
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnderstanding the effects of binder dissolution dynamics on the chemistry and performance of lithium–sulfur batteries†
Saheed A.
Lateef
a,
John
Chmiola
b,
Fabio
Albano
b,
William E.
Mustain
 a and
Golareh
Jalilvand
*a
a and
Golareh
Jalilvand
*a
aDepartment of Chemical Engineering, University of South Carolina, Columbia, SC 29208, USA. E-mail: jalilvand@sc.edu
bNantG Power LLC, El Segundo, CA 90245, USA
First published on 23rd June 2025
Abstract
Lithium–sulfur batteries (LSBs) are promising next-generation energy storage devices due to their higher theoretical specific energy and lower cost compared to conventional Lithium-ion batteries. However, their practical implementation has been hindered by severe performance degradation during extended cycling, primarily driven by shuttling of soluble sulfur discharge products (polysulfides) between the cathode and anode leading to capacity loss. In this work, we investigate the impact of selected binders and solvents, highlighting the effect of the dissolution process of the binder in solvent, on the structural properties and electrochemical performance of sulfur cathode. It is demonstrated for a variety of binders that tailoring the dissolution process of binders within solvents can result in a specific binder morphology around sulfur particles that aids in trapping the polysulfides, controlling the shuttling phenomenon. The best combination of binder and solvent with optimized dissolution process results in outstanding capacity retention of 84% retention over 1000 cycles at C/10.
Broader contextLithium–sulfur batteries (LSBs) are widely regarded as a next-generation energy storage solution due to their high theoretical energy density and the natural abundance of sulfur. Despite their promise, LSB commercialization has remained elusive due to persistent issues such as polysulfide dissolution and cathode degradation, which lead to rapid capacity fade. Research efforts have primarily focused on introducing new materials or designing complex electrode architectures to address these challenges, yet many of these approaches rely on intricate, unscalable synthesis techniques. This study demonstrates that optimizing conventional cathode processing methods—specifically, controlling binder dissolution parameters—can significantly improve the electrochemical performance of LSBs without requiring novel material development. By systematically investigating the influence of binder dissolution duration, solvent/binder ratio, and molecular interactions on cathode structure and performance, this work establishes a practical framework for stabilizing sulfur cathodes. The findings show that subtle modifications in slurry processing can yield remarkable improvements in capacity retention over long-term cycling. These insights provide a scalable and cost-effective pathway for advancing LSB technology, bridging the gap between fundamental research and real-world implementation, while also offering valuable lessons applicable to other emerging battery chemistries. |
1. Introduction
The emergence of long-range electric vehicles, smart grids, and high-energy power electronics are increasing the demand for batteries that are cheaper and lighter while being denser in energy and power. Modern lithium-ion battery (LIB) electrode materials, with intercalation charge and discharge mechanism, offer limited achievable specific energy of 265 Wh kg−1.1 Thus, there is a need to utilize alternative high specific energy electrode materials.2,3 Sulfur is an attractive alternative to the traditional LIB cathode materials due to its high theoretical specific energy when paired with metallic Li anode (2600 Wh kg−1), forming a Li-sulfur battery (LSB). Sulfur also has the advantages of low cost, high natural abundance, and environmental friendliness.4,5 Despite these promising benefits, the practical application of LSBs is affected by several intrinsic challenges. One is the insulating nature of sulfur and its discharge products, so-called Li-polysulfides (LiPSs), that can cause poor discharge kinetics.6 Another concern is the dissolution of the long-chain LiPSs in conventional ether-based electrolytes, which allows the LiPSs to freely travel between the anode and cathode – a phenomenon known as “shuttling”. The polysulfide shuttling results in the loss of active material, low coulombic efficiency, and severe compositional and structural changes in both the anode and cathode, resulting in poor cycle life.7,8 Lastly, the sulfur cathode experiences significant volumetric variation during cycling that leads to structural destruction, loss of structural cohesion, and ultimately battery failure.9To date, a significant amount of research has focused on addressing the challenges in LSBs, through specialized cathode structures. Various approaches have been explored, including forming sulfur composites with conductive materials such as metal–organic frameworks, and carbonaceous materials,10–15 physically confining sulfur within conductive 2D and 3D nano-architectures16 or encapsulating sulfur within protective layers,17–19 scaffolds,20 hollow spheres,21 and yolk–shell structures.22 Generally, these approaches enhance the achievable capacity and cyclability of sulfur by introducing new materials for various electrode components or by developing novel electrode architectures. However, most approaches require highly complex and unscalable synthesis and/or integration to cathode procedures hindering the practical deployment of LSBs.
An effective approach to enhancing LSB performance is optimizing the cathode structure through its processing method, preferably using conventional materials rather than introducing complex new components. Among cathode components, binders play a crucial role in defining electrode structure and performance by ensuring strong adhesion between the active material and conductive species. While binders serve a similar function in LIB electrodes, the >80% volume change of sulfur during cycling amplifies their importance in maintaining electrode integrity and performance. Thus, the choice of binder and solvent, along with the processing method used during electrode fabrication, is critical for achieving distinct structural features and durable, high-performance LSB cathodes.
Although numerous studies have investigated various binder materials to address challenges in LSBs, the majority have concentrated on enhancing chemical interactions of binders with polysulfides through polymerization or functionalization to introduce polar or polysulfide-affinitive groups.23–34 Yet, there is not enough focus on the effect of binder on microstructural characteristics of electrodes and their consequent electrochemical performance. Given the extensive knowledge of conventional binders, refining their processing during electrode fabrication offers a practical route to improving LSBs. Binder-solvent interactions play a crucial role in defining the final electrode structure and performance, yet systematic studies on these effects remain scarce. Key parameters—such as solvent-to-binder ratio, dissolution time, agitation force, stirring temperature, and drying conditions—are often overlooked despite their significant impact. Notably, the solvent-to-binder ratio is typically selected based on a target slurry viscosity, while finer aspects of the dissolution process, including duration, agitation intensity, and drying conditions, are rarely reported or rigorously investigated.
This work systematically investigates the impact of binder dissolution process on the structure and electrochemical performance of sulfur cathodes for LSBs. Three conventional binders—carboxymethyl cellulose (CMC), polyacrylic acid (PAA), and polyvinylpyrrolidone (PVP)—are examined in an organic solvent to evaluate how binder-to-solvent mass ratio and dissolution duration influence binder solubility and resulting electrode morphology. While most prior studies have focused on chemically modifying binders to mitigate polysulfide dissolution, this study presents a fundamentally different strategy, tailoring the microstructure of binders to physically confine polysulfides by engineering the electrode fabrication process. To the best of our knowledge, this is the first demonstration of achieving effective polysulfide confinement solely through the optimization of binder dissolution process, without any chemical modification, using low-cost, commercially available binders. The optimized processing conditions result in the formation of binder-derived shell structures that suppress polysulfide migration, and enhance cycling stability, as illustrated in Scheme 1. These findings demonstrate that simple and scalable modifications to the cathode fabrication process can yield robust electrode architectures and improved performance, offering a new and practical pathway for advancing LSB technology.
 | ||
| Scheme 1 Schematic illustration of hydrogen bonding interactions between PVP, PAA, and CMC binders that drive the formation of binder shells upon their controlled dissolution in NMP. The optimized binder-solvent interactions enable self-assembly of binder shells around sulfur particles, resulting in a physically confining structure that limits polysulfide dissolution and loss. | ||
2. Experimental
2.1. Materials
Sulfur powder (Sigma Aldrich, 100 mesh particle size, MKBQ24992), conductive carbon (Vulcan XC72R, Fuel cell store, 590106-1; and Ketjen black EC-600JD, MSE Supplies, 21421A2), PVP (40 kDa, Fisher Bioreagents, 90415), Na-CMC (250 kDa, Sigma Aldrich, MKCP1912, degree of substitution of 0.9), PAA (450 kDa, Polysciences, 03312-100), and NMP (99% dry basis, TCI, 872-50-4) were all used as received.2.2. Electrode preparation
The electrode preparation method, described in detail in our previous work35 was used to create three groups of electrodes with various binders, summarized in Table 1. For all electrodes, sulfur![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :binder:carbon was mixed in 70:10:20 wt% ratio, targeting an areal sulfur loading of 1.1–1.3 mgsulfur cm−2. The electrodes’ thickness was maintained at 22 μm ± 4, obtaining porosities 63%–69%. These values were chosen for their repeatable electrochemical performance, enabling isolation of the binder morphology effect—the focus of this study. However, recognizing the necessity of higher sulfur loadings for commercial viability, a 5 mgsulfur cm−2 loading was also tested separately.
:binder:carbon was mixed in 70:10:20 wt% ratio, targeting an areal sulfur loading of 1.1–1.3 mgsulfur cm−2. The electrodes’ thickness was maintained at 22 μm ± 4, obtaining porosities 63%–69%. These values were chosen for their repeatable electrochemical performance, enabling isolation of the binder morphology effect—the focus of this study. However, recognizing the necessity of higher sulfur loadings for commercial viability, a 5 mgsulfur cm−2 loading was also tested separately.
| Group | Cathode | Binder type | Solvent type | Solvent amount per mg of the binder (μL) | Time of stirring (days) |
|---|---|---|---|---|---|
| 1 | PVP-NMP-1 | PVP | NMP | 50 | 1 |
| PAA-NMP-1 | PAA | NMP | 50 | 1 | |
| CMC-NMP-1 | CMC | NMP | 50 | 1 | |
| 2 | PVP-NMP-2 | PVP | NMP | 250 | 1 |
| PAA-NMP-2 | PAA | NMP | 250 | 1 | |
| CMC-NMP-2 | CMC | NMP | 250 | 1 | |
| 3 | PVP-NMP-3 | PVP | NMP | 250 | 7 |
| PAA-NMP-3 | PAA | NMP | 250 | 7 | |
| CMC-NMP-3 | CMC | NMP | 250 | 7 |
The sulfur and binder powders were mixed in a dry state and sonicated for 2 minutes. Next, the carbon powder was added and the whole dry mix was sonicated for another 2 minutes. Then, the solvent was added according to the ratios that are presented in Table 1 to make the cathode ink. The slurries from each group were then stirred for 5–168 hours (0.2–7 days) at 800 RPM agitation speed. The naming of the cells follows the B–S–X format, where B refers to the binder material, S refers to the solvent material (NMP or water), and X stands for the group number. Next, the slurries were deposited on an aluminum (Al) foil current collector using an Iwata spray gun. The deposited electrodes were then dried under vacuum at room temperature for 48 hours. All materials and processing parameters except the ones identified in Table 1 were held constant across all electrodes in all groups.
2.3. Coin cell assembly
2032 coin cells were assembled using cell casings provided by Hohsen battery materials company. All coin cell assembly was done inside of an MBraun Labmaster glovebox under ultra-high purity Ar with oxygen and humidity level <0.1ppm. The coin cells were assembled using a 16 mm diameter Li metal foil (99.9%, MTI corporation, MF-Li25) as the anode, and a 16 mm diameter sulfur cathode. The separator was a 19mm diameter Celgard 2320 tri-layer polymer disk. The electrolyte was 1.0 M LiTFSI (Sigma-Aldrich, USA, MKCS0286) and 0.2 M LiNO3 (Alfa Aesar, 13405)36 dissolved in a 1:1 volumetric ratio of 1,2-dimethoxyethane (DME, 99%, Sigma-Aldrich, SHBL3025) and 1,3-dioxolane (DOL, 99%, Alfa Aesar, 42899). 10 μL mg(sulfur)−1 of electrolyte was added to all cells.
2.4. Structural and electrochemical characterization
The morphologies of the three groups of electrodes were investigated by Scanning Electron Microscopy (SEM) using a Zeiss Gemini500. All microscopy was performed around 8–12 mm working distance using a 15 kV acceleration voltage for the electron beam and a secondary electron detector. The electrodes were fixed onto the specimen stubs using double-sided carbon tape. Fourier transform infrared spectroscopy (FT-IR) was used to assess the chemical bonds in different binders upon dissolution. The IR spectra of PVP, PAA, and CMC binder were recorded in the dry and NMP-dissolved state after 1 and 7 days using an Agilent Cary 630 FT-IR Spectrometer with micro-ATR (diamond crystal), sampled within a 4000–1000 cm−1 range, averaged from 10 scans, and obtained with at least 16 cm−1 resolution.The cycling performance of coin cells was evaluated using an Arbin battery cycler Version 3, Build 7.29. The cells were cycled galvanostatically at C/10 between 1.8–2.8 V. The applied current was calculated based on the amount of active sulfur loaded into each electrode and the reported discharge capacity was normalized to the amount of sulfur in the electrode. The experiments were done in triplicates, and the reported capacities are representative of the three measurements with only about ±9 mAh g−1 deviation.
Electrochemical impedance spectroscopy (EIS) was carried out using Biologic VMP3 system over a frequency range from 10 mHz to 1 MHz under potentiostatic mode using 5 mV amplitude. The data were analyzed with the use of EC-Lab software and the resistances were determined by fitting the data to an equivalent circuit.
Moreover, a self-discharge experiment was carried out to investigate the effect of electrode morphology on active material loss. The cells were first fully discharged at C/10 and then fully charged at the same rate. Next, the cells were left idle at the 100% state-of-charge (SOC) for 24 h. This process was repeated 5 times and the change in the capacity of the high potential plateau was recorded.
2.5. In situ Raman spectroscopy
To investigate the chemical evolution of sulfur cathodes with different binder morphologies, in situ Raman spectroscopy was conducted. Thermo-Fisher Scientific DXR3 Raman microscope with a laser radiation at wavelength of 532 nm, energy of 1 mW, frequency of 100–600 cm−1 at a sample exposure time of 2 s was utilized. The electrodes were sealed in an ECC-opto-10 EL-cell in oxygen- and moisture-free glovebox. The separator and Li metal anode were punched centrally and assembled face-to-face with the sulfur electrode such that the laser beam could hit the surface of the sulfur cathode without the interference of other cell components. The cell was properly sealed to protect the components from atmospheric oxygen after removing from the glovebox. After assembly, the EL-cell was placed on the Raman microscope stage and connected to a biologic VSP-3e potentiostat. The cells were discharged galvanostatically at C/10 while Raman spectroscopy was carried out at 2.32 V, 2.15 V, 2.08 V and 1.82 V to study the evolution of the polysulfides during discharge. During the Raman data collection, the cycling was held at the selected potential for 1 min. The potentials were selected according to their key roles in the stepwise process of polysulfides formation and conversion.3. Results and discussion
3.1. Effect of binder dissolution process on electrode microstructure
The electrode microstructure is expected to change with different binder dissolution processes, which directly governs the binder arrangement relative to the active material particles, carbon host, and current collector. Fig. 1 shows the SEM images of Group 1–3 electrodes with PVP, PAA and CMC binders. All cathodes comprised agglomerates ranging from 10–50 μm in size. However, Group 1 electrodes (Fig. 1a–c) with 50 μL mg(binder)−1 of NMP solvent and a stirring time of 1 day at 800 RPM, exhibited a distinct binder shell morphology around the sulfur agglomerates, especially in the PVP- and CMC-based electrodes. This behavior is attributed to limited binder dissolution, governed by controlled parameters during the dissolution process, including duration, agitation, and binder-to-solvent ratio, which maintain the polymer binder in a swollen state – an intermediate dissolution state between the pure polymer and fully solvated state.35,37 Among the three electrodes in Group 1, the binder shells in the PVP-NMP-1 and CMC-NMP-1 electrodes (Fig. 1a and c) were visibly thicker and morphologically uniform (solid line arrows). In contrast, the binder shells in the PAA-based electrode (Fig. 1b) were considerably thinner and exhibited a non-uniform coral-like morphology, suggesting that the binder had dissolved beyond the swollen state, yet not fully dissolved (transitional state-dashed arrows). The zoomed-in views of the binder shells are shown in the insets of Fig. 1a–c. A statistical analysis of binder shell thicknesses across 30 particles revealed that PVP-NMP-1 exhibited a median shell thickness of 0.92 μm, compared to 0.49 μm for PAA-NMP-1 and 1.19 μm for CMC-NMP-1 (Fig. S1†). Among the three, CMC-NMP-1 showed the highest variability, with a standard deviation of 0.32 μm, indicating the least uniform shell formation. In contrast, PAA-NMP-1 exhibited the most uniform shell morphology, with a standard deviation of 0.15 μm. PVP-NMP-1 achieved a favorable balance, forming relatively thick and uniformly distributed binder shells across the electrode, suggesting its suitability for promoting consistent electrode architecture. | ||
| Fig. 1 The SEM images of the precycled Group 1 (a–c), 2 (d–f), and 3 (g–i) electrodes with PVP, PAA, and CMC binder dissolved in NMP solvent. The solid-line arrows point to the binder shells around sulfur particles that are formed due to controlled dissolution and maintaining the swollen state of the polymer binder. The dashed-line arrows point at binder shells transitioning beyond the swollen state toward complete dissolution, forming an inhomogeneous coral-like binder morphology around sulfur particles. | ||
Comparing the electrodes in Group 1, all electrode inks were prepared with a similar solid mixture composition and an equal amount of solvent. The sole difference between those electrodes was the chemistry of the binders, which resulted in different binder morphologies around the sulfur particles. Therefore, the variation in the binder shell thickness and morphology is ascribed to the differences in the interaction/solubility of the binder in NMP, which is determined by the bonding of the functional groups of each binder with NMP molecule. PVP with a carbonyl functional group, PAA with a carboxyl group, and CMC with a carboxylate and hydroxyl groups are polar materials, thus readily soluble in polar NMP. However, the distinction in their solubility comes from their (lack of) protic nature which determines their readiness to participate in hydrogen bonding. The higher readiness of the carboxyl groups in PAA to form hydrogen bonds with the carbonyl group in NMP results in its faster dissolution,38 which is manifested in the coral-like morphology or transitional state of binder shells around sulfur particles in the PAA-based electrode.
The interaction between the binders and NMP was investigated using attenuated total reflectance FTIR (ATR-FTIR), as shown in Fig. 2a–c. The FTIR spectra of the pure polymer revealed characteristic carbonyl (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) stretching peaks at 1658 cm−1 for PVP, and 1703 cm−1 for PAA, while CMC exhibited an asymmetric stretching vibration of the carboxylate group (COO−) at 1591 cm−1.39–41 Additionally, all three binders displayed broad O–H stretching bands above 3200 cm−1, indicative of hydroxyl functionalities.42 Upon addition of NMP and stirring for 24 hours, carbonyl peak shifts were observed in the FTIR spectra for all three binders, indicating interactions between the N and O atoms of NMP and the carbonyl, carboxylate, or hydroxyl functional groups present in the binders. These spectral shifts reflect modifications in hydrogen bonding environments, as the stretching frequencies of these functional groups are highly sensitive to hydrogen bond formation and disruption.43 With prolonged stirring (1 week), further shifts in the carbonyl peaks were observed, along with broadening of the O–H bands, providing additional evidence for hydrogen bonding. These interactions were also reflected in the spectral features of NMP itself, particularly in the carbonyl region around 1673 cm−1, where peak shifts were detected.
O) stretching peaks at 1658 cm−1 for PVP, and 1703 cm−1 for PAA, while CMC exhibited an asymmetric stretching vibration of the carboxylate group (COO−) at 1591 cm−1.39–41 Additionally, all three binders displayed broad O–H stretching bands above 3200 cm−1, indicative of hydroxyl functionalities.42 Upon addition of NMP and stirring for 24 hours, carbonyl peak shifts were observed in the FTIR spectra for all three binders, indicating interactions between the N and O atoms of NMP and the carbonyl, carboxylate, or hydroxyl functional groups present in the binders. These spectral shifts reflect modifications in hydrogen bonding environments, as the stretching frequencies of these functional groups are highly sensitive to hydrogen bond formation and disruption.43 With prolonged stirring (1 week), further shifts in the carbonyl peaks were observed, along with broadening of the O–H bands, providing additional evidence for hydrogen bonding. These interactions were also reflected in the spectral features of NMP itself, particularly in the carbonyl region around 1673 cm−1, where peak shifts were detected.
 | ||
| Fig. 2 FTIR spectra of the binders upon dissolution in NMP (a) PVP, (b) PAA and (c) CMC suggesting stronger hydrogen bond activities in PAA than PVP and CMC. | ||
The FTIR results also reveal distinct binder-specific solvation behaviors in NMP, which correlate with their ability to form binder shell structures around sulfur particles. For PVP, the CO stretching peak shifted from 1658 cm−1 (dry) to 1647 cm−1 after 1 day of stirring and further to 1644 cm−1 after 7 days, indicating a progressive redshift (i.e., shift to lower frequency) consistent with increasing hydrogen bonding between the carbonyl groups and NMP molecules. This gradual solvation supports the evolution of binder shell morphologies from PVP-NMP-1 to PVP-NMP-3 observed in Fig. 1. In the case of PAA, the carbonyl stretching peak shifted from 1703 cm−1 (dry) to 1700 cm−1 (1 day), with much more significant drop to 1643 cm−1 after 7 days, suggesting rapid and extensive hydrogen bonding interactions due to the protic nature of the carboxylic acid groups of PAA. This strong and fast interaction results in nearly complete dissolution of PAA, disrupting shell formation and leading to coral-like, non-uniform morphologies that are apparent even after only 1 day of stirring. CMC, on the other hand, exhibited a blue shift (i.e., shift to higher frequency) from 1591 cm−1 (dry) to 1595 cm−1 (1 day), followed by further shift to 1640 cm−1 after 7 days. The blue shift suggests strong intra-chain hydrogen bonding in CMC, which results in a compact and rigid polymer conformation.44 This limits chain mobility and hinders uniform adsorption onto particle surfaces, leading to heterogeneous/delayed binder dissolution which is manifested in the binder shells. These solvation dynamics are consistent with the SEM observations (Fig. 1 and S1†), where PVP produces relatively uniform binder shells, PAA shows disrupted shell structures due to over-solvation, and CMC results in thicker but heterogeneous or partial shells due to limited chain flexibility. Table 2 provides a detailed comparison of the observed peak positions with literature-reported values.
To study the effect of degree of dilution on dissolution of different binder materials, and their consequent morphology and contribution to the electrode microstructure and electrochemical performance, Group 2 electrodes were prepared with excess solvent, specifically with a 5× increase over Group 1 (Table 1), while maintaining the 24-hour stirring time and 800 RPM agitation. SEM images of the resulting PVP-NMP-2, PAA-NMP-2 and CMC-NMP-2 cells are presented in Fig. 1d–f. The thin and coral-like shells observed in PAA-NMP-1 almost completely disappeared by increasing the solvent/binder mass ratio in PAA-NMP-2, leaving only traces of the coral-like binder morphology around sulfur particles. Moreover, more binder shells in PVP-NMP-2 and CMC-NMP-2 became coral-like and inhomogeneous, suggesting a transitional state to complete binder dissolution. This indicates that increasing the volume of the solvent formed an overly diluted environment beyond the optimal “controlled dissolution” condition for the binders35 and increased the tendency of the binder shells to dissolve beyond the swollen state, but not complete dissolution.
Furthermore, by increasing the solvent/binder ratio, variations in the binder shell morphology in PVP-NMP-2 and CMC-NMP-2 electrodes were observed, where some particles partially maintained their binder shells, some experienced the transition to dissolution, and some had no shell remains due to complete dissolution of binder. This further confirms that over-diluting is necessary but not enough for reaching homogeneity in binder dissolution.
Considering the severe inhomogeneity in Group 2 electrodes, to understand the clear and isolated effect of binder (shell or coral-like) morphology on the electrochemical characteristics of electrodes with different binder materials, it was essential to manipulate the binder dissolution process to achieve an un-shelled structure for reference. For that, the binder/solvent exposure duration was extended. Specifically, Group 3 electrodes were prepared using the same solvent as Groups 1 and 2 and the same solvent amount as Group 2, but with an increased slurry stirring duration from 1 day to 7 days. SEM images of Group 3 electrodes (Fig. 1g–i) revealed that the binder shells, which initially encapsulated the sulfur particles, completely dissolved and disappeared in all cases, regardless of the binder material. This observation underscored the critical role of slurry processing parameters, particularly stirring time which is often overlooked in the literature.45
3.2. Effect of binder morphology on electrochemical performance
The morphology of binder significantly impacts on electronic and ionic conductivity, as well as mass transport in the electrode. To further understand the effect of the binder shell morphology that was observed in Group 1 electrodes, their electrochemical performance was explored and compared to that of Group 3 electrodes with no binder shell morphology. Coin cells with Groups 1 and 3 cathodes were examined by EIS before cycling and after 20 discharge/charge cycles. Measurements were conducted at a fully charged state under potentiostatic conditions with a 5 mV amplitude, with the resulting Nyquist plots shown in Fig. 3a and b. Before cycling, the Nyquist plot displayed a single depressed semicircle, representing the charge transfer resistance (Rct), followed by an inclined line at low frequencies. After cycling, Rct decreased due to the stripping of thin contamination layer on Li metal surface, and a small, depressed semicircle appeared at low frequencies. This additional semicircle reflects the anode's charge transfer resistance, largely due to the resistance caused by the deposition/reduction of polysulfides on the anode (Rp).46,47 The EIS spectra were fitted to an equivalent circuit (inset in Fig. 3b), which includes a constant phase element (Q) to account for non-ideal capacitive behavior and semicircle depression, and a Warburg element (W) to represent ion diffusion limitations within the porous electrode structure and electrolyte. | ||
| Fig. 3 Nyquist plots of the LSB cells from Group 1 (with binder shell morphology) and Group 3 (shell-less binder morphology) (a) before cycling, and (b) after 20 cycles. The resistances associated with polysulfide shuttling increased more in Group 3 electrodes compared to Group 1, suggesting that the presence of binder shell morphology in Group 1 electrodes suppressed the polysulfide loss. | ||
Table 3 summarizes the resistance values obtained from the circuit fitting. The Rb values before cycling were similar, confirming a comparable status for the electrolyte and separator for all cells before cycling. Regarding the charge transfer resistance, PAA-NMP-1 electrode presented the lowest value, which is attributed to its relatively higher ionic conductivity (PAA (2.5 mS cm−1)48 > CMC (2.07 mS cm−1)49 > PVP (0.14 mS cm−1)50) than CMC and PVP, which assists with transport of ions to the active surface area, thereby enhanced reactions. CMC-based electrode showed the largest overall resistance, which is linked to its inelastic/rigid mechanical properties, limiting the compactness and connectivity of the particles.51 CMC binder is also shown to have subpar adhesive properties resulting in substandard adhesion between the electrode and current collector, manifesting in its high charge transfer resistance.52
| Cells | Before cycling | After 30 cycles | W s (Ω √S−1) | |||
|---|---|---|---|---|---|---|
| R b (Ω) | R ct (Ω) | R b (Ω) | R ct (Ω) | R p (Ω) | ||
| PVP-NMP-1 | 9.31 | 68.69 | 10.84 | 9.10 | 1.6 | 5.60 |
| PAA-NMP-1 | 8.32 | 44.78 | 9.81 | 7.79 | 1.5 | 4.24 |
| CMC-NMP-1 | 9.28 | 104.52 | 10.56 | 21.90 | 1.2 | 3.62 |
| PVP-NMP-3 | 8.12 | 67.58 | 15.63 | 11.08 | 3.7 | 5.90 |
| PAA-NMP-3 | 9.50 | 46.22 | 12.93 | 6.45 | 3.1 | 4.27 |
| CMC-NMP-3 | 8.25 | 106.66 | 13.97 | 23.11 | 5.4 | 4.02 |
The pre-cycling resistances were primarily impacted by the intrinsic properties of the binder. However, changes in resistance upon cycling reflect the effect of binder on resistances associated with LiPSs dissolution and shuttling. Notable differences were observed among cells with different binder morphologies after cycling.
Specifically, at the 20th cycle, Rp increased from Group 1 to Group 3 regardless of the binder type. Simultaneously, the electrolyte bulk resistance (Rb) increased in all cells, with a more pronounced rise in Group 3 cells. This increase in both Rb and Rp is attributed to free polysulfide dissolution in the electrolyte in the Group 3 electrodes which was suppressed in Group 1 electrodes.
To understand the effect of binders on LiPSs dissolution and shuttle, both physical and chemical interactions between the two must be considered. PVP, PAA, and CMC are polar materials, with PVP containing carbonyl functional groups, while PAA and CMC contain both carboxyl and hydroxyl groups. The interactions between the polysulfides and binders’ functional groups have been previously modeled, and the polysulfide-trapping capability of these binders has been explored by calculating the binding energies between long chain LiPSs and the oxygen or other heteroatoms in the binders’ functional groups.
The formation of Li–O bonds between the polar LiPS species and polar binders has been a primary focus. In those studies, the binding energies of polysulfide species to amide and carboxyl functional groups were found to be similar, ranging from 1.23 to 1.26 eV.53 At such low binding energies, it is unlikely that the binder functional groups chemically bound the polysulfides.54 To the best of our knowledge, there are no reports of PVP, PAA, or CMC chemical binding ability with polysulfides without specific surface modifications. Therefore, the physical entrapment of polysulfides by the binder morphology is the only possible mechanism of suppressing the shuttling of LiPSs in Group 1 electrodes.
Differential capacity analysis (dQ/dV) was conducted based on a galvanostatic charge–discharge test, to further elucidate the influence of binder morphology on the electrochemical performance of electrodes. The dQ/dV results shown in Fig. 4 reveal two discharge peaks and one charge peak. The discharge peak at 2.25 V–2.27 V corresponds to the conversion of liquid S8 to Li2S4, while the second peak at 2.08 V–1.9 V indicates the transition from liquid Li2S4 to solid Li2S. During charge, a single skewed peak is observed due to the merger of two peaks associated with the conversion of Li2S to long-chain polysulfides and ultimately to sulfur. The two charge peaks are in close proximity,55 and typically merged due to slow redox kinetics, which is the case with all three binders used in this study. However, as polysulfide shuttling and major performance losses in LSB cells primarily occur during discharge, the distinction of dQ/dV discharge peaks remains valuable.
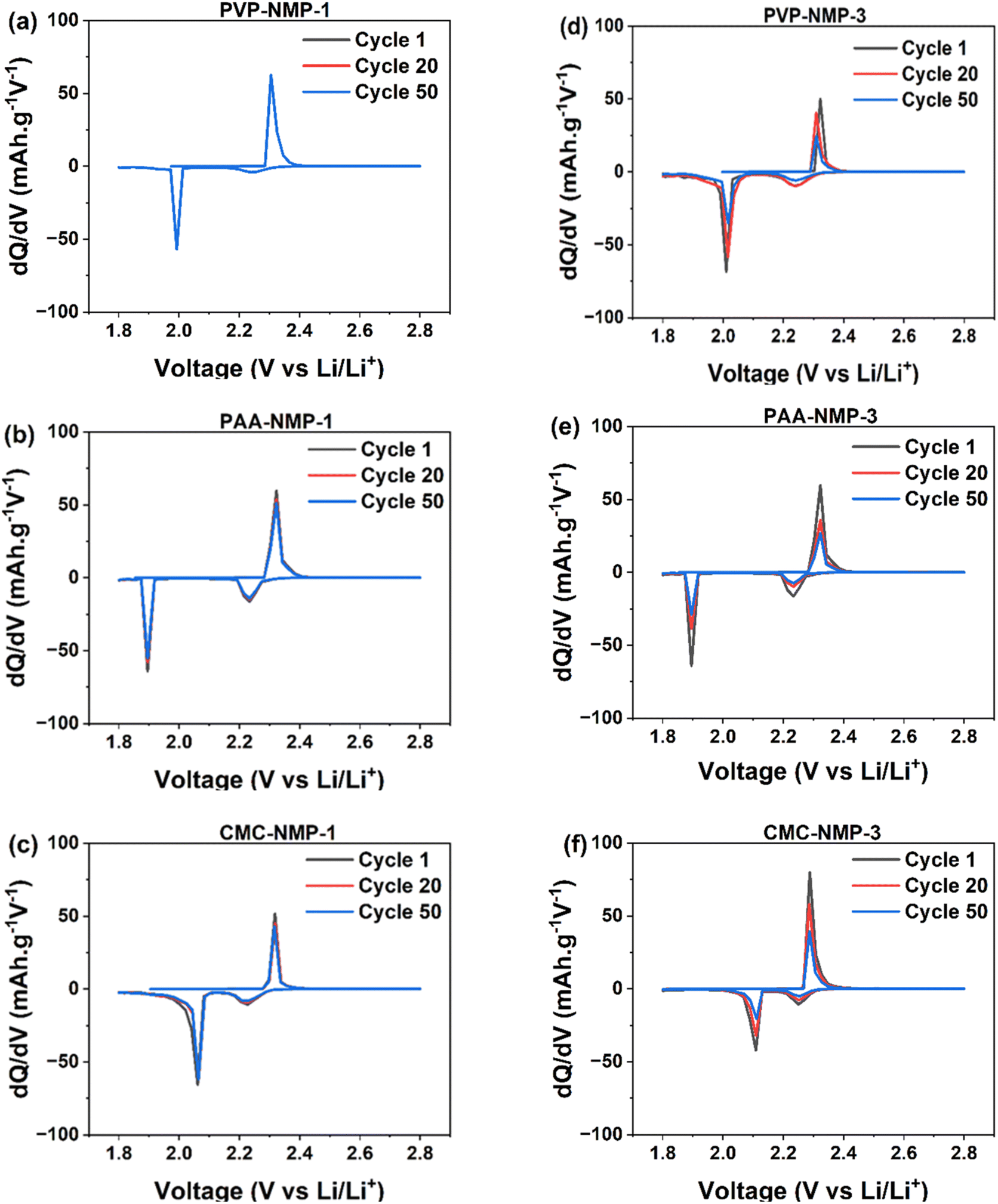 | ||
| Fig. 4 Differential capacity profiles of the cathodes in (a–c) Group 1 and (d–f) Group 3 electrodes. Group 1 electrodes show minimal to no increase in peak area suggesting their controlled LiPSs loss due to their binder shell morphology, compared to the un-shelled Group 3 electrodes. | ||
The decrease in the dQ/dV peak area and peak intensity is attributed to polysulfide loss.56,57 Accordingly, the comparison of dQ/dV curves revealed notable differences among electrodes with varying binder morphologies. In Group 1 cells with binder shell morphology, no shift in peak positions is observed (Fig. 4a–c). A slight decrease in peak areas from the 1st to the 50th cycle, only observed in PAA-NMP-1 and CMC-NMP-1 cells, indicates minimal material loss. In contrast, Group 3 cells (Fig. 4d–f) exhibit a significant reduction in peak areas upon cycling, irrespective of the binder material, suggesting greater sulfur loss. Although a slight shift towards higher potentials in the discharge curve and lower potentials in the charge curve suggests a decrease in charge/discharge overpotential, the substantial loss of coulombic charge, as indicated by the peak area reduction, suggests that Group 3 electrodes are expected to experience greater performance degradation. In essence, there were no substantial differences in the peak shift positions among the electrodes in Groups 1 and 3, indicating similar redox kinetics across the two binders. As summarized in Table S1,† the overpotential (ΔV) between the charge and discharge peaks varied by only about ±2 mV, suggesting minimal impact of binder processing on charge-transfer polarization. Furthermore, the Warburg coefficient values in Group 1 cells were comparable to those in Group 3, as shown in Table 3, indicating that ionic diffusion through the electrode structure remained largely unaffected by changes in the binder dissolution protocol. These observations imply that the capacity fade observed in Group 3 cells is primarily attributable to active material loss—evidenced by the decreasing peak area—rather than to limitations in ion transport or access to electrochemically active regions.
The suggestions by dQ/dV analysis of Group 1 and 3 electrodes were further evaluated by long duration galvanostatic charge–discharge cycling up to 400 cycles, the results of which are shown in Fig. 5. The cycling performance of Group 2 electrodes is displayed in Fig. S3.† A primary mechanism for capacity fading in LSBs is the dissolution of LiPSs into the electrolyte and their diffusion toward the anode, where they deposit onto the metallic Li.58,59 A significant variation in capacity retention was observed among cells within the same group and between Groups 1 and 3. The PVP-NMP-1 electrode offered the highest 400th cycle capacity retention, followed by PAA-NMP-1 and CMC-NMP-1 electrodes, with values of 92%, 81%, and 67%, respectively (Fig. 5a). Despite the thinner shells observed in the PAA-NMP-1 cell compared to the CMC-NMP-1 cell, the higher capacity retention in PAA-NMP-1 may be attributed to its more uniformly distributed shells across the electrode, as indicated by a lower standard deviation in shell thickness (0.15 for PAA vs. 0.32 for CMC; Fig. S1†). With the excellent capacity retention of the Group 1 cells, their long-term capacity retention up to 1000 cycles was also investigated (Fig. S2†), where the PVP-NMP-1, PAA-NMP-1 and CMC-NMP-1 cathodes showed 84%, 60% and 48% capacity retention, respectively. The PAA-NMP-1 electrode showed an undulation in capacity throughout the cycling test, where its capacity dropped initially until the 50th cycle, then increased and peaked after 160 cycles, then dropped back to lower capacities. Such unpredictability in overall capacity in addition to the oscillation in capacity values beyond 150 cycles is attributed to the inhomogeneous morphology of the binder shells surrounding its sulfur particles (Fig. 1b), resulting in varying temporal (and likely spatial) utilization of sulfur active material. The CMC-NMP-1 cell, similar to the PVP-NMP-1 exhibited a reasonably predictable capacity, especially after 100 cycles. The stability in achieved capacity in the PVP-NMP-1 and CMC-NMP-1 cells is in line with their uniform binder shell morphology.
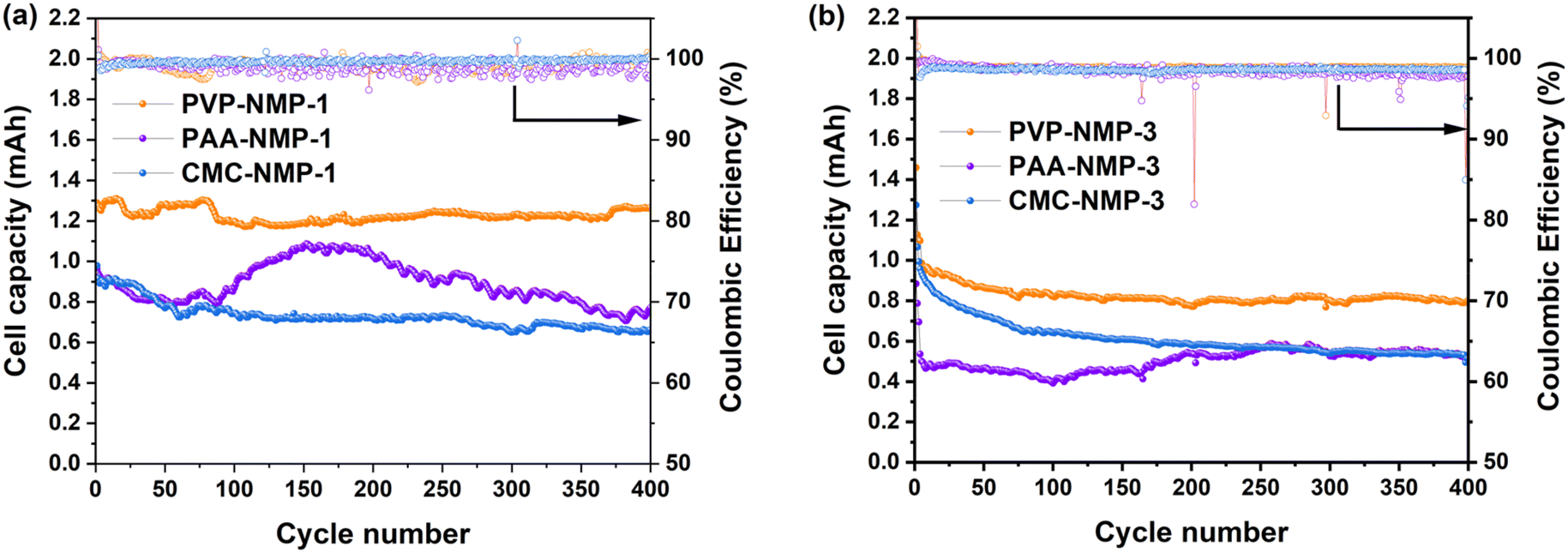 | ||
| Fig. 5 Galvanostatic discharge capacity of the cells cycled at C/10. (a) Group 1, and (b) Group 3. | ||
In Group 3 on the other hand, the galvanostatic cycling results showed that all three binders exhibited high initial capacities (Fig. 5b). The first discharge capacities were 1.45 mAh for PVP-NMP-3, 1.27 mAh for CMC-NMP-3, and 0.88 mAh for PAA-NMP-3. These values exceeded those observed in Group 1, which is attributed to the absence of the less conductive binder shell in Group 3 electrodes (Fig. 1g–i). Without the binder shells, sulfur is more readily accessible, whereas the presence of a polymer binder shell in Group 1 may insulate the sulfur surface, reducing its initial utilization. Despite their high initial values, Group 3 capacities continuously faded as the cycling proceeded, with the most drastic decrease in the first few cycles, resulting in significantly lower capacity retentions for Group 3 electrodes compared to their Group 1 counterparts. The most extreme change happened with PVP, where the 400th cycle capacity retention of the PVP-based cell dropped from 92% to 55% by losing the binder shells around their sulfur particles. Similarly, the capacity retention of the PAA cells dropped from 81% to 56% and the CMC cells dropped from 67% to 40%, all confirming that the binder shell morphology played a significant role in impeding the polysulfide dissolution and capacity loss. The inferior capacity retention of the Group 3 cells is also reflected in their lower coulombic efficiency (CE). As shown in Fig. 5 and Table 4, Group 1 cells maintained a CE exceeding 99%, whereas Group 3 cells exhibited a lower CE of approximately 98%, which continued to decline with further cycling. In both Groups 1 and 3, PVP exhibited the highest initial capacity, while PAA and CMC showed lower and similar values, attributed to their comparable adhesive properties, which are inferior to those of PVP. The higher capacity achieved in PVP-based electrode leads to increased LiPSs formation, which could accelerate capacity decay.
| Electrodes | Group 1 electrodes | Group 3 electrodes | ||||||
|---|---|---|---|---|---|---|---|---|
| Initial capacity | 400th cycle | Retention (%) | CE (%) | Initial capacity | 400th cycle | Retention (%) | CE (%) | |
| PVP-NMP | 1.29 | 1.18 | 92 | 99.99 | 1.43 | 0.79 | 55 | 98.01 |
| PAA-NMP | 0.95 | 0.77 | 81 | 98.64 | 0.87 | 0.49 | 56 | 98.56 |
| CMC-NMP | 0.98 | 0.66 | 67 | 99.75 | 1.27 | 0.51 | 40 | 97.82 |
However, this was not observed in Group 1, highlighting the importance and effectiveness of the combined influence of binder shell morphology and good adhesion of PVP on the longevity of LSB cathodes.
The loss of active material in LSBs can be further evaluated from the well-known plateaus in the electrochemical discharge profile of LSB cathode. The conversion of soluble long-chain polysulfides to insoluble short-chain polysulfides is a slow process contributing to the accumulation of polysulfides, which can inadvertently lead to polysulfide shuttling.60 The ratio of the upper plateau discharge capacity (QH) to the lower plateau discharge capacity (QL) directly quantifies the tendency of the dissolved LiPSs to transform to final discharge products.31,61 Fig. S4 and Table S2† investigate the QH/QL ratio at different cycles to study the utilization and loss of sulfur in electrodes with different binder morphologies, particularly the binder shell morphology that controls the loss of intermediate polysulfides and improves reversibility and capacity retention.
3.3. Effect of binder morphology on polysulfide dissolution
While the electrochemical measurements discussed so far provided useful insights into the loss of polysulfide and capacity fading within the cells, they did not provide direct evidence of the polysulfides shuttling impediment by the binder shells. Raman spectroscopy offers a valuable approach for detecting polysulfides within LSB cells; however, ex situ Raman, which is the more common technique, lacks the capability to capture the time-dependent phenomenon of the LiPSs dissolution and shuttling, limiting its utility in studying such a dynamic process. Therefore, in situ Raman spectroscopy was employed to provide insights into the polysulfide's behavior in electrodes with different morphologies under operational conditions. To validate/invalidate the polysulfide trapping capability of the binder shells around sulfur particles, in situ Raman spectroscopy was carried out on Group 1 and Group 3 electrodes with and without binder shells surrounding their sulfur particles. To assure a reliable detection of polysulfides, sulfur cathodes with higher loadings of 3.05 ± 0.1 mg cm−2, and higher electrolyte/S ratio of 30 μL were used in the in situ cell to promote LiPSs formation and dissolution. The measurement was carried out as described in Section 2.5 with the configuration of the in situ cell components shown in Fig. S5a.† The representative galvanostatic discharge profile of the cells showing the potentials at which the Raman experiment was carried out and the polysulfide species are displayed in Fig. 6a. The representative Raman spectrum of the electrodes before cycling is shown in Fig. 6b, while the Raman spectra of all Group 1 and 3 electrodes before cycling are presented with intensified peaks in Fig. S5b.† The shape and location of the peaks in Fig. 6b are in agreement with the Raman-active peaks of octatomic sulfur (S8). The vibrational modes at ∼152 cm−1 and ∼219 cm−1 represent asymmetric S–S bending and symmetric bending, respectively, while symmetric S–S stretching is represented by the peak at ∼473 cm−1.62 The peak identifications confirm the orthorhombic form of sulfur before cycling63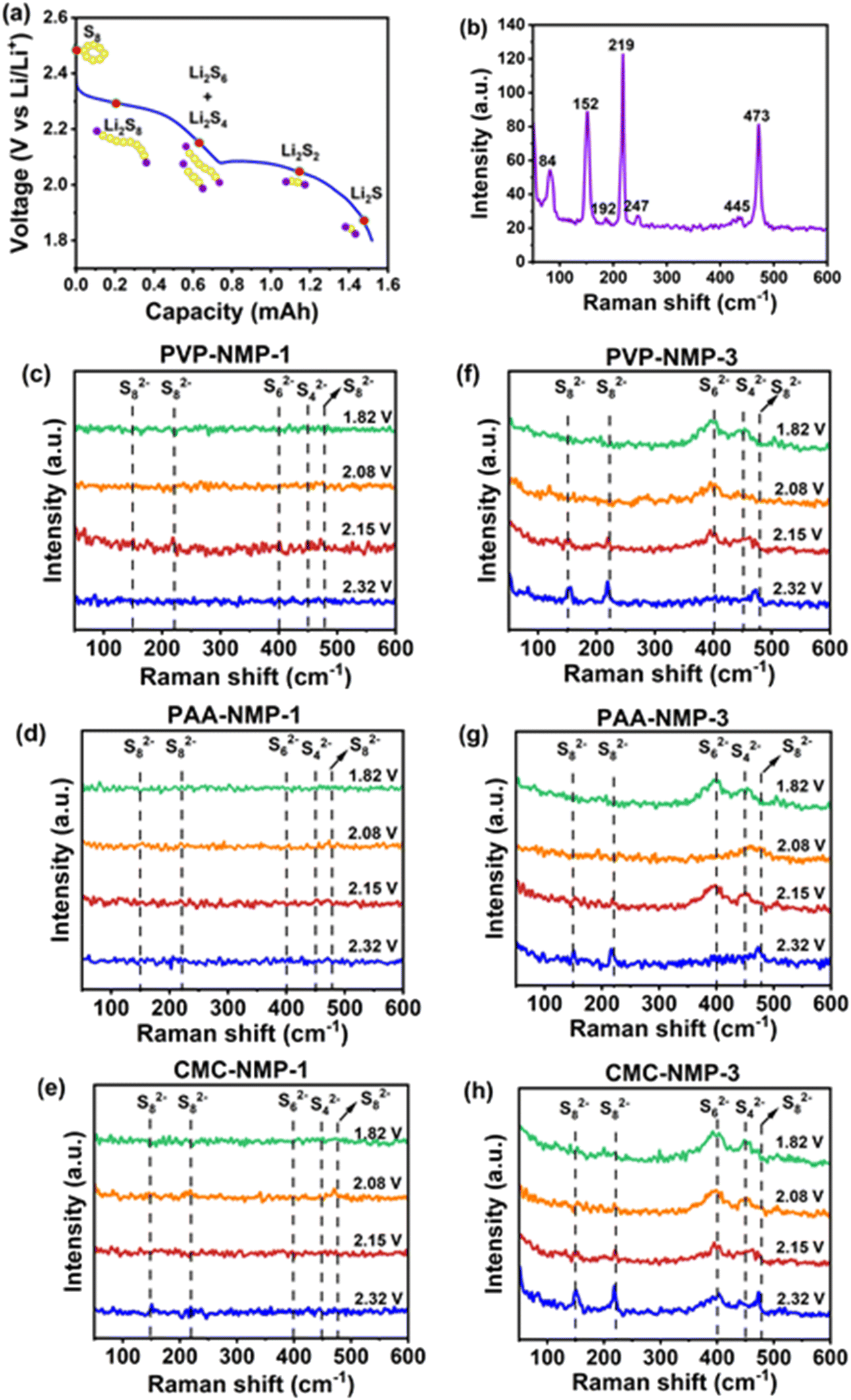 | ||
| Fig. 6 (a) Representative galvanostatic discharge profile of the cells during in situ Raman measurement with key voltages marked by red circles, (b) representative Raman spectrum of the electrodes before cycling, (c–e) Raman spectra of Group 1 cells and (f–h) Raman spectra of Group 3 cells, at marked discharge potentials, showing notably larger presence of solvated long chain LiPSs on un-shelled Group 3 electrodes. | ||
Fig. 6c–h shows the evolution of Raman signals during the first discharge of cathodes with and without binder shells. In Group 3 electrodes with no binder shells (Fig. 6f–h), a similar evolution of polysulfides to what is commonly reported was observed. From the onset of the discharge test, the S8 peaks were depressed, and sharp peaks at 154 cm−1, 222 cm−1 and 474 cm−1 wavenumbers appeared at 2.32 V, suggesting the presence of S82−, which indicate the formation of Li2S8.27,30,60,64,65 This suggests that sulfur S8 rings opened and took part in the reduction reaction to form LiPSs. Dissolution of S8 is also possible, however, as argued by Blanchard, the rapid disappearance of the S8 peak suggests that the electrochemical transformation of sulfur is more plausible than chemical dissolution.63 As the discharge continued, at 2.15 V, corresponding to the onset of the lower voltage plateau, the signals of S82− weakened and finally disappeared at 2.08 V. Simultaneously, two peaks appeared at 401–403 cm−1 and 456–457 cm−1, which are ascribed to S62−, or S42−, or a mixture of both, indicating the formation of Li2S6 and Li2S4.27,30 Raman peak positions are compared and tabulated against literature assignments in Table S3.† The short chain polysulfides (Li2S) peak expected at 375 cm−1 (ref. 66) was not observed due to its knowingly low intensity.67 Yet, the notable presence (intense peaks) of the long chain polysulfides sufficed in suggesting the abundance of polysulfides at the surface of the Group 3 electrodes or the electrolyte near the electrode surface. By conducting a similar measurement on Group 1 electrodes with binder shell around sulfur particles, all peaks associated with polysulfides were significantly suppressed (Fig. 6c–e).
The obscure peaks in Group 1 electrodes correspond to the small amount of polysulfides present in the electrode surface or dissolved in the electrolyte, suggesting their effective containment within the electrodes. This result is also in agreement with the optical images of the electrodes (Fig. S6†) where there is a significant change in the appearance of the Group 3 electrodes as the discharge continued, while less visible changes in Group 1 electrodes were observed.
3.4. Effect of binder morphology on self-discharge
Self-discharge measurement has been previously used to investigate the polysulfide shuttling in LSBs.68,69 Mikhaylik and Akridge have shown that the capacity at the high potential plateau is the appropriate measure of determining self-discharge LSB.70 Accordingly, the self-discharge coefficient, ks, can be calculated using (1): | (1) |
 | ||
| Fig. 7 The change in the capacity of the high potential plateau vs. idle time for the cells with (a) binder shell morphology and (b) un-shelled morphology, exhibiting significantly higher self-discharge rates in Group 3 electrodes and confirming the effectiveness of binder shell morphology in controlling the polysulfide dissolution and loss. | ||
With PVP offering the best discharge capacity, further post-cycling structural and chemical studies were conducted on PVP-based cathodes with binder shells (PVP-NMP-1) and without binder shells (PVP-NMP-3). The SEM characterization was conducted on electrodes in pristine and cycled (fully discharged state) and rinsed, shown in Fig. 8. Fig. 8a and c show the surface of the pre-cycled electrodes, and Fig. 8b and d show the electrodes cycled 20 times and rinsed with DME after disassembly. DME is a good solvent for LiPSs, so treating the electrode with DME is expected to leave the remnant of the host structure, including the binder shells, but remove the sulfur discharge products. The arrows in Fig. 8b point out the remaining binder shells that initially housed the sulfur particles and contained the LiPSs.
 | ||
| Fig. 8 Post cycling SEM images of PVP-based electrodes (a and c) before cycling, and (b and d) after 20 cycles and rinsing with DME. Top row images: Electrodes with binder shell morphology (PVP-NMP-1). Bottom row images: Electrodes without binder shells (PVP-NMP-3). The binder shells in PVP-NMP-1 electrode remain intact during cycling confirming their stability under operation. | ||
Such remnants of binder shells after DME rinsing are absent in PVP-NMP-3. The retention of these self-structured features after cycling, as shown in Fig. 8b, confirms their stability despite the drastic volume variations of sulfur particles during charge/discharge cycles.74 This suggests that these binder structures not only help mitigate LiPS shuttling but also contribute to mechanical stability by creating a flexible space to accommodate the expanded sulfur discharge products. It is worth noting that polysulfides do not necessarily return back to their original location within the binder shells. Nevertheless, because the binder shells are distributed throughout the electrode (Fig. 8), they are still expected to serve as localized polysulfide adsorption domains along diffusion pathways within the electrode matrix. This spatial distribution reinforces their continued role in mitigating polysulfide shuttling during extended cycling.
Building on these insights, a key question emerged: would controlling the dissolution process yield similar effects when using water as the solvent? To explore this, a parallel study was conducted by replacing NMP with water, with results presented in the ESI (Fig. S8–S10†). It was observed that controlled binder dissolution in water produced similar binder shell morphologies (Fig. S8d–f†) and led to improved capacity retention (Fig. S10†). In contrast, prolonged stirring disrupted the binder shell structures (Fig. S8a–c†), resulting in inferior capacity retention (Fig. S9†).
3.5. Industrial relevance
The battery industry requires a sulfur loading of >4 mg cm−2 and achieved capacity of >800 mAh g(sulfur)−1 for practical and commercially viable sulfur cathodes. Among all electrodes studied in this work, the PVP-NMP-1 electrode showed the best capacity retention. The obtained capacity was, however, only a third of the achievable capacity of sulfur, and at low loadings, which questions the commercial viability. One major factor responsible for the poor performance is the choice of Vulcan as the conductive carbon. Vulcan was particularly selected for the binder morphology study because of the advantages it offers in microscopy imaging due to its low microporosity, which minimizes interference with solvent and binder dissolution process. This characteristic enabled clear observation of the isolated effect of binder structure with its fine morphology details – in this case the binder shells around sulfur particles. However, Vulcan has relatively low electronic conductivity and surface area. As a result, only a fraction of the sulfur is reacted, causing low discharge capacity.75 Given that the capacity constraint associated with lower conductivity becomes more pronounced at higher sulfur loadings, an alternative carbon is needed for high loading electrodes to boost the achievable capacity. Several high surface areas carbons have shown to increase the electrochemical performance of various LIB systems,76–81 among which Ketjen Black stands out due to its five times larger surface area than Vulcan, high electronic conductivity, low cost and easy synthesis.82–84 Both low and high loading Ketjen based electrodes were made using the same approach as the PVP-NMP-1 electrodes. It should be noted due to the high surface area of Ketjen black compared to Vulcan, the electrode slurry containing Ketjen black needed higher amounts of solvent. As a result, an optimized amount of solvent (twice the amount of NMP that was used for Vulcan-based slurry), was utilized in Ketjen black-based slurry, to bring the slurries to a similar viscosity. The sulfur loadings were 1.12 mgsulfur cm−2 and 5.28 mgsulfur cm−2 for low and high loading Ketjen black-based cathodes, named as PVP-KJ-NMP-L and PVP-KJ-NMP-H, respectively.Structural examination of the electrodes shows the presence of self-structured binder shells surrounding the sulfur particles at both loadings, as revealed in Fig. 9a and b, suggesting that choosing another carbon type does not lead to a significant difference in the binder shell morphology. This means that the slurry making process explored in this work is extendable to electrodes with other carbon materials and higher loadings. The electrochemical performance of these cells was also studied, the results of which are shown in Fig. 9c. The cell with 1.12 mgsulfur cm−2 showed a capacity greater than 800 mAh g−1 (1.58 mAh) and was stable until 250 cycles – an excellent performance for LSBs using simple carbon black and common PVP binder. Compared with the PVP-NMP-1 whose average capacity was ∼500 mAh g−1 (1.3 mAh), replacing Vulcan with Ketjen black as the conductive carbon brought a significant capacity improvement due to the better properties of Ketjen black as mentioned above, resulting in increased sulfur utilization. The cycling stability of the PVP-KJ-NMP electrodes was also investigated through their QH/QL ratio and were found to be constant across 50th and 100th cycles, as shown in Table S4.† The QH/QL for PVP-NMP-1, using Vulcan black as carbon, was unchanged at 1.01 from the 50th to 100th cycles (Fig. S4†). For a similar cycle range in PVP-KJ-NMP-L using Ketjen black, a fairly constant QH/QL is observed as well (slight change from 0.51 to 0.49, as presented in Table S4†). Considering the theoretical capacity of sulfur at each plateau, for a 100% sulfur utilization a QH/QL ∼1/3 is expected. The enhanced QH/QL of PVP-NMP-KJ-L electrode over the PVP-NMP-1 is due to the enhanced redox kinetic offered by higher surface area of Ketjen black carbon.85,86
 | ||
| Fig. 9 SEM images of the Ketjen-based electrodes with PVP as the binder. (a) Low sulfur loading (PVP-KJ-NMP-L) (b) High sulfur loading (PVP-KJ-NMP-H). The binder shells are pointed by arrows and shown in inset images. (c) Capacity retention plot of the LSB cells at C/10, showing applicability of the learnings from this work in cells with different carbon materials as well as more commercially viable sulfur loadings. | ||
To study the effect of areal sulfur loading – a requirement for higher specific energy in LSBs, a similar analysis was conducted on PVP-KJ-NMP-H with the areal sulfur loading of 5.28 mgsulfur cm−2. Initial instabilities were observed in their cycle performance, which is common in thick electrodes due to delayed electrolyte wetting, high tortuosity and longer transport length for the ions.87–90 The initial capacity of ∼524 mAh g−1 (4.86 mAh) was observed, which decreased to ∼494 mAh g−1 (4.59 mAh) at the 8th cycle before the capacity increased to around 715 mAh g−1 (6.64 mAh) at the 50th cycle. Positively, the capacity retention for this electrode was ca. 79% over 250 cycles. As shown in Table S4,† the QH/QL was also retained up to the 100th cycle at 0.5. These combined results suggest the relevance of the findings in this work for more practical applications requiring >4 mg cm−2 sulfur loading in the electrodes. Most importantly it suggests that the binder dissolution can be engineered across various systems with diverse binder and solvent chemistries, carbon types, and active material loadings (energy content), thereby promising facile adaptation of manufacturing processes for LSBs. The results obtained from this work are compared with other reports, commonly focused on complex chemical and/or functional modification of binders in Table S5,† suggesting that simple, scalable adjustments to the cathode fabrication process can yield competent electrochemical performance, offering a new route toward practical and cost-effective LSB design.
4. Conclusion
This work presents a systematic investigation into the effect of binder dissolution during slurry preparation and electrode processing on the structure and electrochemical performance of sulfur cathodes. The findings demonstrate that by controlling the slurry preparation process, sulfur particles can be encapsulated by self-structured binder shells, effectively mitigating LiPSs shuttling and enhancing the performance of LSB cathodes. The ratio of solvent molecules to solid components in the sulfur cathode slurry, along with the duration of slurry mixing, was found to govern binder dissolution, thereby influencing binder morphology and overall electrode structure. Electrodes prepared with 50 μL of NMP per mg of PVP binder, stirred for one day, produced well-defined binder shells around sulfur particles, leading to good adhesion as well as high sulfur utilization and minimal polysulfide loss. Through in situ Raman spectroscopy and self-discharge testing, the polysulfide-trapping capability of these binder shells was confirmed, preventing the loss of active material from the cathode. Additionally, the results demonstrate the versatility of this approach across various binder/solvent chemistries. While NMP yielded the best electrochemical performance, water also successfully generated the binder shell morphology around sulfur particles, indicating that binder dissolution engineering can be applied to both organic and aqueous battery slurries. Given the differing dissolution strengths of water and organic solvents for various binders, customized binder dissolution parameters are recommended, along with further studies on water-binder interactions based on the methods established here. The effectiveness of the achieved binder morphology through controlled binder dissolution process was proven in sulfur cathodes with high loading confirming the industrial relevance of the findings in this work. The insights into the effect of binder dissolution regimes on electrode morphology and electrochemical performance offer a pathway to developing more robust cathode slurry processing methodologies for LSBs and potentially other battery systems.Conflicts of interest
Two of the authors (Fabio Albano, and John Chmiola) have been employed by NantG Power LLC.Data availability
The data that support the findings of this study are not publicly available due to commercial restrictions and to mitigate risks to NantG Power LLC's product development. Access to the data may be considered on a case-by-case basis upon reasonable request and subject to confidentiality agreements.Acknowledgements
This work was financially supported by NantG Power LLC [account #USCIP10012012].References
- N. Nitta, F. Wu, J. T. Lee and G. Yushin, Mater. Today, 2015, 18, 252–264 CrossRef CAS.
- G. E. Blomgren, J. Electrochem. Soc., 2017, 164, A5019–A5025 CrossRef CAS.
- B. Zhu, X. Wang, P. Yao, J. Li and J. Zhu, Chem. Sci., 2019, 10, 7132–7148 RSC.
- A. Manthiram, Y. Fu, S.-H. Chung, C. Zu and Y.-S. Su, Chem. Rev., 2014, 114, 11751–11787 CrossRef CAS PubMed.
- Y.-X. Yin, S. Xin, Y.-G. Guo and L.-J. Wan, Angew. Chem., Int. Ed., 2013, 52, 13186–13200 CrossRef CAS PubMed.
- A. Manthiram, Y. Fu and Y.-S. Su, Acc. Chem. Res., 2013, 46, 1125–1134 CrossRef CAS PubMed.
- D. Liu, C. Zhang, G. Zhou, W. Lv, G. Ling, L. Zhi and Q.-H. Yang, Adv. Sci., 2018, 5, 1700270 CrossRef PubMed.
- H. Shi, W. Lv, C. Zhang, D.-W. Wang, G. Ling, Y. He, F. Kang and Q.-H. Yang, Adv. Funct. Mater., 2018, 28, 1800508 CrossRef.
- X. Liu, J.-Q. Huang, Q. Zhang and L. Mai, Adv. Mater., 2017, 29, 1601759 CrossRef PubMed.
- Q. Sun, B. Xi, J.-Y. Li, H. Mao, X. Ma, J. Liang, J. Feng and S. Xiong, Adv. Energy Mater., 2018, 8, 1800595 CrossRef.
- V. Marangon, E. Scaduti, V. F. Vinci and J. Hassoun, ChemElectroChem, 2022, 9, e202200374 CrossRef CAS.
- X. Liang, C. Y. Kwok, F. Lodi-Marzano, Q. Pang, M. Cuisinier, H. Huang, C. J. Hart, D. Houtarde, K. Kaup, H. Sommer, T. Brezesinski, J. Janek and L. F. Nazar, Adv. Energy Mater., 2016, 6, 1501636 CrossRef.
- N. H. H. Phuc, M. Takaki, H. Muto, M. Reiko, H. Kazuhiro and A. Matsuda, ACS Appl. Energy Mater., 2020, 3, 1569–1573 CrossRef CAS.
- M. S. Kim, M.-S. Kim, V. Do, Y. R. Lim, I. W. Nah, L. A. Archer and W. I. Cho, Nano Energy, 2017, 41, 573–582 CrossRef CAS.
- A. Sakuda, Y. Sato, A. Hayashi and M. Tatsumisago, Energy Technol., 2019, 7, 1900077 CrossRef CAS.
- A. Raulo and G. Jalilvand, Nano Energy, 2024, 122, 109265 CrossRef CAS.
- S. A. Lateef, M. Manjum, W. E. Mustain and G. Jalilvand, ECS Meet. Abstr., 2022, MA2022-02, 628 CrossRef.
- M. Manjum, S. A. Lateef, H. A. McRay, W. E. Mustain and G. Jalilvand, ECS Meet. Abstr., 2022, MA2022-02, 588 CrossRef.
- S. A. Lateef, M. Manjum, J. Chmiola, F. Albano, W. E. Mustain and G. Jalilvand, ECS Meet. Abstr., 2023, MA2023-01, 614 CrossRef.
- X. Zhou, X. Ma, C. Ding, W. Meng, S. Xu, L. Chen, D. Duan and S. Liu, Mater. Lett., 2019, 251, 180–183 CrossRef CAS.
- P. Li, J. Deng, J. Li, M. Zeng, L. Wang and J. Guo, J. Mater. Sci., 2020, 55, 3964–3973 CrossRef CAS.
- W. Zhou, Y. Yu, H. Chen, F. J. DiSalvo and H. D. Abruña, J. Am. Chem. Soc., 2013, 135, 16736–16743 CrossRef CAS PubMed.
- H. Su, C. Fu, Y. Zhao, D. Long, L. Ling, B. M. Wong, J. Lu and J. Guo, ACS Energy Lett., 2017, 2, 2591–2597 CrossRef CAS.
- J. Liao and Z. Ye, Electrochim. Acta, 2018, 259, 626–636 CrossRef CAS.
- J. Liao, Z. Liu, X. Liu and Z. Ye, J. Phys. Chem. C, 2018, 122, 25917–25929 CrossRef CAS.
- M. Ling, L. Zhang, T. Zheng, J. Feng, J. Guo, L. Mai and G. Liu, Nano Energy, 2017, 38, 82–90 CrossRef CAS.
- C. Wang, P. Chen, Y. Wang, T. Chen, M. Liu, M. Zhang, Y. Fu, J. Xu and J. Fu, Adv. Funct. Mater., 2022, 32, 2204451 CrossRef CAS.
- Z. Huang, L. Wang, Y. Xu, L. Fang, H. Li, B. Zhu and Y. Song, Chem. Eng. J., 2022, 443, 136347 CrossRef CAS.
- Y. Jiao, W. Chen, T. Lei, L. Dai, B. Chen, C. Wu and J. Xiong, Nanoscale Res. Lett., 2017, 12, 195 CrossRef PubMed.
- W. Chen, T. Lei, T. Qian, W. Lv, W. He, C. Wu, X. Liu, J. Liu, B. Chen, C. Yan and J. Xiong, Adv. Energy Mater., 2018, 8, 1702889 CrossRef.
- S. Tu, X. Chen, X. Zhao, M. Cheng, P. Xiong, Y. He, Q. Zhang and Y. Xu, Adv. Mater., 2018, 30, 1804581 CrossRef.
- H. Wang, Y. Wang, P. Zheng, Y. Yang, Y. Chen, Y. Cao, Y. Deng and C. Wang, ACS Sustainable Chem. Eng., 2020, 8, 12799–12808 CrossRef CAS.
- G. Zhou, K. Liu, Y. Fan, M. Yuan, B. Liu, W. Liu, F. Shi, Y. Liu, W. Chen, J. Lopez, D. Zhuo, J. Zhao, Y. Tsao, X. Huang, Q. Zhang and Y. Cui, ACS Cent. Sci., 2018, 4, 260–267 CrossRef CAS PubMed.
- H. Schneider, A. Garsuch, A. Panchenko, O. Gronwald, N. Janssen and P. Novák, J. Power Sources, 2012, 205, 420–425 CrossRef CAS.
- S. A. Lateef, M. Manjum, H. A. McRay, W. E. Mustain and G. Jalilvand, ACS Appl. Energy Mater., 2023, 6, 9307–9317 CrossRef CAS.
- J. Tan, M. Ye and J. Shen, Mater. Horiz., 2022, 9, 2325–2334 RSC.
- J. E. Marshall, A. Zhenova, S. Roberts, T. Petchey, P. Zhu, C. E. J. Dancer, C. R. McElroy, E. Kendrick and V. Goodship, Polymers, 2021, 13, 1354 CrossRef CAS PubMed.
- M. A. Morsi, A. Rajeh and A. A. Menazea, J. Mater. Sci.: Mater. Electron., 2019, 30, 2693–2705 CrossRef CAS.
- I. A. Safo, M. Werheid, C. Dosche and M. Oezaslan, Nanoscale Adv., 2019, 1, 3095–3106 RSC.
- C.-W. Liew, H. M. Ng, A. Numan and S. Ramesh, Polymers, 2016, 8, 179 CrossRef PubMed.
- B. Kumar, R. Priyadarshi, Sauraj, F. Deeba, A. Kulshreshtha, K. K. Gaikwad, J. Kim, A. Kumar and Y. S. Negi, Gels, 2020, 6, 49 CrossRef CAS PubMed.
- F. Dai, Q. Zhuang, G. Huang, H. Deng and X. Zhang, ACS Omega, 2023, 8, 17064–17076 CrossRef CAS PubMed.
- Y. Borodko, S. E. Habas, M. Koebel, P. Yang, H. Frei and G. A. Somorjai, J. Phys. Chem. B, 2006, 110, 23052–23059 CrossRef CAS PubMed.
- H. S. Wahyuni, S. Yuliasmi, H. S. Aisyah and D. Riati, Open Access Maced. J. Med. Sci., 2019, 7, 3878–3881 CrossRef PubMed.
- M. Wang, D. Dang, A. Meyer, R. Arsenault and Y.-T. Cheng, J. Electrochem. Soc., 2020, 167, 100518 CrossRef CAS.
- S. Waluś, C. Barchasz, R. Bouchet and F. Alloin, Electrochim. Acta, 2020, 359, 136944 CrossRef.
- S. A. Lateef, W. E. Mustain and G. Jalilvand, ECS Meet. Abstr., 2022, MA2022–02, 154 CrossRef.
- R. Zhu, S. Liu, T. Li and D. Yang, ChemElectroChem, 2024, 11, e202400326 CrossRef CAS.
- B.-R. Lee and E.-S. Oh, J. Phys. Chem. C, 2013, 117, 4404–4409 CrossRef CAS.
- M. Ravi, K. Kiran Kumar, V. Madhu Mohan and V. V. R. Narasimha Rao, Polym. Test., 2014, 33, 152–160 CrossRef CAS.
- C. Hwang, S. Joo, N.-R. Kang, U. Lee, T.-H. Kim, Y. Jeon, J. Kim, Y.-J. Kim, J.-Y. Kim, S.-K. Kwak and H.-K. Song, Sci. Rep., 2015, 5, 14433 CrossRef CAS PubMed.
- Y. Hwa and E. J. Cairns, Electrochim. Acta, 2018, 271, 103–109 CrossRef CAS.
- Z. W. Seh, Q. Zhang, W. Li, G. Zheng, H. Yao and Y. Cui, Chem. Sci., 2013, 4, 3673 RSC.
- Y. Chu, X. Cui, W. Kong, K. Du, L. Zhen and L. Wang, Chem. Eng. J., 2022, 427, 130844 CrossRef CAS.
- E. Peled, M. Goor, I. Schektman, T. Mukra, Y. Shoval and D. Golodnitsky, J. Electrochem. Soc., 2017, 164, A5001–A5007 CrossRef CAS.
- J. Chen, M. N. Marlow, Q. Jiang and B. Wu, J. Energy Storage, 2022, 45, 103669 CrossRef.
- L. Spitthoff, P. J. S. Vie, M. S. Wahl, J. Wind and O. S. Burheim, J. Electroanal. Chem., 2023, 944, 117627 CrossRef CAS.
- H. Yi, T. Lan, Y. Yang, H. Zeng, T. Zhang, T. Tang, C. Wang and Y. Deng, Energy Storage Mater., 2019, 21, 61–68 CrossRef.
- J. Yan, X. Liu and B. Li, Adv. Sci., 2016, 3, 1600101 CrossRef PubMed.
- L. Peng, Z. Wei, C. Wan, J. Li, Z. Chen, D. Zhu, D. Baumann, H. Liu, C. S. Allen, X. Xu, A. I. Kirkland, I. Shakir, Z. Almutairi, S. Tolbert, B. Dunn, Y. Huang, P. Sautet and X. Duan, Nat. Catal., 2020, 3, 762–770 CrossRef CAS.
- Z.-Z. Pan, W. Lv, Y.-B. He, Y. Zhao, G. Zhou, L. Dong, S. Niu, C. Zhang, R. Lyu, C. Wang, H. Shi, W. Zhang, F. Kang, H. Nishihara and Q.-H. Yang, Adv. Sci., 2018, 5, 1800384 CrossRef PubMed.
- C. Nims, B. Cron, M. Wetherington, J. Macalady and J. Cosmidis, Sci. Rep., 2019, 9, 7971 CrossRef PubMed.
- D. Blanchard and M. Slagter, J. Phys. Energy, 2021, 3, 044003 CrossRef CAS.
- J. Xia, W. Hua, L. Wang, Y. Sun, C. Geng, C. Zhang, W. Wang, Y. Wan and Q.-H. Yang, Adv. Funct. Mater., 2021, 31, 2101980 CrossRef CAS.
- T. Lei, W. Chen, W. Lv, J. Huang, J. Zhu, J. Chu, C. Yan, C. Wu, Y. Yan, W. He, J. Xiong, Y. Li, C. Yan, J. B. Goodenough and X. Duan, Joule, 2018, 2, 2091–2104 CrossRef CAS.
- O. El Jaroudi, E. Picquenard, N. Gobeltz, A. Demortier and J. Corset, Inorg. Chem., 1999, 38, 2917–2923 CrossRef CAS PubMed.
- H.-L. Wu, L. A. Huff and A. A. Gewirth, ACS Appl. Mater. Interfaces, 2015, 7, 1709–1719 CrossRef CAS PubMed.
- V. Knap, D.-I. Stroe, M. Swierczynski, R. Teodorescu and E. Schaltz, J. Electrochem. Soc., 2016, 163, A911 CrossRef CAS.
- S.-H. Chung and A. Manthiram, ACS Energy Lett., 2017, 2, 1056–1061 CrossRef CAS.
- Y. V. Mikhaylik and J. R. Akridge, J. Electrochem. Soc., 2004, 151, A1969 CrossRef CAS.
- D. Moy, A. Manivannan and S. R. Narayanan, J. Electrochem. Soc., 2014, 162, A1 CrossRef.
- H. S. Ryu, H. J. Ahn, K. W. Kim, J. H. Ahn, K. K. Cho and T. H. Nam, Electrochim. Acta, 2006, 52, 1563–1566 CrossRef CAS.
- H. S. Ryu, H. J. Ahn, K. W. Kim, J. H. Ahn, J. Y. Lee and E. J. Cairns, J. Power Sources, 2005, 140, 365–369 CrossRef CAS.
- M. Shaibani, M. S. Mirshekarloo, R. Singh, C. D. Easton, M. C. D. Cooray, N. Eshraghi, T. Abendroth, S. Dörfler, H. Althues, S. Kaskel, A. F. Hollenkamp, M. R. Hill and M. Majumder, Sci. Adv., 2020, 6, eaay2757 CrossRef CAS PubMed.
- D. A. Dornbusch, R. Hilton, M. J. Gordon and G. J. Suppes, J. Ind. Eng. Chem., 2013, 19, 1968–1972 CrossRef CAS.
- Y. You, W. Zeng, Y.-X. Yin, J. Zhang, C.-P. Yang, Y. Zhu and Y.-G. Guo, J. Mater. Chem. A, 2015, 3, 4799–4802 RSC.
- J. Zheng, M. Gu, M. J. Wagner, K. A. Hays, X. Li, P. Zuo, C. Wang, J.-G. Zhang, J. Liu and J. Xiao, J. Electrochem. Soc., 2013, 160, A1624 CrossRef CAS.
- Y. Kee, F. Bardé and P. M. Vereecken, ChemSusChem, 2019, 12, 3967–3970 CrossRef CAS PubMed.
- L. Li, C. Nan, J. Lu, Q. Peng and Y. Li, Chem. Commun., 2012, 48, 6945–6947 RSC.
- F. Wang, P. K. Kahol, R. Gupta and X. Li, J. Electrochem. Energy Convers. Storage, 2019, 16, 041007 CrossRef CAS.
- S. A. Lateef, A. Raulo, J. Chmiola, W. E. Mustain, F. Albano and G. Jalilvand, J. Electrochem. Soc., 2024, 171, 120518 CrossRef CAS.
- Y. Zhang, H. Dong, H. Zhang, Y. Liu, M. Ji, Y. Xu, Q. Wang and L. Luo, Electrochim. Acta, 2016, 201, 179–186 CrossRef CAS.
- G. Hu, X. Sun, H. Liu, Y. Xu, L. Liao, D. Guo, X. Liu and A. Qin, Nanomaterials, 2022, 12, 692 CrossRef CAS PubMed.
- D. Zhao, X. Qian, L. Jin, X. Yang, S. Wang, X. Shen, S. Yao, D. Rao, Y. Zhou and X. Xi, RSC Adv., 2016, 6, 13680–13685 RSC.
- B. Wang, T. Li, X. Qian, L. Jin, S. Yao, X. Shen and S. Qin, J. Solid State Electrochem., 2021, 25, 1579–1590 CrossRef CAS.
- N. Jiang, G. Jiang, D. Niu, J. Mao, M. Chen, K. Li and Y. Li, J. Energy Chem., 2020, 51, 207–215 CrossRef.
- Z. Nie, S. Ong, D. S. Hussey, J. M. LaManna, D. L. Jacobson and G. M. Koenig, Mol. Syst. Des. Eng., 2020, 5, 245–256 RSC.
- J. Wu, Z. Ju, X. Zhang, X. Xu, K. J. Takeuchi, A. C. Marschilok, E. S. Takeuchi and G. Yu, ACS Nano, 2022, 16, 4805–4812 CrossRef CAS PubMed.
- Z. Nie, R. Parai, C. Cai, D. Ghosh and G. M. Koenig, Mol. Syst. Des. Eng., 2021, 6, 708–712 RSC.
- K.-Y. Park, J.-W. Park, W. M. Seong, K. Yoon, T.-H. Hwang, K.-H. Ko, J.-H. Han, Y. Jaedong and K. Kang, J. Power Sources, 2020, 468, 228369 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Image analysis of the binder shell thickness surrounding sulfur particles; polarization of Groups 1 and 3 cells at cycles 1, 20 and 50; long-term capacity retention of PVP-, PAA- and CMC with NMP and water as solvents; ratios of QH/QL for Groups 1–5 cathodes as well as the low and high sulfur loading PVP-KJ-NMP at 50th and 100th cycles; schematic arrangement of the cell components in the in situ Raman test set up and the Raman spectra of Group 1 vs. Group 3 cells before cycling; optical images of top surface in Group 1 and Group 3 electrodes during in situ Raman measurement; and discharge curves of Group 1 and Group 3 cathodes showing the self-discharge behaviors. See DOI: https://doi.org/10.1039/d5eb00062a |
| This journal is © The Royal Society of Chemistry 2025 |