
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Insights into chemical substitution of metal halide solid-state electrolytes for all-solid-state lithium batteries
Chao
Wu
 a,
Zhen
Wang
a,
Zhanhui
Jia
b,
Jiawu
Cui
a,
Chengyong
Shu
a,
Xiaowei
Wang
*c,
Yuping
Wu
d and
Wei
Tang
*a
a,
Zhen
Wang
a,
Zhanhui
Jia
b,
Jiawu
Cui
a,
Chengyong
Shu
a,
Xiaowei
Wang
*c,
Yuping
Wu
d and
Wei
Tang
*a
aSchool of Chemical Engineering and Technology, Xi'an Jiaotong University, Xi'an, 710049, China. E-mail: tangw2018@xjtu.edu.cn
bSchool of Materials Science and Engineering, Xi'an Jiaotong University, Xi'an, 710049, China
cChemical Sciences and Engineering Division, Argonne National Laboratory, Lemont, IL 60439, USA. E-mail: xiaowei.wang@anl.gov
dSchool of Energy and Environment, Southeast University, Nanjing 210096, China
First published on 17th March 2025
Abstract
Over a long period of time, frequent safety incidents in electric vehicles and portable electronics have raised concerns about modern energy storage devices, particularly lithium-ion batteries. However, the emergence of solid-state electrolytes (SSEs) with good thermal stability has eliminated potential safety hazards of conventional lithium-ion batteries, such as liquid electrolyte leakage and explosions, allowing all-solid-state batteries to attract intensive attention. Among all types of SSEs, halide SSEs have gained research focus owing to their high ionic conductivity, good mechanical malleability, and excellent chemical/electrochemical stability. They have risen to the forefront of SSE research within just a few years. This paper firstly summarizes state-of-the-art halide SSEs by briefly introducing various synthesis methods of halide SSEs and comparing their advantages and disadvantages. Secondly, it introduces the composition, structural types, and ionic conduction mechanisms of halide SSEs, analyzing their effects on ionic transport behavior mainly from three perspectives: anion polarizability, cation disorder and stacking faults. Primarily, it not only reviews typical substitution types for current halide SSEs, explaining how each type optimizes ion transport kinetics, but also focuses on chemical substitution strategies to improve the inherent thermodynamic stability window of halide SSEs and the complex electrode/SSE interface. Additionally, this work proposes potential future research directions to address the challenges in the development of halide SSEs. Overall, the review aims to provide fundamental understanding for designing new halide SSEs and their structural characterization.
Broader contextGiven the increasing global demand for clean energy and efficient energy storage technologies, the development of all-solid-state lithium-ion batteries (ASSLBs) with excellent performance and high safety has become a hot topic in scientific research. In particular, the research on solid-state electrolytes (SSEs) has become critical to achieve higher energy density, longer cycle life and wider operating temperature range. In recent years, halide SSEs have attracted much attention due to their outstanding physicochemical properties. High room-temperature ionic conductivity implies fast lithium-ion transport kinetics, and good chemical stability and mechanical strength guarantee battery safety. In this review, the current research status on halide SSEs and the diverse synthesis methods are first briefly described. Next, the basic structural framework and ion transport mechanism of halide SSEs are outlined, and the factors affecting the ion transport behavior are summarized. It then focuses on chemical substitution and explores the specific mechanisms by which it enhances lithium-ion transport kinetics and improves the stability of the electrode/SSE interface by optimizing the electrolyte structure and composition. In addition, this paper also proposes key breakthrough directions for future halide SSEs, which is expected to provide a more solid theoretical foundation for subsequent research work. |
1. Introduction
As the energy storage market evolves, all-solid-state lithium-ion batteries (ASSLBs) are gradually replacing traditional liquid lithium-ion batteries, becoming the cornerstone of large-scale energy storage systems. These batteries address the growing demand for high-energy and high-power storage in power grids and public utilities. The primary advantage of ASSLBs lies in their potential to surpass the energy density limits of liquid lithium-ion batteries, aiming for a high energy density of 500 Wh kg−1.1–4 Additionally, the use of solid-state electrolytes (SSEs) with strong thermal stability eliminates the safety risks, such as thermal runaway and explosions, associated with flammable organic electrolytes. SSEs, a critical component of ASSLBs, have been extensively researched and come in various forms, including polymers, oxides, sulfides and halides. Generally, polymeric SSEs, including poly(ethylene oxide) (PEO), poly(vinylidene fluoride) (PVDF), polyacrylonitrile (PAN), and poly(methyl methacrylate) (PMMA), exhibit better mechanical flexibility, lower mass density, and straightforward synthesis methods. However, their intrinsic ionic conductivity is slow, which is commonly addressed by incorporating ceramic fillers, ionic liquids or metal–organic frameworks (MOFs) to form composite polymeric SSEs.5–9 Oxide SSEs provide good environmental and electrochemical oxidation stability but require high sintering temperatures (over 1000 °C) and lack flexibility, resulting in high fabrication costs.10,11 Sulfide SSEs boast high ionic conductivity with excellent mechanical machinability, but they are highly sensitive to water, producing toxic H2S gas upon hydrolysis, which quickly deteriorates their ionic conductivity.12–14 Additionally, their poor electrochemical oxidation stability limits compatibility with conventional 4 V cathode materials. In contrast, halide SSEs have garnered significant attention in ASSLBs research owing to their high ionic conductivity (∼10−3 S cm−1) at room temperature (RT), wide electrochemical stability window, excellent environmental tolerance and diverse synthetic routes.15–17 These properties allow halide SSEs to overcome many limitations of other SSEs.The development of halide SSEs has undergone significant changes, as illustrated in Fig. 1, which highlights key milestones chronologically. Generally, the evolution of halide SSEs can be divided into two phases, with a pivotal shift occurring in 2018. Before 2018, halide SSEs were held back by their low ionic conductivity, which limited their potential for practical use. At the same time, SSEs like oxides and sulfides were advancing rapidly, causing halide SSEs to attract less interest and resulting in minimal experimental and theoretical exploration during this period. However, a breakthrough came in 2018 when Asano et al. successfully synthesized two halide SSEs with high ionic conductivity, i.e., Li3YCl6 (5.1 × 10−4 S cm−1) and Li3YBr6 (1.7 × 10−3 S cm−1).22 This marked a major milestone in halide SSEs development, as it was the first time their ionic conductivity surpassed the 1 × 10−3 S cm−1 threshold. Following this discovery, research interest in halide SSEs surged, leading to the development of various high-ionic-conductivity halide SSEs in just five to six years, such as Li3AlF6, Li3GaF6, Li3InCl6, Li3ScCl6, Li3ErCl6, Li2Sc2/3Cl4, Li3HoBr6 and Li3ErI6.25,36–42 More importantly, given that all constituent elements of ternary halide SSEs are exchangeable, the structure and chemical composition of the halide SSEs family have been greatly enriched by chemical substitution of elements, e.g., Li3−xM1−xZrxCl6 (M = Er/Y), Li2+xZr1−xFexCl6 (0 ≤ x ≤ 0.5), Li3Y1−xInxCl6 (0 ≤ x ≤ 1), Li3YBr3Cl3 and Li3InCl4.8F1.2.26,28,29,43,44 These substituted halide SSEs surprisingly demonstrate significantly improved ionic conductivity compared with their original counterparts.
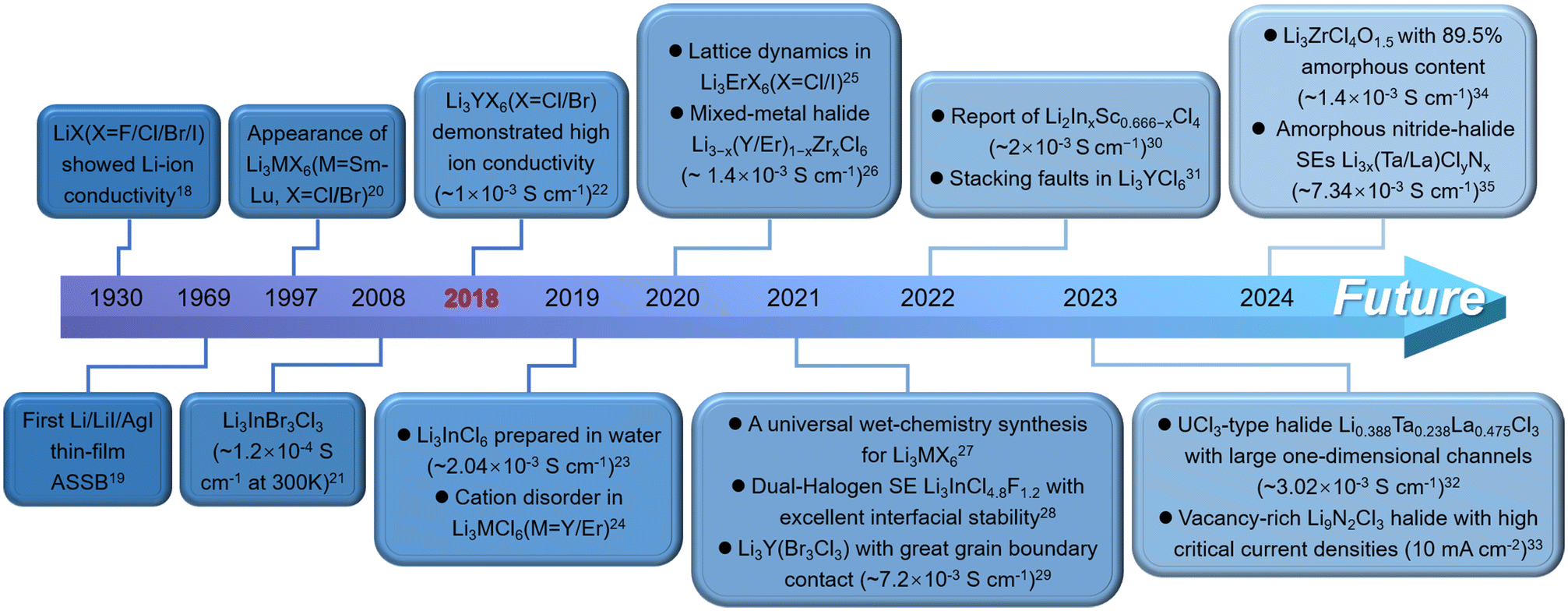 | ||
| Fig. 1 The development history of halide SSEs for ASSLBs with representative achievements.18–35 | ||
It is important to note that halide SSEs encounter interfacial challenges similar to those observed in other SSEs, including significant interfacial side reactions, inadequate interfacial contact and the formation of space charge layers, all of which hinder their further advancement.45,46 To tackle these issues, two well-established interface modification strategies have been extensively explored. The first involves the application of functional coatings (e.g., LiF, Li3PO4) onto the surface of halide SSEs, which serves to mitigate interfacial side reactions and lower interfacial impedance.47–49 The other strategy focuses on the design of nanostructured architectures or the incorporation of a flexible interfacial layer on the halide SSEs surface, effectively improving the physical contact between halide SSEs and electrodes.50–53 Nevertheless, these approaches fail to address the intrinsic electrochemical stability of halide SSEs. To achieve significant interface modification, it remains crucial to modify the surface chemistry of halide SSEs through chemical substitution. Specifically, anionic substitution can regulate the reactivity of the halide SSEs’ surface, thereby facilitating the formation of a stable interface with the electrode. Furthermore, the chemical substitution strategy helps optimize the mechanical properties of halide SSEs, thereby minimizing the formation of cracks.
The synthetic strategies of halide SSEs have also been widely discussed in recent years. Each synthesis route features unique operating conditions and processing parameters that influence the crystal structure and ionic transport behavior of halide SSEs, ultimately affecting the electrochemical properties of ASSLBs. Currently, the most widely used synthesis method is mechanical ball milling, which requires precise control over milling time and speed.15,17 In this process, the mechanical energy generated during high-energy ball milling is converted into the activation energy needed for precursor chemical reactions, resulting in metastable halide SSEs with low crystallinity. Alternatively, solid-phase sintering can be used to increase the crystallinity of halide SSEs and achieve a more stable structure. This method involves sealing the precursor in a vacuumed quartz tube and continuously heating it at elevated temperatures (typically above 350 °C). However, both ball milling and solid-phase sintering are energy-intensive and time-consuming processes, often producing samples that are not homogeneous and that can form impurity phases due to element volatilization at high temperatures. Therefore, it is imperative to explore the liquid phase synthesis methods with mild reaction conditions and high-purity products. A pioneering study by Li et al. demonstrated the first aqueous synthesis of halide SSEs through the production of Li3InCl6.23 In this process, raw materials were dissolved in water, naturally dried in air to form Li3InCl6·H2O, and then heated in a dynamic vacuum at 100–200 °C for 4 hours to produce high-purity Li3InCl6. Since then, several liquid-phase synthesis methods have been developed, such as ammonium-assisted wet chemistry, vacuum evaporation-assisted synthesis, ethanol-mediated Li3InCl6 synthesis and freeze-drying techniques.27,54–56 The liquid-phase synthesis method, while offering significant advantages in terms of material homogeneity, controlling the microscopic morphology or size of SSEs, and fabricating electrode sheets, is more complex and costly. More importantly, it imposes higher requirements on the humidity tolerance of the precursor. Therefore, current research on liquid-phase synthesis primarily focuses on Li3InCl6. As such, there is still considerable progress needed before this method can be widely applied.
This review underscores the most recent progress in the realm of halide SSEs and provides a comprehensive understanding of the structure–property relationship for halide SSEs through the lens of chemical substitution. By systematically analyzing the effects of chemical substitution on ionic conductivity and interfacial stability, it elucidates the fundamental linkages between diverse substitution mechanisms and the electrochemical performance of materials, thereby establishing a more resilient analytical framework. Moreover, this study outlines promising avenues for future research on chemical substitution strategies in halide SSEs, encompassing high-throughput screening of substitutional elements and the design of novel halide SSEs driven by machine learning. These insights offer a forward-thinking perspective that can steer the ongoing development and refinement of halide SSEs, with the objective of rapidly increasing their market share in the commercial landscape of ASSLBs.
2. Structure and ion transport in halide SSEs
2.1 Structural composition
The general formula of ternary halide SSEs is LiaMXb (M = Y, Sc, In, La–Lu, etc., X = F, Cl, Br, I), which is a completely new crystal structure formed by doping rare-earth element M based on LiX structure. Since the ionic radii of the halogen elements are larger than those of almost all rare-earth elements, so the ternary halide SSEs take the anion close-packed sublattice as the basic framework. Among them, the radii and polarity of cations and anions have a remarkable effect on the crystal structure. According to the Pauling coordination polyhedron rule, close contact of cations and anions is the prerequisite for the stabilization of crystal structures. Hence, Liang et al. generalized the structures of all halide SSEs and innovatively proposed to classify the crystal structures by using the radius ratio of M cation to X anion (r+/r−), marked as tm/x.57For the currently dominant Li3MX6 and Li2MX4-type SSEs, t = 0.732–0.414, the anion sublattice can be partitioned into hexagonal close packing (hcp) and cubic close packing (ccp), as shown in the Fig. 2.58 Further refinement of these two sublattices based on the t values leads to two conclusions: (1) when t = 0.637–0.599, the close-packed anions are stacked in ABAB mode. Due to the different symmetric distributions of Li+ and non-Li+ cations on the octahedral (Oct) sites, trigonal (space group: P![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m1) and orthorhombic (space group: Pnma) structures are formed, such as Li3YCl6 and Li3YbCl6.22,59,60 (2) With t value decreases, the close-packed anions are stacked in ABCABC mode. Given the difference in cation occupancy, monoclinic structures (space group: C2/m) and spinel structures (space group: Fdm) are formed, such as Li3InCl6 and Li2MgCl4.38,61
m1) and orthorhombic (space group: Pnma) structures are formed, such as Li3YCl6 and Li3YbCl6.22,59,60 (2) With t value decreases, the close-packed anions are stacked in ABCABC mode. Given the difference in cation occupancy, monoclinic structures (space group: C2/m) and spinel structures (space group: Fdm) are formed, such as Li3InCl6 and Li2MgCl4.38,61
 | ||
| Fig. 2 Common crystal structures of halide SSEs. (a) Trigonal structure. (b) Orthorhombic structure. Reproduced with permissions from ref. 60. Copyright 2021, Elsevier. (c) Monoclinic structure. Reproduced with permissions from ref. 38. Copyright 2019, The Royal Society of Chemistry. (d) Spinel structure. Reproduced with permissions from ref. 61. Copyright 2024, American Chemical Society. | ||
Furthermore, the rare-earth halide UCl3 (U = La–Sm), featuring a non-close-packed anion lattice, exhibits a unique and fascinating structural framework.32,62,63 In contrast to conventional halide SSEs, the metal cations in UCl3-type structures (space group: P63/m) adopt ninefold coordination, forming tricapped trigonal prismatic polyhedra. This high-coordination-number structure not only stabilizes the metal cations but also generates abundant one-dimensional Lithium-ion migration pathways.64,65 Consequently, UCl3-type materials are widely acknowledged as promising contenders for lithium superionic conductors, holding substantial potential for advanced ASSLBs applications.
2.2 Li+ conduction mechanism
Li+ migration in halide SSEs is realized by hopping motion, and the hopping sites of Li+ depend on the type of the anion sublattice, resulting in different migration pathways.22,66 For the hcp structure, Li+ hops among adjacent face-sharing Oct sites (Oct–Oct) along the c-axis, forming rapid diffusion 1D channels, whereas in the ab plane, there exist vast tetrahedral (Tet) interstitial sites, which lie between the two edge-sharing octahedra. They connect all the 1D channels (Oct–Tet–Oct) and a 3D anisotropic diffusion network is ultimately formed (Fig. 3a). However, Li+ migration in these 1D fast channels is prone to be blocked, resulting in ionic conductivities lower than the theoretically calculated values. For the ccp structure, Li+ migration between different Oct sites along all directions requires the assistance of Tet interstitial sites (Oct–Tet–Oct), yielding a 3D isotropic diffusion network (Fig. 3b), which is similar to the migration path along the ab plane in the hcp structure. For the two ccp structures, the Oct sites for Li+ hopping are usually occupied by non-lithium cations in the spinel structure, which also hinders the diffusion of Li+.16 Therefore, the monoclinic phase structure may be the most favorable structure for Li+ migration in halide SSEs, which has also been demonstrated by experiments and theoretical calculations.39,59,67 | ||
| Fig. 3 (a and b) The Li-ion migration pathways in hcp anion lattice and ccp anion lattice. Reproduced with permissions from ref. 66. Copyright 2019, Wiley-VCH. (c) The schematic of the “Li-rich” environment. Reproduced with permissions from ref. 71. Copyright 2022, Elsevier. (d and e) Li+ diffusion and corresponding energy barriers in close-packed anion sublattice by vacancy clusters (divacancies in the layered form). Reproduced with permissions from ref. 69. Copyright 2012, American Chemical Society. | ||
In addition to the long-range diffusion characteristics, short-range diffusion caused by defects (vacancies and interstitial sites) also affects the migration behavior of Li+. The possibility of carriers jumping between neighboring Octahedral sites depends on the content of the surrounding active Li+ and vacancy concentrations. As suggested by Famprikis et al., the diffusion mechanism mediated by vacancy clusters is the main ionic conduction mechanism.68 Higher vacancy concentration and lower adjacent Li+ content would be more favorable for Li+ transport. Van Der Ven has calculated the migration barriers of Li+ in different close-packed lattices (Fig. 3d and e), which confirmed the increase of vacancies number around the Tet interstitial sites favors the reduction of the Li+ migration barrier.69 Although this migration pathway requires passing through Tet interstitial sites, it provides the flattest energy landscape. For Li3MX6-type, M3+ replaces Li+ at three Oct sites, and two vacancies are created based on charge balance. Among all Oct sites of the anion sublattice, the ratio of Li+, M3+, vacancies is 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:2. Therefore, the vacancies occupy 33% of the Oct sites, providing vast available hopping sites for Li+, which is necessary to achieve high ionic conductivity.15,70
:1:2. Therefore, the vacancies occupy 33% of the Oct sites, providing vast available hopping sites for Li+, which is necessary to achieve high ionic conductivity.15,70
Indeed, the realization of ions’ hopping motion between different sites requires overcoming the inter-site energy difference, which is related to the occupation of ions in the surrounding sites. It should be noted that the Tetrahedral interstitial sites act as the bridge in ionic conduction of halide SSEs. When their adjacent Octahedral sites are occupied only by Li+ or vacancies, this prevents strong electrostatic interactions between cations and narrows the energy difference between sites.71 Then the “Li-rich” environment is formed (Fig. 3c). In addition, differences in Li+ concentration and cation configuration are also responsible for the different ionic conductivity. Mo et al. systematically studied a sequence of halide SSEs through ab initio molecular dynamics (AIMD) simulations and concluded that low Li+ content (approximately 40–60%), sparse cation arrangement, and low cation concentration make ion migration easier.72
2.3 Factors affecting ion migration dynamics
In addition to the migration pathways mentioned above, ion transport kinetics can also be affected by many factors, including lattice dynamics, cation disorder, and stacking faults, etc.Lattice dynamics refers to the vibrations of crystal atoms at equilibrium positions, which affects the magnitude of the activation barriers for ion migration.57 Reduction of Li+ vibrational frequency contributes to enhancement of fast Li+ transport performance. This can be explained by the softness of the anion structural frameworks. When the more polarized anion is bound to Li+, the binding effect of Li+ during the migration is weakened due to the longer bond lengths, i.e., a lower Li+ vibrational frequency is obtained (Fig. 4a), which leads to a higher ionic conductivity.25 Therefore, superionic conductors tend to possess low Li+ vibrational frequencies, and their distinctive structural features are expressed as softer and more polarized anion sublattices.40,73,74 As shown in Fig. 4b, by replacing all the anions in Li3ErCl6 with the more polarizable I-, the polarization of the anion sublattice was elevated, obtaining the lower activation barrier and higher ionic conductivity. However, with the addition of more polarizable anions, the average vibrational frequency of the anion phonon band centers was also degraded and the electrochemical oxidation stability started to deteriorate (Fig. 4c).75 Muy attributed this phenomenon to the reaction kinetics, arguing that the reduction in vibrational frequency of the phonon band centers diminished the anion migration enthalpy and facilitated the oxidation reaction.76 Consequently, it is necessary to balance the ionic conductivity and electrochemical stability when probing new Lithium-ion conductors.
 | ||
| Fig. 4 Factors affecting Li-ion transport behavior. (a) The diagram of anion polarizability affecting the activation barrier by changing the ion vibrational frequency. (b) Plots of σ0, Ea and σ as a function of the Debye frequency for Li3ErX6 (X = Cl, I). Reproduced with permissions from ref. 25. Copyright 2020, American Chemical Society. (c) Correlation curves of the oxidative potential limit with the anion phonon band centre for LiAlX4 (X = Cl, Br, I). Reproduced with permissions from ref. 75. Copyright 2022, The Royal Society of Chemistry. (d) Influence of cation disorder on crystal structure. (e) Change of the Er2–Er3 disorder as revealed by Rietveld refinements (open squares) and G(r) fits (open circles). Reproduced with permissions from ref. 24. Copyright 2019, Wiley-VCH. (f) Schematic diagram of stacking faults. (g) Predicted Li+ migration barriers in four model structures. Reproduced with permissions from ref. 31. Copyright 2022, American Chemical Society. | ||
In halide SSEs systems with the same composition, cation order–disordered arrangement can be caused by changing the synthesis method and annealing temperature. Samples typically synthesized using mechanochemical methods have higher cation disorder, favoring the formation of migration channels with flat energy landscapes.77,78 This was also confirmed by Schlem et al. on the arrangement of cation sites in Li3MCl6 (M = Er/Y).24 They found that Li3MCl6 prepared by high-energy ball milling has the highest degree of cation disorder between the Er/Y sites (Fig. 4e), i.e., many M2 sites are swapped to M3 sites. High M2–M3 disorder caused the change of the repulsion force against the surrounding Li+, prompting Li+ sublattice rearrangement (Fig. 4d), which dramatically reduced the energy barrier for Li+ migration along the c-axis. At the same time, the reordered cation sublattices also led to local structural distortions, which benefited the increase of the polyhedral transition areas and the expansion of the bottlenecks for Li+ migration. A similar phenomenon was observed in the study of Ito et al., where β-Li3YCl6 with more disordered Y3+ sublattice exhibited stronger ion transport behavior compared with α-Li3YCl6.79 Overall, cation sites’ disorder in the crystal structure affects the arrangement of Li+ sites, volume changes and local distortions of the polyhedral, thus modifying the Li+ migration path and ionic conductivity.
Consistent with most layered oxide cathodes, stacking faults may be prevalent in halide SSEs. However, the correlation between stacking faults and ion transport was not effectively demonstrated until 2022, when Sebti et al. first demonstrated the presence of stacking faults in Li3YCl6, by using high-resolution synchrotron X-ray diffraction and DFT calculations, which optimized Li+ conduction by generating additional interlayer channels for Li+ migration and lowering the migration barrier (Fig. 4f).31 Specifically, these planar defects altered the distribution of Y3+, forming face-shared YCl63− octahedra and other sparse Y3+ distribution regions. The presence of stronger Coulomb repulsion near these YCl63− octahedra made the Li+ diffusion channel along the c-axis locally disconnected, whereas these disconnected Li sites were reconnected by loops to form the new diffusion channel with lower migration barriers. And in the sparse Y3+ distribution area, Li+ were subject to weakened Coulomb repulsion by Y3+ and the migration barrier was reduced. Fig. 4g shows the Li+ migration barriers predicted by the four different models, and it could be found that the stacking fault model had the smallest migration barriers. This provided important data support for the fact that high concentration stacking faults can boost the Li+ migration in Li3YCl6. It is worth mentioning that samples synthesized by mechanochemical methods tend to exhibit both stacking faults and cation disorder due to the lower energy required for the occurrence of stacking faults.80,81
In addition, the blocking effect of M3+, grain boundaries and impurities have also been shown to be important factors affecting ion migration.29,39,82 Only comprehensive consideration of the influence mechanisms of all factors can help us understand ion migration more deeply.
3. Enhanced ion transport by chemical substitution
To date, although Li3ScCl6 exhibits the highest ionic conductivity (σ = 3 × 10−3 S cm−1) among all ternary halide SSEs, it is still not comparable to the excellent ionic transport properties of conventional organic electrolytes.39 In order to facilitate the practical application of halide SSEs, chemical substitution is considered to be an effective method to enhance ionic conductivity, which optimizes the components through elements with different charges and radii to influence the structural framework and vacancy concentration. Table 1 summarizes the crystal structure and ionic conductivity changes of typical halide SSEs before and after various chemical substitutions. Chemical substitution can be categorized into three categories: (1) aliovalent cation substitution; (2) isovalent cation substitution; (3) anion substitution. It is worth noting that the influence of each substitution type on the ion transport mechanism is completely different and will be discussed in detail below.| Original materials | Crystal structures | σ (S cm−1) | Modified materials | Crystal structures | σ (S cm−1) | T (°C) | Ref. |
|---|---|---|---|---|---|---|---|
| Li3ErCl6 | hcp (Pm1) |
8.7 × 10−5 | Li2.633Er0.633Zr0.367Cl6 | hcp (pnma II) | 1.1 × 10−3 | 25 | 26 |
| Li3YCl6 | hcp (pnma) | 6.08 × 10−5 | Li3Y0.5In0.5Cl6 | ccp (C2/m) | 1.51 × 10−3 | 25 | 44 |
| Li3YCl6 | hcp (Pm1) |
1.39 × 10−4 | Li2.4Y0.4Hf0.6Cl4 | hcp (Pm1) |
1.49 × 10−3 | RT | 83 |
| Li3YCl6 | hcp (Pm1) |
— | Li3YBr3Cl3 | ccp (C2/m) | 7.2 × 10−3 | RT | 29 |
| Li3YbCl6 | hcp (Pm1) |
1.9 × 10−4 | Li2.6Yb0.6Zr0.4Cl6 | ccp (C2/m) | 1.5 × 10−3 | 30 | 59 |
| Li3LuCl6 | hcp (pnma I) | 4.0 × 10−4 | Li2.5Lu0.5Zr0.5Cl6 | hcp (pnma II) | 1.5 × 10−3 | RT | 84 |
| Li3HoCl6 | hcp (Pm1) |
1.0 × 10−4 | Li2.6Ho0.6Zr0.4Cl6 | hcp (pnma II) | 1.8 × 10−3 | RT | 85 |
| Li3HoCl6 | hcp (Pm1) |
1.2 × 10−4 | Li3HoCl4Br2 | ccp (C2/m) | 1.24 × 10−3 | 25 | 86 |
| Li3InCl6 | ccp (C2/m) | 8.8 × 10−4 | Li2.9In0.9Zr0.1Cl6 | ccp (C2/m) | 1.54 × 10−3 | 20 | 87 |
| Li3InCl6 | ccp (C2/m) | 4.7 × 10−4 | Li2.6In0.6Zr0.4Cl6 | ccp (C2/m) | 1.25 × 10−3 | RT | 78 |
| Li3InCl6 | ccp (C2/m) | 6.7 × 10−4 | Li2.7In0.7Hf0.3Cl6 | ccp (C2/m) | 1.28 × 10−3 | 25 | 88 |
| Li3InCl6 | ccp (C2/m) | 9.7 × 10−4 | Li2.6In0.8Ta0.2Cl6 | ccp (C2/m) | 4.47 × 10−3 | 30 | 89 |
| Li3InCl6 | ccp (C2/m) | 8.49 × 10−4 | Li2.75Y0.16Er0.16Yb0.16In0.25Zr0.25Cl6 | ccp (C2/m) | 1.17 × 10−3 | RT | 90 |
| Li3InCl6 | ccp (C2/m) | 9.8 × 10−4 | Li2.9In0.75Zr0.1Sc0.05Er0.05Y0.05Cl6 | ccp (C2/m) | 2.18 × 10−3 | 30 | 91 |
| Li3ScCl6 | ccp (C2/m) | 6.7 × 10−4 | Li2.5Sc0.5Zr0.5Cl6 | ccp (C2/m) | 2.23 × 10−3 | 25 | 92 |
| Li3ScCl6 | ccp (C2/m) | — | Li2.375Sc0.375Zr0.625Cl6 | ccp (C2/m) | 2.2 × 10−3 | 25 | 93 |
| Li2Sc2/3Cl4 | hcp (pnma) | 1.5 × 10−3 | Li2Sc0.222In0.444Cl6 | hcp (Fdm) |
2.0 × 10−3 | 25 | 30 |
| Li2ZrCl6 | hcp (Pm1) |
4.0 × 10−4 | Li2.25Zr0.75Fe0.25Cl6 | hcp (Pm1) |
9.8 × 10−4 | 30 | 43 |
| Li2ZrCl6 | ccp (C2/m) | 7.1 × 10−6 | Li2.7Zr0.3(In/Sc)0.7Cl6 | ccp (C2/m) | 2.1 × 10−3 | 30 | 94 |
| Li2ZrCl6 | hcp (Pm1) |
5.7 × 10−6 | Li2.5Zr0.5Y0.5Cl6 | hcp (Pm1) |
1.19 × 10−3 | RT | 95 |
| Li2ZrCl6 | hcp (Pm1) |
1.2 × 10−4 | Li2.25Zr0.75Al0.25Cl6 | hcp (Pm1) |
1.13 × 10−3 | 25 | 96 |
| Li2ZrCl6 | hcp (Pm1) |
3.0 × 10−4 | Li2.1Zr0.95Mg0.05Cl6 | hcp (Pm1) |
6.2 × 10−4 | 25 | 97 |
| Li2ZrCl6 | hcp (Pm1) |
4.0 × 10−4 | Li2.1Zr0.95Mn0.05Cl6 | hcp (Pm1) |
8.0 × 10−4 | RT | 98 |
| Li2ZrCl6 | hcp (Pm1) |
3.3 × 10−4 | Li3.1ZrCl4.9O1.1 | ccp (C2/m) | 1.3 × 10−3 | 25 | 99 |
| Li2ZrCl6 | hcp (Pm1) |
3.97 × 10−4 | Li3.4ZrCl4.6O1.4 | ccp (C2/m) | 1.46 × 10−3 | 25 | 100 |
| Li2ZrF6 | hcp (Pm1) |
— | Li2ZrF5Cl | hcp (Pm1) |
5.5 × 10−7 | 25 | 101 |
| Li2HfCl6 | hcp (Pm1) |
3.98 × 10−4 | Li2.3Hf0.7In0.3Cl6 | ccp (C2/m) | 1.05 × 10−3 | 30 | 102 |
3.1 Aliovalent cation substitution
The enhancement of the Lithium-ion transport properties in halide SSEs by aliovalent substitution cannot be separated from many factors. In addition to the increase in vacancy concentration due to the introduction of aliovalent cations, changes in Li+ concentration and distribution as well as the evolution of the local structural framework are also crucial. For trivalent metal halides, tetravalent Zr4+ and Hf4+ are often used as aliovalent cations.26,85,93,103,104 Because of their abundant reserves, low cost, suitable sizes and good redox stability, they are expected to achieve large-scale commercial application of halide SSEs.As the packing style of the anion sublattice depends on the cation–anion radius ratio, aliovalent substitution leads to the transformation of the crystal structure or rearrangement of the Li+ sublattice, which results in a significant change for the carrier diffusion path. Park et al. successfully achieved structural modulation and ionic conductivity enhancement by introducing Zr4+ into Li3MCl6 (M = Er/Y).26 The crystal structure experienced evolution from the trigonal phase (phase-I) to the orthorhombic I phase (phase-II) to the orthorhombic II phase (phase-III) with the addition of Zr4+ (Fig. 5a). Among them, the orthorhombic II phase structure exhibited the highest ionic conductivity (σ = 1.1 × 10−3 S cm−1), which was attributed to the tilting of the (Er/Zr)Cl6 octahedra and the creation of additional Tet interstitial Li3 sites (Fig. 5b), strengthening the 3D Li+ diffusion path. As shown in Fig. 5c, the ASSLB using Li2.633Er0.633Zr0.367Cl6 as the cathode electrolyte exhibited higher discharge capacity (more than 110 mAh g−1) and coulombic efficiency (96.4%), superior to that of the Li3PS4. In fact, this phase transition process is also related to the sample preparation temperature. In another study, Park et al. achieved the phase transition of Li3−xYb1−xHfxCl6 from the trigonal phase to the monoclinic phase at lower annealing temperature (Fig. 5d), exhibiting higher ionic conductivity.59 It was attributed to the fact that the presence of more Tet interstitial sites in the monoclinic phase structure played an active role in the establishment of the fast 3D diffusion network. However, as shown in Fig. 5e, the trend of ionic conductivity with doping amount is not linear. Moderate Hf4+ doping increases the vacancy concentration and provides more available sites for Li+ hopping, whereas excessive Hf4+ doping causes a dramatic decrease in carrier concentration and contraction of lattice spacing, which in turn inhibits the Li+ transport behavior. Therefore, designing a reasonable doping scheme to balance the vacancy content and carrier content is the key to modulating ion transport behavior. This is evidenced in the study of Wang et al., who concluded that the optimal ion transport behavior can only be realized when the vacancy concentration is equal to the carrier concentration (Fig. 5f).84
 | ||
| Fig. 5 Effect of aliovalent substitution on halide phase evolution and ionic conductivity. (a) Phase evolution and corresponding ionic conductivities for Li3−xEr1−xZrxCl6. (b) Li+ connection along the (100) direction. (c) Charge–discharge voltage profiles of ASSLBs using different SSEs. Reproduced with permissions from ref. 26. Copyright 2020, American Chemical Society. (d) Phase evolution of Li3−xYb1−xHfxCl6 annealed at different temperatures. (e) Changes in ionic conductivity at different temperatures for Li3−xYb1−xHfxCl6. Reproduced with permissions from ref. 59. Copyright 2021, Elsevier. (f) Effect of the correlation between vacancy concentration and Li-ion content on ion transport. Reproduced with permissions from ref. 84. Copyright 2023, The Royal Society of Chemistry. | ||
It is worth mentioning that not all aliovalent substitutions cause the transformation of intrinsic structure such as Li3−xIn1−x(Zr/Hf)xCl6 and Li3−xSc1−x(Zr/Hf)xCl6, which maintain the ccp structure throughout the solid solution range.87,88,105,106 Because their ionic radii are very similar, doping does not make the average ionic radius of the central metal change significantly. For these compounds, aliovalent substitution enhances ionic conductivity mainly by the synergistic effect of the Li+ sublattice rearrangement and migration path optimization. In the case of Li3InCl6 with a typical layered structure (Fig. 6a), In3+ is only distributed in the (001) plane, while Li+ occupies both the (001) and (002) plane.78 After doping Zr4+, the Li+ on the Tet interstitial site Li3 disappears, and the Li+ content on the M2/Li4 site increases, implying that Li+ is more preferred in occupying the M2/Li4 site with the mixed cation distribution. The former provides more vacancies for Li+ migration along the ab-plane, the latter raises Li+ diffusion rate along the c-axis due to the weakened coulombic repulsion from high-valence cations. The redistribution of Li+ sites leads to the formation of migration channels (Li1/Li2–Li3–M2/Li4–Li3–Li1/Li2) with lower activation barriers (Fig. 6b), enhancing the 3D Li+ diffusion. Similarly, the enhanced 3D diffusivity can also be explained by the change in the preferred orientation of lattice plane.87 For example, by doping Zr4+ in Li3ScCl6, Li et al. made the original random orientation ab planes tend to be aligned in a uniform orientation (Fig. 6c), which reduced the migration resistance of Li+ conduction in all directions (Fig. 6d), especially in the ab plane.92 This optimized ion migration behavior was also reflected by the excellent rate capability of the battery (Fig. 6e). Notably, it is not difficult to find in Fig. 6d that the Li+ conduction along the c-axis was the speed control step, which determined the overall Li+ migration rate. Therefore, the effect of Zr4+ substitution on the change of c-axis channel size in Li3ScCl6 was carefully considered. The researchers found that the triangular bottleneck area for Li+ migration along the c-axis was increased with the increase of Zr4+ (Fig. 6f), which lowered the migration barrier of the c-axis, obtaining a higher ionic conductivity.93
 | ||
| Fig. 6 Cation site distribution and migration pathway affected by aliovalent substitution. (a) Crystal structure of Li3InCl6. (b) Optimal Li+ diffusion pathway from bond valence sum calculations. Reproduced with permissions from ref. 78. Copyright 2021, American Chemical Society. (c) Schematic illustration of Zr4+ substitution changing the (001) plane preferred orientation. (d) Comparison of activation energy through different migration pathways before and after Zr4+ doping. (e) Rate capability of batteries after Zr4+ doping. Reproduced with permissions from ref. 92. Copyright 2022, Elsevier. (f) Change in the bottleneck area and migration barrier for Li+ diffusion along the c-axis. The inset depicts the bottleneck area for Li+ migration along the c-axis. Reproduced with permissions from ref. 93. Copyright 2024, American Chemical Society. (g) Correlation curves of lattice parameter with In3+ doping amount. Reproduced with permissions from ref. 94. Copyright 2022, Elsevier. | ||
Recently, Li2ZrCl6 has attracted much attention due to its cost-effectiveness and good environmental stability, but the lower ionic conductivity restricts its further development, and is related to its insufficient number of carriers.107–109 In view of this, Kwak and his coworkers doped In3+/Sc3+ in Li2ZrCl6 to increase the carrier content in the (002) plane, favoring the more convenient Li+ migration along the ab plane.94 At the same time, due to the difference in electronegativity of the central metal elements, mixed ionic-covalent bonding is formed in the crystal structure and local anisotropic lattice expansion is triggered (Fig. 6g). The existence of mixed ion–covalent bonding is the signature characteristic of rapid Li+ transport. In addition, attempts to dope trivalent low-cost metal elements (such as Fe3+, Al3+) and divalent metal elements (such as Mg2+, Mn2+) into Li2ZrCl6 have also been shown to effectively improve ionic conductivity by increasing carrier contents and broadening migration channels.43,96–98
3.2 Isovalent cation substitution
Compared with aliovalent substitution, the isovalent substitution of the central metal cation does not affect the carrier content and vacancy content. Instead, it enhances the ionic conductivity mainly by changing the basic structural framework or migration channel size due to the difference in ionic radii. Li et al. elevated the ionic conductivity of Li3YCl6 by doping In3+, and its structure was gradually converted from hcp structure to ccp monoclinic phase structure (Fig. 7a).44 In particular, the ionic conductivity of all samples reached 1 × 10−3 S cm−1 when the doping amount exceeded 50%, which confirms that the monoclinic phase structure is the most favorable structure for Li+ conduction. It is worth mentioning that In3+ doping also significantly improves the humidity stability of Li3Y1−xInxCl6, which is attributed to the formation of Li3Y1−xInxCl6·xH2O intermediates to prevent the hydrolysis of Li3YCl6 (Fig. 7b). Inspired by Li3Y1−xInxCl6, Zhou et al. synthesized the chlorospinels Li2Sc0.666−xInxCl6 (0 ≤ x ≤ 0.666), which exhibited high ionic conductivity (1.83–2.03 × 10−3 S cm−1) over the entire compositional range.30 It was associated with the low occupancy of Oct sites and Tet interstitial sites, providing more vacancy sites and forming the 3D Li+ diffusion channel with a lower migration barrier (Fig. 7c and d). Particularly, the ASSLB assembled with Li2In1/3Sc1/3Cl6 also displayed favorable charge–discharge behavior and rate capability when matched with LiNi0.85Co0.1Mn0.05O2 (NCM85), as shown in Fig. 7e. In addition, although some computational simulations have shown that La3+ has the potential to enhance ionic conductivity by increasing the lattice size and broadening the migration channels due to the larger ionic radius, further experiments are needed for validation.110,111 | ||
| Fig. 7 Influence of isovalent substitution of central metal cation on chemical/electrochemical stability. (a) Li+ conductivity at 25 °C for Li3Y1−xInxCl6 and corresponding phase transitions. (b) Comparison of humidity stability between Li3Y1−xInxCl6 and Li3YCl6. Reproduced with permissions from ref. 44. Copyright 2020, American Chemical Society. (c) The main 3D Li+ diffusion channels of Li2Sc0.666−xInxCl6. (d) Enlarged pathways of Li+ diffusion between tetrahedra and octahedra. (e) Charge–discharge curves of the ASSLB loaded with Li2In1/3Sc1/3Cl6 at different rates. Reproduced with permissions from ref. 30. Copyright 2022, Springer Nature. | ||
3.3 Anion substitution
As the skeleton ions constituting the bulk structure of halide SSEs, the local coordination environment of halogen ions plays a crucial role in the ion transport rate. By changing the polarization of the Li–X bond, modifications in the lattice parameters and even anion sublattice stacking style can be triggered.Based on the fact that the more polarizable anion sublattice is more beneficial for Li+ migration, Tomita and his group explored the effect of different halogen doping on the crystal structure and ionic conductivity in Li3InBr6.21,112 As expected, F− doping caused the contraction of the lattice parameter and the decrease in ionic conductivity, while I− doping led to the expansion of the lattice parameter and the increase in ionic conductivity. Surprisingly, because Cl− doping is ordered enough to compensate for the negative effects of lattice shrinkage, the ionic conductivity was also ultimately improved. The substitution mechanism was further explained in studies regarding Li3HoCl6−xBrx, Li3HoBr6−xIx and Li2ZrF6−xClx.80,86,101 For example, Plass et al. obtained faster Li+ diffusion channels with low migration barriers by doping Li3HoBr6 with I−, which has a larger ionic radius and lower electronegativity. This effect originated from the increase in interplanar distance and coordination polyhedral volume, which led to the widening of Li+ diffusion channels. Meanwhile, the weaker bond strength between Li+ and X− led to the weakening of the binding force of the skeleton ion against Li+. It should be noted that the doping amount of I− should not be excessive, as this affects the mixed distribution of cation sites in the lattice and increases the electrostatic repulsion, thus inhibiting the rapid Li+ diffusion and lowering the ionic conductivity.
Besides lattice expansion and lattice softening, anion substitution can also induce a change in the anion sublattice stacking style. Liu et al. prepared a new halide family Li3MBr3Cl3 (M = Y/Er), by mechanochemical milling and hot-pressing (HP) treatment, which exhibited the same ccp monoclinic phase structure as the endmember Li3YBr6.29 Li3YBr3Cl3 achieved ultrahigh ionic conductivity (∼7.2 × 10−3 S cm−1), which was the result of synergistic modulation of the two factors. On the one hand, due to the mixed distribution of Li+ in the Oct and Tet interstitial sites, more vacancies were generated in the Oct site and the energy barrier was lowered (Fig. 8a and b), thus optimizing the 3D Li+ diffusion channel. On the other hand, the hot-pressing-treated samples formed denser blocks and better grain boundary contacts, which greatly reduced the grain boundary impedance and benefited the overall ionic conductivity. The high conductivity of Li3YBr3Cl3 was further explained by a first-principles study.113 Intralayer vacancy diffusion in the Li layer promotes interlayer concerted diffusion between different metal layers, which accelerates the Li+ transport rate.
 | ||
| Fig. 8 Modification of crystal structure and Li+ site distribution by anion substitution. (a) Mixed Tet–Oct distribution of Li+ revealed via Fourier difference map from neutron diffraction of LYBC-HP. (b) Optimized Li+ 3D diffusion pathway due to increased vacancy content at Oct sites. Reproduced with permissions from ref. 29. Copyright 2020, American Chemical Society. (c) Crystal structure of LiNbOCl4. Reproduced with permissions from ref. 114. Copyright 2023, Wiley-VCH. (d) Effect of O2− doping on carrier concentration and migration channels. Reproduced with permissions from ref. 100. Copyright 2024, Elsevier. | ||
Significantly, although it is widely believed that O2− with high electronegativity is not favorable for fast ionic conduction, many recent studies have demonstrated that O2− doping possesses unique advantages in enhancing ionic conductivity.115,116 As shown in Fig. 8c, Tanaka et al. reported a new oxyhalide SSE, LiMOCl4 (M = Nb/Ta), belonging to the orthorhombic structure (space group: Cmc21), in which O2− is only responsible for connecting the octahedra together.114 The addition of O2− broadened the bottleneck size of the Li+ diffusion channel (1.939 Å), achieving an ultrahigh ionic conductivity of 1.07 × 10−2 S cm−1, which is comparable to those of liquid electrolytes. In addition, O2− doping can also affect the ionic conductivity by changing the crystal structure and Li+ site distribution. Park and Cheng et al. achieved a structural transition from the hcp trigonal phase to the ccp monoclinic phase by increasing the amount of O2− doping in Li2ZrCl6.99,100 Accompanied by the stabilization of Li interstitial sites and the enrichment of carrier concentration (Fig. 8d), the ion transport channel was broadened and the ionic conductivity was successfully raised above 1 × 10−3 S cm−1. Interestingly, O2− doping seems to enhance the amorphous proportion of halide SSEs, which is extremely important for inducing polyhedrons distortions and lowering the migration barriers.34,117,118 Similar amorphous characteristics have been observed in recent studies on nitrogen doping. The Li3xTaCl5Nx synthesized by Hong et al. exhibits a highly amorphous structure, demonstrating exceptional ionic conductivity (up to 7.34 × 10−3 S cm−1), significantly exceeding that of most conventional halide SSEs.35 This remarkable performance was attributed to the incorporation of nitrogen, which alters the local coordination environments of both cations and anions, thereby establishing a unique and efficient Li+ migration mechanism within the amorphous structure.
3.4 Double-doping strategy
In order to provide halide SSEs with both high conductivity and excellent interfacial stability, double-doping strategies have emerged as a concern. For example, Subramanian et al. prepared Li2.4Y0.4Zr0.6Cl5.85F0.15 SSE through the high-energy ball milling method.119 Among them, Zr4+ doping increased the vacancy concentration and cation disorder, and a high ionic conductivity of 1.45 × 10−3 S cm−1 was obtained, whereas F− doping contributed to the formation of stable Li+-conducting cathode–electrolyte interphase (CEI), and the electrochemical window was widened to 1.29–3.9 V. In addition, structural modulation by the double-doping strategy was also observed in a UCl3-type LaCl3 SSE. Hao et al. achieved high ionic conductivity and superior compatibility with Li metal by doping both Zr4+ and O2− in a LaCl3-based SSE.64 It was attributed to the Zr4+–O2− co-doping strategy that makes the anion sublattice more stable and smoothes the 1D Li+ transport channel. Indeed, such a double-doping strategy can be further extended to multi-ion doping.91,120 Song and his group synthesized the high-entropy halide SSE Li2.75Y0.16Er0.16Yb0.16In0.25Zr0.25Cl6 (HE-LIC) by replacing In3+ in LIC with various metal elements.90 The local lattice distortion triggered by the high entropy effect effectively improved the diffusion kinetics of Li+ and Cl−, which enhanced the Li+ conductivity and oxidation stability.In summary, the chemical substitution of halide SSEs can tune the vacancy concentration and modify the Li+ migration channel, thereby attaining aliostructural/isostructural alloy halides with higher ionic conductivity. Nevertheless, more efforts are expected to explore the feasibility of multi-element doping in order to further enrich the halide SSEs family to improve its competitiveness in the SSEs field.
4. Interface modification by chemical substitution
In addition to the ionic conductivity of SSEs, interfacial issues between SSEs/electrodes should not be neglected, as they significantly affect the coulombic efficiency, rate capability and cycle life of ASSLBs.45,121–124 In order to construct high-energy and long-life ASSLBs, it is necessary to investigate the electrochemical stability of halide SSEs and effectively regulate the interface behavior through modification strategies.4.1 Intrinsic electrochemical stability
The electrochemical stability of SSEs is closely related to the electrochemical stability window, the upper and lower limits of which represent the plating/stripping potentials, respectively, in which SSEs do not suffer from any redox reactions. And SSE with a wide electrochemical stability window can be better matched with high-voltage cathode active materials and high specific capacity Li metal anode to maximize the energy density of ASSLBs and achieve stable cycling. As shown in Fig. 9a, Mo et al. evaluated the thermodynamic equilibrium voltage curves and phase equilibria of Li3YCl6 and Li3YBr6 by theoretical calculations, both of which have a wide electrochemical window (Li3YCl6: 0.62–4.21 V; Li3YBr6: 0.59–3.15 V), hoping to achieve great contact with the electrodes.66 The relationship between the thermodynamic intrinsic electrochemical window and composition of halide SSEs is specified in Fig. 9b, where the F− and Cl−-based halides indeed exhibit high oxidation potentials. However, the calculation results of the mutual energy between chloride SSEs and cathode materials show that chloride SSEs exhibit high mutual energy (∼100 meV) when matched with the high-pressure ternary system NCM (Fig. 9c).125 It is well above the threshold for the interfacial reaction to occur, implying that the severe interfacial reaction is occurring. As for the reduction stability at low potentials, even though the group III elements represented by the Sc, Y, and La–Lu exhibit the lowest reduction potentials (0.41–0.92 V vs. Li+/Li), it is still higher relative to the Li metal anode.66 | ||
| Fig. 9 Intrinsic electrochemical stability of halide SSEs. (a) The thermodynamic equilibrium voltage profile and phase equilibria for Li3YCl6 and Li3YBr6. (b) Calculated thermodynamics of inherent electrochemical windows for Li–M–X ternary compounds (M = cation, X = F, Cl, Br, I, O, S). Reproduced with permissions from ref. 66. Copyright 2019, Wiley-VCH. (c) Mutual reaction energy between common cathodes and halide SSEs. Reproduced with permissions from ref. 125. Copyright 2021, American Chemical Society. (d) The percentage volume changes caused by the SSEs/cathode interfacial reaction. Compounds are mechanically stable with a percent change in volume of less than 10%. Reproduced with permissions from ref. 130. Copyright 2023, The Royal Society of Chemistry. | ||
In order to improve the high-voltage stability and reduction stability of halide SSEs, current modification measures include adding a coating or buffer layer between the electrode materials and SSEs.27,126–129 However, this increases the assembly cost of ASSLBs and is not suitable for commercial production. Chemical substitution can affect the degree of electron localization between M and X due to the difference in electronegativity, thus determining the oxidation and reduction potentials. Tham et al. explored the influence of different substitution types on the electrochemical stability of halide SSEs with the help of first-principles calculations.130 They suggested that doping halogen ions with higher electronegativity yields higher oxidation potentials, while doping cations with lower electronegativity obtains lower reduction potentials due to the weakened attraction against electrons. This provides design guidelines for the experimental synthesis of halide SSEs with excellent electrochemical stability. Furthermore, the volume change at the SSEs/electrode interface is also crucial for interfacial compatibility; in particular, the Ni-based cathode material has serious volume shrinkage under high pressure, resulting in cracks and contact loss at the interface, which decays the cycle performance.131–133 The partially substituted halides shown in Fig. 9d demonstrate excellent mechanical stability and are expected to be key components in high-voltage batteries.130 The battery configurations and electrochemical performances of ASSLBs using halide SSEs are summarized in Table 2.
| Battery configurations | T (°C) | Voltage range (V vs. Li+/Li) | Electrochemical performances (mA h g−1) | Ref. | |
|---|---|---|---|---|---|
| Current density/specific capacity/initial coulombic efficiency (ICE) | Current density/cycle number/specific capacity/capacity retention | ||||
| Li–In//Li3YCl6//LCO | 25 | 2.5–4.2 | 0.44C/∼117/94.8% | 0.1C/100/∼114/98% | 22 |
| Li11Sn6//Li3PS4//LCO@Li2.633Er0.633Zr0.367Cl6 | RT | 3–4.3 | 0.1C/∼112/96.4% | 0.5C/200/∼80/— | 26 |
| Li–In//Li6PS5Cl//Li2.4Y0.4Hf0.6Cl6//LCO | RT | 2.9–4.2 | 0.1C/120/93% | 0.1C/100/∼80/70% | 83 |
| Li//Li5.3PS4.3Cl1.0Br0.7//Li2.4Y0.6Zr0.4Cl5.85F0.15//NCM811 | RT | 2.5–4.25 | 0.1C/190/87% | 0.5C/250/128/95.5% | 119 |
| Li//Li3YBr6//LCO@LIC | RT | 2.5–4.2 | 0.14 mA cm−2/126.7/89% | 0.14 mA cm−2/70/∼15/12% | 136 |
| Li//Li3YBr5.7F0.3//LCO@LIC | RT | 2.5–4.2 | 0.14 mA cm−2/121.6/90% | 0.14 mA cm−2/70/∼73/60% | 136 |
| Li–In//Li6.7Si0.7Sb0.3S5I//Li2.7Yb0.7Zr0.3Cl6//NCM622 | RT | 2.8–4.3 | 0.2C/170/— | 0.2C/150/136/80% | 103 |
| Li–In//Li10GeP2S12//Li2.556Yb0.492Zr0.492Cl6//LCO | 25 | 2.5–4.5 | 0.1C/193.9/93.3% | 0.3C/50/149.7/82.1% | 60 |
| In//Li10GeP2S12//Li3InCl6//NCM811 | 25 | 2.5–4.4 | 0.13 mA cm−2/154/84.2% | 0.13 mA cm−2/70/150/97.4% | 23 |
| Li–In//Li6PS5Cl//Li3InCl6//LCO | RT | 3–4.2 | 0.1C/104.4/92.2% | 0.1C/50/∼57/54.5% | 88 |
| Li–In//Li6PS5Cl//Li2.7In0.7Hf0.3Cl6//LCO | RT | 3–4.2 | 0.1C/108.1/96.8% | 0.1C/50/∼7871.7% | 88 |
| Li–In//Li6PS5Cl//Li2.6In0.8Ta0.2Cl6//LCO | 30 | 3–4.6 | 4C/∼135/— | 4C/1400/∼95/70% | 89 |
| In//Li6PS5Cl//Li3InCl6//LCO@Li3InCl4.8F1.2 | RT | 2.6–4.47 | 0.063 mA cm−2/160.6/92% | 0.125 mA cm−2/70/102/— | 28 |
| In//Li3ScCl6//LCO | 25 | 2.5–4.2 | 0.13 mA cm−2/126.2/90.3% | 0.13 mA cm−2/160/104.5/82.8% | 39 |
| Li–In//Li6PS5Cl//Li2.5Sc0.5Zr0.5Cl6//NCM811 | RT | 2.8–4.3 | 0.1C/203.6/89.6% | 0.2C/200/174.5/90% | 92 |
| Li–In//Li6.7Si0.7Sb0.3S5I//Li2Sc2/3Cl4//NCM622 | RT | 2.8–4.3 | 0.5C/∼170/— | 0.1C/110/170/— | 41 |
| Li–In//Li6.7Si0.7Sb0.3S5I//Li2Sc1/3In1/3Cl4//NCM85 | RT | 2.8–4.8 | 0.2C/∼215/— | 0.2C/110/∼205/95% | 30 |
| Li–In//Li6PS5Cl//Li2ZrCl6//LCO | 25 | 2.5–4.2 | 14 mA g−1/137/97.9% | 70 mA g−1/100/114/— | 107 |
| Li–In//Li6PS5Cl//Li2ZrCl6//NCM811 | 25 | 2.98–4.18 | 0.1C/152.3/84.3% | 0.1C/100/∼133/87.4% | 96 |
| Li–In//Li6PS5Cl//Li2.25Zr0.75Al0.25Cl6//NCM811 | 25 | 2.98–4.18 | 0.1C/166.8/87.2% | 0.1C/100/∼157/94.3% | 96 |
| Li–In//Li6PS5Cl//Li2.8Zr0.2In0.8Cl6//NCM811 | 25 | 2.82–4.42 | 0.2C/174.5/87.5% | 1C/500/∼110/74% | 137 |
| Li–In//Li6PS5Cl//Li2.1Zr0.95Mg0.05Cl6//LCO | 25 | 3–4.3 | 0.1C/147/96.6% | 0.3C/100/121.3/∼87.1% | 97 |
| Li–In//LZCl5.6F0.4//LCO | 30 | 2.52–4.32 | 0.1C/132/95.57% | 0.5C/70/86.4/76% | 138 |
| Li–In//Li6PS5Cl//Li1.75ZrCl4.75O0.5//LCO | 25 | 2.48–4.18 | 14 mA g−1/137.5/98.28% | 700 mA g−1/150/102/— | 116 |
| Li–In//Li6PS5Cl//Li2.5ZrCl5F0.5O0.5//NCM955 | RT | 2.5–4.35 | 0.1C/207.1/— | 0.5C/500/125.8/81.2% | 139 |
| Li//Li0.388Ta0.238La0.475Cl3//NCM523 | 30 | 2.20–4.35 | 0.44C/163/84.96% | 0.44C/100/∼133.8/81.6% | 32 |
| Li–In//Li3YCl6//Li1.25TaCl5N0.42//NCM811 | 25 | 3–4.3 | 0.5C/∼130/— | 0.5C/350/124.4/95.47% | 35 |
| Li–In//Li6PS5Cl//LiAlOCl-981//LCO | 25 | 2.5–4.3 | 0.1C/145.85/97.3% | 0.5C/300/∼117/86.95% | 118 |
| Li–In//Li6PS5Cl//HE-LIC//LCO | 25 | 2.5–4.2 | 0.1C/144/97% | 0.5C/500/∼115/88.9% | 90 |
4.2 Stability of halide SSEs towards Li metal anode
In view of the poor reduction stability of halide SSEs, when they are directly matched with Li metal anode, continuous interfacial reaction and lithium puncture will occur, shortening battery life and even causing safety problems. For example, the contact of Li3MCl6 with the Li metal anode leads to the occurrence of a reduction reaction, in which M3+ is reduced to M0.134,135 M0 and another reaction product, LiCl, form a mixed ion–electron conduction (MIEC) interface (Fig. 10a), where successive electrons pass through the interface to drive the continuation of the thermodynamically favorable reduction reaction. The continued accumulation of reaction products rapidly increases the interfacial impedance and eventually gives rise to battery failure as Li3MCl6 or Li metal is depleted. This was confirmed by Riegger et al. in their study on the interfacial compatibility between Li3InCl6 and Li metal anode.135 As shown in Fig. 10b, the interfacial impedance increased with time, which was attributed to the non-self-limiting interfacial reaction between the Li3InCl6 and the Li metal anode.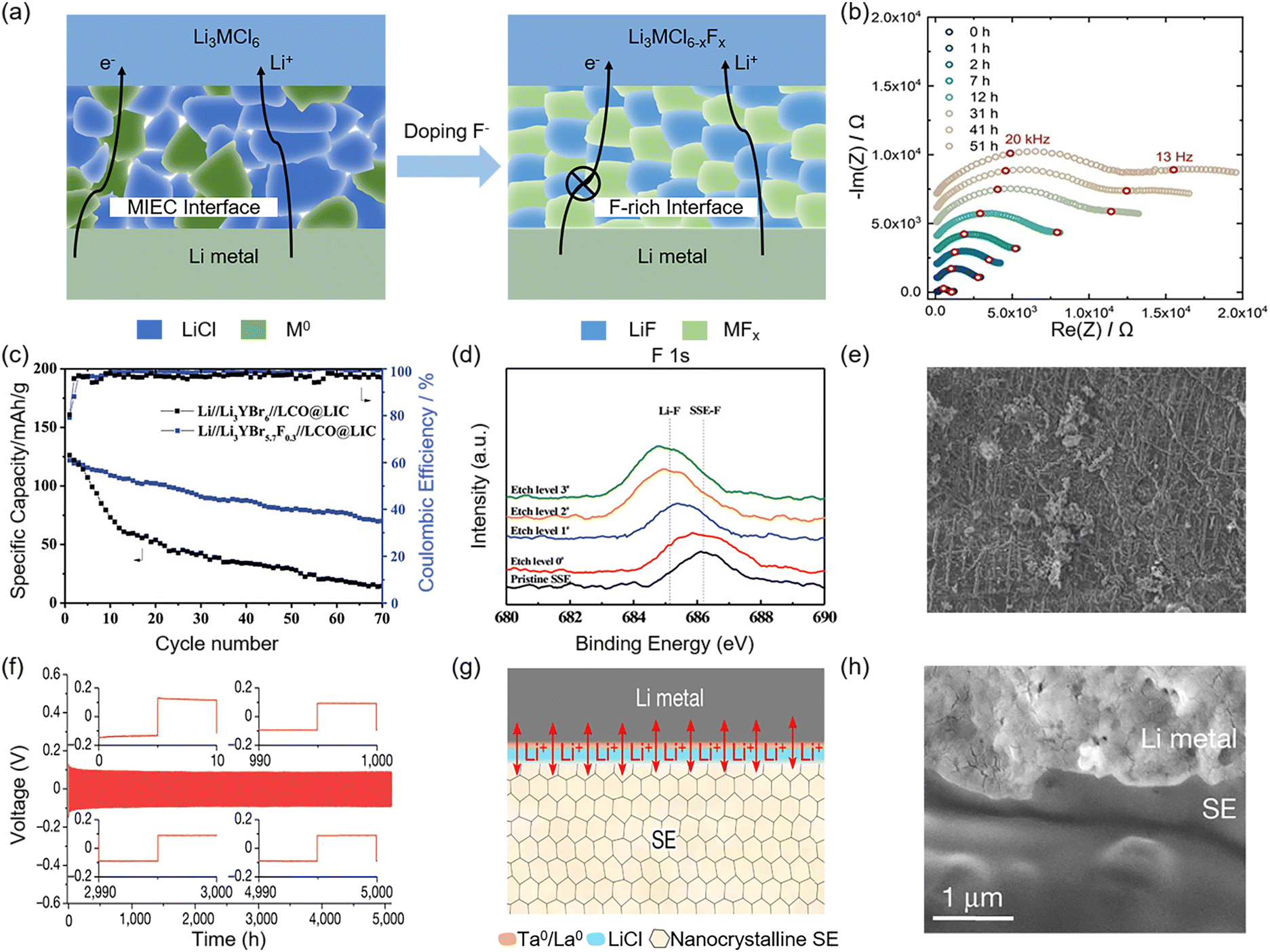 | ||
| Fig. 10 Stability between halide SSEs and Li metal anode. (a) Interfacial comparison of Li3MCl6 and Li3MCl6−xFx in contact with Li metal anode. (b) Evolution of interfacial impedance with time extension of Li//Li3InCl6//Li symmetric battery. Reproduced with permissions from ref. 135. Copyright 2020, Wiley-VCH. (c) Cycling stability for Li//Li3YBr5.7F0.3//LCO@LIC battery. (d) Overall XPS spectra evolution of F 1s at the Li3YBr5.7F0.3/Li interface under different etch levels. (e) SEM of the Li metal anode surface morphology after cycling 1000 h with Li3YBr5.7F0.3 SSE direct matching. Reproduced with permissions from ref. 136. Copyright 2021, Wiley-VCH. (f) Li plating/stripping voltage profiles for Li//Li0.388Ta0.238La0.475Cl3//Li symmetric battery. (g) Schematic of the gradient insulation passivation layer. The red double arrows represent Li+ fluxes. (h) Cross-sectional SEM of Li/Li0.388Ta0.238La0.475Cl3/Li metal interface after cycling 50 h. Reproduced with permissions from ref. 32. Copyright 2023, Springer Nature. | ||
In liquid batteries, LiF is the key component for stabilizing SEI membranes due to its wide electrochemical window, excellent ionic conductivity and electronic insulation as well as suitable elastic modulus.140–142 Correspondingly, constructing an in situ-formed F-rich interfacial layer at the SSEs/Li metal anode interface can effectively inhibit the formation of the MIEC interface and lithium dendrite growth (Fig. 10a).143–145 Yu et al. attempted to dope F− into Li3YBr6 to improve the degradation of Li3YBr6 in contact with the Li metal anode.136 The assembled Li//Li3YBr5.7F0.3//LCO@LIC battery maintained a higher discharge capacity even after 70 cycles (Fig. 10c), which was attributed to the findings from the investigation of the SSE/Li interface post-cycling. XPS depth profiling results revealed that the characteristic peak of the Li–F bond at ≈684.7 eV gradually increased with increasing etching level, indicating the formation of abundant LiF at the interface (Fig. 10d). This was corroborated by the SEM image of the SSE/Li interface, which showed a dense and homogeneous F-rich interfacial layer with a reticular structure (Fig. 10e). The in situ F-rich interfacial layer not only passivated side reactions at the interface but also mitigated the excessive stress caused by inhomogeneous Li deposition, thereby inhibiting the growth of lithium dendrites.
Interestingly, the amorphous content of the material also seems to influence the reduction potential. For example, the reduction potential of amorphous and crystalline Li0.35La0.55TiO3 differs by 0.5 V.146 Hu et al. obtained materials with more amorphous content by doping O2− in Li2ZrCl6; as a result Li1.75ZrCl4.75O0.5 exhibited a reduction potential of 0.37 V lower than that of Li2ZrCl6.116 However, the magnitude of this reduction was not sufficient to achieve direct compatibility between Li1.75ZrCl4.75O0.5 and the bare Li metal anode, as demonstrated by the high overpotential (∼0.7 V) observed during cycling of the assembled Li//Li1.75ZrCl4.75O0.5//Li symmetric battery. Therefore, the incorporation of an intermediate layer, such as LPSC, between Li1.75ZrCl4.75O0.5 and the Li metal anode remains crucial to alleviate severe interfacial side reactions and guarantee stable electrochemical performance. In addition, the LaCl3-based SSE developed by Yin et al., Li0.388Ta0.238La0.475Cl3, could achieve close contact with the Li metal anode. At a current density of 1 mA h cm−2, the lithium symmetric battery could maintain stability for over 5000 hours (Fig. 10f), primarily due to the formation of a gradient insulating passivation layer composed of electron-insulating LiCl at the interface, which promoted uniform Li plating/stripping behavior (Fig. 10g). In particular, the dense nanocrystalline characteristics of Li0.388Ta0.238La0.475Cl3 also contributed to enhancing the SSE/Li interfacial stability, as demonstrated by the close contact between Li0.388Ta0.238La0.475Cl3 and the Li metal anode, which persisted even after 50 hours of cycling (Fig. 10h).
4.3 Stability of halide SSEs towards high-voltage cathodes
Although most halide SSEs display high oxidation potentials (>4 V vs. Li+/Li), they are still insufficient to directly match some high-voltage Ni-rich layered LiMO2 (M = Ni, Co, Mn, and Al) cathodes. In the case of Cl-based halides, Cl− is oxidized to form the Li-deficient metal chloride MCl3 above 4.3 V.66 In particular, when Li3InCl6 comes into contact with high-capacity Li1.15Mn0.53Ni0.265Co0.055O2 (LRM), the Li3InCl6 suffers a severe oxidation reaction to form In2O3 at a high voltage of 4.8 V.147 These Li-deficient metal compounds block the Li+ transport path in the composite cathode and continuously decompose the SSE, thus deteriorating the cycling stability of the ASSLBs. Hence, the search for SSEs with both high-voltage stability and high ionic conductivity is crucial for the construction of high energy density ASSLBs.The F-based halide is predicted to have the oxidation potential of more than 6 V, so the oxidation reaction under high voltage can be inhibited by in situ formation of F-rich CEI at the cathode/SSEs interface (Fig. 11a). A dual-halogen SSE, Li3InCl4.8F1.2, was developed by Zhang et al.28 Compared with the Li3InCl6, the ASSLB with bare LiCoO2 and Li3InCl4.8F1.2 could deliver higher reversible capacity (203.7 mAh g−1) and initial coulombic efficiency (89.2%) at a cut-off voltage of 4.8 V (Fig. 11c). According to the first principles computation result, this stemmed from the generation of F-containing compounds (including LiF, LiInF4, and InF3) after Li3InCl4.8F1.2 exceeded the theoretical oxidation limit of 4.42 V (Fig. 11b), which formed a passivated interphase that prevented the SSE from further decomposition. This was further confirmed by the F K-edge XAS spectra of the cycled LCO/SSE composite cathodes, both of which exhibited a high-energy shift compared with the pristine SSE and a low-energy shift in absorption edges compared with standard LiF (Fig. 11d). This implied the formation of a LiF-rich passivating cathode-electrolyte interphase (CEI) after the first cycle, and subsequent scanning transmission X-ray microscopy (STXM) mapping of a single cycled LCO particle also showed a uniform distribution of F on the surface (Fig. 11e).
 | ||
| Fig. 11 Stability between halide SSEs and high-voltage cathode. (a) Formation of F-rich CEI prevents blockage of Li+ conduction path in composite cathode. (b) Calculated phase equilibria of Li3InCl4.8F1.2 at varous Li+/Li potentials. (c) Charge–discharge voltage profiles comparison of different cathode SSE batteries in the voltage range of 2.6–4.8 V. (d) F K-edge XAS spectra of the cycled LCO/SSE composite cathodes. (e) STXM mapping image of a single cycled LCO particle. Reproduced with permissions from ref. 28. Copyright 2021, Wiley-VCH. (f) TEM image of cycled NCM with LZCFO. (g) Comparison of NiF2−, ClO−, ClO2− content in cyclic NCM with LZCFO and LZC from ToF-SIMS. (h) Difference in ClOx− content from the deconvoluted Cl 2p XPS spectrum. Reproduced with permissions from ref. 139. Copyright 2024, Wiley-VCH. (i) Long-term cycling of the NCM85 ASSB with Li2In1/3Sc1/3Cl4 between 2.8 V and 4.8 V. Reproduced with permissions from ref. 30. Copyright 2022, Springer Nature. | ||
Similarly, Shen et al. succeeded in raising the practical oxidation limit of Li2ZrCl6 to 4.87 V with excellent ionic conductivity (∼1.13 × 10−3 S cm−1) by employing the F−/O2− co-doping strategy.139 The TEM image of the cycled NCM with LZCFO, shown in Fig. 11f, indicated that a flat CEI layer was uniformly generated on the surface, covering the particle, which was responsible for the enhanced oxidation stability. The chemical composition of the CEI layer for the cycled composite cathode was further revealed by ToF-SIMS and XPS (Fig. 11g). The concentration of ClOx− for the cycled NCM with LZCFO was significantly lower, demonstrating that the decomposition of the SSE was effectively inhibited by the formation of the F-rich CEI. Surprisingly, unlike F− doping, the mixed metal–chlorine spinel halide Li2In1/3Sc1/3Cl4 prepared by Zhou et al. was able to achieve stable cycling at a cut-off voltage of 4.8 V when matched with bare NCM85 (Fig. 11i).30 It was benefited by the low electronic conductivity of Li2In1/3Sc1/3Cl4 (4.7 × 10−10 S cm−1) leading to minimal reaction with NCM85. In addition, related studies have suggested that the oxidation stability of halide SSEs may also be related to the stability degree of the valence electron structure for the central metal element. As described by Xu et al., Li3YbCl6 exhibits good oxidation stability at over 4.5 V due to the fully occupied atomic orbitals of Yb, suggesting that Yb3+ has the potential to enhance the high-voltage stability of halide SSEs.60
In summary, chemical substitution has been shown to broaden the intrinsic electrochemical window of halide SSEs, making an important contribution for driving the high energy density of halide-based ASSLBs.
5. Summary and outlook
As opposed to other types of SSEs, it is the intrinsic characteristics of halide SSEs that have enabled them to become the potential stock for the energy storage field of high-performance ASSLBs in recent years. The first is that the lower valence of halogen ions gives the halide SSEs weaker bond cooperation against Li+, resulting in lower energy barrier migration for Li+ in the skeleton structure. Secondly, the larger ionic radii and higher polarizability of halogen ions facilitate the formation of wider Li+ transport channels and higher ion mobility, which ultimately enables halide SSEs to display excellent ion transport kinetics and good deformability. Even the stringent requirements of ASSLBs for the assembly environment are reduced due to the stronger stability of halide SSEs in air/humidity.In this review, starting from the structural framework and ionic conduction mechanism, the basic concepts of halide SSEs were discussed and the implications of anion polarizability, cation disorder and stacking faults on Li-ion migration were specifically analyzed. Immediately thereafter, the common substitution types were summarized and it was shown that optimized crystal structures and Li+ migration kinetics can be obtained by selecting suitable substituent elements and contents for the chemical modification of halide SSEs. In addition, the mechanism of severe interfacial reactions between halide SSEs and high-voltage cathodes or Li metal anode has been revealed, and the strategy of inhibiting interfacial side reaction by using chemical substitution has been proposed to achieve excellent electrochemical compatibility with common electrodes. Although halide SSEs have made impressive achievements in just a few years, shortcomings caused by the physicochemical nature of halogen ions still prevent them from being commercially promoted. Some potential future research directions are as follows:
(1) In-depth exploration of the relationship between local structure evolution and ion transport. Currently, the RT ionic conductivity of most halide SSEs has not yet reached 10−3 S cm−1; in particular, the development of fluoride SSEs is seriously limited, with the maximum of only 10−5 S cm−1, which is far from the theoretically calculated value. The ionic conductivity of halide SSEs is related to their overall/local structure, so it is necessary to have a comprehensive and thorough understanding of the crystal structure and ion migration path of halide SSEs, with the focus on how the local structural evolution shapes the ion transport behavior in halide SSEs. In addition, while amorphous halide SSEs demonstrate great potential in terms of reducing ion migration barrier, the role played by amorphous degree in achieving high ionic conductivity remains unclear, necessitating further exploration into their formation mechanisms. The mechanical knowledge of structure–property relationships can provide materials design principles for engineering strategies such as regulating the trade-off between vacancy concentration and Li+ content, modulating cation disorder and optimizing synthesis parameters, ultimately giving rise to the quantum leap in the ionic conductivity of halide SSEs.
(2) Advanced theoretical calculations can guide the discovery of novel halide SSEs with excellent ionic conductivity and electrochemical stability. Boosting the ion transport performance of halide SSEs through elemental substitution has been widely recognized as an effective tool, but the consideration of substitution elements for ternary halide SSEs has traditionally been obtained through a large number of experimental studies, which is both time-consuming and labor-intensive. With the development of computer technology, the advent of high-throughput simulation helps to quickly screen and identify appropriate substituent ions and content, which greatly elevates the efficiency of preliminary experiments and shortens the experiment period. In addition, advanced computational methods, including AIMD and DFT, can be used to evaluate the chemical/electrochemical stability of halide SSEs, which in turn can be used to select mild operating conditions and high specific energy electrode materials for matching.
(3) In situ/operational observation of interface problems using fine characterization techniques. The degree of interfacial reaction between ASSLBs components is so important that it can determine the overall ion transport rate of ASSLBs. Yet, using conventional interfacial characterization techniques is difficult to accurately reflect the dynamic evolution of the charge/discharge process, which makes the exploration of the interfacial problems always ambiguous. Therefore, there is an urgent need for high-precision characterization techniques, such as transmission X-ray microscopy and X-ray computed tomography, to continuously monitor the interfacial contact between components at high resolution without damaging the interface. And the theoretical model established with the aid of computational simulation can provide clear insight into the structure and function of various surfaces, so as to better guide the formulation of strategies for improving the interfacial stability.
To conclude, only by combining advanced theoretical calculation and characterization methods to clarify the basic research content of structure and composition can we promote the commercialization progress of halide SSEs in high energy density ASSLBs, and better satisfy the wide temperature range and long lifetime required in practical working conditions.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We gratefully acknowledge support from the National Key R&D Program of China (2021YFB2400400, 2022YFB2404600), the National Natural Science Foundation of China (Grant No. 22379120), the Key Research and Development Plan of Shanxi Province (China, Grant No. 2021JLM-36), “Young Talent Support Plan” of Xi'an Jiaotong University (71211201010723), “Young Talent Support Plan” of Xi'an Jiaotong University (HG6J003), “1000-Plan program” of Shaanxi Province and the Higher Education Institution Academic Discipline Innovation and Talent Introduction Plan (“111 Plan”) (No. B23025).References
- A. Manthiram, X. Yu and S. Wang, Nat. Rev. Mater., 2017, 2, 16103 CrossRef CAS.
- C. Sun, J. Liu, Y. Gong, D. P. Wilkinson and J. Zhang, Nano Energy, 2017, 33, 363–386 CrossRef CAS.
- Q. Zhao, S. Stalin, C.-Z. Zhao and L. A. Archer, Nat. Rev. Mater., 2020, 5, 229–252 CrossRef CAS.
- L. Zhao, A. E. Lakraychi, Z. Chen, Y. Liang and Y. Yao, ACS Energy Lett., 2021, 6, 3287–3306 Search PubMed.
- H. Jamal, F. Khan, S. Hyun, S. W. Min and J. H. Kim, J. Mater. Chem. A, 2021, 9, 4126–4137 Search PubMed.
- H. Jamal, F. Khan, H.-R. Si and J. H. Kim, J. Mater. Chem. A, 2021, 9, 27304–27319 Search PubMed.
- E. Kim, H. Jamal, I. Jeon, F. Khan, S.-E. Chun and J. H. Kim, Adv. Energy Mater., 2023, 13, 2301674 Search PubMed.
- Z. Li, J. Fu, X. Zhou, S. Gui, L. Wei, H. Yang, H. Li and X. Guo, Adv. Sci., 2023, 10, 2201718 Search PubMed.
- J. Tan, Z. Wang, J. Cui, Z. Jia, W. Tian, C. Wu, C. Peng, C. Shu, K. Yang and W. Tang, J. Energy Chem., 2024, 95, 288–295 CrossRef CAS.
- K. J. Kim, M. Balaish, M. Wadaguchi, L. Kong and J. L. M. Rupp, Adv. Energy Mater., 2021, 11, 2002689 CrossRef CAS.
- Z. Jia, H. Shen, J. Kou, T. Zhang, Z. Wang, W. Tang, M. Doeff, C.-Y. Chiang and K. Chen, Adv. Mater., 2024, 36, 2309019 Search PubMed.
- Q. Zhang, D. Cao, Y. Ma, A. Natan, P. Aurora and H. Zhu, Adv. Mater., 2019, 31, 1901131 Search PubMed.
- J. Wu, S. Liu, F. Han, X. Yao and C. Wang, Adv. Mater., 2021, 33, 2000751 CrossRef CAS PubMed.
- C. Wang, J. Liang, Y. Zhao, M. Zheng, X. Li and X. Sun, Energy Environ. Sci., 2021, 14, 2577–2619 Search PubMed.
- X. Li, J. Liang, X. Yang, K. R. Adair, C. Wang, F. Zhao and X. Sun, Energy Environ. Sci., 2020, 13, 1429–1461 RSC.
- H. Kwak, S. Wang, J. Park, Y. Liu, K. T. Kim, Y. Choi, Y. Mo and Y. S. Jung, ACS Energy Lett., 2022, 7, 1776–1805 CrossRef CAS.
- K. Tuo, C. Sun and S. Liu, Electrochem. Energy Rev., 2023, 6, 17 Search PubMed.
- D. C. Ginnings and T. E. Phipps, J. Am. Chem. Soc., 1930, 52, 1340–1345 CrossRef CAS.
- C. C. Liang, J. Epstein and G. H. Boyle, J. Electrochem. Soc., 1969, 116, 1452 CrossRef CAS.
- A. Bohnsack, G. Balzer, M. S. Wickleder, H.-U. Güdel and G. Meyer, Z. Anorg. Allg. Chem., 1997, 623, 1352–1356 CrossRef CAS.
- Y. Tomita, H. Matsushita, K. Kobayashi, Y. Maeda and K. Yamada, Solid State Ionics, 2008, 179, 867–870 CrossRef CAS.
- T. Asano, A. Sakai, S. Ouchi, M. Sakaida, A. Miyazaki and S. Hasegawa, Adv. Mater., 2018, 30, 1803075 CrossRef PubMed.
- X. Li, J. Liang, N. Chen, J. Luo, K. R. Adair, C. Wang, M. N. Banis, T. Sham, L. Zhang, S. Zhao, S. Lu, H. Huang, R. Li and X. Sun, Angew. Chem., Int. Ed., 2019, 58, 16427–16432 Search PubMed.
- R. Schlem, S. Muy, N. Prinz, A. Banik, Y. Shao-Horn, M. Zobel and W. G. Zeier, Adv. Energy Mater., 2020, 10, 1903719 Search PubMed.
- R. Schlem, T. Bernges, C. Li, M. A. Kraft, N. Minafra and W. G. Zeier, ACS Appl. Energy Mater., 2020, 3, 3684–3691 Search PubMed.
- K.-H. Park, K. Kaup, A. Assoud, Q. Zhang, X. Wu and L. F. Nazar, ACS Energy Lett., 2020, 5, 533–539 CrossRef CAS.
- C. Wang, J. Liang, J. Luo, J. Liu, X. Li, F. Zhao, R. Li, H. Huang, S. Zhao, L. Zhang, J. Wang and X. Sun, Sci. Adv., 2021, 7, eabh1896 Search PubMed.
- S. Zhang, F. Zhao, S. Wang, J. Liang, J. Wang, C. Wang, H. Zhang, K. Adair, W. Li, M. Li, H. Duan, Y. Zhao, R. Yu, R. Li, H. Huang, L. Zhang, S. Zhao, S. Lu, T.-K. Sham, Y. Mo and X. Sun, Adv. Energy Mater., 2021, 11, 2100836 Search PubMed.
- Z. Liu, S. Ma, J. Liu, S. Xiong, Y. Ma and H. Chen, ACS Energy Lett., 2021, 6, 298–304 CrossRef CAS.
- L. Zhou, T.-T. Zuo, C. Y. Kwok, S. Y. Kim, A. Assoud, Q. Zhang, J. Janek and L. F. Nazar, Nat. Energy, 2022, 7, 83–93 Search PubMed.
- E. Sebti, H. A. Evans, H. Chen, P. M. Richardson, K. M. White, R. Giovine, K. P. Koirala, Y. Xu, E. Gonzalez-Correa, C. Wang, C. M. Brown, A. K. Cheetham, P. Canepa and R. J. Clément, J. Am. Chem. Soc., 2022, 144, 5795–5811 CrossRef CAS PubMed.
- Y.-C. Yin, J.-T. Yang, J.-D. Luo, G.-X. Lu, Z. Huang, J.-P. Wang, P. Li, F. Li, Y.-C. Wu, T. Tian, Y.-F. Meng, H.-S. Mo, Y.-H. Song, J.-N. Yang, L.-Z. Feng, T. Ma, W. Wen, K. Gong, L.-J. Wang, H.-X. Ju, Y. Xiao, Z. Li, X. Tao and H.-B. Yao, Nature, 2023, 616, 77–83 Search PubMed.
- W. Li, M. Li, P.-H. Chien, S. Wang, C. Yu, G. King, Y. Hu, Q. Xiao, M. Shakouri, R. Feng, B. Fu, H. Abdolvand, A. Fraser, R. Li, Y. Huang, J. Liu, Y. Mo, T.-K. Sham and X. Sun, Sci. Adv., 2023, 9, eadh4626 Search PubMed.
- S. Zhang, F. Zhao, L.-Y. Chang, Y.-C. Chuang, Z. Zhang, Y. Zhu, X. Hao, J. Fu, J. Chen, J. Luo, M. Li, Y. Gao, Y. Huang, T.-K. Sham, M. D. Gu, Y. Zhang, G. King and X. Sun, J. Am. Chem. Soc., 2024, 146, 2977–2985 CrossRef CAS PubMed.
- B. Hong, L. Gao, P. Nan, Y. Li, M. Liu, R. Zou, J. Gu, Q. Xu, J. Zhu and S. Han, Angew. Chem., Int. Ed., 2024, e202415847 Search PubMed.
- J. Hu, K. Chen and C. Li, ACS Appl. Mater. Interfaces, 2018, 10, 34322–34331 CrossRef CAS PubMed.
- J. Hu, Z. Yao, K. Chen and C. Li, Energy Storage Mater., 2020, 28, 37–46 CrossRef.
- X. Li, J. Liang, J. Luo, M. Norouzi Banis, C. Wang, W. Li, S. Deng, C. Yu, F. Zhao, Y. Hu, T.-K. Sham, L. Zhang, S. Zhao, S. Lu, H. Huang, R. Li, K. R. Adair and X. Sun, Energy Environ. Sci., 2019, 12, 2665–2671 Search PubMed.
- J. Liang, X. Li, S. Wang, K. R. Adair, W. Li, Y. Zhao, C. Wang, Y. Hu, L. Zhang, S. Zhao, S. Lu, H. Huang, R. Li, Y. Mo and X. Sun, J. Am. Chem. Soc., 2020, 142, 7012–7022 CrossRef CAS PubMed.
- S. Muy, J. Voss, R. Schlem, R. Koerver, S. J. Sedlmaier, F. Maglia, P. Lamp, W. G. Zeier and Y. Shao-Horn, iScience, 2019, 16, 270–282 Search PubMed.
- L. Zhou, C. Y. Kwok, A. Shyamsunder, Q. Zhang, X. Wu and L. F. Nazar, Energy Environ. Sci., 2020, 13, 2056–2063 RSC.
- X. Shi, Z. Zeng, M. Sun, B. Huang, H. Zhang, W. Luo, Y. Huang, Y. Du and C. Yan, Nano Lett., 2021, 21, 9325–9331 CrossRef CAS PubMed.
- H. Kwak, D. Han, J. Lyoo, J. Park, S. H. Jung, Y. Han, G. Kwon, H. Kim, S.-T. Hong, K.-W. Nam and Y. S. Jung, Adv. Energy Mater., 2021, 11, 2003190 Search PubMed.
- X. Li, J. Liang, K. R. Adair, J. Li, W. Li, F. Zhao, Y. Hu, T.-K. Sham, L. Zhang, S. Zhao, S. Lu, H. Huang, R. Li, N. Chen and X. Sun, Nano Lett., 2020, 20, 4384–4392 CrossRef CAS PubMed.
- F. Liu, L. Gao, Z. Zhang, L. Zhang, N. Deng, Y. Zhao and W. Kang, Energy Storage Mater., 2024, 64, 103072 Search PubMed.
- L. Guo, J. Zheng, L. Zhao and Y. Yao, MRS Bull., 2023, 48, 1247–1256 Search PubMed.
- C. Zhang, H. Zhuang, Z. Qi, X. Liu and Y. Ren, Mater. Today Commun., 2023, 35, 105764 CrossRef CAS.
- H. Liu, D. Li, C. Dong, Y. Li, H. Yuan, D. Yu, L. Gao, P. Ding, Y. Li, Z. Qin, Y. Liang, H. Luo, L. Li, Y. Ren, L. Fan and C. Nan, Adv. Energy Mater., 2024, 14, 2402064 CrossRef CAS.
- D. Lee, Z. Cui, J. B. Goodenough and A. Manthiram, Small, 2024, 20, 2306053 CrossRef CAS PubMed.
- C. Wang, X. Li, Y. Zhao, M. N. Banis, J. Liang, X. Li, Y. Sun, K. R. Adair, Q. Sun, Y. Liu, F. Zhao, S. Deng, X. Lin, R. Li, Y. Hu, T. Sham, H. Huang, L. Zhang, R. Yang, S. Lu and X. Sun, Small Methods, 2019, 3, 1900261 Search PubMed.
- Y. Shi, Q. Li, X. Hu, Y. Liao, W. Li, Y. Xu, B. Zhao, J. Zhang and Y. Jiang, Nano Energy, 2024, 120, 109150 CrossRef CAS.
- T. Duan, H. Cheng, Y. Liu, Q. Sun, W. Nie, X. Lu, P. Dong and M.-K. Song, Energy Storage Mater., 2024, 65, 103091 Search PubMed.
- Y.-C. Lan, P.-H. Lai, B. D. Vogt and E. D. Gomez, ACS Energy Lett., 2024, 9, 3324–3334 CrossRef CAS.
- X. Shi, Z. Zeng, H. Zhang, B. Huang, M. Sun, H. H. Wong, Q. Lu, W. Luo, Y. Huang, Y. Du and C.-H. Yan, Small Methods, 2021, 5, 2101002 Search PubMed.
- X. Luo, D. Cai, X. Wang, X. Xia, C. Gu and J. Tu, ACS Appl. Mater. Interfaces, 2022, 14, 29844–29855 Search PubMed.
- T. Ma, Z. Wang, D. Wu, P. Lu, X. Zhu, M. Yang, J. Peng, L. Chen, H. Li and F. Wu, Energy Environ. Sci., 2023, 16, 2142–2152 Search PubMed.
- J. Liang, X. Li, K. R. Adair and X. Sun, Acc. Chem. Res., 2021, 54, 1023–1033 Search PubMed.
- C. Wang, J. Liang, J. T. Kim and X. Sun, Sci. Adv., 2022, 8, eadc9516 Search PubMed.
- J. Park, D. Han, H. Kwak, Y. Han, Y. J. Choi, K.-W. Nam and Y. S. Jung, Chem. Eng. J., 2021, 425, 130630 CrossRef CAS.
- G. Xu, L. Luo, J. Liang, S. Zhao, R. Yang, C. Wang, T. Yu, L. Wang, W. Xiao, J. Wang, J. Yu and X. Sun, Nano Energy, 2022, 92, 106674 CrossRef CAS.
- J. Pan, L. Gao, X. Zhang, D. Huang, J. Zhu, L. Wang, Y. Wei, W. Yin, Y. Xia, R. Zou, Y. Zhao and S. Han, Inorg. Chem., 2024, 63, 3418–3427 Search PubMed.
- J. Fu, S. Wang, J. Liang, S. H. Alahakoon, D. Wu, J. Luo, H. Duan, S. Zhang, F. Zhao, W. Li, M. Li, X. Hao, X. Li, J. Chen, N. Chen, G. King, L.-Y. Chang, R. Li, Y. Huang, M. Gu, T.-K. Sham, Y. Mo and X. Sun, J. Am. Chem. Soc., 2023, 145, 2183–2194 Search PubMed.
- J.-D. Luo, Y. Zhang, X. Cheng, F. Li, H.-Y. Tan, M.-Y. Zhou, Z.-W. Wang, X.-D. Hao, Y.-C. Yin, B. Jiang and H.-B. Yao, Angew. Chem., Int. Ed., 2024, 63, e202400424 CrossRef CAS PubMed.
- X. Hao, K. Chen, M. Jiang, Y. Tang, Y. Liu and K. Cai, J. Mater. Chem. A, 2024, 12, 18459–18468 Search PubMed.
- K. Chen, X. Hao, M. Jiang and Y. Tang, J. Alloys Compd., 2024, 997, 174945 CrossRef CAS.
- S. Wang, Q. Bai, A. M. Nolan, Y. Liu, S. Gong, Q. Sun and Y. Mo, Angew. Chem., Int. Ed., 2019, 58, 8039–8043 Search PubMed.
- S. R. Combs, P. K. Todd, P. Gorai and A. E. Maughan, J. Electrochem. Soc., 2022, 169, 040551 Search PubMed.
- T. Famprikis, P. Canepa, J. A. Dawson, M. S. Islam and C. Masquelier, Nat. Mater., 2019, 18, 1278–1291 CrossRef CAS PubMed.
- A. Van Der Ven, J. Bhattacharya and A. A. Belak, Acc. Chem. Res., 2013, 46, 1216–1225 CrossRef CAS PubMed.
- B. He, F. Zhang, Y. Xin, C. Xu, X. Hu, X. Wu, Y. Yang and H. Tian, Nat. Rev. Chem., 2023, 7, 826–842 Search PubMed.
- G. Yang, X. Liang, S. Zheng, H. Chen, W. Zhang, S. Li and F. Pan, eScience, 2022, 2, 79–86 CrossRef.
- Y. Liu, S. Wang, A. M. Nolan, C. Ling and Y. Mo, Adv. Energy Mater., 2020, 10, 2002356 CrossRef CAS.
- R. Kanno and M. Murayama, J. Electrochem. Soc., 2001, 148, A742 CrossRef CAS.
- Z. Xu, X. Chen, K. Liu, R. Chen, X. Zeng and H. Zhu, Chem. Mater., 2019, 31, 7425–7433 CrossRef CAS.
- N. Flores-González, M. López, N. Minafra, J. Bohnenberger, F. Viñes, S. Rudić, I. Krossing, W. G. Zeier, F. Illas and D. H. Gregory, J. Mater. Chem. A, 2022, 10, 13467–13475 RSC.
- S. Muy, J. C. Bachman, L. Giordano, H.-H. Chang, D. L. Abernathy, D. Bansal, O. Delaire, S. Hori, R. Kanno, F. Maglia, S. Lupart, P. Lamp and Y. Shao-Horn, Energy Environ. Sci., 2018, 11, 850–859 RSC.
- R. Schlem, A. Banik, S. Ohno, E. Suard and W. G. Zeier, Chem. Mater., 2021, 33, 327–337 CrossRef CAS.
- B. Helm, R. Schlem, B. Wankmiller, A. Banik, A. Gautam, J. Ruhl, C. Li, M. R. Hansen and W. G. Zeier, Chem. Mater., 2021, 33, 4773–4782 Search PubMed.
- H. Ito, K. Shitara, Y. Wang, K. Fujii, M. Yashima, Y. Goto, C. Moriyoshi, N. C. Rosero-Navarro, A. Miura and K. Tadanaga, Adv. Sci., 2021, 8, 2101413 Search PubMed.
- M. A. Plass, S. Bette, R. E. Dinnebier and B. V. Lotsch, Chem. Mater., 2022, 34, 3227–3235 Search PubMed.
- M. A. Plass, S. Bette, N. Philipp, I. Moundrakovski, K. Küster, R. E. Dinnebier and B. V. Lotsch, J. Mater. Chem. A, 2023, 11, 13027–13038 Search PubMed.
- H. J. Lee, B. Darminto, S. Narayanan, M. Diaz-Lopez, A. W. Xiao, Y. Chart, J. H. Lee, J. A. Dawson and M. Pasta, J. Mater. Chem. A, 2022, 10, 11574–11586 Search PubMed.
- K. Tuo, C. Sun, C. A. López, M. T. Fernández-Díaz and J. A. Alonso, J. Mater. Chem. A, 2023, 11, 15651–15662 Search PubMed.
- C. Wang, S. Wang, X. Liu, Y. Wu, R. Yu, H. Duan, J. T. Kim, H. Huang, J. Wang, Y. Mo and X. Sun, Energy Environ. Sci., 2023, 16, 5136–5143 Search PubMed.
- L. Zhou, T. Zuo, C. Li, Q. Zhang, J. Janek and L. F. Nazar, ACS Energy Lett., 2023, 8, 3102–3111 Search PubMed.
- J. Peng, L. Xian and L.-B. Kong, Ionics, 2023, 29, 2657–2664 CrossRef CAS.
- X. Luo, X. Wu, J. Xiang, D. Cai, M. Li, X. Wang, X. Xia, C. Gu and J. Tu, ACS Appl. Mater. Interfaces, 2021, 13, 47610–47618 CrossRef CAS PubMed.
- H. Wang, Y. Li, Y. Tang, D. Ye, T. He, H. Zhao and J. Zhang, ACS Appl. Mater. Interfaces, 2023, 15, 5504–5511 CrossRef CAS PubMed.
- Y. Ye, J. Geng, D. Zuo, K. Niu, D. Chen, J. Lin, X. Chen, H. J. Woo, Y. Zhu and J. Wan, ACS Nano, 2024, 18, 18368–18378 CrossRef CAS PubMed.
- Z. Song, T. Wang, H. Yang, W. H. Kan, Y. Chen, Q. Yu, L. Wang, Y. Zhang, Y. Dai, H. Chen, W. Yin, T. Honda, M. Avdeev, H. Xu, J. Ma, Y. Huang and W. Luo, Nat. Commun., 2024, 15, 1481 CrossRef CAS PubMed.
- D. Li, D. Yu, G. Zhang, A. Du, Z. Ye, Y. Jia, W. Hou, T. Xu, F. Li, S. Chi, Y. Zhu and C. Yang, Angew. Chem., Int. Ed., 2024, e202419735 Search PubMed.
- W. Li, Z. Chen, Y. Chen, W. Duan, G. Liu, Y. Lv, H. Yang and L. Yao, Chem. Eng. J., 2023, 455, 140509 Search PubMed.
- R. Li, P. Lu, X. Liang, L. Liu, M. Avdeev, Z. Deng, S. Li, K. Xu, J. Feng, R. Si, F. Wu, Z. Zhang and Y.-S. Hu, ACS Energy Lett., 2024, 9, 1043–1052 CrossRef CAS.
- H. Kwak, D. Han, J. P. Son, J. S. Kim, J. Park, K.-W. Nam, H. Kim and Y. S. Jung, Chem. Eng. J., 2022, 437, 135413 CrossRef CAS.
- S. Chen, C. Yu, C. Wei, Z. Jiang, Z. Zhang, L. Peng, S. Cheng and J. Xie, Energy Mater. Adv., 2023, 4, 0019 Search PubMed.
- K.-N. Gao, F. Bai, Z. Sun and T. Zhang, Energy Storage Mater., 2024, 70, 103444 CrossRef.
- H. Zhang, Z. Yu, H. Chen, Y. Zhou, X. Huang and B. Tian, J. Energy Chem., 2023, 79, 348–356 Search PubMed.
- H.-J. Jeon, Y. Subramanian and K.-S. Ryu, J. Power Sources, 2024, 602, 234343 CrossRef CAS.
- K.-H. Park, S. Y. Kim, M. Jung, S.-B. Lee, M.-J. Kim, I.-J. Yang, J.-H. Hwang, W. Cho, G. Chen, K. Kim and J. Yu, ACS Appl. Mater. Interfaces, 2023, 15, 58367–58376 CrossRef CAS PubMed.
- J. Cheng, H. Zhang, Z. Wang, Y. Zhou, K. Yu, Y. Cheng, Z. Yu, X. Huang and B. Tian, J. Energy Storage, 2024, 89, 111700 CrossRef.
- E. Umeshbabu, S. Maddukuri, Y. Hu, M. Fichtner and A. R. Munnangi, ACS Appl. Mater. Interfaces, 2022, 14, 25448–25456 CrossRef CAS PubMed.
- K. Tuo, F. Yin, F. Mi and C. Sun, J. Energy Chem., 2023, 87, 12–23 CrossRef CAS.
- S. Y. Kim, K. Kaup, K.-H. Park, A. Assoud, L. Zhou, J. Liu, X. Wu and L. F. Nazar, ACS Mater. Lett., 2021, 3, 930–938 CrossRef CAS.
- S. Yu, J. Noh, B. Kim, J.-H. Song, K. Oh, J. Yoo, S. Lee, S.-O. Park, W. Kim, B. Kang, D. Kil and K. Kang, Science, 2023, 382, 573–579 CrossRef CAS PubMed.
- H. Zhang, Z. Zeng, X. Shi, C.-H. Wang and Y. Du, EcoMat, 2023, 5, e12315 CrossRef CAS.
- J. Fu, S. Yang, J. Hou, L. Azhari, Z. Yao, X. Ma, Y. Liu, P. Vanaphuti, Z. Meng, Z. Yang, Y. Zhong and Y. Wang, J. Power Sources, 2023, 556, 232465 CrossRef CAS.
- K. Wang, Q. Ren, Z. Gu, C. Duan, J. Wang, F. Zhu, Y. Fu, J. Hao, J. Zhu, L. He, C.-W. Wang, Y. Lu, J. Ma and C. Ma, Nat. Commun., 2021, 12, 4410 CrossRef CAS PubMed.
- F. Li, X. Cheng, L.-L. Lu, Y.-C. Yin, J.-D. Luo, G. Lu, Y.-F. Meng, H. Mo, T. Tian, J.-T. Yang, W. Wen, Z.-P. Liu, G. Zhang, C. Shang and H.-B. Yao, Nano Lett., 2022, 22, 2461–2469 CrossRef CAS PubMed.
- X. Luo, Y. Zhong, X. Wang, X. Xia, C. Gu and J. Tu, ACS Appl. Mater. Interfaces, 2022, 14, 49839–49846 CrossRef CAS PubMed.
- T. H. Wan and F. Ciucci, ACS Appl. Energy Mater., 2021, 4, 7930–7941 CrossRef CAS.
- Y. Ren, J. Liu, C. Zhang, H. Zhuang, C. Sun, G. Cai, X. Tan and S. Sun, Chem. Eng. J. Adv., 2022, 12, 100377 CrossRef CAS.
- Y. Tomita, H. Nishiyama, K. Kobayashi, Y. Kohno, Y. Maeda and K. Yamada, ECS Trans., 2009, 16, 137 CrossRef CAS.
- T. Jeon and S. C. Jung, J. Mater. Chem. A, 2023, 11, 4334–4344 RSC.
- Y. Tanaka, K. Ueno, K. Mizuno, K. Takeuchi, T. Asano and A. Sakai, Angew. Chem., Int. Ed., 2023, 62, e202217581 Search PubMed.
- H. Kwak, J.-S. Kim, D. Han, J. S. Kim, J. Park, G. Kwon, S.-M. Bak, U. Heo, C. Park, H.-W. Lee, K.-W. Nam, D.-H. Seo and Y. S. Jung, Nat. Commun., 2023, 14, 2459 CrossRef CAS PubMed.
- L. Hu, J. Wang, K. Wang, Z. Gu, Z. Xi, H. Li, F. Chen, Y. Wang, Z. Li and C. Ma, Nat. Commun., 2023, 14, 3807 CrossRef CAS PubMed.
- S. Zhang, F. Zhao, J. Chen, J. Fu, J. Luo, S. H. Alahakoon, L.-Y. Chang, R. Feng, M. Shakouri, J. Liang, Y. Zhao, X. Li, L. He, Y. Huang, T.-K. Sham and X. Sun, Nat. Commun., 2023, 14, 3780 CrossRef PubMed.
- G. Wang, S. Zhang, H. Wu, M. Zheng, C. Zhao, J. Liang, L. Zhou, J. Yue, X. Zhu, Y. Xu, N. Zhang, T. Pang, J. Fu, W. Li, Y. Xia, W. Yin, X. Sun and X. Li, Adv. Mater., 2025, 37, 2410402 CrossRef CAS PubMed.
- Y. Subramanian, R. Rajagopal and K.-S. Ryu, ACS Appl. Mater. Interfaces, 2024, 16, 24534–24546 CrossRef CAS PubMed.
- X. Li, Y. Xu, C. Zhao, D. Wu, L. Wang, M. Zheng, X. Han, S. Zhang, J. Yue, B. Xiao, W. Xiao, L. Wang, T. Mei, M. Gu, J. Liang and X. Sun, Angew. Chem., Int. Ed., 2023, 62, e202306433 CrossRef CAS PubMed.
- W. D. Richards, L. J. Miara, Y. Wang, J. C. Kim and G. Ceder, Chem. Mater., 2016, 28, 266–273 Search PubMed.
- Z. Wang, J. Tan, Z. Jia, J. Cui, X. Wang, C. Shu, X. Gao, Y. Wu and W. Tang, ACS Energy Lett., 2024, 9, 4485–4492 CrossRef CAS.
- C. Wang, S. Hwang, M. Jiang, J. Liang, Y. Sun, K. Adair, M. Zheng, S. Mukherjee, X. Li, R. Li, H. Huang, S. Zhao, L. Zhang, S. Lu, J. Wang, C. V. Singh, D. Su and X. Sun, Adv. Energy Mater., 2021, 11, 2100210 CrossRef CAS.
- C. Wang, K. Adair and X. Sun, Acc. Mater. Res., 2022, 3, 21–32 CrossRef CAS.
- G. H. Chun, J. H. Shim and S. Yu, ACS Appl. Mater. Interfaces, 2022, 14, 1241–1248 Search PubMed.
- W. Ji, D. Zheng, X. Zhang, T. Ding and D. Qu, J. Mater. Chem. A, 2021, 9, 15012–15018 RSC.
- X. Xu, G. Du, C. Cui, J. Liang, C. Zeng, S. Wang, Y. Ma and H. Li, ACS Appl. Mater. Interfaces, 2022, 14, 39951–39958 CrossRef CAS PubMed.
- Z. Deng, Z. Jin, D. Chen, D. Ni, M. Tian, Y. Zhan, S. Li, Y. Sun, X. Huang and Y. Zhao, ACS Appl. Mater. Interfaces, 2022, 14, 48619–48626 Search PubMed.
- H. Kobayashi, G. Yuan, Y. Gambe and I. Honma, ACS Appl. Energy Mater., 2021, 4, 9866–9870 CrossRef CAS.
- B. T. Tham, M.-S. Park, J. H. Kim and J. Moon, J. Mater. Chem. A, 2023, 11, 15968–15978 RSC.
- Y. Pang, J. Pan, J. Yang, S. Zheng and C. Wang, Electrochem. Energy Rev., 2021, 4, 169–193 CrossRef CAS.
- P. Minnmann, F. Strauss, A. Bielefeld, R. Ruess, P. Adelhelm, S. Burkhardt, S. L. Dreyer, E. Trevisanello, H. Ehrenberg, T. Brezesinski, F. H. Richter and J. Janek, Adv. Energy Mater., 2022, 12, 2201425 CrossRef CAS.
- J. Yun, H. R. Shin, T. D. Hoang, S. Kim, J. H. Choi, B. Kim, H. Jung, J. Moon and J.-W. Lee, Energy Storage Mater., 2023, 59, 102787 Search PubMed.
- Y. Fu and C. Ma, Sci. China Mater., 2021, 64, 1378–1385 CrossRef CAS.
- L. M. Riegger, R. Schlem, J. Sann, W. G. Zeier and J. Janek, Angew. Chem., Int. Ed., 2021, 60, 6718–6723 CrossRef CAS PubMed.
- T. Yu, J. Liang, L. Luo, L. Wang, F. Zhao, G. Xu, X. Bai, R. Yang, S. Zhao, J. Wang, J. Yu and X. Sun, Adv. Energy Mater., 2021, 11, 2101915 CrossRef CAS.
- K. Wang, Z. Gu, H. Liu, L. Hu, Y. Wu, J. Xu and C. Ma, Adv. Sci., 2024, 11, 2305394 Search PubMed.
- W. Tang, W. Xia, F. Hussain, J. Zhu, S. Han, W. Yin, P. Yu, J. Lei, D. S. Butenko, L. Wang and Y. Zhao, J. Power Sources, 2023, 568, 232992 CrossRef CAS.
- L. Shen, J. Li, W. Kong, C. Bi, P. Xu, X. Huang, W. Huang, F. Fu, Y. Le, C. Zhao, H. Yuan, J. Huang and Q. Zhang, Adv. Funct. Mater., 2024, 34, 2408571 CrossRef CAS.
- Z. Li, L. Wang, X. Huang and X. He, Small, 2024, 20, 2305429 CrossRef CAS PubMed.
- Y. Xiao, R. Xu, L. Xu, J.-F. Ding and J.-Q. Huang, Energy Mater., 2021, 1, 10013 Search PubMed.
- Z. Pan, H. Chen, Y. Zeng, Y. Ding, X. Pu and Z. Chen, Energy Mater., 2023, 3, 300054 CAS.
- X. Chen, Z. Jia, H. Lv, C. Wang, N. Zhao and X. Guo, J. Power Sources, 2022, 545, 231939 CrossRef CAS.
- X. Luo, X. He, H. Su, Y. Zhong, X. Wang and J. Tu, Chem. Eng. J., 2023, 465, 143036 CrossRef CAS.
- P. Ganesan, M. Soans, M. A. Cambaz, R. Zimmermanns, R. Gond, S. Fuchs, Y. Hu, S. Baumgart, M. Sotoudeh, D. Stepien, H. Stein, A. Groß, D. Bresser, A. Varzi and M. Fichtner, ACS Appl. Mater. Interfaces, 2023, 15, 38391–38402 CrossRef CAS PubMed.
- Z. Zheng, H. Fang, Z. Liu and Y. Wang, J. Electrochem. Soc., 2014, 162, A244 CrossRef.
- A. Zhang, J. Wang, R. Yu, H. Zhuo, C. Wang, Z. Ren and J. Wang, ACS Appl. Mater. Interfaces, 2023, 15, 8190–8199 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2025 |