
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStable cycling of high-mass loaded MnO2 electrodes for sodium-ion batteries†
Yunkai
Luo
 a,
Bintao
Hu
a,
Swetha
Chandrasekaran
b,
Megan C.
Freyman
b,
Dun
Lin
c,
Yat
Li
c,
Marcus
Worsley
b and
Bruce
Dunn
*a
a,
Bintao
Hu
a,
Swetha
Chandrasekaran
b,
Megan C.
Freyman
b,
Dun
Lin
c,
Yat
Li
c,
Marcus
Worsley
b and
Bruce
Dunn
*a
aDepartment of Materials Science and Engineering, University of California, Los Angeles, California 90095, USA. E-mail: bdunn@ucla.edu
bLawrence Livermore National Laboratory, California, 94550, USA
cDepartment of Chemistry and Biochemistry, University of California, Santa Cruz, California 95064, USA
First published on 4th June 2025
Abstract
Achieving cost-effective, sustainable solutions for large-scale energy storage are critical for advancing the global clean energy transition. In view of the challenges posed by limited lithium reserves, low-cost sodium-ion batteries (SIBs) have emerged as a promising direction, especially for grid-level energy storage. Among the various battery electrode materials, manganese dioxide (MnO2) stands out as a favorable choice for such large-scale applications due to its earth abundance, cost-effectiveness, and non-toxic nature. Although MnO2 is known as a pseudocapacitive material with superior cycling stability in aqueous electrolytes, its dissolution in non-aqueous electrolytes has restricted its use in long-lifetime batteries. In this study, we address two issues which have limited the use of MnO2 electrodes in non-aqueous electrolytes. First, using electrochemical quartz crystal microbalance measurements in combination with other electrochemical methods, we demonstrate that diglyme (bis(2-methoxyethyl) ether) electrolyte can achieve stable cycling of electrodeposited ε-MnO2. These results enable us to tackle a second objective, that is increasing the mass loading of the MnO2 electrode, since achieving high areal energy density is a significant factor in reducing manufacturing costs. Using 3D printed graphene aerogel (GA) as a scaffold, our studies show that the electrodeposited MnO2/GA electrodes possess scalable properties with mass loadings from 20 to 80 mg cm−2. The resulting electrodes exhibit areal energy densities as high as 4.4 mA h cm−2 at a current density of 10 mA cm−2. The high mass loaded MnO2 electrodes were incorporated as a cathode in a SIB which used TiO2 as the anode. The SIB device exhibited excellent performance with power densities in excess of 70 mW cm−2. These studies highlight the promise of MnO2 electrodes for use in a low-cost technology for large-scale energy storage.
Broader contextAchieving cost-effective, sustainable solutions for large-scale energy storage is critical for advancing the global clean energy transition. MnO2 is an earth-abundant electrode material with superior cycling stability in aqueous electrolytes. However, its dissolution in non-aqueous electrolytes has restricted its use in long-lifetime batteries. In this manuscript, we address two important issues which have limited the use of MnO2 electrodes in energy storage devices involving non-aqueous electrolytes. First, using a variety of chemical and electrochemical methods, we demonstrate that diglyme (bis(2-methoxyethyl) ether) electrolyte suppresses dissolution and achieves stable cycling of electrodeposited ε-MnO2. These results enable us to tackle a second objective, that is, increasing the mass loading of the MnO2 electrode well beyond what is possible with traditional cast electrodes. High mass loading is critical in achieving the high areal energy density required for reducing manufacturing costs. Using 3D printed graphene aerogel (GA) as a scaffold, our studies show that the electrodeposited MnO2/GA electrodes possess scalable properties with mass loadings up to 80 mg cm−2. In addition to characterizing the properties of high mass loaded MnO2 electrodes, we demonstrate their effectiveness in a sodium ion battery and achieve power densities in excess of 70 mW cm−2. These studies highlight the promise of MnO2 electrodes for use in a low-cost technology for large-scale energy storage. |
1. Introduction
In order to fulfill the increasing demands for electricity and achieve netzero emissions over the next few decades, developing renewable power sources, such as solar and wind, require reliable large-scale grid-level energy storage capabilities.1 Due to the intermittent nature of such power sources, lithium-ion batteries (LIBs), which possess long cycle life along with high energy and power densities, are highly desirable for grid-level energy storage applications.2 However, the availability of lithium, the use of certain transition metals such as Ni and Co, and the increasing demands for LIBs from electric vehicles and portable electronics make the utilization of LIBs in grid-level energy storage functions economically less attractive.3–5 For this reason, exploring other low-cost rechargeable metal-ion battery alternatives is imperative.Sodium, because of its greater abundance than lithium, has a significantly lower cost and thus is much more suitable for large-scale energy storage applications.6 Na-based batteries are not unique as over a half century ago, the high-temperature Na–S battery invented by researchers at Ford was commercialized for grid-level energy storage.2,4 Although Na–S batteries met the low-cost criteria, the high operating temperature (above 300 °C) and the reliability of the sodium β-alumina ceramic present significant safety concerns.7 Moreover, the lower energy density of Na–S batteries is not ideal. Nonetheless, this prior experience shows that the development of high areal and volumetric energy density sodium-ion batteries (SIBs) is a viable alternative to LIBs.8
Manganese dioxide (MnO2) possesses a long history in the energy storage field and remains ubiquitous in our daily lives.9 The Leclanché cell, composed of Zn and MnO2 has been known for over 150 years and is still used in powering small-size portable electronics.10,11 In 1999, Goodenough et al. reported the capacitive behavior of amorphous MnO2 in aqueous electrolyte. Its high specific capacitance of 200 F g−1 opened new directions for utilizing MnO2 in electrochemical energy storage.12 Due to the surface pseudocapacitance exhibited by MnO2 and its impressive theoretical capacitance of 1370 F g−1, MnO2 has become an important electrode material for high-power energy storage devices in aqueous electrolytes.13–15 The integration of MnO2 onto conductive substrates such as carbon and conducting polymers has gained considerable attention by addressing the inherent low electrical conductivity of MnO2 that leads to diminished performance of MnO2 electrodes when mass loading and electrode thickness increase.16–19 Another significant feature is that MnO2 electrodeposition is well-established and used to synthesize MnO2 on free-standing conductive scaffolds. A number of 2D free-standing conductive scaffolds including carbon nanotube (CNT) films and carbon cloth have been widely utilized to improve the electrochemical performance of electrodeposited MnO2.20,21 An important research direction in this field is to increase the areal energy density of the MnO2 electrode as both mass loading and thickness are significant factors in reducing manufacturing costs.22 Among the most successful aqueous results is the work reported by Li et al. who used a 3D-printed graphene aerogel (GA) scaffold to improve the performance of electrodeposited MnO2 electrodes with high mass loading. The gravimetric capacity of MnO2 remained almost unchanged when the mass loading of the electrode increased from 2 mg cm−2 to 45 mg cm−2.23 Additionally, the areal capacity scaled with the thickness of the electrode as a result of the high electrical conductivity of the graphene aerogel scaffold and rapid ion diffusion from channels present in 3D-printed GA. A high areal capacity of 9.8 mA h cm−2 was achieved.23,24 The successful results with the aqueous electrolyte influenced our decision to use the graphene aerogel electrode architecture for our study of MnO2 in non-aqueous electrolyte.
Although pseudocapacitive MnO2 exhibits stable performance and superior cycle life in aqueous electrolytes, Mn-based materials are susceptible to capacity fade in non-aqueous electrolytes.25–27 Due to their high redox potential, Mn-based materials have been widely investigated as cathode materials for lithium-ion batteries.28,29 However, the significant capacity fade during cycling, caused by Mn dissolution into the electrolyte, has limited the widespread application of Mn-based materials, especially for long-lifetime batteries.30 The Jahn–Teller distortion upon deep discharge as well as the Mn3+ disproportionation reaction were identified as the main bottlenecks.30,31 Consequently, there are only a limited number of studies employing MnO2 as a cathode material for SIBs in non-aqueous electrolytes because of its poor cycling stability. For example, Su et al. reported that β-MnO2 with a 1 × 1 tunnel structure exhibited drastic capacity fade (42%) in the first 100 cycles when using 1 M NaClO4 in EC/PC as electrolyte.32 Pandit et al. showed a similarly serious capacity fade (∼60% in 800 cycles) in alpha-type MnO2 with a 2 × 2 tunnel structure upon using NaPF6 in EC/DMC with 5% FEC.33 This poor cycling stability of MnO2 can be attributed to Mn2+ ion dissolution into the electrolyte which is triggered by the disproportionation reaction where two Mn3+ ions tend to form one soluble Mn2+ ion plus MnO2 with a +4 oxidation state.31,34 It is important to note that in these previous non-aqueous studies, only carbonate-based electrolytes were used for cycling the MnO2 electrode.35–41
In the current study, we addressed two key challenges limiting the application of MnO2 electrodes in sodium-ion batteries. First, we demonstrated that diglyme (bis(2-methoxyethyl) ether) electrolyte effectively suppresses Mn dissolution, thus enabling stable cycling performance in a non-aqueous electrolyte. Second, we were able to achieve high areal capacity by fabricating high-mass-loaded MnO2 electrodes, up to 80 mg cm−2. Upon using the ether-based electrolyte, a significant increase in areal energy density was obtained because the device voltage was not limited by water splitting as occurs in aqueous electrolytes.25 The high mass loaded electrodes were prepared by electrodeposition of MnO2 onto graphene aerogel scaffolds which were adapted from the earlier study with aqueous electrolyte.23 A full SIB was fabricated using highly loaded MnO2/GA as the cathode and TiO2 on Cu foam as the anode. This SIB full-cell device showed both a high areal energy density of 6.2 mW h cm−2 and a high areal power density of 70.7 mW cm−2. These results show the promise of using MnO2 as a cathode in SIBs for large scale energy storage applications.
2. Experimental
Preparation of 250–400-2 mm 3D printed graphene aerogel
The fabrication process for 3D printed graphene aerogel in this study is similar to that reported in a previous study.23 The process involves the synthesis of a carbon ink, 3D printing of the ink using the direct ink write method, and finally a carbonization process to obtain 3D printed electrodes with varied thickness. A graphene oxide-based ink was used for preparing the 3D graphene aerogel (3D GA). Specifically, single layer graphene oxide sheets (GO) purchased from Cheaptubes Inc. were used. The GO suspension was prepared by ultra-sonicating 0.8 g of GO in 20 g of DI water for 24 h in a sonication bath which is maintained at a constant temperature of 12 °C to avoid heating during the process. This combination yields a GO concentration of 40 mg ml−1. The suspension is then mixed with 7 wt% of hydroxypropyl methylcellulose (DOW Chemical) GO-ink in a planetary Thinky mixer at 2000 rpm for 5 minutes. To avoid the formation of agglomerates, the ink was frozen in dry ice in between each mixing step, to ensure the complete dissolution of cellulose in the ink.The ink was then loaded into a 10 ml syringe barrel (EFD) and centrifuged at 4500 rpm for one minute to remove air bubbles. Subsequently, the ink was extruded through a nozzle (250 μm diameter) to pattern 3D structures on a glass substrate coated with vaseline. For direct ink writing (DIW), the syringe was attached by a luer-lock to a smooth-flow tapered nozzle whose inner diameter(d) is 250 μm. The ink was then extruded by means of an air-powered fluid dispenser (Ultimus V, EFD) which provides an appropriate pressure (in the range of 15–20 psi) for writing. The writing speed was kept at 10 mm s−1 for all the 3D printed GA scaffolds. Although it is not mandatory to change the PTFE nozzle tip between the prints, for the fabrication of 3D GA, a new tip was used for every sample. Simple cubic lattices with multiple orthogonal layers of parallel cylindrical rods were printed alternately. The diameter of the cylindrical rods equals the diameter of the nozzle and the center-to-center rod spacing of 400 μm (for 250 μm nozzle). The height of the printed sample is 2 mm and the layers were stacked on the structure such that each layer has a z-spacing of 0.18 mm. To avoid cracking or drying due to the evaporation of water, the printed samples were immediately immersed in liquid nitrogen after the printing process and freeze-dried for 48 h under vacuum to form aerogels. The printed aerogels were then heated in a tube furnace under a nitrogen atmosphere at 1050 °C for 3 h with a heating and cooling rate of 2 °C min−1 to form the 3D GA scaffold. The graphene aerogel with 250 μm nozzle, 400 μm enter-to-center rod spacing, and 2 mm height or thickness was defined in this study as 250–400-2 mm 3D GA.
Electrodeposition of MnO2 and calcination process
The 250–400-2 mm 3D GA was transferred into an air plasma cleaner for 6 min for surface treatment and better wetting in the electrolyte. A three-electrode system was fabricated for MnO2 electrodeposition on 3D GA, where 3D GA was the working electrode, graphite paper was the counter electrode, and a saturated calomel electrode (SCE) was used as a reference electrode. A 0.2 M manganese acetate (Sigma, 99.99% trace metal basis) solution was prepared and added to the three-electrode cell. The three-electrode cell was then transferred to a desiccator and evacuated for 15 min to remove air bubbles and ensure uniform electrodeposition. In the deposition process, a constant current of 5 mA cm−2 was applied for 2 min followed by a rest period of 20 s to enable ion diffusion. The 2-minute electrodeposition and 20 s resting process are considered as 1 cycle. After 60, 120, 180, and 240 cycles of electrodeposition, the resulting mass loadings on the MnO2/GAs electrodes was approximately 20, 40, 60, and 80 mg cm−2, respectively. Upon completing the electrodeposition, the MnO2/GA electrode was first rinsed with DI water and then dried at 80 °C overnight. In a separate series of experiments, the MnO2 was electrodeposited on a carbon cloth substrate in a similar fashion. The average mass loading of MnO2 on carbon cloth was around 3 mg cm−2. All samples, regardless of loading, were subjected to heat treatment by transferring the MnO2/GA electrodes into a box furnace and heating to 300 °C in air for 6 h with a 5 °C min−1 ramp rate. The MnO2/GA electrodes were cooled by natural convection to room temperature.Electrochemical cycling
All electrochemical measurements were performed using a three-electrode system in an Argon-filled glovebox (H2O < 0.1 ppm, O2 < 0.1 ppm). Over-capacitive activated carbon was used as the counter electrode and Ag/AgNO3 non-aqueous electrode as the reference electrode. The Ag/AgNO3 non-aqueous reference electrode was calibrated by Na metal, where 0 V in Ag/AgNO3 non-aqueous is roughly equivalent to 3.2 V vs. Na/Na+. The cycling potential window for the MnO2 electrode is between −1.4 V to 1 V vs. Ag/AgNO3 and for dip-coating TiO2 on Cu is between −3 V to −1.4 V vs. Ag/AgNO3. The dip-coated TiO2 on Cu was activated by using cyclic voltammetry at a sweep rate of 0.4 mV s−1 for 3 cycles to complete the surface amorphization and solid electrolyte interface formation process. In the current study, 1 M NaClO4 in diglyme (bis(2-methoxyethyl) ether) was used as an electrolyte for achieving better cycling stability, and 1 M NaClO4 in propylene carbonate (PC) was used to compare the cycling stability of the MnO2 electrode. Before assembling as a full battery, the MnO2/GA cathode was discharged to −1.4 V and the dip-coated TiO2 on Cu anode was charged to −1.4 V at 10 mA cm−2 current density.Characterization techniques
X-ray powder diffraction (XRD) was carried out using a PANalytical X'Pert Pro diffractometer. X'Celerator detector with Cu-Kα1–Kα2 (λ = 1.54060, 1.54439 Å) radiation was used and diffraction patterns were collected between 10° and 80° (2θ). Scanning electron microscopy (SEM; FEI Nova NanoSEM230) images were obtained by using a 10 keV accelerating voltage with a 5 mm working distance. The energy dispersive X-ray (EDX) elemental map was collected for 5 minutes per sample. X-ray photoelectron spectroscopy (XPS; Kratos Axis Ultra) of Mn 3s was collected with a monochromatic aluminum X-ray source by using a voltage of 12 kV and emission current of 15 mA. A pass energy of 20 eV and a dwell time of 800 ms for each element with an average of 60 scans were used for the detailed scans. Thermogravimetric analysis (TGA; Instrument Specialists Inc. TGA 1000) was performed by heating as-deposited MnO2/GA from room temperature to 500 °C in static air with 5 °C min−1 ramp rate.Electrochemical quartz crystal microbalance (EQCM)
The EQCM tests were carried out using BluQCM from BioLogic. The EQCM test was conducted in an Argon-filled glovebox with an oxygen level lower than 0.5 ppm. The MnO2 was electrodeposited onto a gold QCM sensor (fundamental frequency of 10 MHz). The electrodeposition time is 60 s and the applied current is 0.5 mA. The coated sensor was then heat treated at 300 °C for 6 h. The electrodeposition and heat treatment process are the same as that used for MnO2/GA electrodes. The mass loading of MnO2 was estimated by using the theoretical charge transfer number. The Ag/AgNO3 non-aqueous reference electrode was the same as that used for MnO2/GA electrodes and the counter was platinum mesh.Preparation of dip-coated TiO2@Cu foam anode
TiO2 powder (Alfa, anatase, 99% metal basis) of 10 nm particle size was mixed with Ketjen carbon black (KJB), carbon nanofiber (CNF), carboxymethyl cellulose (CMC) binder, and styrene–butadiene rubber (SBR) binder with a mass ratio of 80% active material, 8% KJB, 4% CNF, 4% CMC, and 4% SBR. The appropriate amount of DI water was added and after mixing for 10 minutes with mortar and pestle, a 1 × 2 cm piece of copper foam with a thickness of 2 mm was dipped into the slurry and slowly removed. The dip-coated TiO2@Cu anode was dried at room temperature overnight and transferred into 80 °C oven for 4 h.Calculation
The areal capacity of a single MnO2/GA electrode reported in this study is calculated based on the geometric deposition area of MnO2 and volumetric capacity was calculated by using areal capacity divided by the thickness of MnO2/GA electrode (0.2 cm). The gravimetric capacity of a single MnO2/GA electrode is calculated based on the mass of MnO2 only. In the TiO2–MnO2 full-cell device, the geometric deposition area of the MnO2/GA electrodes is used for the areal capacity (CA, mA h cm−2) calculation. Thus, the areal energy density (EA, mW h cm−2) and power density (PA, mW cm−2) for the TiO2–MnO2 device were calculated with eqn (1) and (2) shown below, where V representes the device voltage output excluding IR drop and t is the discharge time of the device. | (1) |
 | (2) |
3. Results and discussion
Two separate series of studies were carried out. The initial study focused on identifying a Na-ion nonaqueous electrolyte that would enable stable cycling of the MnO2. This research involved using electrodeposited MnO2 with low mass loading (3 mg cm−2) on a commercial carbon cloth scaffold. These results informed a second series of experiments in which the electrochemical properties of high mass loaded MnO2 electrodes on a 3D graphene aerogel (3D GA) scaffold were determined. Based on these results, we fabricated a sodium-ion battery (SIB) which utilized the diglyme electrolyte discussed below and incorporated a high mass loaded MnO2 cathode that was paired with a TiO2 anode.The use of diglyme (bis(2-methoxyethyl) ether) electrolyte to limit MnO2 dissolution
The poor stability of Mn-based cathode materials in non-aqueous electrolytes was previously reported for both LIBs and SIBs, where a dissolution–migration–deposition mechanism was proposed to be the main cause.31 Xu et al. reported that strong solvation from oxygen in the carbonyl group of the ester solvent to Mn2+ leads to the acceleration of Mn dissolution in the electrolyte.42 Chen et al. recently reported that the stable cycling of MnO2 in a lithium metal battery using ether-based electrolyte was due to limited proton generation.43 For these reasons, it was hypothesized that using an ether-based solvent might reduce the Mn dissolution from the electrode surface into the electrolyte.The appropriate cycling potential window for MnO2 in the non-aqueous electrolyte was determined by conducting a sweep voltammetry window opening experiment in combination with the Trasatti analysis (see Fig. S1†). These experiments along with the ones discussed in this section, were carried out with electrodeposited MnO2 on carbon cloth (Fig. S2†). The improved cycling of the ether-based electrolyte was shown in a series of galvanostatic experiments where electrodeposited MnO2 (∼3 mg cm−2) was cycled at a specific current of 1 A g−1 for 1000 cycles. The electrodeposited material was identified by X-ray diffraction as ε-MnO2 as described in the following section. The high potential end was reduced from 1 V to 0.4 V vs. Ag/AgNO3 to avoid possible oxygen evolution reaction induced by the carbon cloth substrate while the low potential end was kept constant at −1.4 V vs. Ag/AgNO3. The frequently used ester-based electrolyte, 1 M NaClO4 in EC/PC, was compared to 1 M NaClO4 in ether-based diglyme.32 The cyclic voltammograms (CVs) of MnO2 on carbon cloth for those two electrolytes are shown in Fig. S3.† The galvanostatic results shown in Fig. 1a indicate that the MnO2 electrode in diglyme electrolyte has 75% capacity retention over 1000 cycles. This response is much better than that of the ester-based electrolyte, 1 M NaClO4 in EC/PC (58% retention after 1000 cycles; Fig. 1a). Recent work suggested that the presence of a looser solvation structure may inhibit Mn dissolution, which might explain the better capacity retention obtained with diglyme electrolyte.44 Additionally, the MnO2 shows a higher specific capacity when cycled in 1 M NaClO4 in diglyme (∼30 mA h−1 after 1000 cycles) compared to 1 M NaClO4 in EC/PC (∼15 mA h g−1 after 1000 cycles). These values are consistent with published work.33 Those findings highlight the strong electrolyte effect on the cycling stability and electrochemical performance of MnO2 in non-aqueous electrolytes.
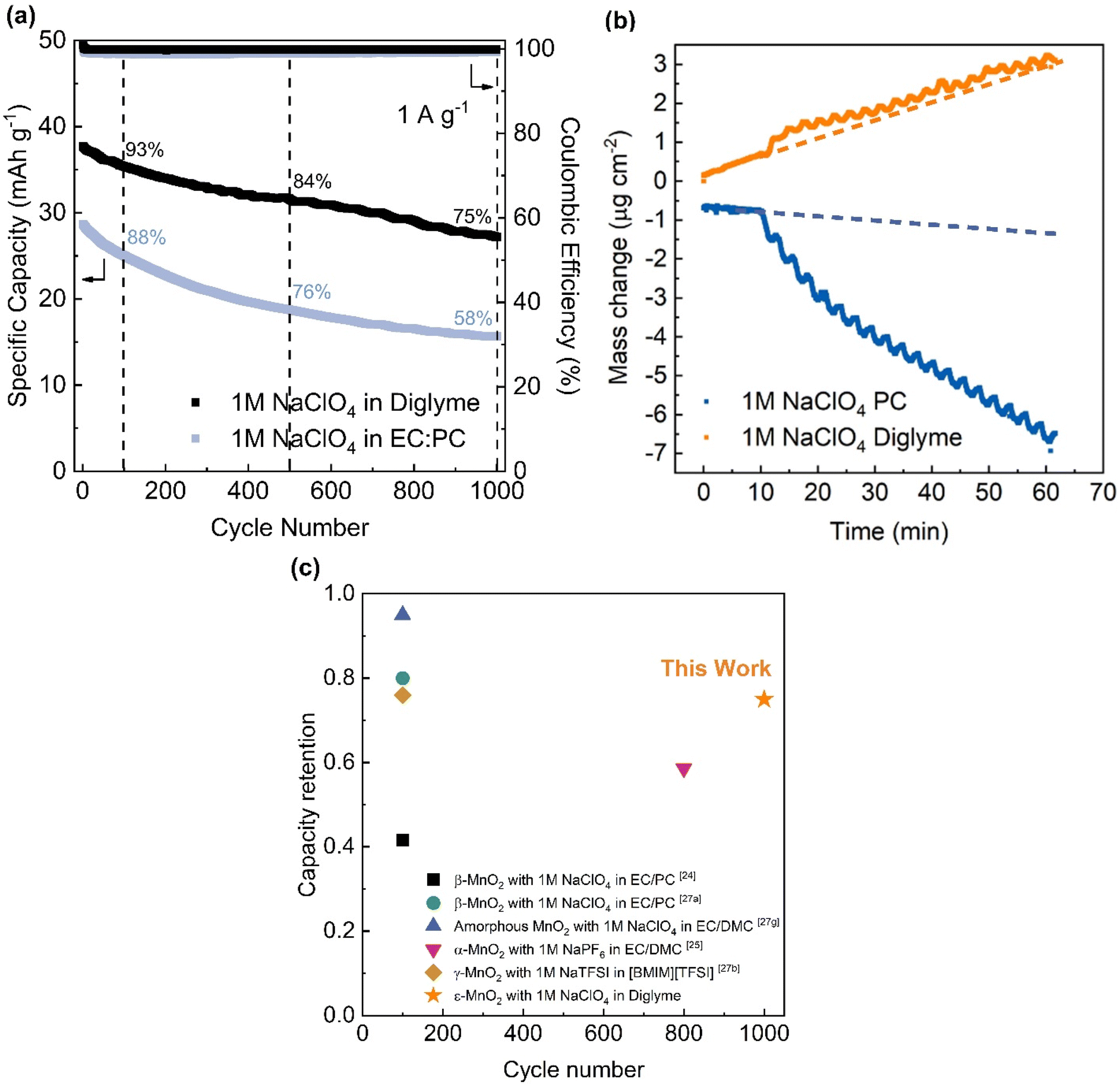 | ||
| Fig. 1 (a) The long-term cycling stability comparison of MnO2 on carbon cloth substrate in 1 M NaClO4 in diglyme and 1 M NaClO4 in EC/PC electrolyte. (b) EQCM results showing the mass change for MnO2 in diglyme and PC electrolyte during OCV (initial 10 minutes) and during CV measurements. The diglyme electrolyte shows effectively no mass change (except baseline mass change) while the PC electrolyte exhibits a continuous decrease in mass. (c) Capacity retention as a function of cycling for MnO2 in various electrolytes. | ||
Electrochemical quartz crystal microbalance (EQCM) measurements were carried out to characterize the mass change of the MnO2 electrodes during the electrochemical charge–discharge process.45 In the EQCM experiments, a representative ester-based electrolyte, 1 M NaClO4 in propylene carbonate (PC), was compared with that of 1 M NaClO4 in diglyme. Prior to the voltammetry measurements, the open circuit potential (OCV) of MnO2 was held for 10 min and the mass change was measured. As shown in Fig. 1b the electrode mass increased slightly or remained constant over the first 10 minutes. The fact that there is virtually no change in mass indicates that no MnO2 dissolution is induced by chemical reaction with either electrolyte. During this initial period, the small (<1 μg) baseline mass increase for the MnO2 electrode in the diglyme electrolyte arises from an increase in electrolyte viscosity at the electrolyte–electrode interface which was noted previously.46,47 This increase in electrolyte viscosity at the electrolyte–electrode interface continues after the OCV period, as indicated by the orange dashed line in Fig. 1b. The oscillations with both electrolytes after the OCV period are due to the mass changes occurring during the CV measurements (10 mV s−1 for 30 cycles). These results show that in the PC electrolyte, the mass of MnO2 deviates from the OCV background (blue dashed line) and decreases continuously as the sample is cycled. The deviation is attributed to the dissolution of MnO2 into the PC electrolyte and contributes to the poor cycling stability of MnO2 (Fig. 1a). In contrast, after the OCV period, the diglyme electrolyte has only a slight increase in the mass of MnO2 above the orange dashed line in Fig. 1b, indicating its stability. In these EQCM experiments, the potential was varied from −0.4 V to 0.4 V versus Ag/Ag+, which is comparable to the potential window where MnO2 exhibits surface pseudocapacitance in aqueous electrolyte. An explanation for using this potential range is presented in Fig. S4.† The potential versus time curve for MnO2 combined with the corresponding mass change in diglyme electrolyte from cycles 3 to 6 is shown in Fig. S5.† For each cycle, the point of maximum mass is located at the low potential end (−0.4 V), indicating that the mass change is a cation-related process.48 Assuming that the mass change during the EQCM measurement comes entirely from sodium ions, this corresponds to approximately 0.05 sodium ions per MnO2. This amount of charge storage is what one expects for a surface-controlled redox reaction and is consistent with the results of the Trasatti analysis in Fig. S4.† The fact that the MnO2 electrode experiences only a small change in mass during the electrochemical measurement is also consistent with the hypothesis that the dissolution of MnO2 in diglyme electrolyte is negligible and the superior cycling stability of the diglyme electrolyte comes from the limited dissolution of MnO2. The EQCM measurement further confirms that the dissolution of MnO2 is responsible for the poor cycling stability reported previously. In Fig. 1c, the cycling stability of the diglyme electrolyte is compared with other literature in which MnO2 serves as the cathode for sodium-ion electrolytes. Although different phases of MnO2 might exert an impact on cycling stability, the results of these studies clearly establish the importance of using ether-based electrolytes for achieving stable MnO2 cycling in non-aqueous electrolytes.35
High mass loaded MnO2 electrodes on graphene aerogel scaffolds
The development of electrodes with high mass loading, which produces high areal capacity, is a critical parameter for energy storage devices in general and grid level storage in particular.49,50 Thus, a key issue addressed in the current work is fabricating high mass loaded MnO2 electrodes with scalable levels of mass loading. One of the more attractive approaches to high mass loading has been achieved via electrodeposition onto carefully designed conductive scaffolds. In the present research, we utilized a previously reported graphene aerogel (GA) scaffold which, in aqueous electrolyte, achieved MnO2 loadings greater than 150 mg cm−2. This electrode architecture is also effective in characterizing electrochemical properties because the channels in the scaffold enable the electrolyte to access the redox-active MnO2 while the high conductivity of the scaffold provides the electronic conduction required for redox-based charge storage. The use of the diglyme electrolyte in the current work benefits the energy density since it means that the voltage is not limited by water dissociation.The fabrication process for the MnO2/GA electrode is depicted in Fig. 2. The GA scaffold is printed by a direct ink writing process as described in the Experimental Methods. The ‘stacked logs’ geometry involving multiple orthogonal layers of parallel cylindrical rods gives this scaffold a prominent three-dimensional configuration. This 3D structure is comprised of 250 μm thick ligaments, with center-to-center spacing of 400 μm and a total thickness of 2 mm. As shown in Fig. 3a, the 3D GA scaffold contains large channels which benefit electrolyte penetration, an advantage for both electrodeposition and electrochemical measurements. In a previous publication, a BET surface area of 27 m2 g−1 was reported for an identical GA scaffold using a similar ink chemistry and the same 3D printer.23 The magnified image of GA in Fig. 3b shows that the sheet-like graphene structure is retained in the scaffold. SEM cross-sections show interconnected ribbon-like graphene sheets inside porous ligaments (Fig. S6a–S6c†).23 Mass loading of the electrodeposited MnO2 ranges from 21.6 mg cm−2 to nearly 80 mg cm−2 depending upon the number of electrodeposition cycles. The SEM image of the 21.6 mg cm−2 MnO2/GA electrode indicates the conformality of the MnO2 film, shown in Fig. 3c and d. A more magnified image, shown in Fig. S7a and S7b,† indicates that the electrodeposited MnO2 has a nanoflake morphology with a thickness of ∼30 nm. The morphologies for the different mass loadings of MnO2 ranging from 37.4 to 57.2 mg cm−2 are presented in Fig. S8.† Fig. S6d–S6m† show that the ligaments of the 3D electrode are conformally coated by MnO2. We consider this feature to be important in maintaining high power capabilities of the high mass loaded electrodes. Energy dispersive X-ray (EDX) analysis of the 80 mg cm−2 electrode (Fig. S9†) indicates a uniform deposition process for this high mass loaded 3D electrode. Above 80 mg cm−2, the MnO2 deposition is no longer uniform as a dense MnO2 layer of ∼15 μm is formed which leads to an increase in electrode resistance and sluggish kinetics (Fig. S10†).
 | ||
| Fig. 2 The schematic illustration of the preparation process of 250–400-2 mm 3D MnO2/GA electrode. | ||
 | ||
| Fig. 3 (a) and (b) The planar view SEM image of pure 3D GA. (c) and (d) The planar view SEM image of 21.6 mg cm−2 MnO2/GA and morphology of deposited MnO2. (e) The XRD patterns of 3D GA, as-deposited MnOx, 3D MnO2/GA after heat treatment and the standard XRD pattern (AMCSD #0017794) of ε-MnO2 (f) The XPS spectra of Mn 3s for as-deposited MnOx and 3D MnO2/GA. | ||
X-ray diffraction scans of 3D GA, as deposited MnO2/GA, and MnO2/GA after calcination are shown in Fig. 3e. The 3D GA exhibits only a broad peak located at 26 degrees (2Θ) while the as-deposited MnOx exhibits a small peak at 37 degrees. After heat treatment, phase pure ε-MnO2, which consists of an intergrowth of [MnO6] octahedra of 1 × 1 and 1 × 2 tunnel structures could be identified, and its XRD pattern matches well with standard ε-MnO2 (AMCSD #0017794).51 The Mn oxidation state of as-deposited MnOx and crystalline ε-MnO2 was determined by using X-ray photoelectron spectroscopy (XPS). The distance of peak splitting of the Mn 3s orbital was utilized to establish the average oxidation state of Mn, where 4.7 eV represents Mn4+.23,52 In Fig. 3f, the distance of peak splitting in ε-MnO2 after heat treatment was consistent with Mn4+ while the as-deposited MnOx has a lesser degree of oxidation. Thermogravimetric analysis (TGA) results shown in Fig. S11† indicate that there is a plateau region ranging from 250 to 400 °C where the as-deposited MnOx was completely oxidized to ε-MnO2.14 The decrease in mass between room temperature and 250 °C corresponds to the loss of physically absorbed moisture and crystalline water during heating, and above 400 °C, the mass decrease might be attributed to the beginning of the transformation of MnO2 to Mn2O3 since Mn3+ is thermodynamically more stable than Mn4+ at high temperature.43,53–55
The electrochemical properties of 3D MnO2/GA were determined using cyclic voltammetry (CV) and galvanostatic cycling (GV). A three-electrode arrangement was used (Fig. S12†) where MnO2/GA with various loadings served as the working electrode, overcapacitive carbon was the counter electrode with a Ag/AgNO3 reference electrode. As indicated previously, the electrolyte was 1 M NaClO4 in diglyme (bis(2-methoxyethyl) ether). The potential range for the electrochemical measurements was determined by the window opening experiment for this electrolyte (Fig. S1†) and, for most experiments, varies from 1.8 V to 4.2 V (vs. Na/Na+). The CV results at 2 mV s−1 for the pure GA electrode and the series of MnO2/GA electrodes are shown in Fig. 4a. The nearly box-like shape of the CV is typical for electrodeposited MnO2 and is consistent with the pseudocapacitive nature of the material. From −1.4 V to 1.0 V, the currents at a given potential scale inversely with the lowest loading, with the 14.4 mg cm−2 exhibiting the highest current. The CV results were also analyzed by the Trasatti method using the CV curves for 14.4 mg cm−2 loading at various sweep rates (Fig. S13†). The Trasatti method separates the contribution of total charge (Qtotal) into the outer charge and the inner charge, where the outer charge (Qouter) is independent of the scan rate and the inner charge (Qinner) is associated with charge storage being limited by semi-infinite diffusion.56,57 The details of the analysis are reviewed in the ESI.† For a sample with 14.4 mg cm−2 loading (Fig. 4b and c), we are able to determine the Qouter and Qinner, respectively. As shown in Fig. S14,† the analysis indicates that most of the capacity (76%) can be attributed to surface-controlled currents, which is consistent with the box-like shape of the CV (Fig. 4a). The extrapolated infinite sweep rate specific capacitance (Fig. 4b) of ∼100 mA h g−1 is in good agreement with the gravimetric capacity of ∼80 mA h g−1 determined by galvanostatic measurements discussed below. The gravimetric capacity of pure GA calculated from the CV curve in Fig. 4a is around 3.1 mA h g−1 and accounts for only ∼3% of the total capacity for the 14.4 mg cm−2 mass loaded 3D MnO2/GA electrode. Thus, an overwhelming majority of the capacity from the 3D MnO2/GA electrode is from MnO2 with only a minor amount coming from the GA scaffold.
 | ||
| Fig. 4 (a) The box-like CV curves of pure GA and MnO2/GA electrodes with mass loading of 14.4, 21.6, 37.4, 57.2, and 79.6 mg cm−2 at a scan rate of 2 mV s−1. (b) The specific capacity as a function of ν−0.5 for the 14.4 mg cm−2 MnO2/GA. (c) The reciprocal of specific capacity as a function of ν0.5 for the 14.4 mg cm−2 MnO2/GA. | ||
The galvanostatic (GV) results at current densities of 10 and 40 mA cm−2 for the various loadings of MnO2/GA electrodes are shown in Fig. 5a and b. The GV responses for the other current densities are shown in Fig. S15.† The nearly linear voltage profiles from −1.4 V to 1.0 V are consistent with the box-like CV in Fig. 4a. The increase in areal energy density with loading (Fig. 5a) for a given current density is expected as more material is added per unit area to the electrode. A second significant feature is that the magnitude of the areal energy density, at a given amount of loading, decreases as the current density increases (Fig. 5c). Thus, the areal energy densities at 40 mA cm−2 (Fig. 5b) are less than those at 10 mA cm−2 for the same level of loading. Both of these results can be explained based on the scaling properties of the MnO2/GA electrode.
 | ||
| Fig. 5 (a) The GV curves of 21.6, 37.4, 57.2, and 79.6 mg cm−2 MnO2/GA electrodes at a current density of 10 mA cm−2. (b) The GV curves of 21.6, 37.4, 57.2, and 79.6 mg cm−2 MnO2/GA electrodes at a current density of 40 mA cm−2. (c) The areal capacity and volumetric capacity of MnO2/GA at current densities from 10 mA cm−2 to 60 mA cm−2 for 21.6, 37.4, 57.2, and 79.6 mg cm−2 MnO2/GA electrodes. (d) The overpotentials at current densities from 10 mA cm−2 to 60 mA cm−2 for 21.6, 37.4, 57.2, and 79.6 mg cm−2 MnO2/GA electrodes. (e) The long-term cycling performance of 21.6 and 79.6 mg cm−2 MnO2/GA electrodes at a current density of 40 mA cm−2 for 1000 cycles. | ||
Our discussion of scaling is based on the GV experiments in which the areal capacity is determined as a function of loading and current. Fig. 5c characterizes this effect by showing the variation in areal energy density as a function of current density for different levels of loading. The nearly linear behavior at 10 mA cm−2 gives a good indication that the added MnO2 continues to contribute to the areal capacity. The slope is such that we can estimate that a 4× increase in loading from 20 to 80 mg cm−2, leads to a nearly 3× increase in areal energy density. The higher current densities are not as effective in terms of the added loading. For example, the 4× increase in loading for the 40 mA cm−2 current density, produces only a ⅓ increase in areal energy density. At 60 mA cm−2, the areal capacity at the highest loading (80 mg cm−2) is less than that at the lowest loading of 20 mg cm−2.
The overpotential (see ESI† for details) plays a significant role in determining the scaling at different current densities. As shown in Fig. 5d, the overpotential increases significantly with current density for a given loading. In contrast, the effect of loading on the overpotential at a given current density is relatively small, with the exception of the highest loading of nearly 80 mg cm−2 where there is a clear increase compared to the other loadings. The small change in overpotential as a function of loading at 10 mA cm−2 is one reason why electrodes operating at this current density can be scaled effectively up to 80 mg cm−2. Reasonably effective scaling can be achieved up to 40 mA cm−2 and 60 mg cm−2, although the areal capacities are reduced accordingly. The reduced capacity at 80 mg cm−2 implies the mass loading is close to the limit available for the GA scaffold. In a previous study using electrodeposited VO2, a similar GA was shown to have a mass loading limit of around 70 mg cm−2. Beyond the limit, the coating was less uniform and cracks were observed which are detrimental to electrochemical performance.58
The long-term cycling performance of MnO2/GA electrodes was determined at a current density of 40 mA cm−2. At a loading of 80 mg cm−2, the MnO2/GA displayed excellent capacity retention of 84% after 1000 cycles (Fig. 5e). The capacity initially increases for the first 30 cycles due to the activation process and the capacity retention stayed above 90% for 500 cycles. The capacity retention for MnO2/GA electrodes loaded at 20 mg cm−2 is also very stable although the areal capacity is less because of the lower loading level, (Fig. 5e). These results demonstrate the effectiveness of the long-term cycling properties of MnO2/GA electrodes. Moreover, this level of cycling stability significantly outperforms the results of previously reported MnO2 cathodes used in SIBs.32,33,35,36,39,41
A high mass loaded MnO2/GA electrode was incorporated as a cathode in a sodium-ion battery (SIB). In this case the anode was anatase TiO2 which exhibits a surface-controlled redox reaction and fast-charging capability upon the insertion of sodium ions.59 The anatase TiO2 becomes amorphous upon sodiation and, up to 1.5 A g−1, the properties of amorphous TiO2 (a-TiO2) electrodes dip coated on a Cu foam current collector (Fig. S16a†) compare well with prior results of cast electrodes despite the higher loading (Fig. S16b†). The fabrication process and testing procedures for a full-cell SIB composed of 58 mg cm−2 MnO2/GA as cathode and 16 mg cm−2 a-TiO2 as anode are detailed in the ESI.† The device was cycled in a two-electrode beaker cell, illustrated in Fig. S17.† The CV and GV plots of the SIB device with 4 V voltage output are shown in Fig. 6(a) and 6(b). The relatively low coulombic efficiency of MnO2|a-TiO2 device at the rate of 10 mA cm−2 might come from slightly unmatched capacity between MnO2 and TiO2, leading to extra SEI layer formation. In this study, the proof-of-concept MnO2|a-TiO2 SIB exhibited excellent long-term cycling performance (Fig. 6(c)), achieving approximately 85% capacity retention after 500 cycles at a current density of 40 mA cm−2. The highest areal energy density of 6.2 mW h cm−2 was achieved at 10 mA cm−2 current density (using discharge capacity only) and the maximum areal power density could reach somewhat greater than 70 mW cm−2 at an areal energy density of 0.6 mW h cm−2. As shown in Fig. 6d, this SIB outperformed the areal power density of other devices with 3D electrode structures, including SIBs, sodium-ion capacitors, lithium ion capacitors, and aqueous capacitors.23,48,60–63 These results highlight the advantage of incorporating the MnO2/GA electrode in electrochemical energy storage devices and becoming a promising low-cost technology for large-scale energy storage.
 | ||
| Fig. 6 (a) The CV curve for a sodium ion battery device with 58 mg cm−2 3D MnO2/GA as cathode and 16 mg cm−2 dip-coated TiO2 as anode from scan rate of 2 mV s−1 to 50 mV s−1. (b) The GV curve for a sodium ion battery device with 58 mg cm−2 3D MnO2/GA as cathode and 16 mg cm−2 dip-coated TiO2 as anode from 10 mA cm−2 to 60 mA cm−2. (c) The long-term cycling performance of 3D MnO2|TiO2 device at 40 mA cm−2 for 500 cycles. (d) The Ragone plot of areal energy density versus areal power density of 3D MnO2|TiO2 device and the performance comparison with reported lithium-ion batteries and sodium-ion batteries devices with 3D electrodes. | ||
4. Conclusions
This research addressed two issues which have limited the use of MnO2 as an electrode in non-aqueous SIBs: Mn dissolution in non-aqueous electrolytes and achieving high areal capacity MnO2 electrodes. For the former, an ether-based electrolyte, diglyme, was shown to overcome the dissolution problems which plague MnO2 electrodes in non-aqueous electrolytes. A combination of long-term cycling experiments and EQCM studies established that stable cycling is obtained with capacity retention over 80% after 1000 cycles. The high areal capacity is enabled by the prominent mass loading of MnO2. The conformal and uniform electrodeposition of MnO2 on the graphene aerogel scaffold is the key to achieving excellent electrochemical performance with small overpotential for high mass loaded electrodes. With this electrode architecture, the electrochemical properties of MnO2 are scalable up to a loading of 80 mg cm−2. Moreover, by using the diglyme non-aqueous electrolyte, a wider electrochemical window of 2.4 V is available that boosts the areal energy density to levels well beyond what is achievable with aqueous electrolytes. An important motivation for scaling such mass loading levels is the prospect of reduced manufacturing costs. The highly loaded MnO2/GA electrodes achieve an areal energy density up to 4.4 mA h cm−2 at a current density of 10 mA cm−2. The high mass loaded MnO2 electrodes were incorporated as a cathode in a 4 V MnO2|a-TiO2 SIB device that exhibits a high areal energy density and an areal power density up to 70 mW cm−2.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The research was supported by Lawrence Livermore National Laboratory under the auspices of the U.S. Department of Energy (DE-AC52-07NA27344). Partial support was provided by the Office of Naval Research (N00014-23-1-2667). The work was partially supported by Laboratory Directed Research and Development award 23-SI-002.References
- T. M. Gür, Energy Environ. Sci., 2018, 11, 2696 RSC.
- B. Dunn, H. Kamath and J.-M. Tarascon, Science, 2011, 334, 928 CrossRef CAS PubMed.
- G. Berckmans, M. Messagie, J. Smekens, N. Omar, L. Vanhaverbeke and J. Van Mierlo, Energies, 2017, 10, 1314 CrossRef.
- X. Fan, B. Liu, J. Liu, J. Ding, X. Han, Y. Deng, X. Lv, Y. Xie, B. Chen, W. Hu and C. Zhong, Trans. Tianjin Univ., 2020, 26, 92 CrossRef.
- J. Duan, X. Tang, H. Dai, Y. Yang, W. Wu, X. Wei and Y. Huang, Electrochem. Energy Rev., 2019, 3, 1 Search PubMed.
- P. K. Nayak, L. Yang, W. Brehm and P. Adelhelm, Angew. Chem., Int. Ed., 2018, 57, 102 CrossRef CAS.
- Y. Li, Y. Lu, C. Zhao, Y.-S. Hu, M.-M. Titirici, H. Li, X. Huang and L. Chen, Energy Storage Mater., 2017, 7, 130 CrossRef.
- H. S. Hirsh, Y. Li, D. H. S. Tan, M. Zhang, E. Zhao and Y. S. Meng, Adv. Energy Mater., 2020, 10, 2001274 CrossRef CAS.
- J. S. Bora and Y. Chellam, Microelectron. Reliab., 1979, 19, 281 CrossRef.
- Z. Rogulski and A. Czerwiński, J. Solid State Chem., 2003, 7, 118 CAS.
- C. L. Quintanilha, J. C. Afonso, C. A. Vianna, V. Gante and J. L. Mantovano, J. Power Sources, 2014, 248, 596 CrossRef CAS.
- H. Y. Lee and J. B. Goodenough, J. Solid State Chem., 1999, 144, 220 CrossRef CAS.
- C. Choi, D. S. Ashby, D. M. Butts, R. H. DeBlock, Q. Wei, J. Lau and B. Dunn, Nat. Rev. Mater., 2019, 5, 5 CrossRef.
- H. Zhao, G. Han, Y. Chang, M. Li and Y. Li, Electrochim. Acta, 2013, 91, 50 Search PubMed.
- Z. J. Han, Z. Bo, D. H. Seo, S. Pineda, Y. Wang, H. Y. Yang and K. K. Ostrikov, ChemSusChem, 2016, 9, 1020 CrossRef CAS PubMed.
- S. Yang, X. Song, P. Zhang and L. Gao, ACS Appl. Mater. Interfaces, 2013, 5, 3317 CrossRef CAS PubMed.
- A. Sumboja, C. Y. Foo, J. Yan, C. Yan, R. K. Gupta and P. S. Lee, J. Mater. Chem., 2012, 22, 2392 RSC.
- C. Shen, X. Wang, S. Li, J. G. Wang, W. Zhang and F. Kang, J. Power Sources, 2013, 234, 30 CrossRef.
- X. Li, L. Xiang, X. Xie, C. Zhang, S. Liu, Z. Li and J. Shen, Nanotechnology, 2020, 31, 215406 CrossRef CAS PubMed.
- T. Bordjiba and D. Bélanger, J. Electrochem. Soc., 2009, 156, A378 CrossRef CAS.
- H. Wang, C. Xu, Y. Chen and Y. Wang, Energy Storage Mater., 2017, 8, 127 CrossRef.
- A. M. Boyce, D. J. Cumming, C. Huang, S. P. Zankowski, P. S. Grant, D. J. L. Brett and P. R. Shearing, ACS Nano, 2021, 15, 18624 CrossRef CAS PubMed.
- B. Yao, S. Chandrasekaran, J. Zhang, W. Xiao, F. Qian, C. Zhu, E. B. Duoss, C. M. Spadaccini, M. A. Worsley and Y. Li, Joule, 2019, 3, 459 Search PubMed.
- B. Yao, S. Chandrasekaran, H. Zhang, A. Ma, J. Kang, L. Zhang, X. Lu, F. Qian, C. Zhu, E. B. Duoss, C. M. Spadaccini, M. A. Worsley and Y. Li, Adv. Mater., 2020, 32, e1906652 CrossRef.
- H. Zhang, X. Liu, H. Li, I. Hasa and S. Passerini, Angew. Chem., Int. Ed., 2021, 60, 598 CrossRef CAS.
- T. Brousse, P.-L. Taberna, O. Crosnier, R. Dugas, P. Guillemet, Y. Scudeller, Y. Zhou, F. Favier, D. Bélanger and P. Simon, J. Power Sources, 2007, 173, 633 CrossRef CAS.
- E. Moazzen, K. Kucuk, S. Aryal, E. V. Timofeeva and C. U. Segre, J. Power Sources, 2020, 448, 227374 Search PubMed.
- J. Abou-Rjeily, I. Bezza, N. A. Laziz, C. Autret-Lambert, M. T. Sougrati and F. Ghamouss, Energy Storage Mater., 2020, 26, 423 CrossRef.
- M. M. Thackeray, W. I. F. David, P. G. Bruce and J. B. Goodenough, Mater. Res. Bull., 1983, 18, 461 CrossRef CAS.
- R. Sheil, D. Butts, K. Jungjohann, J. Yoo, B. Dunn and J. P. Chang, J. Vac. Sci. Technol., A, 2021, 39, 012408 CrossRef CAS.
- C. Zhan, T. Wu, J. Lu and K. Amine, Energy Environ. Sci., 2018, 11, 243 RSC.
- D. Su, H.-J. Ahn and G. Wang, NPG Asia Mater., 2013, 5, e70 CrossRef CAS.
- B. Pandit, S. R. Rondiya, N. Y. Dzade, S. F. Shaikh, N. Kumar, E. S. Goda, A. A. Al-Kahtani, R. S. Mane, S. Mathur and R. R. Salunkhe, ACS Appl. Mater. Interfaces, 2021, 13, 11433 CrossRef CAS.
- H. Lv, Y. Song, Z. Qin, M. Zhang, D. Yang, Q. Pan, Z. Wang, X. Mu, J. Meng, X. Sun and X.-X. Liu, J. Chem. Eng., 2022, 430, 133064 CrossRef CAS.
- D. Su, H.-J. Ahn and G. Wang, J. Mater. Chem. A, 2013, 1, 4845 Search PubMed.
- H. Si, L. Li, W. Hao, L. Seidl, X. Cheng, H. Xu, G. Jia, O. Schneider, S. An and X. Qiu, ACS Appl. Energy Mater., 2019, 2, 5050 Search PubMed.
- F. Liu, Y. Xiao, Y. Liu, P. Han and G. Qin, Chem. Eng. J., 2020, 380, 122487 CrossRef CAS.
- Z. Wang, X. Yan, F. Wang, T. Xiong, M. S. Balogun, H. Zhou and J. Deng, Carbon, 2021, 174, 556 CrossRef CAS.
- W. Zhang, H. Jin, Y. Du, Y. Zhang, Z. Wang and J. Zhang, ChemistrySelect, 2020, 5, 12481 CrossRef CAS.
- Z. Zhang, X. Zhao and J. Li, ChemNanoMat, 2016, 2, 196 Search PubMed.
- Y. Zhou, T. Chen, J. Zhang, Y. Liu and P. Ren, Chin. J. Chem., 2017, 35, 1294 CrossRef CAS.
- C. Wang, L. Xing, J. Vatamanu, Z. Chen, G. Lan, W. Li and K. Xu, Nat. Commun., 2019, 10, 3423 CrossRef PubMed.
- D. Xia, H. Gao, M. Li, J. Holoubek, Q. Yan, Y. Yin, P. Xu and Z. Chen, SmartMat, 2023, 4, e1208 CrossRef CAS.
- Y. Ugata, T. Kuriyama and N. Yabuuchi, Chem. Commun., 2025, 61, 338–341 Search PubMed.
- X. Ye, D. Han, G. Jiang, C. Cui, Y. Guo, Y. Wang, Z. Zhang, Z. Weng and Q.-H. Yang, Energy Environ. Sci., 2023, 16, 1016 Search PubMed.
- K. Kwon and J. W. Evans, Electrochem. Solid-State Lett., 2002, 5, A59 Search PubMed.
- K. Kwon and J. W. Evans, Electrochim. Acta, 2004, 49, 867 CrossRef CAS.
- M. D. Levi, G. Salitra, N. Levy, D. Aurbach and J. Maier, Nat. Mater., 2009, 8, 872 Search PubMed.
- S.-H. Park, P. J. King, R. Tian, C. S. Boland, J. Coelho, C. Zhang, P. McBean, N. McEvoy, M. P. Kremer, D. Daly, J. N. Coleman and V. Nicolosi, Nat. Energy, 2019, 4, 560 Search PubMed.
- K. G. Gallagher, S. E. Trask, C. Bauer, T. Woehrle, S. F. Lux, M. Tschech, P. Lamp, B. J. Polzin, S. Ha, B. Long, Q. Wu, W. Lu, D. W. Dees and A. N. Jansen, J. Electrochem. Soc., 2015, 163, A138 Search PubMed.
- D. E. Simon, R. W. Morton and J. J. Gislason, Adv. X-Ray Anal., 2004, 47, 267 Search PubMed.
- M. Toupin, T. Brousse and D. Bélanger, Chem. Mater., 2002, 14, 3946 Search PubMed.
- L. Lan, Q. Li, G. Gu, H. Zhang and B. Liu, J. Alloys Compd., 2015, 644, 430 Search PubMed.
- M. Liu, G. J. Zhang, Z. R. Shen, P. C. Sun, D. T. Ding and T. H. Chen, Solid State Sci., 2009, 11, 11 Search PubMed.
- T. Patra, A. Mohanty, L. Singh, S. Muduli, P. K. Parhi and T. R. Sahoo, Chemosphere, 2022, 288, 132472 CrossRef CAS PubMed.
- S. Ardizzone, G. Fregonara and S. Trasatti, Electrochim. Acta, 1990, 35, 263 Search PubMed.
- Y. Luo, E. Le Calvez, Y. Zhou, É. Gautron, É. Quarez, M. Preefer, O. Crosnier, J. N. Weker, L. Pilon, T. Brousse and B. Dunn, Chem. Mater., 2023, 35, 8675 Search PubMed.
- Y. Luo, S. Chandrasekaran, B. Hu, R. Chen, M. Worsley and B. Dunn, Nano Res., 2024, 17, 8809–8818 Search PubMed.
- Q. Wei, X. Chang, D. Butts, R. DeBlock, K. Lan, J. Li, D. Chao, D. L. Peng and B. Dunn, Nat. Commun., 2023, 14, 7 Search PubMed.
- Z. Fan, C. Wei, L. Yu, Z. Xia, J. Cai, Z. Tian, G. Zou, S. X. Dou and J. Sun, ACS Nano, 2020, 14, 867 Search PubMed.
- J. Ding, K. Shen, Z. Du, B. Li and S. Yang, ACS Appl. Mater. Interfaces, 2017, 9, 41871 CrossRef CAS PubMed.
- S. Zheng, H. Huang, Y. Dong, S. Wang, F. Zhou, J. Qin, C. Sun, Y. Yu, Z. S. Wu and X. Bao, Energy Environ. Sci., 2020, 13, 82 Search PubMed.
- J. Ma, S. Zheng, L. Chi, Y. Liu, Y. Zhang, K. Wang and Z. S. Wu, Adv. Mater., 2022, 34, 2205569 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Determining the potential window for cyclic voltammetry of MnO2, determining the potential window for conducting EQCM studies, SEM/EDX images for 3D MnO2/GA for a wide variety of loadings, TGA study for characterizing the MnO2 heat treatment, complete CV/GV cycling of 3D MnO2/GA electrodes, a schematic of the beaker cell used for electrochemical measurements and performance of the dip-coated TiO2 electrode. See DOI: https://doi.org/10.1039/d5eb00048c |
| This journal is © The Royal Society of Chemistry 2025 |