J. Xu and C. Wang, Nat. Res., 2024, 8, 30–44 CAS.
- M. Kerner, N. Plylahan, J. Scheers and P. Johansson, Phys. Chem. Chem. Phys., 2015, 17, 19569–19581 RSC.
- T. Ma, Y. Ni, Q. Wang, W. Zhang, S. Jin, S. Zheng, X. Yang, Y. Hou, Z. Tao and J. Chen, Angew. Chem., Int. Ed., 2022, 61(39), e202207927 CrossRef CAS PubMed.
- Y. Yamada and A. Yamada, J. Electrochem. Soc., 2015, 162, A2406–A2423 CrossRef CAS.
- J. Zheng, J. A. Lochala, A. Kwok, Z. D. Deng and J. Xiao, Adv. Sci., 2017, 4(8), 1700032 CrossRef PubMed.
- Y. Hu, Z. Wang, H. Li, X. Huang and L. Chen, J. Electrochem. Soc., 2004, 151, A1424 CrossRef CAS.
- J. Hu, Y. Ji, G. Zheng, W. Huang, Y. Lin, L. Yang and F. Pan, Aggregate, 2022, 3(1), e153 CrossRef CAS.
- J. Ye, Y. C. Wu, K. Xu, K. Ni, N. Shu, P. L. Taberna, Y. Zhu and P. Simon, J. Am. Chem. Soc., 2019, 141, 16559–16563 CrossRef CAS PubMed.
- J. Wu, Chem. Rev., 2022, 122(12), 10821–10859 CrossRef CAS PubMed.
- J. W. Zhang, J. L. Sun, D. N. Zhao, Y. J. Zhao, X. Y. Hu, Y. N. Wang, Y. J. Yao, N. S. Zhang, L. J. Zhang, C. L. Li, P. Wang, S. Y. Li and X. L. Cui, Energy Storage Mater., 2024, 72, 103698 CrossRef.
- E. O. Nachaki and D. G. Kuroda, J. Phys. Chem. B, 2024, 128, 3408–3415 CrossRef CAS PubMed.
- M. Callsen, K. Sodeyama, Z. Futera, Y. Tateyama and I. Hamada, J. Phys. Chem. B, 2017, 121, 180–188 CrossRef CAS PubMed.
- Y. Xie, H. Xiang, P. Shi, J. Guo and H. Wang, J. Membr. Sci., 2017, 524, 315–320 CrossRef CAS.
- Y. Xie, H. Zou, H. Xiang, R. Xia, D. Liang, P. Shi, S. Dai and H. Wang, J. Membr. Sci., 2016, 503, 25–30 CrossRef CAS.
- L. Zhu, Y. Wang, J. Chen, W. Li, T. Wang, J. Wu, S. Han, Y. Xia, Y. Wu, M. Wu, F. Wang, Y. Zheng, L. Peng, J. Liu, L. Chen and W. Tang, Sci. Adv., 2022, 8, eabj7698 CrossRef CAS PubMed.
- T. Frömling, M. Kunze, M. Schönhoff, J. Sundermeyer and B. Roling, J. Phys. Chem. B, 2008, 112, 12985–12990 CrossRef PubMed.
- P. G. Bruce, J. Evans and C. A. Vincent, Solid State Ionics, 1988, 28–30(part 2), 918–922 CrossRef.
- T. Chakraborty, A. Hens, S. Kulashrestha, N. Chandra Murmu and P. Banerjee, Phys. E, 2015, 69, 371–377 CrossRef CAS.
- J. Bośko and J. Rybicki, Solid State Ionics, 2003, 157(1–4), 227–232 CrossRef.
- H. Zheng, L. Chai, X. Song and V. Battaglia, Electrochim. Acta, 2012, 62, 256–262 CrossRef CAS.
- D. Aurbach, J. Power Sources, 2005, 146, 71–78 CrossRef CAS.
- D. Aurbach, M. D. Levi and E. Levi, Solid State Ionics, 2008, 179, 742–751 CrossRef CAS.
- B. D. Adams, J. Zheng, X. Ren, W. Xu and J. G. Zhang, Adv. Energy Mater., 2018, 8(7), 1702097 CrossRef.
- S. S. Zhang, InfoMat, 2021, 3, 125–130 CrossRef CAS.
- Z. Yu, N. P. Balsara, O. Borodin, A. A. Gewirth, N. T. Hahn, E. J. Maginn, K. A. Persson, V. Srinivasan, M. F. Toney, K. Xu, K. R. Zavadil, L. A. Curtiss and L. Cheng, ACS Energy Lett., 2022, 7(1), 461–470 CrossRef CAS.
- Z. Wang, Z. Sun, J. Li, Y. Shi, C. Sun, B. An, H. M. Cheng and F. Li, Chem. Soc. Rev., 2021, 50, 3178–3210 RSC.
- S. Xue, Y. Zhou, Z. Zhang, K. Hou, M. He and X. Liu, J. Energy Storage, 2024, 95, 112558 CrossRef.
- E. Peled and S. Menkin, J. Electrochem. Soc., 2017, 164, A1703–A1719 CrossRef CAS.
- W. Fang, Z. Wen, L. Chen, Z. Qin, J. Li, Z. Zheng, Z. Weng, G. Wu, N. Zhang, X. Liu, X. Yuan and G. Chen, Nano Energy, 2022, 104(part B), 107881 CrossRef CAS.
- S. Liu, X. Ji, N. Piao, J. Chen, N. Eidson, J. Xu, P. Wang, L. Chen, J. Zhang, T. Deng, S. Hou, T. Jin, H. Wan, J. Li, J. Tu and C. Wang, Angew. Chem., Int. Ed., 2021, 60, 3661–3671 CrossRef CAS PubMed.
- C. F. J. Francis, I. L. Kyratzis and A. S. Best, Adv. Mater., 2020, 32(18), 1904205 CrossRef CAS PubMed.
- B. Wu, J. Lochala, T. Taverne and J. Xiao, Nano Energy, 2017, 40, 34–41 CrossRef CAS.
- Y. Li, J. Long, Y. Liang and J. Hu, Ionics, 2023, 29, 4933–4938 CrossRef CAS.
- J. Huang, Electrochim. Acta, 2018, 281, 170–188 CrossRef CAS.
- D. P. Abraham, M. M. Furczon, S. H. Kang, D. W. Dees and A. N. Jansen, J. Power Sources, 2008, 180, 612–620 CrossRef CAS.
- G. Homann, L. Stolz, J. Nair, I. C. Laskovic, M. Winter and J. Kasnatscheew, Sci. Rep., 2020, 10, 4390 CrossRef CAS PubMed.
- P. E. Blochl, Phys. Rev. B:Condens. Matter Mater. Phys., 1994, 50, 17953 CrossRef PubMed.
- G. Kresse and D. Joubert, Phys. Rev. B:Condens. Matter Mater. Phys., 1999, 59, 1758 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1997, 77, 3865 CrossRef PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1997, 78, 1396 CrossRef CAS.
- W. G. Hoover, Phys. Rev. A:At., Mol., Opt. Phys., 1985, 31, 1695 CrossRef PubMed.
- V. Varshney, S. S. Patnaik, A. K. Roy and B. L. Farmer, Macromolecules, 2008, 41, 6837–6842 CrossRef CAS.
- V. Varshney, S. S. Patnaik, A. K. Roy and B. L. Farmer, J. Phys. Chem. C, 2010, 114, 16223–16228 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ee00119f |
| ‡ These authors (Jemin Lee and Wonwoo Choi) contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Wonwoo
Choi‡
a,
Wonwoo
Choi‡













