
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOver 12% efficiency solar-powered green hydrogen production from seawater†
Xuanjie
Wang
ab,
Jintong
Gao
c,
Yipu
Wang
 c,
Yayuan
Liu
*d,
Xinyue
Liu
*ae and
Lenan
Zhang
*ac
c,
Yayuan
Liu
*d,
Xinyue
Liu
*ae and
Lenan
Zhang
*ac
aDepartment of Mechanical Engineering, Massachusetts Institute of Technology, Cambridge, MA 02139, USA
bDepartment of Mechanical Engineering and Mechanics, Lehigh University, Bethlehem, PA 18015, USA
cSibley School of Mechanical and Aerospace Engineering, Cornell University, Ithaca, NY 14853, USA. E-mail: lzhang@cornell.edu
dDepartment of Chemical and Biomolecular Engineering, Johns Hopkins University, Baltimore, MD 21218, USA. E-mail: yayuanliu@jhu.edu
eDepartment of Chemical Engineering and Materials Science, Michigan State University, East Lansing, MI 48824, USA. E-mail: xyliu@msu.edu
First published on 25th March 2025
Abstract
Solar-powered water electrolysis holds significant promise for the mass production of green hydrogen. However, the substantial water consumption associated with electrolysis not only increases the cost of green hydrogen but also raises critical concerns about accelerating water scarcity. Although seawater can serve as an infinite water supply for green hydrogen production, its complex composition poses substantial challenges to efficient and reliable electrolysis. Here, we demonstrate a high-efficiency solar-powered green hydrogen production from seawater. Our approach takes advantage of the full-spectrum utilization of solar energy. Photovoltaic electricity is used to drive the electrolysis, whereas the waste heat from solar cells is harnessed to produce clean water through seawater distillation. With natural sunlight and real seawater as the sole inputs, we experimentally demonstrate a 12.6% solar-to-hydrogen conversion efficiency and a 35.9 L m−2 h−1 production rate of green hydrogen under one-sun illumination, where additional 1.2 L m−2 h−1 clean water is obtained as a byproduct. By reducing reliance on clean water and electricity supplies, this work provides a fully sustainable strategy to access green hydrogen with favorable energy efficiency and technoeconomic feasibility.
Broader contextHydrogen produced by renewable energy through water electrolysis is known as green hydrogen, which plays a vital role in deep decarbonization of hard-to-abate sectors. Sustainable development of green hydrogen technologies is limited by significant water consumption, because producing one kilogram of hydrogen at least requires nine kilograms of water. Considering that two thirds of global population is facing severe water scarcity, producing green hydrogen by consuming clean water poses a critical challenge at the water–energy nexus toward sustainability. Here, we leverage the most abundant and accessible resources on the Earth, natural sunlight and seawater, to unlock a sustainable pathway: “seawater + sunlight = green hydrogen + clean water”. Our approach relies on full-spectrum utilization of solar energy, where photovoltaic electricity is used to produce hydrogen through electrolysis and photothermal energy is harnessed to purify seawater through distillation. We demonstrate the potential of our approach using a hybrid solar distillation–water electrolysis (HSD-WE) device, where over 12% solar-to-hydrogen conversion efficiency was achieved with additional 1.2 L m−2 h−1 clean water as a byproduct. By exploiting the full potential of solar energy and seawater, our approach reduces the reliance on clean water and electricity supplies, promising sustainable green hydrogen production with high efficiency and low cost. |
Introduction
Green hydrogen, a clean alternative to fossil fuels, plays an increasingly important role in realizing deep decarbonization of energy systems and achieving net zero emissions by 2050.1–4 Solar energy is a carbon-neutral source of electricity, which has been recognized as a promising means to power water electrolysis for green hydrogen production.5–14 However, in addition to electricity, conventional electrolysis also requires a considerable amount of high-purity water as the input. Theoretically, producing one kilogram of hydrogen requires at least nine kilograms of water. To meet the increasing demand for green hydrogen in 2050 (>500 Mt),1 water electrolysis will consume more than 4.5 trillion liters of clean water per year, equal to the annual drinking water demand of more than 3.5 billion people. Considering that four billion people are experiencing severe water shortage,15 limited access to sustainable and reliable clean water supplies has become one of the major bottlenecks to the worldwide adoption of green hydrogen technologies through electrolysis.16–19Seawater, on the other hand, is one of the most abundant and accessible resources on our planet, which can be an infinite water supply for solar-powered green hydrogen production.20–22 Despite the huge potential, directly splitting natural seawater can be particularly challenging due to its complex composition. As a result, state-of-the-art electrocatalysts for direct seawater electrolysis typically exhibit limited activity and stability due to severe fouling and corrosion, impeding its immediate implementation to meet the urgent need for green hydrogen.23–26 Alternatively, indirect seawater electrolysis, which integrates a conventional electrolyzer with external seawater desalination and subsequent deionization, can be a more reliable and practical strategy. Although seawater desalination, such as reverse osmosis, has become a mature technology to produce water with very high energy efficiency,27–29 producing high-purity water to meet the standard of electrolysis requires further deionization, which induces additional energy consumption and considerable cost for installation and operation.30 Furthermore, the maximum hydrogen productivity of indirect seawater electrolysis is fundamentally limited by the capacity of seawater desalination. The rapidly increasing demand of green hydrogen has posed a huge pressure on existing desalination facilities, where most of the produced water is now used for living purposes and agriculture operations.15,31–34
A recent advance by introducing in situ water purification into an electrolyzer has provided a promising pathway to harness seawater by combining the advantages of both direct and indirect seawater electrolysis technologies.35 With a membrane distillation driven by the vapor pressure difference between alkaline electrolyte and seawater, clean water can be directly supplied to the electrolyzer to facilitate continuous evolution of green hydrogen. As a result, this technique not only avoids the direct contact between seawater and electrocatalysts but also enables an all-in-one approach without the installation of external desalination and deionization facilities. Despite the significant progress, this approach can be inherently limited by the water production rate due to the low vapor pressure difference across the membrane. More importantly, it is still unclear how to adapt the in situ water purification strategy to solar-powered green hydrogen and achieve the optimal solar-to-hydrogen (STH) efficiency.
In this work, we demonstrate a solar-powered approach to produce green hydrogen directly from seawater with high STH efficiency and low cost. This method takes advantage of the full-spectrum utilization of solar energy by combining photovoltaic (PV) and photothermal (PT) effects (Fig. 1a). High-energy photons are converted to electricity through a PV panel to drive electrolysis, whereas the rest of the absorbed solar energy is converted to heat to produce clean water through interfacial thermal distillation. By exploiting the full potential of waste heat produced by the PV panel, we achieve in situ water purification to address the critical fouling and corrosion of electrodes without consuming electricity. All electricity produced by the PV panel is used for electrolysis, promising a high STH efficiency toward the fundamental limit dictated by the solar-to-electricity conversion. More notably, owing to the passive operation nature, we can minimize the cost associated with water purification and electricity supply, which contributes primarily to the operational expenditure (OPEX) of existing water electrolysis.36–40 To prove our concept, we developed a hybrid solar distillation–water electrolysis (HSD-WE) prototype, which integrates a proton exchange membrane (PEM) electrolyzer with a PV panel and an interfacial thermal distillation device. With natural sunlight and seawater as the sole inputs, we demonstrate green hydrogen production with 12.6% STH efficiency using a 17.3% efficiency silicon (Si) PV panel. Under one-sun illumination (1000 W m−2), we achieved a 35.9 L m−2 h−1 production rate of dry hydrogen with 1.2 L m−2 h−1 distilled water continuously fed into the PEM through interfacial thermal distillation. Due to the simple architecture and solar-powered passive operation, the technoeconomic analysis shows that the cost of green hydrogen production with our approach is expected to be $5 per kg with three-year operation and $1 per kg with 15-year operation.
 | ||
| Fig. 1 Solar-powered green hydrogen production from seawater. (a) Full-spectrum utilization enabling in situ water purification and uncompromised electricity production. High-energy photons above the band gap of photovoltaic (PV) panels are converted to electricity (green shadow), whereas waste heat produced by the PV panel due to the photothermal (PT) effect (yellow shadow) is used to purify seawater through interfacial thermal distillation. Electricity produced by the PV panel is used to power hydrogen production with PT distilled water fed into the electrolyzer. (b) Schematic of the hybrid solar distillation–water electrolysis (HSD-WE) device. An interfacial thermal evaporator is integrated on the backside of the PV panel to enable PV cooling and in situ water purification. A unidirectional flow driven by the siphon effect is introduced into the evaporator to avoid salt accumulation. The evaporator and electrolyzer are separated by an air gap, which avoids the direct contact between seawater and electrocatalysts. Distilled vapor directly condenses on the anode side and feeds into the electrolyzer. With electricity produced by the PV panel, hydrogen is generated from the cathode of the PEM electrolyzer. Latent heat released during condensation is transported to the electrolyzer, which elevates the temperature and increases the efficiency of electrolysis. | ||
Results and discussion
Design of the hybrid solar distillation–water electrolysis device
Fig. 1b shows the working principle of the HSD-WE device, which harnesses natural sunlight and seawater and produces clean water and green hydrogen. A PV panel is facing the solar illumination, converting solar energy to electricity and heat. An interfacial thermal evaporator is attached to the backside of the PV panel. The evaporator has a wicking structure, enabling a passive supply of seawater due to the capillary effect. Waste heat is transferred from the PV panel to the evaporator, driving the seawater distillation. The evaporator and PEM electrolyzer are separated by an air gap, which avoids the direct contact between seawater and electrocatalysts to minimize fouling and corrosion while allowing the distilled vapor to transport from the evaporator to the electrolyzer. Purified water is fed into the anode of the electrolyzer through vapor condensation. Meanwhile, electricity produced by the PV panel is supplied to the electrolyzer to drive hydrogen production from the cathode.In addition to the general working principle, we discuss four key features that facilitate high STH efficiency and reliable operation of the HSD-WE device. Firstly, the interfacial thermal evaporator not only enables seawater distillation but also provides efficient cooling for the PV panel (① in Fig. 1b), which reduces the temperature of the PV panel and increases the solar-to-electricity conversion efficiency. Secondly, the compact integration of the interfacial thermal evaporator and electrolyzer enables in situ water purification without the need of external water treatment, collection, and transportation facilities (② in Fig. 1b). More notably, compared with the low vapor pressure difference between seawater and electrolyte shown in the recent in situ water purification approach (<3 kPa),35 the thermal effect can create a much larger vapor pressure difference (>10 kPa), facilitating a higher production rate of distilled water. Thirdly, since vapor condensation directly occurs on the anode, the latent heat due to the vapor-to-liquid phase change is released to the electrolyzer to elevate the operating temperature and improve the efficiency of hydrogen production (③ in Fig. 1b). Finally, to mitigate salt accumulation and potential fouling inside the interfacial thermal evaporator, we introduced a unidirectional flow across the evaporator to accelerate salt rejection through convection (Fig. 1b). The unidirectional flow is driven by the siphon effect, where no pumping power is required. The effectiveness of unidirectional flow in salt rejection has been demonstrated in recent studies.41,42 Therefore, we highlight that such a hybrid design can bring significant additional benefits to further enhance solar-to-electricity (i.e., PV cooling) and water-to-hydrogen conversion (i.e., condensation heating), which cannot be achieved by simply combining PV panels, water purification facilities, and electrolysis devices.
Fig. 2a shows the detailed design of the HSD-WE device, which comprises a silicon (Si) PV panel (passivated emitter and rear contact solar cells), an interfacial thermal evaporator (capillary wick), a polycarbonate spacer, bipolar plates (BPs), gas diffusion layers (GDLs), a PEM, gaskets, a heat sink, and an end plate (Note S.1, ESI†). These components were stacked into a compact assembly (Fig. 2b). The top surface of the HSD-WE device had an area of 10 × 10 cm2, whereas the total solar absorption area of the PV panel was 7 × 7 cm2. A polycarbonate spacer with an inner aperture area of 5 × 5 cm2 was used to create a 1.5 cm air gap between the interfacial thermal evaporator and the BP of the anode side. Different from conventional BPs, we note that the BP of the anode side also acted as a condenser and a heat sink, which plays a critical role in distilled water feeding and thermal management of the HSD-WE device. Specifically, the BP of the anode side was an L-shaped stainless steel plate with multiple parallel slots (5 cm in length and 1 mm in width) through the surface facing the evaporator (Fig. 2a). Vapor condensation occurred on the BP. Distilled water can directly flow through the slots and wet the GDL. Meanwhile, the latent heat released during condensation was conducted to the PEM through the BP, raising the temperature of electrolysis. The rest of the heat was dissipated through the heat sink to avoid overheating the entire device, which is detrimental to the solar-to-electricity conversion of the PV panel. The PEM was sandwiched by two titanium GDLs. The active regions of the PEM were coated with platinum- (on the cathode side) and iridium-based (on the anode side) electrocatalysts. Both GDLs and the active regions of the PEM have an area of 5 × 5 cm2.
 | ||
| Fig. 2 Design and characterization of the HSD-WE prototype. (a) Structure of the HSD-WE device. The PV panel, capillary wick, polycarbonate spacer, BPs, GDLs, PEM, gaskets, heat sink, and end plate are integrated into a compact assembly. (b) Optical image of a fully assembled HSD-WE device. (c) Effect of evaporative cooling on the current–voltage (I–V) responses of PV panels (A = 49 cm2). Blue curve: I–V curve of the PV panel directly exposed to ambient air without evaporative cooling. Red curve: I–V curve of the PV panel with evaporative cooling. (d) Schematic of the experimental setup to initiate unidirectional flow across the interfacial thermal evaporator. The reservoir at the inlet was elevated by h = 6 cm above the reservoir at the outlet to induce the siphon effect. Salinity increased along the flow direction and reached the maximum at the outlet. (e) Salinity of the outlet reservoir as a function of time. Real seawater concentrated to different salinities was used in the measurements. Blue curve: 3.5 wt% seawater filled in the inlet reservoir. Red curve: 10.5 wt% concentrated seawater filled in the inlet reservoir. (f) Time-lapse images of the conventional capillary wick during continuous distillation of 10.5 wt% concentrated seawater. The right side of the evaporator was connected with the inlet of the saline reservoir, whereas the left side was a dead end. (g) Time-lapse images of the interfacial thermal evaporator with unidirectional flow during continuous distillation of 10.5 wt% concentrated seawater. The right and left sides of the evaporator acted as the inlet and outlet of the unidirectional flow, respectively. No salt crystallization was observed in the evaporator throughout the ten-hour continuous operation. (h) Polarization curves of the PEM electrolyzer (A = 25 cm2) at representative temperatures. Grey dashed line: the PEM electrolyzer performance at 1.65 V, where a significant increase of the current from 278 mA to 495 mA was observed when elevating the operating temperature from 23 °C to 60 °C. | ||
We first performed characterizations on each key component of the HSD-WE device and quantified the impacts of evaporation, unidirectional flow, and temperature rise on PV cooling, salt rejection, and electrolysis, respectively. Fig. 2c shows the current–voltage (I–V) curves of PV panels with (red curve) and without (blue curve) evaporative cooling. In each PV panel, there were four identical solar cells connected in a series circuit (Fig. 2b and Note S.1, ESI†). Two identical PV panels were prepared and placed under ambient conditions. An interfacial thermal evaporator containing seawater was attached to the backside of one of the PV panels, whereas the backside of the other PV panel was directly exposed to ambient air (Note S.2, ESI†). A solar simulator was used to provide a uniform solar flux. Under one-sun illumination, we measured the temperature rise and I–V curves of these two PV panels using thermocouples (T-type) and a potentiostat, respectively. A significant cooling effect was observed, where the steady-state temperature of the PV panel with the interfacial thermal evaporator (40.9 °C) was much lower than that of the PV panel directly exposed to ambient air (55.5 °C) (Fig. 2c and Note S.2, ESI†). The cooling performance is comparable to that demonstrated in state-of-the-art evaporation-based PV cooling approaches.43–45 As a result, the solar-to-electricity conversion efficiency at the maximum power point (MPP) increased from 15.1% (blue curve in Fig. 2c) to 17.3% (red curve in Fig. 2c), which is essential to achieve a high STH efficiency.
To ensure reliable thermal distillation without salt accumulation, we tested the salt rejection performance with unidirectional flow (Note S.3, ESI†). Fig. 2d shows the experimental setup consisting of a PV panel with an interfacial thermal evaporator on the backside. The inlet and outlet of the capillary wick were inserted into two reservoirs, respectively, which contained the saline and collected the brine discharge. To induce the siphon effect, the saline reservoir was placed 6 cm above the brine reservoir (h = 6 cm in Fig. 2d). Driven by the hydraulic head between two reservoirs, there was a unidirectional flow from the inlet to the outlet, carrying the accumulated salt out of the evaporator. Along the flow direction, salinity was building up and reached the maximum at the outlet (Fig. 2d). To mitigate salt fouling due to crystallization, it is critical to ensure the highest salinity within the evaporator much lower than the saturation salinity (i.e., ∼26 wt%). We performed seawater distillation and measured the salinity at the outlet of the evaporator. Real seawater (3.5 wt% salinity) with complex composition was used in our experiments. The salinity was measured using a digital refractometer. Fig. 2e shows the salinity at the outlet as a function of time. For seawater distillation (blue curve in Fig. 2e), there was a slow increase of salinity from 3.5 wt% to 5 wt% during the first five-hour operation. After the fifth hour, the salinity at the outlet became independent with time, indicating a steady state. No salt crystallization was observed because the highest salinity (5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) wt%) at the steady state was much lower than the saturation salinity (26wt%). To understand the salt rejection performance in a more extreme condition, we further concentrated the seawater to 10.5 wt%, reaching the salinity level of waste brine.46,47 Similar to the seawater distillation test, the salinity at the outlet showed a slow increase first and then reached the steady state at 15 wt% (red curve in Fig. 2e), which was still far away from the saturation salinity (26 wt%). We note that the salinity at the beginning of our test was slightly lower than 10 wt%, because the capillary wick was rinsed using 3.5 wt% seawater before the experiment. To further highlight the significance of unidirectional flow to achieve reliable distillation, we compared salt rejection performance with and without introducing the unidirectional flow. Fig. 2f shows the time-lapse images of a conventional capillary wick evaporator,48,49 where the right side was the inlet connected with the saline reservoir (10.5 wt%), while the left side was a dead end. To achieve better imaging contrast, a black capillary wick was adopted in this test. Salt crystallization was observed near the dead end with only a 1.5-hour operation (dashed line in Fig. 2f). After a seven-hour operation, salt crystals covered half of the evaporator, inducing significant fouling. In contrast, no salt crystallization was found on the evaporator throughout a ten-hour operation when the unidirectional flow was initiated (Fig. 2g). Combined with the salinity test (Fig. 2e), we can confirm that our interfacial thermal evaporator is capable of distilling highly concentrated waste brine without salt crystallization.
wt%) at the steady state was much lower than the saturation salinity (26wt%). To understand the salt rejection performance in a more extreme condition, we further concentrated the seawater to 10.5 wt%, reaching the salinity level of waste brine.46,47 Similar to the seawater distillation test, the salinity at the outlet showed a slow increase first and then reached the steady state at 15 wt% (red curve in Fig. 2e), which was still far away from the saturation salinity (26 wt%). We note that the salinity at the beginning of our test was slightly lower than 10 wt%, because the capillary wick was rinsed using 3.5 wt% seawater before the experiment. To further highlight the significance of unidirectional flow to achieve reliable distillation, we compared salt rejection performance with and without introducing the unidirectional flow. Fig. 2f shows the time-lapse images of a conventional capillary wick evaporator,48,49 where the right side was the inlet connected with the saline reservoir (10.5 wt%), while the left side was a dead end. To achieve better imaging contrast, a black capillary wick was adopted in this test. Salt crystallization was observed near the dead end with only a 1.5-hour operation (dashed line in Fig. 2f). After a seven-hour operation, salt crystals covered half of the evaporator, inducing significant fouling. In contrast, no salt crystallization was found on the evaporator throughout a ten-hour operation when the unidirectional flow was initiated (Fig. 2g). Combined with the salinity test (Fig. 2e), we can confirm that our interfacial thermal evaporator is capable of distilling highly concentrated waste brine without salt crystallization.
Fig. 2h shows the polarization curves of the PEM electrolyzer at different operating temperatures, where deionized (DI) water was used in the measurement. The polarization curves were measured using a sourcemeter and the temperature of feed water was increased from 23 °C to 60 °C (Note S.4, ESI†). A moderate increase in temperature led to substantial enhancement of electrolysis performance. For example, when raising the operating temperature from 23 °C to 40 °C, the current at 1.65 V increased from 278 mA to 384 mA (grey dashed line in Fig. 2h), leading to a 38% increase in the hydrogen production rate. Moreover, when the operating temperature increased to 60 °C, the current reached 495 mA, resulting in a 78% increase in the hydrogen production rate compared to the room temperature operation. These results indicate that if a similar temperature rise can be achieved through condensation heating, considerable improvement in STH efficiency can be expected. We further performed a controlled experiment to directly quantify the impact of condensation heating on electrolysis performance (Note S.5, ESI†). The results demonstrated that condensation heating resulted in a more than 15 °C rise in PEM electrolyzer temperature and a 9.4% increase in the hydrogen production rate.
Modeling and optimization
In addition to the design of each component, system-level optimization is of equal importance, as mismatches among components can result in significant energy losses. In particular, the PV panel and PEM electrolyzer have distinct I–V characteristics, where their coupling dictates the amount of electricity that can be extracted for electrolysis. For a fixed PEM electrolyzer and a constant total solar absorption area, the coupling between the PV panel and PEM electrolyzer can be tuned by changing the number of solar cells in a series circuit connection. Fig. 3a shows a schematic illustration of the coupling between the PV panel and PEM electrolyzer, where the intersection of the corresponding I–V curves determines the operation state of the HSD-WE device. For example, when the number of solar cells within the PV panel (N1 in Fig. 3a) is too low, despite the high short circuit current, the current at the intersection point (A in Fig. 3a) can be very low, leading to minimum electricity supplied from the PV panel to the PEM electrolyzer (red shadow in Fig. 3a). However, by varying the number of solar cells (N2 in Fig. 3a), the intersection point (B in Fig. 3a) can be optimized, resulting in a substantial increase in electricity extraction for electrolysis (blue shadow in Fig. 3a).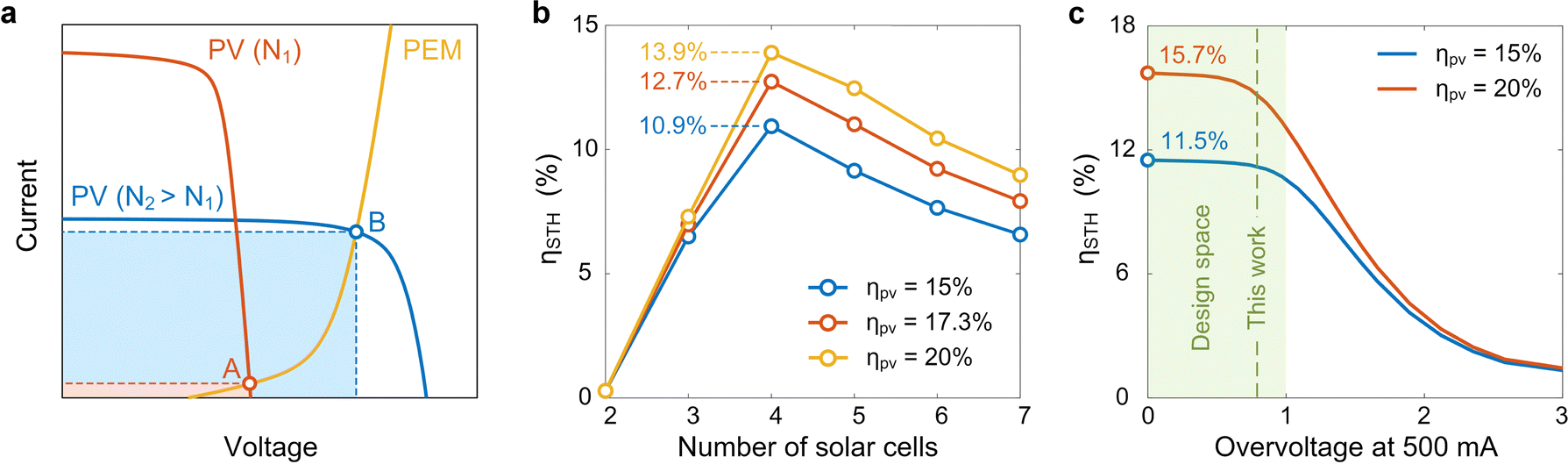 | ||
| Fig. 3 System-level optimization of the coupling between the PV panel and PEM electrolyzer. (a) Schematic current–voltage (I–V) responses of the PV panel and PEM electrolyzer. For the same solar absorption area, increasing the number of solar cells in series circuit connection reduces the short-circuit current while increasing the open-circuit voltage, which changes the intersection point (A and B) of the PEM electrolyzer polarization curve and the PV panel I–V curve. Red and blue curves: PV panel I–V curves with small (N1) and larger (N2) numbers of solar cells in series circuit connection, respectively. Yellow curve: PEM electrolyzer polarization curve. (b) STH efficiency as a function of the number of solar cells. The STH efficiency was calculated based on the higher heating value (HHV). With the increase of the number of solar cells, the STH efficiency first increases and then decreases, resulting in a peak STH efficiency when the number of solar cells is equal to four. (c) STH efficiency as a function of the overvoltage of the PEM electrolyzer. The overvoltage at 500 mA current was chosen as an indicator of the PEM electrolyzer performance. The STH efficiency shows a weak dependence on overvoltage when it is less than 1 V, leading to a design space as indicated in the green shadow. | ||
We performed theoretical modeling to optimize the coupling between the PV panel and PEM electrolyzer. Our model took the I–V characteristics of the PV panel and PEM electrolyzer as the inputs and calculated the STH efficiency of the HSD-WE device (Note S.6, ESI†). Fig. 3b shows the calculated STH efficiency as a function of the number of solar cells within the PV panel. Three modeled Si PV panels with 15%, 17.3%, and 20% solar–electricity conversion efficiencies at the MPP were considered in our analysis (Note S.6, ESI†). The STH efficiency in our calculation was based on the higher heating value (HHV) (Note S.7, ESI†).6,11,37 With the increase of the number of solar cells, the STH efficiency first increased and then decreased, resulting in a peak value when the number of solar cells (N) is equal to four. The initial increase trend when N < 4 can be attributed to the increase of open circuit voltage of the PV panel, which makes the intersection point move toward the high-current region approaching the short circuit current. The subsequent decrease in STH efficiency when N > 4 was due to the reduced short circuit current, which limits the maximum current supplied to the electrolyzer. Therefore, we chose four solar cells with series circuit connection for our design. With the 17.3% efficiency Si PV panel, our model shows that the HSD-WE device can reach 12.7% STH efficiency (red curve in Fig. 3b), where the 4.6% efficiency difference can be attributed to the energy loss within the PEM electrolyzer. In general, for Si PV panels with efficiencies ranging from 15% to 20%, which could be induced by different operating temperatures (Fig. 2c), the corresponding peak STH efficiencies can vary from 10.9% to 13.9% (blue and yellow curves in Fig. 3b).
Our modeling further provided insights into the proper selection of electrocatalysts for the HSD-WE device (Note S.6, ESI†). Fig. 3c shows the theoretical STH efficiency of the HSD-WE device as a function of the PEM electrolyzer overvoltage. The PEM electrolyzer was powered by a PV panel consisting of four solar cells. We chose the overvoltage value at 500 mA current condition, which is close to the short circuit current of the PV panel (Fig. 2c), as an indicator to describe the performance of the PEM electrolyzer, where a higher overvoltage represents a larger energy loss associated with electrochemical conversion. The coupling between the PV panel and PEM electrolyzer led to an interesting dependence of the STH efficiency on overvoltage. Specifically, when the overvoltage at 500 mA was too high (>2 V), the total voltage required to power the PEM electrolyzer became comparable to the open circuit voltage of the PV panel (2.7 V). As a result, the polarization curve intersected with the I–V curve of the PV panel at a point with very low current, leading to an undesirable STH efficiency (<3%). When the overvoltage was sufficiently high (>1 V), even a slight reduction of overvoltage can lead to a substantial increase of the STH efficiency. For example, by reducing the overvoltage from 1.5 V to 1 V, the resulting STH efficiency increased from ∼6.5% to >10% when the PV panel efficiency was 15% (blue curve in Fig. 3c). However, we note that the benefit of pursuing lower overvoltage was diminishing when the overvoltage at 500 mA was less than 1 V (green shadow in Fig. 3c). For example, by further reducing the overvoltage from 1 V to 0 V, the resulting STH efficiency only increased from 10.4% to 11.5% when the PV panel efficiency was 15% (blue curve in Fig. 3c). Our modeling suggested that the performance of electrocatalysts is not a bottleneck to the STH efficiency as long as the overvoltage at 500 mA is below 1 V (green shadow in Fig. 3c), which can be achieved by existing electrocatalysts used in PEM electrolyzers.50 As a result, we chose platinum- and iridium-based electrocatalysts for the cathode and anode reactions, respectively, which showed ∼0.8 V overvoltage under 500 mA current (green dashed line in Fig. 3c).
Laboratory testing
The performance of the HSD-WE prototype was characterized in a laboratory environment (Note S.8, ESI†). Fig. 4a shows a schematic of the experimental setup. One-sun illumination was provided by the solar simulator. The HSD-WE device was horizontally placed on a table. Two reservoirs were used to supply seawater and collect brine discharge, respectively. Real seawater with 3.5 wt% salinity was used in our test. To initiate the unidirectional flow for salt rejection, the reservoir containing seawater was lifted 6 cm using a lab jack (h = 6 cm in Fig. 4a). Two digital balances were used to measure the mass changes of the seawater (Δm1 in Fig. 4a) and waste brine (Δm2 in Fig. 4a) reservoirs, respectively. Due to the conservation of mass, the amount of clean water production through interfacial thermal distillation can be estimated from |Δm1 + Δm2|. Three T-type thermocouples were inserted into the HSD-WE device to measure the temperature responses of the PV panel (T1), air gap (T2), and PEM electrolyzer (T3), respectively (Fig. 4a). Another T-type thermocouple was used to record the ambient temperature (Tamb). The cathode side of the PEM electrolyzer was connected with a gas collection setup through tubing, where the amount of hydrogen inside the cylinder was directly recorded using a camera. Therefore, our experimental setup can measure the hydrogen production rate and STH efficiency using two independent approaches. On the one hand, the hydrogen production rate and STH efficiency can be calculated from the current at the intersection point of the PEM electrolyzer polarization curve and the PV panel I–V curve (Note S.7, ESI†). On the other hand, they can also be determined from the actual hydrogen collection. If there is no leakage of hydrogen during collection, results obtained from these two approaches should be consistent with each other. During the test, electricity and water required for the PEM electrolyzer were supplied by the PV panel and interfacial thermal distillation, respectively. The operating current and voltage were determined by the coupling between the PV panel and the PEM electrolyzer. | ||
| Fig. 4 Performance of the HSD-WE device in a laboratory environment. (a) Schematic of the experimental setup for the laboratory characterization. The HSD-WE device was placed under a solar simulator. Two digital balances were used to measure the mass change of the inlet (Δm1) and outlet (Δm2) reservoirs as a function of time. Hydrogen was collected by a cylinder using the water displacement approach. (b) Measured current–voltage (I–V) response of the HSD-WE device. Blue curve: I–V curve of the PV panel with four solar cells in series circuit connection. Red curve: polarization curve of the PEM electrolyzer when integrated with the interfacial thermal evaporator. Green shadow: total electricity that can be extracted for water electrolysis. The corresponding HHV-based STH efficiency was 12.6%, according to the intersection point of I–V curves. (c) Temperature of the HSD-WE device as a function of time. (d) Seawater distillation and hydrogen production performance of the HSD-WE device. Left axis: total mass change recorded by two digital balances as a function of time, which represents the evaporation rate of seawater during interfacial thermal distillation. Right axis: wet (blue curve) and dry (red curve) hydrogen collections as a function of time obtained from the water displacement approach. (e) Time-lapse images of continuous green hydrogen production from the HSD-WE device. Water inside the gas collection cylinder was marked by the blue shadow. (f) Summary of STH efficiencies of the HSD-WE device. The STH efficiency was determined from the intersection point of I–V curves, dry hydrogen collection, and wet hydrogen collection. Both the HHV (blue bar) and Gibbs free energy-based (red bar) STH efficiencies were calculated. | ||
Fig. 4b shows the PEM electrolyzer polarization curve overlapped with the PV panel I–V curve of the HSD-WE device. The coupling between the PV panel and PEM electrolyzer resulted in an intersection point at ∼2.07 V and ∼417 mA (green spot in Fig. 4b). Compared with the PEM electrolyzer performance before integration (Fig. 2h), the overvoltage of the HSD-WE device at the same current showed an increase, which can be attributed to the additional ohmic resistance induced by the customized BP and condenser (Fig. 2a). Nevertheless, the overvoltage at the intersection point of the PEM electrolyzer polarization curve and the PV panel I–V curve was still less than 1 V (green spot in Fig. 4b). In fact, the intersection point was close to the MPP of the PV panel, indicating an efficient extraction of electricity to power the PEM electrolyzer (green shadow in Fig. 4b). According to the current at the intersection point, we obtained a 12.6% HHV-based STH efficiency of the HSD-WE device (Note S.7, ESI†). Fig. 4c shows the transient temperature response of the HSD-WE device during a 2.5-hour operation. The temperature of the PV panel rapidly increased to 40 °C within 10 minutes and then gradually reached a steady-state temperature of 50 °C (blue curve in Fig. 4c). The air gap (red curve in Fig. 4c) and the PEM electrolyzer (yellow curve in Fig. 4c) were heated sequentially, reaching 37 °C and 31 °C at steady state, respectively. Despite a large thermal resistance of the PEM electrolyzer, thanks to the evaporative cooling, the temperature of the PV panel was maintained within a reasonable range without overheating. In the HSD-WE device, the condensation heating effect on the PEM electrolyzer was significantly stronger than the Joule heating effect (Note S.9, ESI†). The increased temperature of the PEM electrolyzer above the ambient temperature was primarily attributed to the condensation heating effect.
In addition to characterizing the I–V curves, we directly measured the STH efficiency of the HSD-WE device from the actual hydrogen collection. Fig. 4d shows the total mass change of two digital balances (i.e., |Δm1 + Δm2|) (yellow curve) and the amount of hydrogen collection (blue and red curves) as a function of time. The total mass change increased gradually within the first ten minutes due to the transient temperature response and then exhibited a linear dependence with time after the thermal steady state was established, indicating a constant rate of evaporation. With linear fitting, we obtained an evaporation rate of ∼1.2 L m−2 h−1, which was scaled by the total solar absorption area. To validate the purity of the distilled water, we measured the conductivity before and after solar distillation (Note S.10, ESI†). The distilled water exhibited an average conductivity of 3.646 ± 1.614 μS cm−1 through four independent 10-hour distillation tests. This indicates that the distilled water met the American Society of Testing Materials (ASTM) standard of high purity water (<5 μS cm−1), which was feasible for water electrolysis. The corresponding salinity of the distilled water was 0.0002 ± 0.00008 wt% only, which was two orders of magnitude lower than the World Health Organization (WHO) standard for drinking water (200 mg L−1). Meanwhile, notably, we measured a substantial hydrogen production from the gas collection cylinder. Fig. 4e shows the time-lapse images of hydrogen collection using the water displacement approach, where water inside the cylinder was highlighted by blue shadow to enhance the image contrast. The volume of hydrogen within the cylinder linearly increased with time, indicating a highly stable hydrogen production rate (Fig. 4d). Throughout a 2.5-hour operation, ∼450 mL hydrogen was collected into the cylinder (blue curve in Fig. 4d and Video S1, ESI†). With linear fitting, we obtained a hydrogen production rate of ∼188 mL h−1, equivalent to 38.4 L m−2 h−1 by scaling with the solar absorption area. We note that the hydrogen collected through the water displacement approach is known as wet hydrogen because it contains a small amount of water vapor due to the presence of the liquid–gas interface.51 We estimated the amount of water vapor by calculating the saturation vapor pressure at ambient temperature (Note S.8, ESI†). By excluding water vapor from the total gas collection, we obtained the dry hydrogen collection as a function of time (red curve in Fig. 4d), showing a production rate of ∼176 mL h−1 or 35.9 L m−2 h−1 scaled by the solar absorption area. To enable continuous hydrogen production at a rate of 35.9 L m−2 h−1, at least 27 mL m−2 h−1 of clean water should be supplied to the PEM electrolyzer. Owing to the high evaporation rate (i.e., ∼1.2 L m−2 h−1), we note that the amount of clean water produced by the interfacial thermal distillation is highly sufficient to feed the PEM electrolyzer, where the remaining clean water could be collected as a byproduct of the HSD-WE device. Furthermore, the long-term stability of the HSD-WE device was confirmed through a cyclic test using seawater (Note S.11, ESI†). The HSD-WE device exhibited a stable hydrogen production rate of 180 mL h−1 in each cycle.
Fig. 4f summarizes the STH efficiencies of the HSD-WE device calculated based on different approaches (Note S.7, ESI†). In addition to the intersection point of I–V curves, the STH efficiency can also be determined from the hydrogen production rate. The HHV-based STH efficiency estimated from the production rate of dry hydrogen (35.9 L m−2 h−1) shows an excellent agreement with that obtained from the I–V curve approach (12.6%), indicating negligible leakage during the hydrogen collection. In fact, we note that the amount of dry hydrogen shown in this work can be a conservative estimation because we assumed the water vapor inside the gas collection cylinder reached a saturation state, which could lead to an overestimation of the amount of water vapor. To estimate the upper bound of the STH efficiency, we calculated the STH efficiency based on the production rate of wet hydrogen as well (13.5% in Fig. 4f). With several independent approaches, we confirmed that the HHV-based STH efficiency of the HSD-WE device was above 12.5%. In addition to the HHV-based STH efficiency, we also calculated the Gibbs free energy-based STH efficiency (red bars in Fig. 4f), which is commonly used to quantify the performance of electrolysis (Note S.7, ESI†). We demonstrated over 10.5% Gibbs free energy-based STH efficiency of the HSD-WE device. We note that the STH efficiency demonstrated in our work (12.6%) is comparable to or even higher than that of the state-of-the-art solar-powered green hydrogen production techniques, such as Si PV electrolysis (∼10%),13,52 photoelectrochemical water splitting (∼10%),53–56 and photocatalytic water splitting (∼1%),57–59 which are fed by clean water, instead of seawater (Note S.12, ESI†). Therefore, the HSD-WE unlocked an unprecedented opportunity for solar-powered green hydrogen production, i.e., achieving high STH efficiency (>10%) with seawater as the input.
Outdoor testing
To further understand the performance of the HSD-WE device under realistic weather conditions, we conducted an outdoor experiment on a partly sunny day (August 5, 2024). Fig. 5a shows the experimental setup, which was installed on a rooftop at the MIT campus (Cambridge, MA, USA). The HSD-WE device was horizontally placed on a table (Note S.13, ESI†). To avoid solar heating, the experimental setup was covered by reflective aluminum sheets with only the PV panel exposed to sunlight through an aperture. A pyranometer was placed on the side of the HSD-WE device to measure the incident solar flux on the PV panel. Four thermocouples (T-type) were used to measure the temperature of the PV panel (T1), air gap (T2), PEM electrolyzer (T3) and ambient air (Tamb), respectively. The hydrogen collection setup was the same as that used in the laboratory experiment, where the real-time hydrogen collection was recorded using a camera. Real seawater with 3.5 wt% salinity was used as the water source in the outdoor experiment. | ||
| Fig. 5 Outdoor test of the HSD-WE device on a partly sunny day. (a) Image of the experimental setup installed on the rooftop. The outdoor test was performed on a partly sunny day in Cambridge, MA (August 05, 2024). (b) Temperature of the HSD-WE device as a function of time during the outdoor test. (c) Solar flux (red curve) and green hydrogen production rate (blue curve) as a function of time. The hydrogen production rate was determined from the water displacement approach. The average HHV-based STH efficiency of the HSD-WE device during the outdoor test was 12.3%. (d) Time-lapse images of continuous green hydrogen production from the HSD-WE device during the outdoor test. | ||
The experiment started at 11:30 am (local time) and ended at 5:30 pm (local time). After the experiment started, the temperature of the HSD-WE device rose up rapidly within the first one hour (Fig. 5b). We note that the peak temperature of the PV panel was ∼43 °C (blue curve in Fig. 5b), which was only ∼10 °C above the ambient temperature (purple curve in Fig. 5b). Compared with the laboratory experiment, the temperature rise of the PV panel was much lower, which is desirable to achieve high solar-to-electricity conversion efficiency. This is because the wind provided additional convective cooling, and the natural sunlight had a relatively lower solar flux (<800 W m−2) than the one-sun illumination. Owing to the condensation heating, the temperature of the PEM electrolyzer was above 35 °C, which was comparable to that in the laboratory experiment. The weather was sunny during the first one-hour operation and then became increasingly cloudier after 1:00 pm. As a result, the solar flux was stable at around 750 W m−2 from 11:30 am to 1:00 pm and then became highly fluctuating from 200 W m−2 to 800 W m−2 due to clouds (red curve in Fig. 5c). Accordingly, the temperature of the HSD-WE device also showed a fluctuation, where each spike in the measured solar flux (red curve in Fig. 5c) corresponds to a temperature drop-and-recovery cycle (Fig. 5b). Fig. 5d shows the time-lapse images of hydrogen collection during the outdoor test, where water inside the gas collection cylinder was marked by blue shadows (Video S2, ESI†). Due to the stable solar flux from 11:30 am to 1:30 pm (red curve in Fig. 5c), the first two-hour operation showed a highly stable hydrogen production rate around 150 mL h−1 (blue curve in Fig. 5c). At 2:15 pm, the gas collection cylinder was fully filled with 435 mL wet hydrogen (Fig. 5d). At 2:20 pm, we installed a new cylinder to continue the hydrogen collection (Fig. 5d). The hydrogen production rate was maintained above 100 mL h−1 until 3:00 pm. After that, due to the significant reduction of solar flux (<350 W m−2 on average), the hydrogen production rate decreased to around 50 mL h−1 (blue curve in Fig. 5c). By the end of the experiment (5:20 pm), an additional 220 mL wet hydrogen was collected inside the cylinder (Fig. 5d). Throughout the six-hour operation, the HSD-WE device showed an average HHV-based STH efficiency of 12.3% (Fig. 5c) (Note S.13, ESI†). Even on a partly sunny day, more than 655 mL hydrogen was collected in total, indicating a daily hydrogen production rate of ∼133.7 L m−2. To further confirm the reliability of the HSD-WE device, we performed multiple outdoor tests on the other days, and similar STH efficiencies were obtained (Note S.13, ESI†). The STH efficiency shown in the outdoor testing was highly consistent with that demonstrated in the laboratory experiment (Fig. 4f), indicating a stable performance of hydrogen production.
Technoeconomic analysis
We performed a technoeconomic analysis to assess the translational potential of the solar-powered seawater electrolysis for green hydrogen production. Fig. 6a shows the cost of hydrogen production as a function of time, where the HSD-WE device (red curve) was compared with the conventional water electrolysis (WE) based on PEM electrolyzer (blue curve). The general approach of technoeconomic analysis has been demonstrated in our previous works (Note S.14, ESI†).60,61 The capital expenditure (CAPEX) of a baseline PEM electrolyzer was determined from the US Department of Energy (DOE) record in 2020,62,63 whereas the operational expenditure (OPEX) for WE included the cost of electricity and clean water. The CAPEX of the HSD-WE device was estimated by adding the cost of the PV panel and capillary wick to that of the baseline PEM electrolyzer (Note S.14, ESI†). In general, the cost of hydrogen production decreased with the operation time for both conventional WE and HSD-WE (Fig. 6a). This is because the longer the operation time is, the more hydrogen will be produced in total and hence the easier it is to recover the initial capital investment. However, for conventional WE, the cost of hydrogen production is fundamentally limited to OPEX. As a result, after two-year operation, our analysis shows that the hydrogen cost of conventional WE stabilized at $10 per kg, which was consistent with recent reports.10,64 In contrast, the economic feasibility of HSD-WE became significant during a long-term operation (Fig. 6a). Owing to the passive operation nature, the OPEX associated with the HSD-WE device can be minimized. Despite the slightly higher CAPEX due to the adoption of the PV panel and capillary wick, the cost of hydrogen production for HSD-WE became lower than that for conventional WE after the first year (grey dashed line in Fig. 6a), monotonically decreasing with time. Notably, our technoeconomic analysis shows that the cost of hydrogen production for HSD-WE can be reduced to $5 per kg with three-year operation and $1 per kg with 15-year operation (Fig. 6a). To make a fair comparison, we assumed the hydrogen production rate of conventional WE was the same as that of HSD-WE in the above analysis, which might not be always valid, especially considering the high current density commonly applied to conventional WE systems. However, we note that the long-term technoeconomic performance of conventional WE can be weakly dependent on the hydrogen production rate because the OPEX is the predominant source of hydrogen cost (Note S.14, ESI†). | ||
| Fig. 6 Technoeconomic analysis of solar-powered seawater electrolysis for green hydrogen production. (a) Cost of hydrogen production as a function of operation time. Hydrogen production through conventional water electrolysis (WE) is limited by the operational expenditure (OPEX) due to the consumption of clean water and electricity. Hydrogen production through the HSD-WE approach can be cheaper than the conventional WE after one-year operation, reaching $5 per kg with three-year operation and $1 per kg with 15-year operation. (b) Global potential of green hydrogen production using the HSD-WE approach. An average annual green hydrogen production of 233 kWh m−2 was predicted with the demonstrated STH efficiency as the input. Green hydrogen production in selected cities is marked on the global map. The amount of annual green hydrogen production in each city is shown in brackets with a unit of kWh m−2 per year. | ||
We further simulated the global potential of green hydrogen production using the HSD-WE device with the demonstrated STH efficiency and global direct normal irradiance as the inputs (Note S.15, ESI†). Fig. 6b shows the global distribution of annual green hydrogen production. Considering only the inland operation, the total annual production of green hydrogen can reach 3.5 × 107 TWh, equivalent to 8.8 × 105 Mt, which is much higher than the global demand for green hydrogen in 2050 (>500 Mt). We note that our analysis was largely idealized because it is practically impossible to cover the entire land surface with HSD-WE devices. Nevertheless, the above analysis has shown the huge potential of HSD-WE devices for solar-powered seawater electrolysis, because even if only 0.06% of the land surface can be covered by HSD-WE devices, the resulting hydrogen production will satisfy the global demand. With offshore operation, the total green hydrogen production can be further increased. Depending on solar irradiance, the local hydrogen production ranged from 22 kWh m−2 per year to 383 kWh m−2 per year, with an average value of 233 kWh m−2 per year (5.91 kg m−2 per year). For most locations as marked in Fig. 6b, the hydrogen production can be above 200 kWh m−2 per year (5.08 kg m−2 per year). To facilitate large-scale deployment and long-term operation, effective scale-up strategies, detailed degradation mechanisms, and rigorous maintenance protocols should be systematically investigated in future studies.
Conclusions
In summary, we developed a high-efficiency and low-cost solar-powered seawater electrolysis approach for green hydrogen production. Our approach combined PV and PT effects to achieve the full-spectrum utilization of solar energy. Electricity produced by the PV panel was used to power electrolysis, whereas the waste heat of the PV panel was harnessed to distill seawater. We designed an HSD-WE device, which integrated an interfacial thermal evaporator with a PEM electrolyzer, achieving in situ water purification without electricity consumption. We carefully optimized the coupling among the Si PV panel, interfacial thermal evaporator, and PEM electrolyzer to enable high-performance PV cooling, clean water production, salt rejection, and condensation heating, which are critical to facilitate efficient solar-to-hydrogen conversion. With real seawater as the input, we demonstrated more than 12% STH efficiency and up to 35.9 L m−2 h−1 hydrogen production rate in both laboratory and outdoor conditions. With a fully passive operation and low-cost materials, our approach exhibited superior economic feasibility, making less than $1 per kg green hydrogen possible within decades. The developed HSD-WE device can be not only integrated with existing solar farms but also installed in various off-grid and water-stressed areas, significantly increasing the accessibility to green hydrogen. Taking advantage of the most abundant resources on our planet, sunlight and seawater, this work provides a viable solution to address the substantial electricity and clean water consumption requirements associated with electrolysis, promising a sustainable pathway toward low-cost green hydrogen production with record-high energy efficiency.Author contributions
L. Z., X. L., and Y. L. conceived the initial concept. X. W., L. Z., X. L., and Y. L. designed and built the HSD-WE prototype. X. W., L. Z., and X. L. developed the theoretical model and optimized design parameters. X. W., X. L., J. G., Y. W., and L. Z. performed the experiments and processed the experimental data. X. W., L. Z., X. L, J. G., Y. W., and Y. L. interpreted the theoretical and experimental results. L. Z., X. W., X. L., Y. L., J. G., and Y. W. wrote the manuscript. L. Z., X. L., and Y. L. supervised the project.Data availability
The data that support the findings of this study are available in the ESI.† Additional data are available from the corresponding authors on request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Dr Toochukwu Aka for providing technical support during outdoor measurements. The authors gratefully acknowledge the funding support from the National Science Foundation under Grant No. 2401017.References
- Net Zero by 2050: A Roadmap for the Global Energy Sector, International Energy Agency, 2021.
- I. Dincer, Int. J. Hydrogen Energy, 2012, 37, 1954–1971 Search PubMed.
- A. M. Oliveira, R. R. Beswick and Y. Yan, Curr. Opin. Chem. Eng., 2021, 33, 100701 CrossRef.
- M. A. Modestino, D. Fernandez Rivas, S. M. H. Hashemi, J. G. E. Gardeniers and D. Psaltis, Energy Environ. Sci., 2016, 9, 3381–3391 Search PubMed.
- J. Jia, L. C. Seitz, J. D. Benck, Y. Huo, Y. Chen, J. W. D. Ng, T. Bilir, J. S. Harris and T. F. Jaramillo, Nat. Commun., 2016, 7, 1–6 Search PubMed.
- J. H. Kim, D. Hansora, P. Sharma, J. W. Jang and J. S. Lee, Chem. Soc. Rev., 2019, 48, 1908–1971 RSC.
- H. Park, I. J. Park, M. G. Lee, K. C. Kwon, S. P. Hong, D. H. Kim, T. H. Lee, C. Kim and H. W. Jang, ACS Appl. Mater. Interfaces, 2019, 11, 33835–33843 CrossRef CAS PubMed.
- A. M. K. Fehr, A. Agrawal, F. Mandani, C. L. Conrad, Q. Jiang, S. Y. Park, O. Alley, B. Li, S. Sidhik, I. Metcalf, C. Botello, J. L. Young, J. Even, J. C. Blancon, T. G. Deutsch, K. Zhu, S. Albrecht, F. M. Toma, M. Wong and A. D. Mohite, Nat. Commun., 2023, 14, 1–12 Search PubMed.
- Z. Li, S. Fang, H. Sun, R. J. Chung, X. Fang and J. H. He, Adv. Energy Mater., 2023, 13, 2203019 CrossRef CAS.
- A. Sharma, T. Longden, K. Catchpole and F. J. Beck, Energy Environ. Sci., 2023, 16, 4486–4501 RSC.
- I. Holmes-Gentle, S. Tembhurne, C. Suter and S. Haussener, Nat. Energy, 2023, 8, 586–596 CrossRef CAS.
- P. Shi, J. Li, Y. Song, N. Xu and J. Zhu, Nano Lett., 2024, 24, 5673–5682 CrossRef CAS PubMed.
- V. A. Martinez Lopez, H. Ziar, J. W. Haverkort, M. Zeman and O. Isabella, Renewable Sustainable Energy Rev., 2023, 182, 113407 CrossRef CAS.
- S. Ardo, D. F. Rivas, M. A. Modestino, V. S. Greiving, F. F. Abdi, E. A. Llado, V. Artero, K. Ayers, C. Battaglia, J. Becker, D. Bederak, A. Berger, F. Buda, E. Chinello, B. Dam, V. Di Palma, T. Edvinsson, K. Fujii, H. Gardeniers, H. Geerlings, S. M. H. Hashemi, S. Haussener, F. Houle, J. Huskens, B. D. James, K. Konrad, A. Kudo, P. P. Kunturu, D. Lohse, B. Mei, E. L. Miller, G. F. Moore, J. Muller, K. L. Orchard, T. E. Rosser, F. H. Saadi, J. Schüttauf, B. Seger, S. W. Sheehan, W. A. Smith, J. Spurgeon, M. H. Tang, R. van de Krol, P. C. K. Vesborg and P. Westerik, Energy Environ. Sci., 2018, 11, 2768–2783 RSC.
- E. E. Greenwood, T. Lauber, J. van den Hoogen, A. Donmez, R. E. Bain, R. Johnston, T. W. Crowther and T. R. Julian, Science, 2024, 385, 784–790 CrossRef CAS PubMed.
- W. Tong, M. Förster, F. Dionigi, S. Dresp, R. Sadeghi Erami, P. Strasser, A. J. Cowan and P. Farràs, Nat. Energy, 2020, 5, 367–377 CrossRef CAS.
- S. S. Veroneau and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, 1–5 CrossRef PubMed.
- J. Guo, Y. Zhang, A. Zavabeti, K. Chen, Y. Guo, G. Hu, X. Fan and G. K. Li, Nat. Commun., 2022, 13, 5046 CrossRef CAS PubMed.
- C. Qiu, Z. Xu, F. Y. Chen and H. Wang, ACS Catal., 2024, 14, 921–954 CrossRef CAS.
- Y. Kuang, M. J. Kenney, Y. Meng, W. H. Hung, Y. Liu, J. E. Huang, R. Prasanna, P. Li, Y. Li, L. Wang, M. Lin, M. D. McGehee, X. Sun and H. Dai, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 6624–6629 CrossRef CAS PubMed.
- W. Zheng, L. Y. S. Lee and K. Y. Wong, Nanoscale, 2021, 13, 15177–15187 RSC.
- L. Zhang, Z. Wang and J. Qiu, Adv. Mater., 2022, 34, 2109321 CrossRef CAS PubMed.
- X. Lu, J. Pan, E. Lovell, T. H. Tan, Y. H. Ng and R. Amal, Energy Environ. Sci., 2018, 11, 1898–1910 RSC.
- S. Dresp, T. N. Thanh, M. Klingenhof, S. Brückner, P. Hauke and P. Strasser, Energy Environ. Sci., 2020, 13, 1725–1729 RSC.
- H. Jin, X. Wang, C. Tang, A. Vasileff, L. Li, A. Slattery and S. Z. Qiao, Adv. Mater., 2021, 33, 1–8 Search PubMed.
- J. Guo, Y. Zheng, Z. Hu, C. Zheng, J. Mao, K. Du, M. Jaroniec, S. Z. Qiao and T. Ling, Nat. Energy, 2023, 8, 264–272 CAS.
- J. N. Hausmann, R. Schlögl, P. W. Menezes and M. Driess, Energy Environ. Sci., 2021, 14, 3679–3685 RSC.
- P. Farràs, P. Strasser and A. J. Cowan, Joule, 2021, 5, 1921–1923 CrossRef.
- J. N. Hausmann, L. R. Winter, M. A. Khan, M. Elimelech, M. G. Kibria, T. Sontheimer and P. W. Menezes, Joule, 2024, 8, 2436–2442 CrossRef.
- H. Jin, J. Xu, H. Liu, H. Shen, H. Yu, M. Jaroniec, Y. Zheng and S. Z. Qiao, Sci. Adv., 2023, 9, 1–13 Search PubMed.
- A. Boretti and L. Rosa, NPJ Clean Water, 2019, 2, 15 CrossRef.
- Y. Zhang and S. C. Tan, Nat. Sustain., 2022, 5, 554–556 CrossRef.
- P. I. Babb, S. F. Ahmadi, F. Brent, R. Gans, M. A. Lopez, J. Song, Q. Wang, B. Zou, X. Zuo, A. Strom, J. Nolt, T. Susko, K. Fields and Y. Zhu, Cell Rep. Phys. Sci., 2023, 4, 101682 CrossRef CAS.
- P. Poredoš, J. Gao, H. Shan, J. Yu, Z. Shao, Z. Xu and R. Wang, Nat. Commun., 2024, 15, 7890 CrossRef PubMed.
- H. Xie, Z. Zhao, T. Liu, Y. Wu, C. Lan, W. Jiang, L. Zhu, Y. Wang, D. Yang and Z. Shao, Nature, 2022, 612, 673–678 CrossRef CAS PubMed.
- X. Pang, J. T. Davis, A. D. Harvey and D. V. Esposito, Energy Environ. Sci., 2020, 13, 3663–3678 RSC.
- A. Hodges, A. L. Hoang, G. Tsekouras, K. Wagner, C. Y. Lee, G. F. Swiegers and G. G. Wallace, Nat. Commun., 2022, 13, 1304 CrossRef CAS PubMed.
- M. N. I. Salehmin, T. Husaini, J. Goh and A. B. Sulong, Energy Convers. Manag., 2022, 268, 115985 CrossRef CAS.
- L. Zhang, R. Iwata, Z. Lu, X. Wang, C. D. Díaz-Marín and Y. Zhong, Chem. Rev., 2024, 124, 10052–10111 CrossRef CAS PubMed.
- A. Angulo, P. van der Linde, H. Gardeniers, M. Modestino and D. Fernández Rivas, Joule, 2020, 4, 555–579 CrossRef CAS.
- Y. Zhang, H. Zhang, T. Xiong, H. Qu, J. J. Koh, D. K. Nandakumar, J. Wang and S. C. Tan, Energy Environ. Sci., 2020, 13, 4891–4902 RSC.
- M. Sheng, Y. Yang, X. Bin, S. Zhao, C. Pan, F. Nawaz and W. Que, Nano Energy, 2021, 89, 106468 CrossRef CAS.
- W. Wang, Y. Shi, C. Zhang, S. Hong, L. Shi, J. Chang, R. Li, Y. Jin, C. Ong, S. Zhuo and P. Wang, Nat. Commun., 2019, 10, 3012 CrossRef PubMed.
- R. Li, Y. Shi, M. Wu, S. Hong and P. Wang, Nat. Sustain., 2020, 3, 636–643 CrossRef.
- W. Wang, S. Aleid, Y. Shi, C. Zhang, R. Li, M. Wu, S. Zhuo and P. Wang, Joule, 2021, 5, 1873–1887 CrossRef.
- A. Panagopoulos, K. J. Haralambous and M. Loizidou, Sci. Total Environ., 2019, 693, 133545 CrossRef CAS PubMed.
- M. O. Mavukkandy, C. M. Chabib, I. Mustafa, A. Al Ghaferi and F. AlMarzooqi, Desalination, 2019, 472, 114187 CrossRef CAS.
- Z. Xu, L. Zhang, L. Zhao, B. Li, B. Bhatia, C. Wang, K. L. Wilke, Y. Song, O. Labban, J. H. Lienhard, R. Wang and E. N. Wang, Energy Environ. Sci., 2020, 13, 830–839 RSC.
- X. Wang, M.-L. Hsieh, J. A. Bur, S.-Y. Lin and S. Narayanan, Mater. Today Energy, 2020, 17, 100453 CrossRef.
- C. C. L. McCrory, S. Jung, I. M. Ferrer, S. M. Chatman, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2015, 137, 4347–4357 CrossRef CAS PubMed.
- Y. Cengel, M. Boles and M. Kanoglu, Thermodynamics: An Engineering Approach, McGraw-Hill, 2023 Search PubMed.
- C. R. Cox, J. Z. Lee, D. G. Nocera and T. Buonassisi, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 14057–14061 CrossRef CAS PubMed.
- J. W. Ager, M. R. Shaner, K. A. Walczak, I. D. Sharp and S. Ardo, Energy Environ. Sci., 2015, 8, 2811–2824 RSC.
- S. Tembhurne, F. Nandjou and S. Haussener, Nat. Energy, 2019, 4, 399–407 CrossRef CAS.
- W. Yang, R. R. Prabhakar, J. Tan, S. D. Tilley and J. Moon, Chem. Soc. Rev., 2019, 48, 4979–5015 RSC.
- L. Cai, J. Zhao, H. Li, J. Park, I. S. Cho, H. S. Han and X. Zheng, ACS Energy Lett., 2016, 1, 624–632 CrossRef CAS.
- Z. Wang, C. Li and K. Domen, Chem. Soc. Rev., 2019, 48, 2109–2125 RSC.
- H. Nishiyama, T. Yamada, M. Nakabayashi, Y. Maehara, M. Yamaguchi, Y. Kuromiya, Y. Nagatsuma, H. Tokudome, S. Akiyama, T. Watanabe, R. Narushima, S. Okunaka, N. Shibata, T. Takata, T. Hisatomi and K. Domen, Nature, 2021, 598, 304–307 CrossRef CAS PubMed.
- P. Zhou, I. A. Navid, Y. Ma, Y. Xiao, P. Wang, Z. Ye, B. Zhuo, K. Sun and Z. Mi, Nature, 2023, 613, 66–70 CrossRef CAS PubMed.
- L. Zhang, Z. Xu, L. Zhao, B. Bhatia, Y. Zhong, S. Gong and E. N. Wang, Energy Environ. Sci., 2021, 14, 1771–1793 RSC.
- Y. Zhong, L. Zhang, X. Li, B. El Fil, C. D. Díaz-Marín, A. C. Li, X. Liu, A. LaPotin and E. N. Wang, Nat. Rev. Mater., 2024, 9, 681–698 CrossRef.
- Technical Targets for Proton Exchange Membrane Electrolysis. U.S. Department of Energy.
- M. Hubert, D. Peterson, E. Miller, J. Vickers, R. Mow and C. Howe, DOE Hydrogen Program Record 24005: Clean Hydrogen Production Cost Scenarios with PEM Electrolyzer Technology, 2022.
- Green Hydrogen Cost Reduction: Scaling up Electrolysers to Meet the 1.5 °C Climate Goal. International Renewable Energy Agency, 2020.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ee06203e |
| This journal is © The Royal Society of Chemistry 2025 |