
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
High-voltage cycling degradation mechanisms of the NaNi1/3Fe1/3Mn1/3O2 cathode in sodium-ion pouch cells†
Shini
Lin
a,
Wei
Li
a,
Huiya
Yang
a,
Minghui
Chen
a,
Honghao
Xie
a,
Yuan
Qin
a,
Jing
Zeng
*a,
Peng
Zhang
*b and
Jinbao
Zhao
 *a
*a
aState-Province Joint Engineering Laboratory of Power Source Technology for New Energy Vehicle, State Key Laboratory of Physical Chemistry of Solid Surfaces, Engineering Research Center of Electrochemical Technology, Collaborative Innovation Center of Chemistry for Energy Materials, College of Chemistry and Chemical Engineering, Ministry of Education, Xiamen University, Xiamen 361005, China. E-mail: jbzhao@xmu.edu.cn
bCollege of Energy & School of Energy Research, Xiamen University, Xiamen 361102, China
First published on 4th June 2025
Abstract
Sodium-ion batteries (SIBs) utilizing the NaNi1/3Fe1/3Mn1/3O2 (NFM) cathode paired with a hard carbon (HC) anode exhibit a relatively high energy density. To further enhance the energy density, elevating the charging cutoff voltage offers a more universally applicable strategy compared with material approaches, which may encounter limitations due to raw material cost and supply constraints. However, the accelerated degradation mechanisms induced by high-voltage operation severely compromise the cycle life, creating a critical barrier to commercialization. This study reveals the high-voltage degradation mechanism of the NFM cathode at the full battery level, as determined by evaluation of the electrochemical performance at the upper voltage range, profiling of the structural characterization and evolution, tracking of interfacial reactions, analysis of reaction kinetics, and quantification of transition metal dissolution. The HC||NFM battery cycling at a charging cut-off voltage of 4.2 V showed a significantly reduced capacity retention rate (55%, 300 cycles) due to the interfacial side reactions and NFM structural degradation, although it had a considerably higher initial capacity. The cathode underwent an irreversible structural evolution (X and O3′ phases), with frequent cell volume expansion/contraction exacerbating the particle cracking and interfacial parasitic reactions. In contrast, cycling at a 4.0 V upper voltage could maintain a good balance between a high capacity and long cycle life due to the simple reversible structural evolution of NFM (O3–P3–O3) and the moderate impedance during cycling. Finally, the use of electrolytes with boron-containing additives was demonstrated to be an effective strategy to improve the comprehensive performance of the NFM at high voltage. The mechanistic insights and material modification strategy presented herein can pave the way for engineering high-performance layered oxide cathodes that could concurrently achieve extended cyclability and high energy density in SIBs.
Broader contextDue to the advantages of abundant resources, controllable cost and excellent safety, sodium-ion batteries (SIBs) have become a supplement to lithium-ion batteries. SIBs utilizing a NaNi1/3Fe1/3Mn1/3O2 (NFM) cathode paired with a hard carbon (HC) anode exhibit a relatively high energy density, thus demonstrating promising application prospects in large-scale energy storage systems. Moreover, elevating the charging cutoff voltage offers a more universally applicable strategy to further enhance the energy density compared with material approaches. However, the accelerated degradation mechanisms induced by high-voltage operation severely compromise the cycle life, creating a critical barrier to commercialization. A systematic evaluation and comprehensive understanding of the high-voltage degradation mechanism of NFM//HC batteries is the foundation for the development of a long cycle life with high energy density for SIBs. In this study, the cycling performance and degradation mechanism/source under different upper voltages are explored, and the degradation boundary between normal and abuse working conditions of the battery is clarified. The degradation source quantified by a reverse coin half-battery was found to be mainly from active Na+ loss, while the active NFM loss was significantly aggravated at 4.2 V due to an irreversible X and O3′ phase transition. Finally, a powerful case strategy of electrolytes with boron-containing additives showed an enhanced all-round performance for the NFM cathode with high capacity and a long cycle life. |
Introduction
In recent years, sodium-ion batteries (SIBs) have been increasingly investigated as a supplementary energy-storage technology for coping with the resource scarcity dilemma and in response to the inordinate price fluctuations of lithium-ion batteries (LIBs). They benefit from abundant crustal-marine resources, affordable cost, excellent battery performances, and compatible manufacturing process with LIBs.1–5 However, their energy density is unsatisfactory, limiting their further application. Thus, the growing demand for SIBs with higher energy density will be a future mainstream research trend involving material selection and optimization, battery manufacturing technology, and pack structure designs.6–9The mature layered transition metal oxide NaNi1/3Fe1/3Mn1/3O2 (NFM) has become one of the most promising cathode materials emerging from long-term research, due to its high capacity, high voltage, environmental friendliness and ease of production.10–12 Hard carbon (HC), characterized by high capacity, low voltage plateau and extensive abundance of source raw materials, has brilliant prospects as an anode.13–18 Consequently, the HC||NFM battery has a relatively high energy density among SIBs and can be a typical model case to be further researched and developed. However, the poor cycling stability of this electrochemical system hinders its practical application lifespan. This weakness can be ascribed to a great extent to the instability of the NFM cathode.19 In practical application scenarios, in order to achieve the higher energy density goal, increasing the charging cut-off voltage can be a universal strategy.20 Elevating the charge cut-off voltage can facilitate enhanced Na+ deintercalation from the layered cathode framework, enabling greater active ion participation in redox reactions and thereby achieving concurrent improvements in both the operating voltage and reversible capacity. However, as a trade-off, the cycling stability weakness will be further magnified because a higher cut-off voltage poses a greater challenge to the structural stability of the cathode.11,21 The cathode can experience a severe phenomenon of crystal phase evolution, particle cracking, side reactions at the interface with the electrolyte, and transition metal (TM) dissolution.
Multidimensional methods for improving the cycling stability of the NFM cathode under normal conditions or high charging cut-off voltages have been widely researched. For example, a doping and mixed phase design can inhibit the harmful O3′/OP2 phase transition, while surface coating and electrolyte modification can enhance the interface stability under high voltages.22–28 However, most of the work to date has been only focused on the material level and has not investigated prolonging the battery level or the actual working conditions. Due to the potential difference between the Na metal anode and HC anode during charge/discharge cycling and the unique sodium-storage mechanism of HC, the degradation mechanism of the NFM cathode at the material level (Na||NFM) and battery level (HC||NFM) cannot be equated. Additionally, there are few reports on elucidating the structural evolution process of the NFM in pouch batteries or dissecting the cycling failure mechanism combined with different charging cut-off voltages. The coin battery system has been the main research object so far, which has left a certain gap in work on appropriate battery architectures for practical industrialization. Therefore, relevant research based on HC||NFM pouch batteries would be helpful to analyze the battery performance failure process dominated by the NFM cathode, especially on the premise of fitting it to actual working conditions, so as to provide a feasible scheme for the cathode modification and battery application.
In this work, a HC||NFM pouch battery was assembled to systematically analyze its high-voltage failure mechanism. At a battery level, combined with a reverse coin half battery, the detailed degradation mechanism of the HC||NFM battery was quantitatively analyzed. The structural evolution of the NFM cathode in the pouch battery was for the first time non-destructively traced by operando XRD, with the original battery architecture. HC||NFM batteries during cycling at different charging cut-off voltages were dissembled for postmortem analysis of the phase transition, particle cracking, interface thickening, and TM dissolution/migration/deposition. At a 4.0 V cut-off voltage, the HC||NFM battery possessed both a high energy density and long cycle stability (capacity retention = 73% after 300 cycles). The performance benefited from the simple reversible O3–P3–O3 structural evolution of the NFM cathode per cycle. In contrast, at a 4.2 V cut-off voltage, the HC||NFM battery showed large capacity fading (capacity retention = 55% after 300 cycles) and severe voltage attenuation despite the high energy density in the initial 100 cycles. The reverse coin half battery tests tell that the performance loss resulted from the presence of interfacial side reactions (impedance: NFM > HC) and cathode structural degradation. During one cycle, NFM undergoes a complex and irreversible O3–P3–X–O3′–P3–O3 structural evolution, with frequent unit cell volume expansion and contraction, resulting in structural instability (phase transition presented in the ex situ XRD and at a 32 mAh g−1 capacity loss). At 4.0 V and 3.8 V upper voltages, the NFM underwent unit cell volume expansion and contraction only once during one cycle. Meanwhile, the NFM at 4.2 V upper voltage experienced earlier particle cracking and severe side reactions with the electrolyte. Through ICP tests, Fe and Mn have severer dissolution than Ni in the electrolyte from NFM. Finally, an improvement strategy was provided based on boron-containing electrolyte additives to optimize the interface and this showed excellent performance. This work reveals the high-voltage failure mechanism of HC||NFM batteries, providing insights for developing structurally stable high-voltage layered cathodes and long-life high-specific-energy SIBs (Scheme 1).
 | ||
| Scheme 1 General model of the degradation and chemical crosstalk of the NFM cathode in a practical sodium-ion full battery. | ||
Results and discussion
Electrochemical degradation source
NFM has a NaFeO2 structure with the R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m space group. It is characterized by an ion-diffusion controlled electrochemical reaction. Here, a large capacity increment of 27 mAh g−1 was observed when the upper voltage was increased from 4.0 V to 4.2 V (Fig. S1–17†). At 4.0 V upper voltage, the NFM showed both high capacity and stable cycling (capacity retention = 86%/81% after 300/500 cycles, Fig. S9†). To benchmark commercial sodium-ion batteries, HC with promising applications was selected as the anode to analyze the degradation mechanism of NFM at a battery level.29 The HC||NFM pouch battery demonstrated a large energy density increment of 26.7 Wh kg−1 when the charging cut-off voltage increased from 4.0 V to 4.2 V, illustrating increasing the charging cut-off voltage is an effective way to improve the energy density of the battery (Fig. S18a†). Different from Na metal and other anodes, HC has a unique sodium-ion-storage mechanism involving adsorption–intercalation–filling (Fig. S18b†). Sodium ion loss on HC through processes such as SEI formation and Na metal deposition will cause less Na+ to go back to the NFM cathode and more intense phase transition (Fig. S18c†). Due to the voltage change experienced by the HC anode during cycling, the NFM cathode may have a different degradation process in a full battery compared to in a half battery (Fig. S18d–f†). Additionally, the voltage of the NFM cathode in a full battery examined by a three-electrode pouch battery was quite different from that in a half battery, especially for the discharge process (Fig. S18g–i and S19†). These reveal that evaluating NFM failure with a full battery is essential and beneficial for battery manufacturing optimization and material modification.
m space group. It is characterized by an ion-diffusion controlled electrochemical reaction. Here, a large capacity increment of 27 mAh g−1 was observed when the upper voltage was increased from 4.0 V to 4.2 V (Fig. S1–17†). At 4.0 V upper voltage, the NFM showed both high capacity and stable cycling (capacity retention = 86%/81% after 300/500 cycles, Fig. S9†). To benchmark commercial sodium-ion batteries, HC with promising applications was selected as the anode to analyze the degradation mechanism of NFM at a battery level.29 The HC||NFM pouch battery demonstrated a large energy density increment of 26.7 Wh kg−1 when the charging cut-off voltage increased from 4.0 V to 4.2 V, illustrating increasing the charging cut-off voltage is an effective way to improve the energy density of the battery (Fig. S18a†). Different from Na metal and other anodes, HC has a unique sodium-ion-storage mechanism involving adsorption–intercalation–filling (Fig. S18b†). Sodium ion loss on HC through processes such as SEI formation and Na metal deposition will cause less Na+ to go back to the NFM cathode and more intense phase transition (Fig. S18c†). Due to the voltage change experienced by the HC anode during cycling, the NFM cathode may have a different degradation process in a full battery compared to in a half battery (Fig. S18d–f†). Additionally, the voltage of the NFM cathode in a full battery examined by a three-electrode pouch battery was quite different from that in a half battery, especially for the discharge process (Fig. S18g–i and S19†). These reveal that evaluating NFM failure with a full battery is essential and beneficial for battery manufacturing optimization and material modification.
A HC||NFM battery (further details of the HC anode are provided in Fig. S20–24†) was assembled and the relevant battery parameters are listed in Table S1.† When the upper voltage increased from 4.0 V to 4.2 V, the capacity retention clearly dropped from 73% to 55%, and the capacity further gradually decreased throughout the 300 cycles, revealing the cycling instability at 4.2 V (Fig. 1a). In addition, with the cycling, more capacity was distributed at a lower voltage range (<3 V), indicating the severe voltage attenuation, while the other two voltages (3.8 V and 4.0 V) showed little change (Fig. 1b–d). The capacity fading and voltage attenuation jointly led to a sharp energy density decline at 4.2 V for the HC||NFM battery despite its early-stage high energy density. In comparison, at 4.0 V upper voltage, the capacity fading was concentrated in the early cycle stage, and the capacity distribution versus voltage remained almost unchanged. On the whole, the energy density through the whole cycling was relatively stable. Thus, the critical charging cut-off voltage that best balances a long-life and high energy density may be 4.0 V. Next, a HC||NFM coin full battery was assembled. Similarly, it was operated at a 4.2 V upper voltage, and demonstrated more significant capacity fading and voltage attenuation than at the other two voltages (Fig. 1e–g and S25, 26†). Also, it was noticeable that the capacity decay and polarization increment were quite small in the early 100 cycles but climbed steeply around 200 cycles. It is speculated that due to the much larger internal resistance and polarization of coin batteries, sodium deposition or dendrite formation may occur on the anode, resulting in a large capacity loss. There was a distinct oxidation peak near 4.2 V in the dQ/dV curve corresponding to the voltage plateau near 4.2 V in the charge–discharge curve (Fig. 1g and j). At 3.8 and 4.0 V upper voltages, the capacity decay and polarization increment were moderate, exhibiting superior and comparable cycling stability (Fig. 1e, f and S26†). Notably, the voltage plateaus around 3.3 V (charge) and 2.5 V (discharge) contributed to the large specific-capacity ratios (Fig. 1h and i). Through EIS analysis of the cycled HC||NFM batteries, it was found that the battery impedance increased with increasing the charging cut-off voltage, and this was especially significant from 3.8 V to 4.0 V (Fig. S27†).
 | ||
| Fig. 1 (a) Cycling performance of a HC||NFM pouch full battery with upper voltages of 3.8, 4.0 and 4.2 V. (b–d) Evolution of capacity distribution versus voltage of HC||NFM pouch full batteries cycling at the upper voltages of 3.8, 4.0, and 4.2 V, respectively. (e–g) Galvanostatic charge/discharge curves and (h–j) increment capacity curves of the HC||NFM full battery cycling at the upper voltages of 3.8, 4.0, and 4.2 V, respectively. | ||
Subsequently, the reverse coin battery was reassembled from the cycled pouch battery to quantitate the capacity degradation sources of the HC||NFM battery at 3.8, 4.0 and 4.2 V upper voltages. Active Na+ loss, active NFM material loss and active HC material loss were the three capacity degradation sources, whereby the first one usually results from the interfacial side reactions, the second was rooted in the cathode structure degradation, and the third was due to the anode structure degradation. During the first cycle of Na||NFM, the charge capacity was generally less than the discharge one, and the difference value was larger than that between the initial discharge capacity and the cycled discharge capacity, indicating the full battery capacity loss was mainly from the loss of active Na+ (Fig. 2a–c and h, i). Besides, the charge/discharge capacity in the following cycle were similar to the discharge one in the first cycle, again proving the result from the loss of active Na+. Through the EIS measurements, it was revealed that the impedance of NFM was significantly larger than that of HC, indicating the ion-diffusion difficulty in the cathode (Fig. 2d and e). Besides, the charging cut-off voltage clearly influenced the impedance of NFM, while it had no obvious effect on the impedance of HC (Fig. 2f). This was presumably related to the different oxidation reactions of the electrolyte on NFM at different upper voltages and the structural degradation of NFM. In the Na||HC reverse coin batteries, the initial discharge capacity showed almost no decrease compared to that of the fresh one, illustrating the structural stability of the HC anode in the full battery (Fig. 2g). In the Na||NFM reverse coin batteries, the difference value between the fresh discharge capacity and the cycled discharge capacity indicates the structural failure. The exact values were 5 mAh g−1 (4.2%), 10 mAh g−1 (7.7%) and 32 mAh g−1 (20.8%) capacity loss for the cathode from the pouch batteries cycled at 3.8, 4.0, and 4.2 V respectively (Fig. 2h and i). The capacity loss at 4.2 V was significantly the largest, indicating the severity of the active NFM loss at high voltage.
 | ||
| Fig. 2 Analysis of the capacity degradation source. Charge/discharge curves of Na||NFM with the cathode from cycled pouch batteries at (a) 3.8, (b) 4.0 and (c) 4.2 V upper voltages. EIS of (d) Na||NFM and (e–f) Na||HC, with the NFM and HC from cycled pouch batteries at 3.8, 4.0 and 4.2 V upper voltages. (g) Discharge curves of HC anode from cycled pouch batteries at 3.8, 4.0 and 4.2 V upper voltages. (h and i) Active Na+ loss and active NFM loss leading to capacity fading. | ||
Structural evolution of NFM when cycling a pouch battery
The structural evolution of NFM in a pouch battery was investigated through operando XRD to explore the reasons for the surge in active cathode loss at high voltage. Notably, the real-time non-destructive detection of the NFM cathode was achieved in the original working environment (Fig. 3a). In the operando XRD results, the peaks of the NFM cathode were stronger than those of the other amorphous components in the pouch battery and could be easily distinguished from those of Al foil (Fig. 3b–d). The structural evolution of the NFM cathode at the 3.8 and 4.0 V upper voltages were similar with the O3, P3, O3 phase transition sequence. In the initial 5 h charge stage, the NFM cathode experienced a solid–solution reaction of the O3 phase with the expansion of the c-axis and cell volume, as indicated by the shift to a lower angle of the (003) peak. The biphasic reaction was quick and not distinct in the subsequent desodiation process (full battery around 2.5 V). Also, in the reverse Na+ intercalation process, the P3–O3 biphasic reaction also appeared around 2.5 V for the full battery. The (003) peak shifts when the full battery was charged to 3.8 and 4.0 V were almost equal, and it could be noticed that the (003) peak for 4.0 V had a tendency to weaken and move to a higher angle, indicating this was the critical point going from the P3 to OP2 phase (Fig. 3e and f). While for the charge process at 4.2 V upper voltage, the (003) peak first shifted to a lower angle and then between 4.0–4.2 V in the reverse direction to a higher angle. Due to its reverse shift, the volume change of the unit cell was more severe than those at 3.8 and 4.0 V upper voltages (Fig. 3d, g and Table S2†). Besides, the XRD pattern when fully charged could not be indexed to any known phase, and was characterized by the higher angle of the (003) peak than the initial (shift = 0.32°, Fig. 3g), the (006) peak vanishing, and both the (101) and (102) peaks shifting to higher angle and overlapping; this is called “X” phase.30 In the reverse process of Na+ intercalation in the NFM cathode, a structural evolution sequence of X, O3′, P3, O3 occurred in NFM, while the other two upper voltages (3.8 and 4.0 V) showed neither the X phase nor O3′ phase. Specifically, when the full battery was discharged from 4.2 V to 3.5 V, the X-phase solid–solution reaction and X–O3′ biphasic reaction happen successively in the NFM cathode, with the (003) peak shifting to a lower angle. From 3.5 to 3.3 V, it underwent a hexagonal O3′-phase solid–solution reaction, with the two significant overlapping peaks of (101) and (102), (107) and (108) peaks, and the (105) peak shifting to 15.6°. In conclusion, the irreversible structural evolution at 4.2 V upper voltage aggravated the accumulation of local stress in NFM particles, thereby reducing the structural stability, which could be largely attributed to the harmful X and O3′ phases with sodium-ion deficiency in NFM.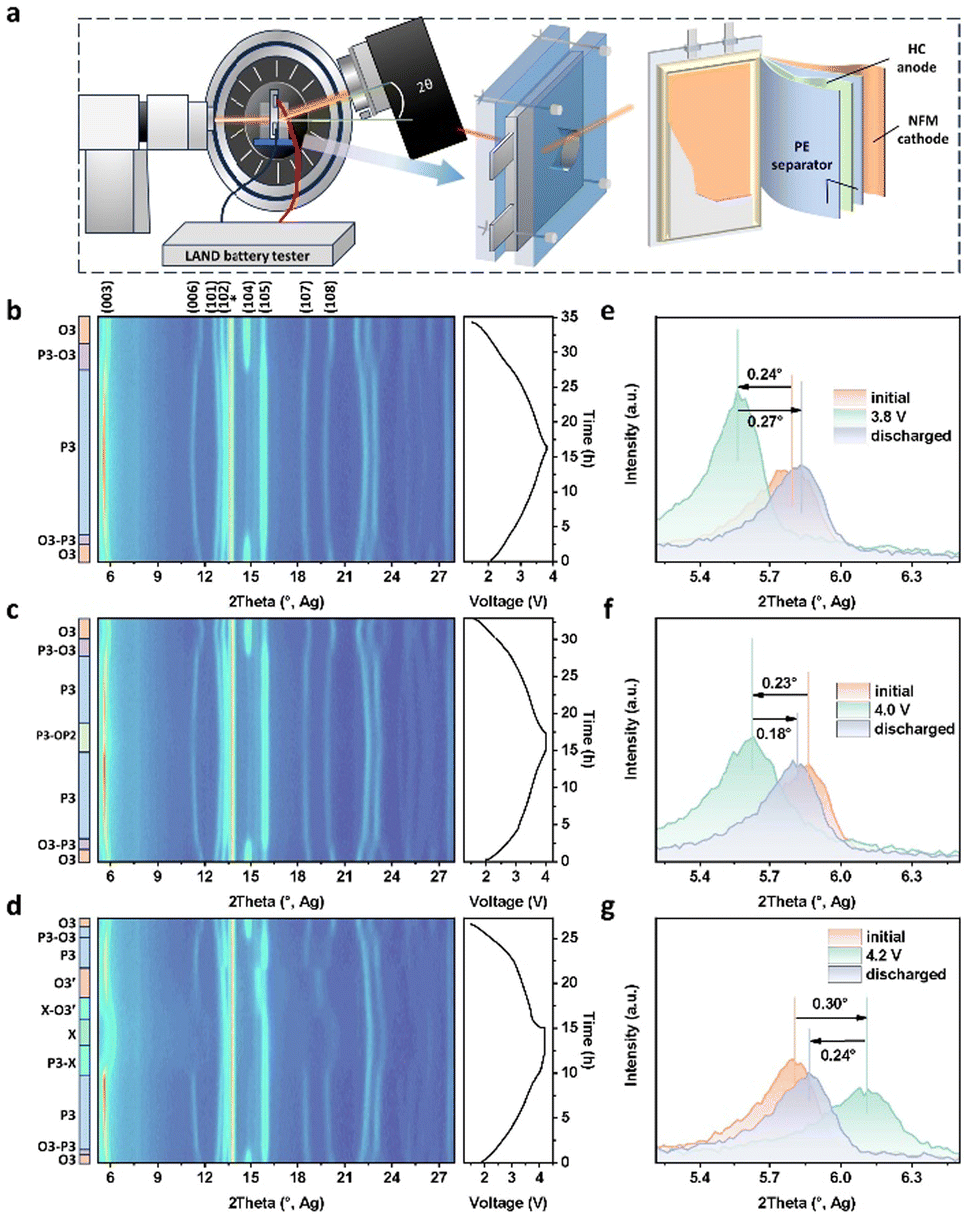 | ||
| Fig. 3 Real-time structural evolution of the NFM cathode in pouch batteries through non-destructive detection. (a) Schematic diagram of the pouch-battery operando XRD measurements. (b–d) Operando XRD test results of the NFM cathode in pouch battery cycling at 3.8, 4.0, and 4.2 V upper voltages, respectively. (e–g) Local (003) peak shift in the initial, fully charged, fully discharged states at 3.8, 4.0, and 4.2 V, respectively. | ||
In general, though the cathode from the pouch battery cycling at 4.2 V upper voltage released more capacity in the early cycles, this was also accompanied with a more intense structural evolution, which resulted in severe structural degradation, cracking, interfacial side reactions and a large capacity loss (active ion loss and active NFM loss >20%).
Bulk structure–interface degradation
To deeply understand the electrochemical degradation mechanism of the NFM material in a full battery arising from its structural evolution as presented above, the NFM cathode was sampled at different cycles of high, normal and low charging cut-off voltages.When cycling at 4.2 V upper voltage, the NFM cathode had an earlier phase transition, as indicated by the vanishing of the (104) peak at the 50th cycle, while those at 3.8 and 4.0 V show better structural stability, as shown by the (104) peak weakening slowly during cycle (Fig. 4a–c and S28–30†). The (003) peak when cycling at 3.8 V gradually shifted to a lower angle accompanied with the unit cell volume expansion, indicating a gentle structural degradation. While the (003) peak with cycling at 4.0 and 4.2 V showed a significant shift in the first cycles, indicating the structural degradation was concentrated in the early cycling. Notably, the (104) peak near 42°, indicating the O3 phase in the NFM cathode, was accompanied with a peak near 16.2°, as can be seen in the cycled cathode at the 3.8 and 4.0 V upper voltages. While for the case at the 4.2 V upper voltage, the (104) peak near 42° disappeared, indicating the scarce O3 phase and the significant structural transition of the cathode. Thus, the concomitant peak near 16.2° also disappeared. Besides, from the operando XRD at the 4.2 V upper voltage, during charge, the (006) peak shifted to a lower angle and then disappeared, while the (101) and (102) peaks shifted to higher angles and only one peak was left. Thus, the two characteristic (101) and (102) peaks between 35°–37.5° in the cycled 4.2 V sample nearly disappeared, with the peak appearing near 38° instead. The NFM cathode with the 3.8 V upper voltage showed obvious cracks in the 300th cycle, while that at 4.0 V showed the same in the 200th cycle, and that at 4.2 V in the 100th cycle, demonstrating the frequent and intense phase transition caused the particles to crack faster (Fig. S31†). When particles crack, the continuing interfacial parasitic reactions between the exposed fresh surface and the electrolyte will make the CEI thicker, which will affect the interfacial kinetics and lead to the loss of capacity (Fig. 4d–f).31–34 Thus, the interfacial kinetics was traced through EIS. The fitted equivalent circuit model of EIS is in Fig. S32.† The ion-diffusion impedance of the cathode/electrolyte interface (RCEI) decreased with the increase in charging cut-off voltage, indicating the interfacial components at high voltage were more beneficial to ion diffusion (Fig. 4g and Fig. S33–35, Table S3†); whereas RCEI increased faster with the cycling at higher upper voltages, indicating the accelerated interfacial degradation at higher voltages. The charge-transfer impedance (Rct) was significantly larger than RCEI, revealing that the electrochemical reaction of the NFM cathode was a more obvious matter affecting the battery performance than the ion diffusion in the interface. Additionally, Rct increased significantly with the charging cut-off voltage increase, especially from 4.0 V to 4.2 V, indicating the electrode reactions would be more sluggish at higher upper voltages. The galvanostatic intermittent titration technique (GITT) was used to quantitatively evaluate the polarization evolution and Na+-diffusion behavior changes in the NFM material during cycling to predict the aging mechanism. The Na+-diffusion coefficient (DNa+) increased from 3.0 V to 3.9 V (≈2 × 10−9 cm2 s−1), but decreased as the voltage continued to rise, well matching the Rct change with the increased charging cut-off voltage in the EIS results above (3.8 V < 4.0 V < 4.2 V) (Fig. S36†). This further proved the sluggish electrochemical reaction kinetics above 4.0 V in the NFM cathode. Through polarization analysis during cycling at various upper voltages, the NFM desodiation/sodiation was proven to be mainly controlled by the activation polarization (Fig. S37 and 38†). Compared with the 3.8 V upper voltage, the activation polarizations of the 4.0 and 4.2 V upper voltages were significantly greater, and their increase with cycling was even more serious. The systematic high-voltage failure mechanism of the NFM cathode is shown in Fig. 4h.
 | ||
| Fig. 4 Structure–interface evolution generating NFM cathode failure during cycling. (a–c) Postmortem XRD of the NFM cathode from fully discharged pouch batteries in different cycles at 3.8, 4.0, and 4.2 V upper voltages. (d–f) TEM images of the CEI of NFM from fully discharged pouch batteries after 300 cycles at 3.8, 4.0, and 4.2 V upper voltages, respectively. (g) Fitted EIS data of the fully charged NFM cathode in different cycles at varying upper voltages. (h) Failure mechanism of the NFM cathode with cycling at high/normal voltages. | ||
TM dissolution in the cathode
The irreversible migration of TM ions to Na+ slabs in the NFM cathode, especially at high voltage, will not only hinder the bulk-phase diffusion of active Na+, but also cause TM ions to dissolve and migrate, driven by the anode potential, and finally be deposited on the HC anode, making the SEI thicker, increasing the interfacial impedance, and catalyzing the electrolyte reduction, thereby reducing the battery life.35,36 To more deeply understand this process, HC anodes from pouch batteries after 300 cycles at 3.8, 4.0 and 4.2 V upper voltages were observed through SEM/EDS. Ni/Fe/Mn deposition was observed on the HC anode, with spotted and mossy by-products attached to the HC surface, obviously degrading the HC structure (Fig. 5a–d and S39†). The three types of HC anode were further reassembled into Na||HC half batteries for EIS measurements, and it was found that the interfacial impedance of the HC anode increased only slightly with the increase in the charging cut-off voltage. Also, the impedance of the HC anode was much smaller than that of the NFM cathode throughout the cycle, indicating the matter concentrating on the cathode in full battery (Fig. S40–42†). However, the interfacial impedance of the HC anode increased significantly with long cycling (Fig. S43†).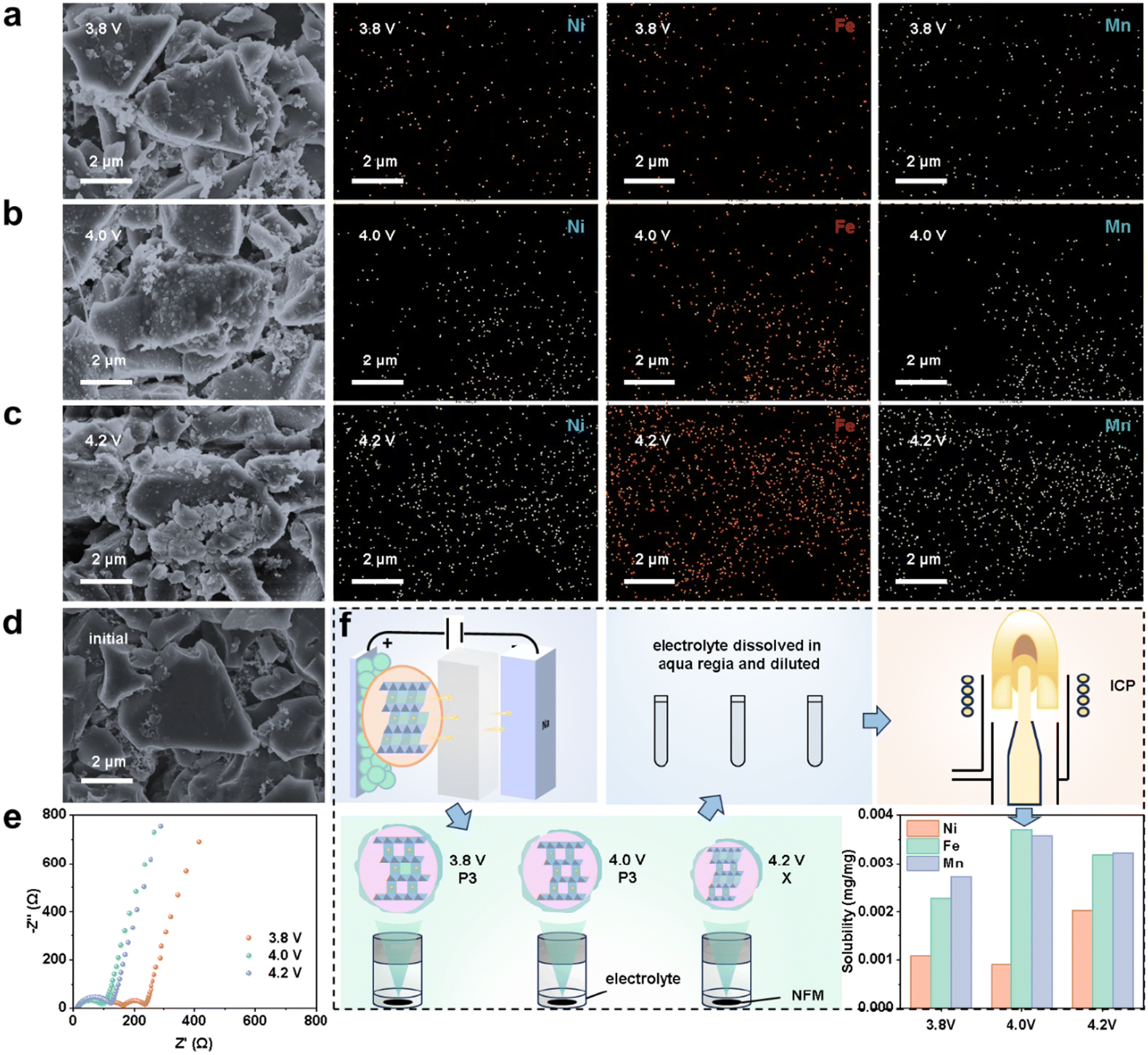 | ||
| Fig. 5 TM ion dissolution, mitigation and deposition on the HC anode. (a–c) SEM and elemental mapping images of the anode from HC||NFM batteries cycled at 3.8, 4.0, and 4.2 V upper voltages, respectively. (d) SEM image of the initial anode. (e) Nyquist plots of Na||HC half batteries whose HC electrodes were taken from HC||NFM pouch batteries after 300 cycles at 3.8, 4.0, and 4.2 V. (f) ICP-OES testing process and results of TM dissolution in the electrolyte from NFM cathodes charging to 3.8, 4.0, and 4.2 V (the unit mg mg−1 means mg dissolved TM per mg NFM). | ||
From analysis of the NFM cathode, the TM dissolution amount was quantitatively characterized by inductively coupled plasma-optical emission spectrometry (ICP-OES). A fresh NFM cathode was assembled into full batteries and charged to 3.8, 4.0, and 4.2 V at 0.1 C. It was then extracted from the batteries, washed with DMC, and dipped in the electrolytes, which were then tested by ICP-OES (Fig. 5f). From the test results, it was found that the Fe and Mn dissolution were more serious than that of Ni, which could be attributed to the ion migration probability increasing by the Jahn–Teller distortion of Fe4+ and the high reaction activity of Mn4+ to soluble Mn2+ under H+ attack.35,37 Besides, with the charging cut-off voltage increasing, the dissolution amount of total TMs from the NFM cathode increased.
Therefore, the TM dissolution, mitigation, and deposition, indicated not only the degraded structure and blocked Na+ migration in the NFM cathode, but also the aggravated side reactions of the electrolyte and damaged structure of the SEI for the HC anode. It was the chemical crosstalk between the electrodes that resulted in the combined failure of the structure and performance during cycling, especially under a high charging cut-off voltage.
Case of high-voltage NFM performance improvement
Since NFM in the full battery cycled at high upper voltage experiences serious particle cracking due to harmful X and O3′ phase transition, it was clear that cathode–electrolyte side reactions continuously occur and a thick CEI was formed on NFM particles. As a preliminary attempt to address this, boron-containing additives (lithium difluoro oxalate borate (LiDFOB) and trimethyl borate (TMB)) were introduced into the electrolyte, with an aim to form a stable CEI and protect the NFM cathode, thus suppressing the capacity degradation from active ion loss and active NFM loss at high voltage.38–43 The structure–performance relationships and improvement mechanism are presented in Fig. 6a. The additives could self-sacrifice oxidate on NFM during charging due to their high HOMO energy,44 which could inhibit the carbonate solvent decomposition, promote more PF6− decomposition, and adsorb the by-products such as HF, thus forming an inorganic-rich CEI. The XPS spectra of the NFM cathode after cycling in the electrolyte with the boron-containing additive demonstrate the achievement of this effect (Fig. 6b). The comprehensive electrochemical performance of NFM, including under high-voltage, high-temperature and fast-charging cycling, was greatly improved (Fig. 6c and S44–46†). The interfacial impedance of NFM with the boron-containing additives was much smaller, indicating that the interface was resilient enough to suppress the interfacial parasitic reactions and exhibited high ion conductivity (Fig. S47†). Besides, the structural stability of NFM was improved (Fig. S48 and 49†). Therefore, the electrolytes with boron-containing additives could suppress the active ion loss and active cathode loss significantly, which suggests a possible improvement strategy for addressing the high-voltage failure of NFM cathodes. | ||
| Fig. 6 (a) Schematic illustration of the CEI growth on NFM particles during high-voltage cycling and the reference case of a stable interface design. (b) Comparison of the XPS spectra of NFM in NPED and NPED-0.2L electrolytes after 500 cycles within the voltage range of 2.0–4.2 V at 60 °C. (c) Long cycling performance of Na||NFM in NPED, NPED-0.8L and NPED-3TMB electrolytes within the voltage range of 2.0–4.2 V at 1.0 C and room temperature. NPED: 1 M NaPF6 in EC/DEC. NPED-0.2L: 0.2 wt% LiDFOB in NPED. NPED-0.8L: 0.8 wt% LiDFOB in NPED. NPED-3TMB: 3 wt% TMB in NPED. | ||
Conclusions
This work reveals the high-voltage degradation mechanism of layered cathode-based SIBs by uncovering the capacity degradation sources through a reverse coin half battery, tracing the structural evolution of the cathode through operando spectroscopy, and characterizing the material failure processes through XRD, electrochemical techniques and electron microscopy. At a 4.0 V upper voltage, HC||NFM full battery could retain both a high specific energy and long cycle life, benefiting from the reversible structural evolution with the O3–P3–O3 phase sequence. At 4.2 V upper voltage, although this battery system had high energy density in the early cycles, its cycling stability dropped dramatically in later cycles, with the capacity loss coming from the interfacial side reactions and structural degradation of the cathode. During cycling at this upper voltage, NFM experienced a complex irreversible structural evolution with the O3–P3–X–O3′–P3–O3 phase sequence, with the unit cell expansion–contraction only occurring during charging, exacerbating the particle cracking and interfacial parasitic reactions. Meanwhile, the reaction kinetics was obviously retarded and the polarization became more than twice as large. Finally, boron-containing additives were introduced for self-sacrificial oxidation on the NFM surface to form an inorganic-rich dense interface, thereby protecting the NFM and allowing obtaining an excellent comprehensive electrochemical performance. This work provides a fundamental insight into the high-voltage failure mechanism of layered cathode-based SIBs, and a powerful reference to aid the design of long-life high-specific-energy SIBs and stable high-voltage NFM cathodes.Experimental
Electron microscopy
The morphology of the materials was observed through scanning electron microscopy (SEM), using Hitachi TM3030 and Zeiss GeminiSEM 500 instruments. The working voltage of the Hitachi TM3030 was 15 kV and that of the Zeiss GeminiSEM 500 was 30 kV. Element mapping of the material surface was achieved by energy dispersion spectroscopy (EDS) using a Zeiss GeminiSEM 500 instrument. The bulk structures of the materials and the cathode–electrolyte interface (CEI) were observed through transmission electron microscopy (TEM, JEOL JEM-2100plus), where the working voltage of the TEM instrument was 200 kV.Operando and postmortem XRD measurements
Operando XRD measurements were conducted to trace the structural evolution of the NFM cathode in a working pouch full cell on a Malvern PANalytical Empyrean diffractometer with Ag Kα radiation. The scanning range was from 5.2° to 28.0° for 17 min. Ex situ XRD measurements were conducted to assess the material structure on a Malvern PANalytical Empyrean diffractometer at a scan rate of 10° min−1 and with Cu Kα radiation.Spectral characterizations
Raman spectroscopy (Horiba LabRAM HR Evolution) was used to characterize the structures of the NFM and HC powders, with a 532 nm wavelength laser and 0.28 mW power. X-Ray photoelectron spectroscopy (XPS, Thermo Scientific ESCALAB Xi+) was used to analyze the surface components of the NFM cathode. The NFM cathodes were taken out from cycled cells, dipped in dimethyl carbonate (DMC) solvent and dried enough to remove the residual electrolyte on the NFM. Inductively coupled plasma-optical emission spectroscopy (ICP-OES, SPECTRO SPECTROBLUE FMX36) was used to identify the transition metal content dissolved from the NFM cathode in to the electrolyte. The NFM cathodes fully charged at 3.8, 4.0, and 4.2 V were dipped in the electrolyte for 24 h, respectively, and then taken out. Subsequently, the electrolytes were dissolved with aqua regia and diluted with ultrapure water for ICP-OES measurements.Electrochemical measurements
Na||NFM and Na||HC half cells were assembled to evaluate their electrochemical performance, including their capacity, cycling stability, rate performance, EIS data and galvanostatic intermittent titration (GITT) data. After being assembled, the coin cells were held for 6 h before testing. The cathode electrode was fabricated with 80 wt% NFM, 10 wt% PVDF and 10 wt% Super P carbon conductive agent, with N-methyl-1,2-pyrrolidone (NMP) as the solvent and then dried at 80 °C for 12 h. They then underwent cycling and cyclic voltammetry tests. A porous glass fiber separator (Whatman, GF/D) was used in the coin cells (CR2016) to well hold the electrolyte during long cycling at 0.1 C, corresponding to a 13 mA g−1 current density. EIS measurements were performed on a Solartron electrochemical workstation and the voltage amplitude was 10 mV with the frequency range from 105 Hz to 0.1 Hz. GITT tests were conducted on a Land battery tester with the voltage ranging from 2.0 V to each selected upper voltage.Author contributions
Shini Lin and Wei Li conceived the idea and collaborated to complete all experiments, tests and writing. Conceptualization: Shini Lin and Wei Li. Experimental design and investigation: Shini Lin. Data analysis: Shini Lin, Wei Li, Huiya Yang, Minghui Chen, Jing Zeng and Peng Zhang. Validation: Shini Lin, Yuan Qin and Honghao Xie. Writing – original draft: Shini Lin. Writing – review & editing: Shini Lin, Wei Li, Peng Zhang and Jinbao Zhao. Funding acquisition & resources: Jinbao Zhao.Conflicts of interest
There are no conflicts to declare.Data availability
The authors confirm that the findings of this study are available within the main text and ESI.†Acknowledgements
The authors gratefully acknowledge financial support from the National Key Research and Development Program of China [2021YFB2400300], Yunnan Natural Science Foundation Project [202202AG050003] and National Natural Science Foundation of China [22021001]. We are grateful to Tan Kah Kee Innovation Laboratory (IKKEM) for their help with TEM and ICP measurements.References
- Y. Gao, H. Zhang, J. Peng, L. Li, Y. Xiao, L. Li, Y. Liu, Y. Qiao and S.-L. Chou, Carbon Energy, 2024, 6, e464 CrossRef.
- C. Vaalma, D. Buchholz, M. Weil and S. Passerini, Nat. Rev. Mater., 2018, 3, 1–11 CrossRef.
- J.-Y. Hwang, S.-T. Myung and Y.-K. Sun, Chem. Soc. Rev., 2017, 46, 3529–3614 RSC.
- H. S. Hirsh, Y. Li, D. H. S. Tan, M. Zhang, E. Zhao and Y. S. Meng, Adv. Energy Mater., 2020, 10, 2070134 CrossRef CAS.
- D. Hou, D. Xia, E. Gabriel, J. A. Russell, K. Graff, Y. Ren, C.-J. Sun, F. Lin, Y. Liu and H. Xiong, ACS Energy Lett., 2021, 6, 4023–4054 CrossRef CAS PubMed.
- J. Liu, Y. Zhang, J. Zhou, Z. Wang, P. Zhu, Y. Cao, Y. Zheng, X. Zhou, C. Yan and T. Qian, Adv. Funct. Mater., 2023, 33, 2302055 CrossRef CAS.
- G. Liu, W. Wan, Q. Nie, C. Zhang, X. Chen, W. Lin, X. Wei, Y. Huang, J. Li and C. Wang, Energy Environ. Sci., 2024, 17, 1163–1174 RSC.
- G. Bree, D. Horstman and C. T. J. Low, J. Energy Storage, 2023, 68, 107852 CrossRef.
- H. Jo, J. H. Park, D. Choi, K. Kim and S. An, Adv. Mater., 2024, 36, 2407719 CrossRef PubMed.
- Y.-J. Guo, R.-X. Jin, M. Fan, W.-P. Wang, S. Xin, L.-J. Wan and Y.-G. Guo, Chem. Soc. Rev., 2024, 53, 7828–7874 RSC.
- G.-L. Xu, R. Amine, Y.-F. Xu, J. Liu, J. Gim, T. Ma, Y. Ren, C.-J. Sun, Y. Liu, X. Zhang, S. M. Heald, A. Solhy, I. Saadoune, W. L. Mattis, S.-G. Sun, Z. Chen and K. Amine, Energy Environ. Sci., 2017, 10, 1677–1693 RSC.
- X. Liang, J.-Y. Hwang and Y.-K. Sun, Adv. Energy Mater., 2023, 13, 2301975 CrossRef CAS.
- R. Dong, L. Zheng, Y. Bai, Q. Ni, Y. Li, F. Wu, H. Ren and C. Wu, Adv. Mater., 2021, 33, 2008810 CrossRef CAS PubMed.
- S. Qiu, L. Xiao, M. L. Sushko, K. S. Han, Y. Shao, M. Yan, X. Liang, L. Mai, J. Feng, Y. Cao, X. Ai, H. Yang and J. Liu, Adv. Energy Mater., 2017, 7, 1700403 CrossRef.
- Z. Wang, X. Feng, Y. Bai, H. Yang, R. Dong, X. Wang, H. Xu, Q. Wang, H. Li, H. Gao and C. Wu, Adv. Energy Mater., 2021, 11, 2003854 CrossRef CAS.
- T. Perveen, M. Siddiq, N. Shahzad, R. Ihsan, A. Ahmad and M. I. Shahzad, Renewable Sustainable Energy Rev., 2020, 119, 109549 CrossRef CAS.
- N. LeGe, X.-X. He, Y.-X. Wang, Y. Lei, Y.-X. Yang, J.-T. Xu, M. Liu, X. Wu, W.-H. Lai and S.-L. Chou, Energy Environ. Sci., 2023, 16, 5688–5720 RSC.
- Y. Li, A. Vasileiadis, Q. Zhou, Y. Lu, Q. Meng, Y. Li, P. Ombrini, J. Zhao, Z. Chen, Y. Niu, X. Qi, F. Xie, R. Van Der Jagt, S. Ganapathy, M.-M. Titirici, H. Li, L. Chen, M. Wagemaker and Y.-S. Hu, Nat. Energy, 2024, 9, 134–142 CrossRef CAS.
- H. Yang, D. Wang, Y. Liu, Y. Liu, B. Zhong, Y. Song, Q. Kong, Z. Wu and X. Guo, Energy Environ. Sci., 2024, 17, 1756–1780 RSC.
- L. Chen, X. Fan, E. Hu, X. Ji, J. Chen, S. Hou, T. Deng, J. Li, D. Su, X. Yang and C. Wang, Chem, 2019, 5, 896–912 CAS.
- Y. Zhao, Q. Liu, X. Zhao, D. Mu, G. Tan, L. Li, R. Chen and F. Wu, Mater. Today, 2023, 62, 271–295 CrossRef CAS.
- S. Feng, C. Zheng, Z. Song, X. Wu, M. Wu, F. Xu and Z. Wen, Chem. Eng. J., 2023, 475, 146090 CrossRef CAS.
- Y. You, S. Xin, H. Y. Asl, W. Li, P.-F. Wang, Y.-G. Guo and A. Manthiram, Chem, 2018, 4, 2124–2139 CAS.
- K. Zhang, Z. Xu, G. Li, R.-J. Luo, C. Ma, Y. Wang, Y.-N. Zhou and Y. Xia, Adv. Energy Mater., 2023, 13, 2302793 CrossRef CAS.
- T.-Y. Yu, J. Kim, G. Oh, M. H. Alfaruqi, J.-Y. Hwang and Y.-K. Sun, Energy Storage Mater., 2023, 61, 102908 CrossRef.
- M. Keller, D. Buchholz and S. Passerini, Adv. Energy Mater., 2016, 6, 1501555 CrossRef PubMed.
- Y. Yu, D. Ning, Q. Li, A. Franz, L. Zheng, N. Zhang, G. Ren, G. Schumacher and X. Liu, Energy Storage Mater., 2021, 38, 130–140 CrossRef.
- N. Li, J. Ren, R. Dang, K. Wu, Y. L. Lee, Z. Hu and X. Xiao, J. Power Sources, 2019, 429, 38–45 CrossRef CAS.
- Y. Li, F. Wu, Y. Li, M. Liu, X. Feng, Y. Bai and C. Wu, Chem. Soc. Rev., 2022, 51, 4484–4536 RSC.
- T.-Y. Yu, J. Kim, G. Oh, M. H. Alfaruqi, J.-Y. Hwang and Y.-K. Sun, Energy Storage Mater., 2023, 61, 102908 CrossRef.
- K. Zhang, Z. Xu, G. Li, R.-J. Luo, C. Ma, Y. Wang, Y.-N. Zhou and Y. Xia, Adv. Energy Mater., 2023, 13, 2302793 CrossRef CAS.
- X. Liu, Y. Wan, M. Jia, H. Zhang, W. Xie, H. Hu, X. Yan and X. Zhang, Energy Storage Mater., 2024, 67, 103313 CrossRef.
- X. Zeng, G.-L. Xu, Y. Li, X. Luo, F. Maglia, C. Bauer, S. F. Lux, O. Paschos, S.-J. Kim, P. Lamp, J. Lu, K. Amine and Z. Chen, ACS Appl. Mater. Interfaces, 2016, 8, 3446–3451 CrossRef CAS PubMed.
- S. Komaba, N. Yabuuchi, T. Nakayama, A. Ogata, T. Ishikawa and I. Nakai, Inorg. Chem., 2012, 51, 6211–6220 CrossRef CAS PubMed.
- S. Chu and S. Guo, Adv. Funct. Mater., 2024, 34, 2313234 CrossRef CAS.
- Y. Liu, Y.-H. Zhang, J. Ma, J. Zhao, X. Li and G. Cui, Chem. Mater., 2024, 36, 54–73 CrossRef CAS.
- M. Jeong, H. Lee, J. Yoon and W.-S. Yoon, J. Power Sources, 2019, 439, 227064 CrossRef CAS.
- K. Ma, Y. Cao, S. Zhang, Y. Zhang, S. Fang, X. Han, F. Jin and J. Sun, Nano Lett., 2024, 24, 8826–8833 CrossRef CAS PubMed.
- Z. Liu, W. Hou, H. Tian, Q. Qiu, I. Ullah, S. Qiu, W. Sun, Q. Yu, J. Yuan, L. Xia and X. Wu, Angew. Chem., Int. Ed., 2024, 63, e202400110 CrossRef CAS PubMed.
- J. Li, J. Yang, Z. Ji, M. Su, H. Li, Y. Wu, X. Su and Z. Zhang, Adv. Energy Mater., 2023, 13, 2301422 CrossRef CAS.
- S. Li, H. Xu, Y. Zhu, Z. Yang, Y. Bao and Y. Chen, J. Mater. Chem. A, 2024, 12, 31347–31361 RSC.
- J. Liu, X. Li, J. Huang, G. Yang and J. Ma, Adv. Funct. Mater., 2024, 34, 2312762 CrossRef CAS.
- Q. Liu, G. Yang, S. Liu, M. Han, Z. Wang and L. Chen, ACS Appl. Mater. Interfaces, 2019, 11, 17435–17443 CrossRef CAS PubMed.
- Y. Zhang, Y. Chen, Q. He, J. Ke, W. Wang, J.-F. Wu, P. Gao, Y. Li and J. Liu, J. Energy Chem., 2024, 92, 639–647 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5eb00077g |
| This journal is © The Royal Society of Chemistry 2025 |