
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBiomass-derived functional additive for highly efficient and stable lead halide perovskite solar cells with built-in lead immobilisation†
Jing
Li
 a,
Xiang
Qiao
c,
Bingchen
He
d,
Yuan
Zhang
b,
Subhajit
Pal
b,
Linchao
Sun
a,
Muhammad
Bilal
b,
Zhenhuang
Su
d,
Xingyu
Gao
d,
Joe
Briscoe
b,
Isaac
Abrahams
a,
Meng
Li
*c,
Zhe
Li
*b and
Yao
Lu
*a
a,
Xiang
Qiao
c,
Bingchen
He
d,
Yuan
Zhang
b,
Subhajit
Pal
b,
Linchao
Sun
a,
Muhammad
Bilal
b,
Zhenhuang
Su
d,
Xingyu
Gao
d,
Joe
Briscoe
b,
Isaac
Abrahams
a,
Meng
Li
*c,
Zhe
Li
*b and
Yao
Lu
*a
aDepartment of Chemistry, School of Physical and Chemical Sciences (SPCS), Queen Mary University of London, London, E1 4NS, UK. E-mail: yao.lu@qmul.ac.uk
bSchool of Engineering and Materials Science (SEMS), Queen Mary University of London, London, E1 4NS, UK. E-mail: zhe.li@qmul.ac.uk
cKey Lab for Special Functional Materials of Ministry of Education, National & Local Joint Engineering Research Center for High-efficiency Display and Lighting Technology, School of Nanoscience and Materials Engineering, and Collaborative Innovation Center of Nano Functional Materials and Applications, Henan University, Kaifeng, 475004, P. R. China. E-mail: mengli@henu.edu.cn
dShanghai Synchrotron Radiation Facility (SSRF), Shanghai Advanced Research Institute Chinese Academy of Sciences, 239 Zhang Heng Road, Shanghai, 201204, P. R. China
First published on 8th May 2025
Abstract
Despite notable progress in the power conversion efficiency (PCE) of lead halide perovskite solar cells (PSCs), their commercial viability remains limited by stability issues and the risk of lead contamination. Uncoordinated lead ions can introduce defects during perovskite crystallization, resulting in reduced stability and potential environmental contamination. Here, we synthesized a biomass-derived tetrabutylammonium alginate (TBA-Alg) polymer that forms a connected network at the perovskite surface and grain boundaries to effectively manage lead ions and passivate defects. The alginate groups anchor unbound lead ions, promoting more ordered crystallization, while the hydrophobic tetrabutylammonium chains enhance moisture resistance. The TBA-Alg-modified inverted p–i–n PSCs achieved a PCE of 25.01% and retained 95.5% of their initial performance after 2000 hours of storage. Under continuous illumination at ∼60% relative humidity (RH) for 1050 hours, the devices retained 80% efficiency. Even under water immersion, the TBA-Alg network effectively protected lead ions from water erosion and suppressed 83% of lead leakage. This strategy simultaneously achieves high PCE and stability of lead halide PSCs, and effectively prevents lead contamination; thereby offering the potential to greatly advance the commercialization of lead halide PSCs.
Broader contextPerovskite solar cells (PSCs) have emerged as a next-generation photovoltaic technology due to their high efficiency, low production costs, and tunable optoelectronic properties. However, the large-scale commercialization of PSCs faces two critical challenges: the instability of perovskite materials under operational conditions and the potential environmental risk of lead leakage. These issues hinder the adoption of PSCs as a sustainable alternative to traditional silicon-based solar cells. To address these challenges, this study introduces a biomass-derived multifunctional polymer complex, tetrabutylammonium alginate (TBA-Alg), which provides dual benefits of stabilizing perovskite structures and immobilizing lead ions. By forming a robust “egg-box” structure with lead ions, TBA-Alg significantly reduces lead leakage, mitigating potential environmental hazards. Simultaneously, TBA-Alg enhances crystallization, reduces defects, and introduces hydrophobic barriers to protect PSCs from moisture-induced degradation. This strategy promotes both long-term operational stability and environmental safety, offering a comprehensive approach to PSC commercialization. This work supports global efforts to develop sustainable and environmentally friendly photovoltaic technologies. The introduction of a scalable, cost-effective, and bio-derived additive facilitates the large-scale deployment of PSCs with improved efficiency, stability, and environmental safety, representing a significant step toward safe and high-performance solar energy solutions. |
Introduction
Organic–inorganic lead halide perovskites (PSCs) have emerged as a promising class of materials for the development of the next generation of solar cells due to their outstanding optoelectronic properties. These include remarkably high absorption coefficients, tunable bandgaps, longer charge-carrier diffusion lengths, and cost-effective manufacturing processes.1,2 To date, the certified power conversion efficiency (PCE) of lead halide perovskite solar cells has reached 26.64%, approaching that of commercial silicon-based solar cells.3–6 Despite considerable advancements, the practical application of PSCs is still facing significant challenges, particularly the instability of perovskites under real-world operating conditions, which remains a barrier to commercialization. This instability reduces the efficiency and overall performance of PSC devices. Furthermore, the large-scale commercialization of PSCs poses risks of lead pollution, a potential threat to both human health and the environment.The instability of lead halide perovskites is usually attributed to deep-level defects within its internal structure, particularly uncoordinated lead ions, as well as defects at the surface and grain boundaries of the perovskite layer. During the fabrication of solution-based perovskite films, rapid phase transitions often result in incomplete coordination between the lead ions and halide ions, disrupting the crystallization process and leading to the formation of defects at the grain boundaries and interfaces of the perovskite films. These defects act as non-radiative recombination centers, impairing charge transport, reducing overall device efficiency, and accelerating device degradation.7,8 Additionally, unbound lead ions in the perovskite layer are susceptible to dissolution and migration in the presence of moisture, leading to lead leakage and posing significant environmental and health risks.9–11 Therefore, to achieve real-world practical applications, it is essential to effectively stabilize lead ions, minimise defect formation, and prevent moisture invasion for enhancing the long-term performance of PSCs.
Additive engineering is a widely used and effective strategy for enhancing the stability of perovskite solar cells.12–15 Lewis base-acid adducts containing N-donors, S-donors or O-donors with lone electron pairs have demonstrated the ability to effectively coordinate with lead ions, contributing to the development of high quality and stable perovskite solar cells.15–17 Biomass-derived polymers such as cellulose, chitosan, and starch have attracted attention due to their electron-donating functional groups.18,19 These biopolymers contain hydroxyl, amino, and ester groups, which can interact with perovskite components through hydrogen bonding and coordination with lead ions.20 Han et al. employed cellulose derivatives, including cellulose acetate butyrate (CAB) and hydroxyethyl cellulose (HEC), as additives to passivate defects through hydrogen bonding and coordination with lead ions, leading to improved crystallinity and stability. CAB-based devices achieved a PCE of 21.5%, maintaining over 90% efficiency after 3300 h aging under 35% RH.21 Vanni et al. used camphor sulfonic-acid-modified chitosan as additive to stabilize the α-phase FAPbI3, improving film uniformity and charge transport; the resulting devices showed a PCE of 20.3% and retained 90% of their initial efficiency after 1000 h aging in air22 Zhang et al. introduced a starch–polyiodide complex as an additive to form a buffer layer that suppressed ion migration and facilitated vacancy healing, achieving a certified PCE of 23.9% and high operational stability.23 Despite these benefits, these additives offer only limited efficiency and stability improvement. Furthermore, few additives have demonstrated their potential to simultaneously reduce the risks of lead leaching, and their resistance to moisture remains insufficiently explored.
Existing studies have demonstrated that tetrabutylammonium ions (TBA+), as hydrophobic materials with alkyl chains, can enhance surface water resistance when used as additives, thereby improving the long-term stability of perovskite solar cells.24,25 Additionally, when incorporated into the perovskite matrix, TBA+ can promote more orderly crystallization, reducing defect sites where degradation is typically initiated.26 However, TBA+ tends to form a layered structure, complicating the uniform embedding within the perovskite structure. Consequently, simultaneously achieving high PCE, long-term stability, and effective lead containment within a single perovskite system remains a significant challenge.
This study introduces a sustainable and low-cost multi-functional polymer complex, tetrabutylammonium alginate (TBA-Alg), derived from biomass sources, into perovskite solar cells to enhance their overall performance, operational durability, and environmental safety. The alginate groups effectively capture and immobilise lead ions by forming a robust “egg-box” structure with lead ions, significantly mitigating the risk of lead leakage during device operation. Unlike conventional lead containment methods, this bio-derived polymer provides dual functionality by simultaneously enhancing device stability and reducing environmental hazards. During the crystallization process, the TBA-Alg chelates lead ions through Lewis base-acid interactions involving C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups, extending the crystallization time and minimizing defects. Furthermore, the TBA-Alg forms a connected network at the grain boundaries and perovskite surfaces, creating hydrophobic barriers that protect the perovskite films from moisture-induced erosion, a critical factor for long-term stability. These unique properties overcome the limitations of existing strategies by integrating lead immobilization with improved structural and environmental stability. This approach enables PSCs with an inverted architecture to achieve a high efficiency of 25.01% and retaining 95.5% of their initial efficiency after 2000 hours of storage. Under continuous illumination at 60% humidity for 1050 hours, the devices maintained 80% of their initial efficiency. TBA-Alg treated PSCs with an n–i–p structure also achieved an impressive PCE of 24.44%, and TBA-Alg treated large-area modules (16.8 cm2) achieved a PCE of 22.17%, thereby confirming the compatibility of the strategy with different PSC device configurations and scalable deposition methods. Furthermore, unencapsulated perovskite films treated with TBA-Alg show significantly reduced lead leakage, even when fully immersed in water for 2 hours. These findings highlight the potential of TBA-Alg as a sustainable and scalable material for advancing the stability and performance of PSCs while preventing lead contamination.
O groups, extending the crystallization time and minimizing defects. Furthermore, the TBA-Alg forms a connected network at the grain boundaries and perovskite surfaces, creating hydrophobic barriers that protect the perovskite films from moisture-induced erosion, a critical factor for long-term stability. These unique properties overcome the limitations of existing strategies by integrating lead immobilization with improved structural and environmental stability. This approach enables PSCs with an inverted architecture to achieve a high efficiency of 25.01% and retaining 95.5% of their initial efficiency after 2000 hours of storage. Under continuous illumination at 60% humidity for 1050 hours, the devices maintained 80% of their initial efficiency. TBA-Alg treated PSCs with an n–i–p structure also achieved an impressive PCE of 24.44%, and TBA-Alg treated large-area modules (16.8 cm2) achieved a PCE of 22.17%, thereby confirming the compatibility of the strategy with different PSC device configurations and scalable deposition methods. Furthermore, unencapsulated perovskite films treated with TBA-Alg show significantly reduced lead leakage, even when fully immersed in water for 2 hours. These findings highlight the potential of TBA-Alg as a sustainable and scalable material for advancing the stability and performance of PSCs while preventing lead contamination.
Results and discussion
To mitigate lead leakage, we selected alginate groups featuring an “egg-box” structure to capture lead ions. However, a key challenge is that alginates do not dissolve well in dimethyl sulfoxide (DMSO) and N,N-dimethylformamide (DMF), which are commonly used precursor solvents for perovskite solar cells, as shown in Fig. S1(a) (ESI†). To overcome this limitation, we synthesized tetrabutylammonium alginate (TBA-Alg) via a simple, cost-effective route by acidifying biomass-derived sodium alginate with HCl in an ethanol/water solution, followed by cation exchange with TBAOH (Fig. S2, ESI†). The resulting TBA-Alg dissolves uniformly in a DMSO/DMF solvent system (Fig. S1(b), ESI†). Its molecular design combined an “egg box” structure from the alginate anchoring groups and a hydrophobic feature from the alkyl arms. This synthesis approach not only ensures functionality but also aligns with the principles of green chemistry, offering an eco-friendly, economically viable pathway for integrating TBA-Alg into perovskite solar cells. Proton nuclear magnetic resonance (1H NMR) analysis confirms that sodium alginate has been effectively modified with tetrabutylammonium as shown in Fig. S3 (ESI†). Fig. S4 (ESI†) shows the Fourier transform infrared (FTIR) spectra of sodium alginate and the target polymer TBA-Alg. The presence of the C–N stretching vibration peak at 1240 cm−1 confirms the successful synthesis of TBA-Alg. The molecular weight of TBA-Alg was determined using the viscometric method (as shown in Fig. S5, ESI†), and the viscosity-average molecular weight (Mn) was 28.7 kDa.The chemical structure of the target polymer TBA-Alg and its hypothesised reaction with lead ions are shown in Fig. 1a. When TBA-Alg is added to the perovskite precursor, its polymer chains are expected to capture lead ions, creating ordered nucleation sites that facilitate a controlled and uniform film crystallization process.27,28Fig. 1b illustrates the mechanism of the nucleation and crystallization process. Without TBA-Alg chains, crystallization proceeded in a disordered manner, leading to the formation of irregularly sized and distributed perovskite grains with numerous defects. In contrast, the presence of TBA-Alg chains significantly affects the crystallization process, promoting the formation of larger and more ordered crystals. Moreover, the TBA-Alg connected at the grain boundaries and surfaces, passivating defects and improving the moisture resistance of the perovskite.
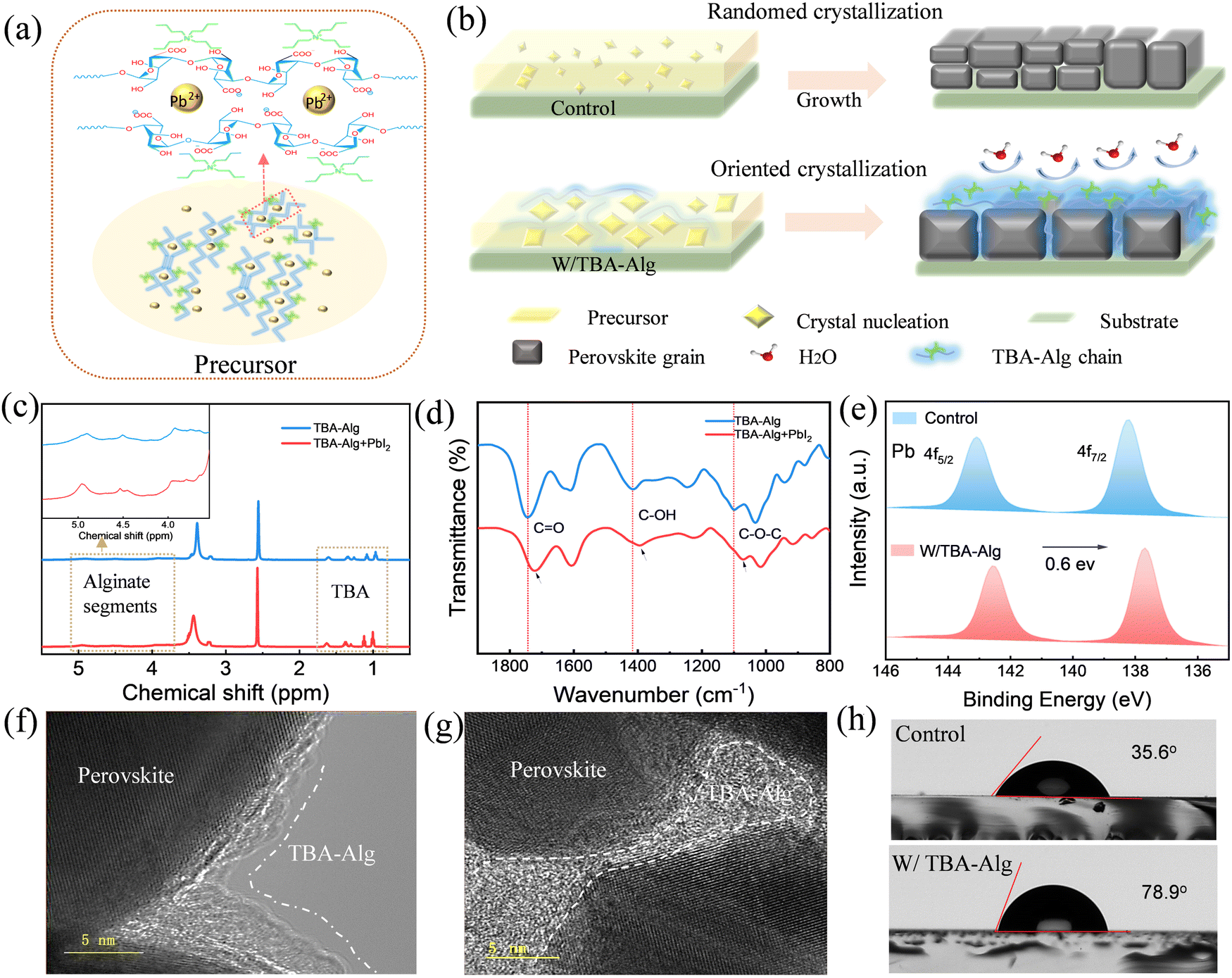 | ||
| Fig. 1 The functional mechanism of TBA-Alg treatment in perovskite films. (a) Molecular interactions within the precursor solution of TBA-Alg-treated samples. (b) Mechanism of nucleation and crystallization processes in control samples compared to those with incorporated TBA-Alg. (c) 1H NMR spectra and (d) FTIR spectra of TBA-Alg and TBA-Alg with PbI2. (e) Pb 4f XPS spectra of the perovskite films for both the control sample and the TBA-Alg-treated sample. (f) and (g) are HRTEM images of the TBA-Alg-treated film, showing the grain surface and grain boundary, respectively. (h) Water contact angle of control and TBA-Alg-treated (W/TBA-Alg) samples. | ||
We subsequently investigate the interaction between the TBA-Alg and the lead ions. First, we performed 1H NMR spectroscopy on pure TBA-Alg and TBA-Alg with PbI2, as shown in Fig. 1c. Upon interaction with lead ions, the resonance signal for the protons adjacent to the CO group exhibits a downfield shift from 4.89 ppm to 4.96 ppm. This shift suggests a decrease in electron cloud density, highlighting the electron-donating nature of the CO group when interacting with uncoordinated lead ions.29 Similarly, Fig. 1d shows the FTIR spectra of pure TBA-Alg and TBA-Alg with PbI2. The results indicate that pure TBA-Alg exhibits stretching vibrations of –OH (3504 cm−1), CO (1745 cm−1), and C–O–C (1097 cm−1) groups.30 Upon the addition of PbI2, the stretching vibration peaks of –OH, CO and C–O–C are shifted to 3471 cm−1, 1743 cm−1, and 1091 cm−1, respectively.31,32 The shift of characteristic peaks indicates an interaction between the O-donor and the uncoordinated lead ions due to Lewis base interactions.33 Additionally, the peaks at 2944 cm−1 and 2886 cm−1, corresponding to –CH3 and –CH2 bending vibrations on the TBA alkyl group respectively, are shifted to 2938 cm−1 and 2882 cm−1 after mixing with PbI2, as shown in Fig. S6 (ESI†). This shift was attributed to the electrostatic interaction between the TBA alkyl group and uncoordinated lead ions.34,35 We used X-ray photoelectron spectroscopy (XPS) to confirm the binding energy of the TBA-Alg-treated perovskite sample that exhibits a noticeable reduction (0.6 eV) compared to that of the control sample (without TBA-Alg), indicating that the CO bonds are strongly coordinated with lead ions,36 consistent with the 1H-NMR and FTIR results. The spectra confirm the presence and binding states of lead ions, indicating the successful incorporation of lead ions into the TBA-Alg matrix through effective coordination. In order to clarify the interaction between the additive TBA-Alg and the lead halide perovskite, we calculated the electrostatic potential (ESP) distribution of the TBA-Alg molecule, as shown in Fig. S7a (ESI†). The positions with lower potential (higher electron density) in the TBA-Alg molecule are mainly concentrated on the carbonyl oxygen atom, indicating that this functional group has a strong Lewis basicity and can coordinate with the uncoordinated ions in the perovskite to achieve defect passivation. We further calculated the binding energy between the carbonyl group and lead ions using density functional theory (DFT), which was found to be −1.13 eV (Fig. S7b, ESI†), indicating a strong interaction between the CO sites and lead atoms, this is consistent with the results of the ESP analysis.
High-resolution transmission electron microscope (HRTEM) was utilized to investigate the distribution of connected network formed by TBA-Alg. Fig. 1f and Fig. S8 (ESI†) show the presence of an amorphous layer (approximately 2–3 nm thick) surrounding the surfaces of perovskite grains, while Fig. 1g reveals that the amorphous polymer has interconnected the crystal boundaries, linking adjacent domains. The TEM results further confirm that the TBA-Alg can form a connected network at the grain boundaries and interfaces, contributing to defect passivation and reducing the number of trap states.37–39 This is consistent with the crystallization mechanism in Fig. 1b. To further confirm the composition of the amorphous layer, HRTEM and corresponding energy-dispersive X-ray spectroscopy (EDS) elemental mapping were performed as shown in Fig. S9 (ESI†). The results indicate that the Pb signal still existed in the marked region, though more diffuse and less intense. Meanwhile, strong signals of C, N, and O elements were detected, indicating that the region is primarily composed of organic components. Therefore, this amorphous layer is more likely to be a TBA-Alg-Pb composite rather than pure TBA-Alg. Additionally, the iodine(I) signal is relatively weak, indicating that this region is not a typical perovskite structure but a complex formed by the interaction between lead ions and TBA-Alg. This further supports the role of TBA-Alg in regulating the perovskite crystallization process by stabilizing lead ions at the grain boundaries, which aligns with the crystallization mechanism shown in Fig. 1b. Fig. 1h shows water contact angle measurements of perovskite films. The significant increase in contact angle from 35.6° to 78.9° after TBA-Alg treatment suggests that the surface has become more hydrophobic, potentially attributed to the alkyl chains on the TBA-Alg. This result indicates that the addition of TBA-Alg is capable of enhancing the resistance of the perovskite film to water infiltration.34
Scanning electron microscopy (SEM), X-ray diffraction (XRD), atomic force microscopy (AFM), and grazing incidence wide-angle X-ray scattering (GIWAXS) were conducted to analyse the crystallization morphology of the films and monitor the film formation process. Fig. 2a and b shows the top-view and cross-sectional SEM images of the control and TBA-Alg-treated perovskite films, respectively. Compared to the control film (Fig. 2a), the modified film in Fig. 2b did not exhibit the white PbI2 phase on its surface and displayed a smoother and larger crystal morphology. The cross-sectional view further revealed that the grains in the modified film were larger and vertically oriented, in contrast to the smaller, irregular grains observed in the control film. Additionally, the grain size distribution of perovskite film was measured, as shown in Fig. S10 (ESI†). The average grain size of the TBA-Alg-treated perovskite film was approximately 668 nm, which is significantly larger than that of the control sample (approximately 261 nm). The enlarged grains and smoother surface of the TBA-Alg films are also evident in the AFM images presented in Fig. S11 (ESI†). The root mean square (RMS) value of the perovskite surface roughness decreased from 13.9 nm in the control sample to 11.9 nm in the treated sample. These improvements are beneficial in reducing the recombination of carriers at grain boundaries and accelerating charge transport.40–42
 | ||
| Fig. 2 Top-view and cross-sectional SEM images of (a) the control film and (b) the film with TBA-Alg treatment. (c) XRD patterns of the control and TBA-Alg-treated perovskite films. Temporal evolution of in situ GIWAXS patterns during annealing (d) control and (e) TBA-Alg-treated perovskite films. (f) Time-resolved integrated peak area intensity for black phases of the control and the TBA-Alg-treated perovskites (q = 9.98 nm−1). | ||
Compared with the XRD patterns of the control sample, the diffraction peak of the modified perovskite film shows a significant increase in the peak intensities of the (001) reflection, with an increase of the intensity ratio between the (001) and (012) peaks from 3.3 (control) to 9.2 (treated) (Fig. 2c). Additionally, the full width at half maximum (FWHM) of the (001) peak is shown to decrease from 0.21 (control) to 0.10 (treated), suggesting enhanced crystallinity and a more pronounced (001) orientation in the modified film.36 Furthermore, small characteristic peaks of PbI2 are observed in the control films, which are absent in the modified film. The findings indicate that the incorporation of TBA-Alg can effectively regulate the growth of perovskite crystals and suppress the formation of PbI2, resulting in higher-quality films.43,44
To elucidate the impact of TBA-Alg on the crystallization kinetics of perovskites films, GIWAXS was employed to monitor the perovskite crystallization process. The perovskite precursor solution was spin-coated onto a glass substrate under ambient air, followed by the addition of an anti-solvent at 30 s. Subsequently, phase transformations during the annealing process at 100 °C were probed with GIWAXS. For the Cs0.05(FA0.95MA0.05)0.95Pb(I0.95Br0.05)3 formulation, both the perovskite phase (q = 9.98 nm−1) and the intermediate phase (q = 5.80 nm−1) were initially present. Over time, the intermediate phase gradually diminished and ultimately disappeared.45 In the control sample, as shown in Fig. 2d, the intermediate phase fully transitions into the perovskite phase within 35 s.
With the introduction of TBA-Alg, the O-donor groups coordinate with lead ions, providing controlled nucleation sites and facilitating the incorporation of more free lead ions into the octahedral framework of perovskite crystals, as illustrated in Fig. S12a (ESI). This interaction stabilizes the intermediate phase, prolonging its existence and allowing more time for thorough crystallization. As a result, the perovskite crystallization process is delayed, significantly extending the phase transition time to 45 s, as shown in Fig. 2e. The GIWAXS profile analysis (Fig. S12b, ESI†) further confirms that TBA-Alg slows down the crystallization process. In the control sample, the intermediate phase disappeared within 35 s, indicating a faster transition. In comparison, with TBA-Alg, the existence of intermediate phase prolonged to 45 s, demonstrating a more gradual crystallization process. This is attributed to the interaction between TBA-Alg and lead ions, which stabilizes the intermediate phase and enables more controlled crystal formation. By extending this intermediate phase, TBA-Alg provides more time for perovskite growth, leading to reduced defects and improved film quality.
The temporal evolution of the q = 9.98 nm−1 peak (Fig. 2d and e), with the peak area plotted in Fig. 2f, reveals enhanced crystallinity in the TBA-Alg-treated sample compared to the control sample. Furthermore, GIWAXS patterns in Fig. S13 (ESI†) indicate that the Debye–Scherrer-like ring (D–S ring) intensities at the (001) and (002) crystal planes are higher in the TBA-Alg-treated film than that in the control film. These GIWAXS results indicate that the incorporation of TBA-Alg effectively extends the duration of the mesophase, providing sufficient time for perovskite growth, which results in increased crystal grain size and improved film quality.45,46 These structural improvements should enable better performance in the optoelectronic properties of perovskite solar cells.
Conductive atomic force microscopy (C-AFM) measurements were taken in both dark and illuminated states to reveal the defect behaviour and photoconductive response of perovskite film, as shown in Fig. 3a–d. The control film in the dark (Fig. 3a) shows large grain boundaries and visible defects. Compared to the control film, the TBA-Alg-treated film (Fig. 3b) in the dark exhibits a much smoother and more uniform surface, with significantly fewer visible defects and grain boundaries.
 | ||
| Fig. 3 Characterization of the control and TBA-Alg treated PSCs. C-AFM images in the dark: (a) control film, (b) TBA-Alg-treated film, and under illumination: (c) control film, (d) TBA-Alg film. (e) Current distribution histograms under dark and light conditions for both the control and TBA-Alg-treated films. (f) and (g) are schematic diagrams showing the surface of the control and TBA-Alg-treated perovskite films. (h) tDOS, (i) time-resolved PL decay curves, and (j) TPV curves of the control and TBA-Alg treated perovskite films. | ||
When exposed to light, the control film (Fig. 3c) exhibits bright regions at the grain boundaries, and a dramatical increase in current at the grain boundaries (seen as bright regions), implying increased defect-related activity under light conditions. In comparison, the TBA-Alg-treated film (Fig. 3d) shows a more uniform current distribution under light. The grain boundaries are less pronounced, suggesting that the TBA-Alg effectively passivates defects and suppresses current leakage at grain boundaries, which is beneficial for high-performance PSCs.47 The histograms shown in Fig. 3e represent the current distribution under dark and light conditions for both the control and TBA-Alg-treated films. The control film under dark (Fig. 3e(I)) shows a narrow current distribution with low current values, indicating low overall conductivity in the absence of light. For the control film under light (Fig. 3e(II)), the current distribution expands significantly, exhibiting a bimodal pattern. A prominent peak appears near 0 pA, along with a broader peak centred around −7 pA, suggesting the presence of photo-inactive regions within the control film, which could limit the photovoltaic performance by reducing the efficiency and uniformity of charge carrier generation and collection.47 In the dark, a narrow current distribution with low current values is observed for the TBA-Alg-treated film (Fig. 3e(III)), indicating stable behaviour in the absence of light. Comparison with the control film, the TBA-Al-treated film under light (Fig. 3e(IV)) shows a narrow current distribution with higher current values, indicating that the film responds more uniformly to light excitation. This suggests that the TBA-Alg treatment can enhance charge carrier generation and transport under illuminated conditions. The schematic in Fig. 3f illustrates the control film, highlighting the high density of defects (represented by circles) at the grain boundaries. These defects are likely responsible for the inhomogeneous current generation under light excitation. The schematic in Fig. 3g highlights the reduced density of defects at grain boundaries after the TBA-Alg treatment, suggesting that TBA-Alg passivates these regions and improve the overall film quality, enabling a more stable performance under light.48
Fig. 3h illustrates the trap density of states (tDOS) for the control and the TBA-Alg-treated sample. The TBA-Alg-treated sample shows lower trap densities compared to the control sample in both the shallow trap region (0.26–0.4 eV) and the deep trap region (0.4–0.5 eV). This reduction indicates that TBA-Alg treatment can effectively passivate defects within the perovskite film, resulting in less non-radiative recombination and improved charge transport properties, ultimately enhancing the efficiency of the solar cells.49
Time-resolved photoluminescence (TRPL) and steady-state photoluminescence (PL) were used to evaluate the charge recombination behaviour in perovskite films. As shown in Fig. 3i and Fig. S14 (ESI†), the perovskite films with TBA-Alg treatment exhibit high PL intensity and a longer carrier lifetime. The detailed fitting parameters for the PL lifetime results are provided in Table S1 (ESI†). A significantly longer carrier lifetime is observed in the perovskite film with TBA-Alg (1085.63 ns) compared to the control sample (886.04 ns). This indicates that the introduction of TBA-Alg treatment can effectively reduce non-radiative recombination by decreasing the defect density.50 A comparison of the UV-vis absorption spectra between the perovskite films with and without TBA-Alg treatment is shown in Fig. S15 (ESI†). The results indicate that TBA-Alg treatment has no significant influence on the optical absorption and bandgap of the perovskite films.
Transient photocurrent/photovoltage (TPC/TPV) measurements were conducted to investigate the impact of TBA-Alg on the carrier recombination and extraction in perovskite solar cells. As shown in Fig. 3j, the TPV decay lifetime of the TBA-Alg-treated sample was 88.18 μs, significantly longer than that of the control sample (45.72 μs), indicating significantly suppressed recombination of the photogenerated carriers.51 The TPC decay lifetimes of the perovskite solar cell with and without TBA-Alg are 0.58 μs and 0.69 μs, respectively (Fig. S16, ESI†), suggesting that TBA-Alg treatment can facilitate the separation and transport of photogenerated carriers.52,53
To futher explore the effect of TBA-Alg treatment on the defect density of perovskite solar cells, devices based on the ITO/SnO2/perovskite/C60/Ag structure were prepared and characterised using space-charge-limited current (SCLC) measurements. In Fig. S17 (ESI†), the VTFL values for the control and treated perovskite devices are 0.69 V and 0.23 V, respectively. The calculated trap density indicates that TBA-Alg treatment can reduce the trap density and suppress charge carrier recombination, consistent with the measured values from the TRPL results.
To further investigate the carrier recombination behaviour in perovskite devices, electrochemical impedance spectroscopy (EIS) measurements were conducted. Fig. S18 (ESI†) shows the Nyquist plots of devices along with the fitting model. An increase in recombination resistance (Rrec) was observed in the TBA-Alg treated perovskite device, indicating a reduction in carrier recombination.54 The dark current curves in Fig. S19 (ESI†) presents the comparison of current–voltage (J–V) characteristics of PSCs with and without TBA-Alg treatment under dark conditions. The TBA-Alg treated perovskite device exhibited a lower dark current compared to the control sample, indicating that TBA-Alg treatment effectively reduced leakage and recombination losses, which is beneficial for device performance.55
To investigate the impact of TBA-Alg treatment on the photovoltaic performance of perovskite solar cells, devices with an ITO/SAM/perovskite/PCBM/BCP/Ag structure were fabricated. Initially, a series of devices with various concentrations of TBA-Alg were tested. The best efficiency was recorded at a concentration of 1 mg mL−1 in the TBA-Alg-treated device, as shown in Fig. S20 and Table S2 (ESI†). This device achieved a power conversion efficiency of 25.01%, significantly higher than that of the control device (22.64%). The corresponding open-circuit voltage (VOC), short-circuit current density (Jsc), and fill factor (FF) were 1.18 V, 25.12 mA cm−2, and 84.18%, respectively, as shown in Fig. 4a. For comparison, the control device without TBA-Alg (Fig. 4b) exhibited a VOC of 1.15 V, a Jsc of 24.8 mA cm−2, and an FF of 78.7%. These results suggest that the TBA-Alg treatment significantly improved photovoltaic performance, particularly in the fill factor, which increased from 78.7% (control) to 84.18%. Furthermore, the statistical distribution of these performance parameters (Fig. S21, ESI†) confirms that the enhancements with TBA-Alg are consistent and reproducible across multiple devices. Fig. 4c shows the external quantum efficiency (EQE) measurements of perovskite solar cells, giving an integrated current density of 23.97 mA cm−2 for the control sample and 24.77 mA cm−2 for the TBA-Alg treated device, consistent with the J–V curves and confirming the enhanced photovoltaic performance after TBA-Alg treatment.
 | ||
| Fig. 4 Performance of TBA-Alg modified and the control perovskite solar cell devices. J–V curve of (a) TBA-Alg treated film and (b) the control film. (c) EQE spectra and corresponding integrated Jsc curve. (d) MPP stability and (e) long term storage stability performance. (f) Stability of devices under continuous illumination conditions at ∼60% humidity for 1050 hours. (g) Leaked Pb concentration following a 120-minute immersion of both control and TBA-Alg treated perovskite films in water. The inset shows optical photographs of the lead leakage test. (h) SEM and EDS images of the control film and the TBA-Alg-treated film after 1 s water immersion tests. | ||
Additionally, we characterised the output stability of the device at the maximum power output point (MPP), as shown in Fig. 4d. The control device exhibited a photocurrent of 22.12 mA cm−2 and a power output efficiency of 22.52%. After TBA-Alg treatment, the photocurrent was stabilized at 23.25 mA cm−2, resulting in a power conversion efficiency of 24.65%. Long-term stability was also tested as shown in Fig. 4e. After 2000 hours of storage in a nitrogen-filled glove box, the TBA-Alg-modified device retained 95.5% of its initial efficiency, compared to 54% for the control device.
Furthermore, we evaluated the device stability under continuous one-sun illumination in ambient conditions with a RH of 60%. The experimental setup and process are shown in Fig. S22 and Video S1 (ESI†), with the corresponding stability results shown in Fig. 4f. After 450 hours, the PCE of the control device declined to 53% of its initial value, whereas the TBA-Alg-treated device retained 80% of its initial efficiency even after 1050 hours, demonstrating the significantly enhanced stability of PSCs with TBA-Alg treatment.36,56,57 This decrease in efficiency refers to the formation of PbI2 in perovskite thin film as shown in Fig. S23 (ESI†). An increased peak at 12.6° can be seen in the XRD patterns of the unencapsulated control film after 30 days of air exposure, indicating the formation of PbI2 from film decomposition.58 In contrast, the TBA-Alg-treated film shows greater resistance to degradation, attributed to the enhanced hydrophobicity and film quality from TBA-Alg modification.
We also fabricated a large-area perovskite module (5 cm × 5 cm) using the TBA-Alg treatment, with an effective area of 16.8 cm2. An optical image of this module (Fig. S24, ESI†) shows uniform coverage of the perovskite layer, indicating a homogeneous protective coating. The corresponding J–V curve reveals that the TBA-Alg-treated module achieved a PCE of 22.17%. The high efficiency achieved in large area devices further demonstrates that the protective layer forms uniformly over large areas, confirming their compatibility with scalable deposition methods.
To validate the TBA-Alg treatment approach, n-i-p structured perovskite devices with an FTO/SnO2/Perovskite/Spiro-MeOTAD/Au architecture were fabricated, as shown in Fig. S25a (ESI†). Fig. S25b (ESI†) presents the J–V curves of TBA-Alg treated devices, achieving a PCE of 24.44%, a VOC of 1.17 V, and an FF exceeding 84%. In contrast, Fig. S25c (ESI†) displays the J–V curves of the control devices, which exhibit a lower PCE of 21.65%, a VOC of 1.12 V, and an FF of 78.92%. Additionally, the statistical distribution plots in Fig. S25d (ESI†) confirm that the photovoltaic efficiency enhancement is consistently observed across multiple devices, further demonstrating the effectiveness and broad applicability of our approach. These results demonstrate that our approach effectively optimizes the properties of perovskite layers in both p–i–n and n–i–p architectures, evidencing their versatility for application in different device configurations.
To evaluate the effectiveness of TBA-Alg treatment in preventing lead ion leakage from the perovskite film under extreme conditions, a water immersion test was performed on unencapsulated perovskite films. The optical images in Fig. S26 (ESI†) show that the TBA-Alg treated film (left) remained black up to 15 minutes immersion in water, while the control film (right) turned yellow immediately after immersion. After 60 minutes, the TBA-Alg treated film remained on the substrate as indicated by its yellow color, whereas the control film became transparent, indicating significant lead leaching into water.59,60 To monitor the leakage of lead ions into water over time, inductively coupled plasma mass spectrometry (ICP-MS) measurements were performed on water samples taken at 5, 10, 30, 60, and 120 minutes, as shown in Fig. 4g. After 120 minutes, the control film exhibited a high lead ion concentration, stabilizing at 60.93 μg cm−2, while the TBA-Alg treated film showed minimal lead leakage at 10.37 μg cm−2, corresponding to 83% reduction in lead leakage. Fig. 4h presents the SEM and EDS analysis of the sample surface after being immersed in water for approximately one second and immediately removed. The TBA-Alg treated film showed a collapsed but dense structure, with EDS confirming lead as the dominant surface element. In contrast, the control film displayed a loose structure with many holes, and EDS revealed a reduction in lead content on the film's surface. This indicates that the TBA-Alg treated film significantly retarded degradation and delayed the loss of structural integrity, showing enhanced resistance to degradation and a more stable perovskite structure under water immersion. Compared to conventional methods, our approach offers superior moisture resistance, enabling the film to maintain stability even after 15 minutes of water immersion. This remarkable moisture-resistant capability is achieved using a low-cost, biomass-derived TBA-Alg polymer, which is produced through a simple preparation process.
In addition, we performed lead leakage tests using ICP-MS at 0 °C, 25 °C, 50 °C, and 75 °C, comparing control samples and TBA-Alg-treated samples after immersion in 40 mL of DI water at these temperatures. Fig. S27 (ESI†) indicates that lead leakage increases with a higher temperature. However, samples treated with TBA-Alg effectively show suppressed lead ion release even at a higher temperature. After 2 hours, the reduction in lead leakage compared to the control samples was 89.2% (0 °C), 80.1% (25 °C), 56.6% (50 °C), and 30.0% (75 °C). We also examined lead leakage under different pH conditions (pH values of 3.2, 5.1, 7.0, and 11.2) using ICP-MS, as presented in Fig. S28 (ESI†). The results show that lead leakage is significantly influenced by pH. At a pH of 3.2 and a pH of 11.2, the highest levels of lead ion release were observed, indicating that both strongly acidic and alkaline environments accelerate perovskite degradation.61 In comparison, moderate leakage occurs at pH values of 5.1 and 7.0. Across all tested conditions, TBA-Alg-treated samples consistently exhibit lower lead leakage than the control sample. Specifically, the reduction in lead ion release achieved by TBA-Alg treatment were 14.5% at pH 3.2, 41.8% at pH 5.1, 80.1% at pH 7.0, and 34.3% at pH 11.2. The most pronounced suppression effect appears at pH 7.0, suggesting that TBA-Alg effectively stabilizes the perovskite structure under near-neutral water conditions. Even under harsh conditions such as pH levels of 3.2 and 11.2, TBA-Alg continues to mitigate lead leakage, although its effectiveness is reduced. Its environmentally sustainable nature, coupled with its successful application in perovskite solar cells, contributes to high efficiency and long-term operational stability.
Our baseline TBA-Alg additive was synthesized from low molecular weight (Mw) sodium alginate (5–20 kDa), which produced a device PCE of 25.01% (Fig. 4a). To further investigate the impact of alginate's molecular weight on device photovoltaic performance, we synthesized three additional TBA-Alg variants from alginates with higher molecular weights (30–100 kDa, 100–200 kDa, and 300–500 kDa). Each polymer additive was incorporated into the perovskite precursor solution for solar cell fabrication. Fig. S29 (ESI†) summarizes the PCE results for devices incorporating these additives. The device with TBA-Alg derived from 30–100 kDa alginate achieved a PCE of 24.56% (Fig. S29a, ESI†), nearly matching the 25.01% from the low-Mw baseline (Fig. 4a). The device using TBA-Alg from a 100–200 kDa alginate showed a slightly lower PCE of 23.45% (Fig. S29b, ESI†), which still exceeded the control device without any additive (22.64%, Fig. 4b). These results suggest that effective defect passivation and improved charge transport are maintained using polymer additives in this moderate molecular weight range (up to ∼200 kDa). However, the device containing TBA-Alg synthesized from higher molecular weight alginate (300–500 kDa) exhibited a notable drop in efficiency to 21.38% PCE (Fig. S29c, ESI†). This value is lower than the control sample's efficiency, indicating that an excessively high polymer molecular weight can minimalize the passivation benefits and reduce device performance. In summary, low to moderate alginate molecular weights (up to ∼200 kDa) enable TBA-Alg to effectively enhance perovskite solar cell efficiency, whereas very high molecular weights (300–500 kDa) of alginate weaken the additive's passivation capability and lead to a lower photovoltaic efficiency (Fig. S29d, ESI†).
To compare the device performance achieved in our work to those employing other types of biomass-derived additives in the literature, we have included a table (Table S3, ESI†).
Conclusions
In summary, we designed a novel dual-functional polymer, tetrabutylammonium alginate (TBA-Alg), derived from biomass resources, and introduced a passivation strategy to regulate lead ions in inverted perovskite solar cells. This approach leverages the sustainable and renewable nature of alginate, aligning with environmentally friendly principles while enhancing device performance. The TBA-Alg features alginate groups and hydrophobic alkyl arms. The alginate chelates lead ions, prolongs crystallization time and reduces defects, while the alkyl arms act as barriers against moisture erosion. The TBA-Alg treatment enables defect passivation and crystalline structure stability to be achieved simultaneously in the perovskite film. As a result, the modified perovskite films exhibit excellent electron transport ability and reduced electron recombination, resulting in outstanding photovoltaic performance and stability. The PCE of the TBA-Alg treated device achieved 25.01% with outstanding operational stability. A large-area module (16.8 cm2) treated with TBA-Alg achieved 22.17% efficiency and n–i–p structured PSCs achieved a PCE of 24.44%, confirming the compatibility of the strategy with state-of-the-art device configurations and scalable deposition methods. Additionally, the unencapsulated device exhibits hydrophobic protection, inhibiting up to 83% lead leakage. This work presents an effective strategy to simultaneously achieve superior PCE while enhancing the long-term stability of devices and preventing lead leakage to the environment, it would greatly contribute to the advancement of PSC commercialization.Author contributions
J. Li, X. Qiao and B. He, contributed equally to this work. Y. Lu, Z. Li, M. Li and J. Li conceived the research idea and designed the experiments. J. Li performed the experiments including material synthesis, perovskite film characterization and solar cell fabrication, and analyzed the experimental data with the assistance of Y. Zhang and I. Abrahams, under the supervision of Y. Lu and Z. Li. X. Qiao optimised the perovskite solar cell, fabricated and characterised the solar module, under the supervision of M. Li. B. He, Z. Su and Y. Gao conducted grazing incidence wide-angle X-ray scattering (GIWAXS) measurements. S. Pal performed C-AFM measurements with guidance from J. Briscoe. J. Li, Y. Lu, Z. Li and M. Li drafted the manuscript, and all authors discussed the results and provided feedback on the manuscript.Data availability
The data supporting the findings of this study, including raw experimental data (J–V curves, stability tests, lead leakage measurements), characterization data (XRD, SEM, TEM images, UV-Vis spectra), and processed results, are available in figshare at https://figshare.com/s/cda6b437d86bcb41187c. Additional supporting information is provided in the ESI.† Further details or specific data can be obtained from the corresponding authors upon reasonable request.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors thank the Shanghai Synchrotron Radiation Facility, particularly the staff at beamlines BL14B1 and BL03HB, for providing essential beam time and technical support. J. Li acknowledge the support of the China Scholarship Council (CSC201906890018) and Queen Mary University of London for a PhD Scholarship. Z. Li and M. Li acknowledge financial support from the AXA research fund. S. Pal and J. Briscoe acknowledge the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation program (Grant Agreement No. 101001626).References
- C. Eames, J. M. Frost, P. R. F. Barnes, B. C. O’Regan, A. Walsh and M. S. Islam, Nat. Commun., 2015, 6, 7497 CrossRef CAS PubMed.
- M. A. Green, A. Ho-Baillie and H. J. Snaith, Nat. Photonics, 2014, 8, 506–514 CrossRef CAS.
- A. Kojima, K. Teshima, Y. Shirai and T. Miyasaka, J. Am. Chem. Soc., 2009, 131, 6050–6051 CrossRef CAS PubMed.
- S. Li, Y. Jiang, J. Xu, D. Wang, Z. Ding, T. Zhu, B. Chen, Y. Yang, M. Wei, R. Guo, Y. Hou, Y. Chen, C. Sun, K. Wei, S. M. H. Qaid, H. Lu, H. Tan, D. Di, J. Chen, M. Grätzel, E. H. Sargent and M. Yuan, Nature, 2024, 635, 82–88 CrossRef CAS PubMed.
- I. Chung, B. Lee, J. He, R. P. H. Chang and M. G. Kanatzidis, Nature, 2012, 485, 486–489 CrossRef CAS PubMed.
- A. Polman, M. Knight, E. C. Garnett, B. Ehrler and W. C. Sinke, Science, 2016, 352, aad4424 CrossRef PubMed.
- T. S. Sherkar, C. Momblona, L. Gil-Escrig, J. Ávila, M. Sessolo, H. J. Bolink and L. J. A. Koster, ACS Energy Lett., 2017, 2, 1214–1222 CrossRef CAS PubMed.
- S. Reichert, Q. An, Y.-W. Woo, A. Walsh, Y. Vaynzof and C. Deibel, Nat. Commun., 2020, 11, 6098 CrossRef CAS PubMed.
- P. Toloueinia, H. Khassaf, A. Shirazi Amin, Z. M. Tobin, S. P. Alpay and S. L. Suib, ACS Appl. Energy Mater., 2020, 3, 8240–8248 CrossRef CAS.
- J. Zhao, Z. Su, J. Pascual, H. Wu, H. Wang, M. H. Aldamasy, Z. Zhou, C. Wang, G. Li, Z. Li, X. Gao, C. S. Hsu and M. Li, Adv. Mater., 2024, 36, 2406246 CrossRef CAS PubMed.
- G. Li, Y. Hu, M. Li, Y. Tang, Z. Zhang, A. Musiienko, Q. Cao, F. Akhundova, J. Li, K. Prashanthan, F. Yang, P. Janasik, A. N. S. Appiah, S. Trofimov, N. Livakas, S. Zuo, L. Wu, L. Wang, Y. Yang, B. Agyei-Tuffour, R. W. MacQueen, B. Naydenov, T. Unold, E. Unger, E. Aktas, S. Eigler and A. Abate, Angew. Chem., Int. Ed., 2023, 62, 202307395 CrossRef PubMed.
- Y. C. Kim, T. Y. Yang, N. J. Jeon, J. Im, S. Jang, T. J. Shin, H. W. Shin, S. Kim, E. Lee, S. Kim, J. H. Noh, S. I. Seok and J. Seo, Energy Environ. Sci., 2017, 10, 2109–2116 RSC.
- L. Meng, J. You and Y. Yang, Nat. Commun., 2018, 9, 5256 CrossRef PubMed.
- F. Wang, S. Bai, W. Tress, A. Hagfeldt and F. Gao, npj Flexible Electron., 2018, 2, 22 CrossRef.
- H. Zhu, S. Teale, M. N. Lintangpradipto, S. Mahesh, B. Chen, M. D. McGehee, E. H. Sargent and O. M. Bakr, Nat. Rev. Mater., 2023, 8, 569–586 CrossRef.
- S. Wang, A. Wang, X. Deng, L. Xie, A. Xiao, C. Li, Y. Xiang, T. Li, L. Ding and F. Hao, J. Mater. Chem. A, 2020, 8, 12201–12225 RSC.
- T. H. Han, J. W. Lee, C. Choi, S. Tan, C. Lee, Y. Zhao, Z. Dai, N. De Marco, S. J. Lee, S. H. Bae, Y. Yuan, H. M. Lee, Y. Huang and Y. Yang, Nat. Commun., 2019, 10, 520 CrossRef CAS PubMed.
- Y. Wang, Y. Xiao, L. Wang, Z. Su, Y. Xu, L. Fan, G. Yao, X. Qian and J.-Y. Lin, J. Power Sources, 2024, 602, 234383 CrossRef CAS.
- X. Qiao, R. Zhu, D. Yan, Z. Su, Z. Zhang, H. Wu, Y. Tan, M. Liang, W. Zuo, J. Zhang, G. Li, X. Gao, M. Saliba and M. Li, Adv. Funct. Mater., 2024, 34, 2409852 CrossRef CAS.
- J. S. Nam, J. M. Choi, J. W. Lee, J. Han, I. Jeon and H. D. Kim, Adv. Energy Mater., 2024, 14, 2304062 CrossRef CAS.
- J. Han, Y. Tian and I. Jeon, Adv. Mater., 2024, 2410327 CrossRef PubMed.
- N. Vanni, A. Giuri, M. Calora, E. Podda, A. P. Caricato, K. Sparnacci, R. Suhonen, M. Ylikunnari, A. Covarelli, L. Gregori, F. De Angelis, G. Marra, P. Biagini, R. Po and A. Rizzo, Sol. RRL, 2024, 8, 2400612 CrossRef CAS.
- Y. Zhang, Q. Song, G. Liu, Y. Chen, Z. Guo, N. Li, X. Niu, Z. Qiu, W. Zhou, Z. Huang, C. Zhu, H. Zai, S. Ma, Y. Bai, Q. Chen, W. Huang, Q. Zhao and H. Zhou, Nat. Photonics, 2023, 17, 1066–1073 CrossRef CAS.
- F. Mafakheri, A. Asakereh, S. Khoee and M. Kamankesh, Sci. Rep., 2023, 13, 10326 CrossRef CAS PubMed.
- S. N. Pawar and K. J. Edgar, Carbohydr. Polym., 2013, 98, 1288–1296 Search PubMed.
- X. Liu, X. Wang, T. Zhang, Y. Miao, Z. Qin, Y. Chen and Y. Zhao, Angew. Chem., Int. Ed., 2021, 60, 12351–12355 Search PubMed.
- S. Chen, Y. Deng, H. Gu, S. Xu, S. Wang, Z. Yu, V. Blum and J. Huang, Nat. Energy, 2020, 5, 1003–1011 CrossRef.
- H. Luo, P. Li, J. Ma, X. Li, H. Zhu, Y. Cheng, Q. Li, Q. Xu, Y. Zhang and Y. Song, Nat. Commun., 2023, 14, 4730 CrossRef CAS PubMed.
- X. Zhu, J. Sheng, T. Ding, J. He and W. Wu, Sol. Energy, 2023, 266, 112182 CrossRef CAS.
- M. M. Thomas Schleeh and D. Roessner, Carbohydr. Polym., 2014, 114, 493–499 CrossRef PubMed.
- K. Manna, P. Patra, A. Roy, R. K. Roy, K. Chaitanya Sunka, S. Dhara, N. Patra and S. Pal, ACS Appl. Polym. Mater., 2022, 4, 4432–4444 CrossRef CAS.
- S. N. Pawar and K. J. Edgar, Biomacromolecules, 2011, 12, 4095–4103 CrossRef CAS PubMed.
- J. Wang, J. Zhang, Y. Zhou, H. Liu, Q. Xue, X. Li, C. C. Chueh, H. L. Yip, Z. Zhu and A. K. Y. Jen, Nat. Commun., 2020, 11, 177 CrossRef CAS PubMed.
- J. Xiong, Z. Dai, S. Zhan, X. Zhang, X. Xue, W. Liu, Z. Zhang, Y. Huang, Q. Dai and J. Zhang, Nano Energy, 2021, 84, 105882 CrossRef CAS.
- J. Lim, A. I. Rafieh, N. Shibayama, J. Xia, J. N. Audinot, T. Wirtz, S. Kinge, S. W. Glunz, Y. Ding, B. Ding, H. Kim, M. Saliba, Z. Fei, P. J. Dyson, M. K. Nazeeruddin and H. Kanda, Energy Environ. Sci., 2024, 17, 8209–8218 RSC.
- Y. Xu, X. Guo, Z. Lin, Q. Wang, J. Su, J. Zhang, Y. Hao, K. Yang and J. Chang, Angew. Chem., Int. Ed., 2023, 62, e202306229 CrossRef CAS PubMed.
- M. Yang, T. Tian, Y. Fang, W.-G. Li, G. Liu, W. Feng, M. Xu and W.-Q. Wu, Nat. Sustain., 2023, 6, 1455–1464 CrossRef.
- Y. Dong, J. Zhang, W. Wang, B. Hu, D. Xia, K. Lin, L. Geng and Y. Yang, Small, 2023, 19, e2301824 CrossRef PubMed.
- C. Tian, B. Li, Y. Rui, H. Xiong, Y. Zhao, X. Han, X. Zhou, Y. Qiu, W. An, K. Li, C. Hou, Y. Li, H. Wang and Q. Zhang, Adv. Funct. Mater., 2023, 33, 2302270 CrossRef CAS.
- J. Zhang, Z. Li, F. Guo, H. Jiang, W. Yan, C. Peng, R. Liu, L. Wang, H. Gao, S. Pang and Z. Zhou, Angew. Chem., Int. Ed., 2023, 62, e202305221 CrossRef PubMed.
- R. Azmi, D. S. Utomo, B. Vishal, S. Zhumagali, P. Dally, A. M. Risqi, A. Prasetio, E. Ugur, F. Cao, I. F. Imran, A. A. Said, A. R. Pininti, A. S. Subbiah, E. Aydin, C. Xiao, S. I. Seok and S. De Wolf, Nature, 2024, 628, 93–98 CrossRef CAS PubMed.
- H. Chen, C. Liu and J. Xu, et al. , Science, 2024, 384, 189–193 CrossRef CAS PubMed.
- M. Li, R. Sun, J. Chang, J. Dong, Q. Tian, H. Wang, Z. Li, P. Yang, H. Shi, C. Yang, Z. Wu, R. Li, Y. Yang, A. Wang, S. Zhang, F. Wang, W. Huang and T. Qin, Nat. Commun., 2023, 14, 573 CrossRef CAS PubMed.
- Q. Cao, T. Wang, J. Yang, Y. Zhang, Y. Li, X. Pu, J. Zhao, H. Chen, X. Li, I. Tojiboyev, J. Chen, L. Etgar and X. Li, Adv. Funct. Mater., 2022, 32, 2201036 CrossRef CAS.
- H. Meng, K. Mao, F. Cai, K. Zhang, S. Yuan, T. Li, F. Cao, Z. Su, Z. Zhu, X. Feng, W. Peng, J. Xu, Y. Gao, W. Chen, C. Xiao, X. Wu, M. D. McGehee and J. Xu, Nat. Energy, 2024, 9, 536–547 CrossRef CAS.
- Z. Wang, X. Duan, J. Zhang, W. Yuan, D. Qu, Y. Chen, L. He, H. Wang, G. Yang, W. Zhang, Y. Bai and H.-M. Cheng, Commun. Mater., 2024, 5, 131 CrossRef CAS.
- J. Song, Y. Zhou, N. P. Padture and B. D. Huey, Nat. Commun., 2020, 11, 3308 CrossRef CAS PubMed.
- C. S. Pathak, B.-J. Chang and S. Song, Dyes Pigm., 2023, 218, 111469 CrossRef CAS.
- J. Wu, R. Zhu, G. Li, Z. Zhang, J. Pascual, H. Wu, M. H. Aldamasy, L. Wang, Z. Su, S. H. Turren-Cruz, R. Roy, F. A. Alharthi, A. Alsalme, J. Zhang, X. Gao, M. Saliba, A. Abate and M. Li, Adv. Mater., 2024, 36, e2407433 CrossRef PubMed.
- X. Zhao, R. Luo, C. Yu, X. Xu, W. Zhu, Y. Min and N. Cai, Adv. Funct. Mater., 2024, 34, 2307568 CrossRef CAS.
- Y. Chang, X. Zhu, Y. Shi, Y. Liu, K. Meng, Y. Li, J. Xue, L. Zhu, J. Zhang, H. Zhou, W. Ma, Z. Wei and K. Lu, Energy Environ. Sci., 2022, 15, 2937–2947 RSC.
- M. Wang, Y. Zhao, X. Jiang, Y. Yin, I. Yavuz, P. Zhu, A. Zhang, G. S. Han, H. S. Jung, Y. Zhou, W. Yang, J. Bian, S. Jin, J.-W. Lee and Y. Yang, Joule, 2022, 6, 1032–1048 CrossRef CAS.
- C. Qiu, L. Wagner, J. Liu, W. Zhang, J. Du, Q. Wang, Y. Hu and H. Han, EcoMat, 2022, 5, e12268 CrossRef.
- H. Wen, Z. Zhang, Y. Guo, W. Luo, S. Si, T. Yin, H. Wu and S. Huang, Adv. Energy Mater., 2023, 13, 2301813 CrossRef CAS.
- Y. Wang, X. Feng, W. Huang, X. He, L. Wang and M. Ouyang, Adv. Energy Mater., 2023, 13, 2203841 CrossRef CAS.
- Y. Yang, Q. Xiong, J. Wu, Y. Tu, T. Sun, G. Li, X. Liu, X. Wang, Y. Du, C. Deng, L. Tan, Y. Wei, Y. Lin, Y. Huang, M. Huang, W. Sun, L. Fan, Y. Xie, J. Lin, Z. Lan, V. Stacchinii, A. Musiienko, Q. Hu, P. Gao, A. Abate and M. K. Nazeeruddin, Adv. Mater., 2024, 36, e2310800 CrossRef PubMed.
- S. Wu, Z. Li, M. Q. Li, Y. Diao, F. Lin, T. Liu, J. Zhang, P. Tieu, W. Gao, F. Qi, X. Pan, Z. Xu, Z. Zhu and A. K. Jen, Nat. Nanotechnol., 2020, 15, 934–940 CrossRef CAS PubMed.
- X. Wei, M. Xiao, B. Wang, C. Wang, Y. Li, J. Dou, Z. Cui, J. Dou, H. Wang, S. Ma, C. Zhu, G. Yuan, N. Yang, T. Song, H. Zhou, H. Chen, Y. Bai and Q. Chen, Angew. Chem., Int. Ed., 2022, 61, e202204314 CrossRef CAS PubMed.
- Y. Liang, P. Song, H. Tian, C. Tian, W. Tian, Z. Nan, Y. Cai, P. Yang, C. Sun, J. Chen, L. Xie, Q. Zhang and Z. Wei, Adv. Funct. Mater., 2021, 32, 2110139 CrossRef.
- K. Ma, X. Li, F. Yang and H. Liu, Coatings, 2023, 13, 1009 CrossRef CAS.
- Y. Chen, N. Li, L. Wang, L. Li, Z. Xu, H. Jiao, P. Liu, C. Zhu, H. Zai, M. Sun, W. Zou, S. Zhang, G. Xing, X. Liu, J. Wang, D. Li, B. Huang, Q. Chen and H. Zhou, Nat. Commun., 2019, 10, 1112 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ee06038e |
| This journal is © The Royal Society of Chemistry 2025 |