
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMicroscopic electrochemical–mechanical coupling in layered cathodes under high-voltage and fast-charging conditions
Ziqi
Wu
a,
Chenchen
Zhang
a,
Mengyi
Zheng
a,
Chaoyuan
Zeng
 c,
Wen
Yang
*b and
Ruiwen
Shao
*ad
c,
Wen
Yang
*b and
Ruiwen
Shao
*ad
aBeijing Advanced Innovation Center for Intelligent Robots and Systems, School of Medical Technology, Beijing Institute of Technology, Beijing 100081, China. E-mail: rwshao@bit.edu.cn
bKey Laboratory of Cluster Science of Ministry of Education, Beijing Key Laboratory of Photoelectronic/Electrophotonic Conversion Materials, School of Chemistry and Chemical Engineering, Beijing Institute of Technology, Beijing 100081, P. R. China. E-mail: wenyang@bit.edu.cn
cSchool of Chemistry and Chemical Engineering, Key Laboratory of Ministry of Education for Advanced Materials in Tropical Island Resources, Collaborative Innovation Center of Ecological Civilization, Hainan University, No. 58, Renmin Avenue, Haikou 570228, China
dAnalysis & Testing Center, Beijing Institute of Technology, Beijing 102488, China
First published on 14th January 2025
Abstract
The operation of modern society and the advancement of the electric vehicle industry are intrinsically linked to high-performance lithium-ion batteries (LIBs). However, the inherent structural instability of traditional cathode materials under high voltage (high capacity) and rapid charging conditions leads to swift degradation of battery voltage and capacity. This significantly impedes the commercialization of LIBs and contributes to issues such as “range anxiety”. Currently, the failure mechanisms of cathode materials remain highly debated, with most research focusing on material modification and interfacial transport kinetics. Recent studies indicate that the accumulation of nanoscale strain and lattice displacement within cathode materials are critical driving forces behind structural degradation and oxygen loss. Despite their fundamental importance, lattice displacements and nanoscale strain remain among the least understood structural characteristics in battery materials. This review highlights the latest research advancements concerning the electrochemical–mechanical coupling mechanisms and advanced modification strategies of cathode materials. We begin by elucidating the electrochemical–mechanical failure mechanisms of cathode materials, spanning from the atomic to the macroscopic scale, and the interconnections among various failure mechanisms, with particular emphasis on lattice defects and microscopic mechanical anisotropy. Subsequently, we discuss advanced strategies and recent progress in modifying layered oxide cathode materials, which hold promise for furthering the development of the new energy industry. Finally, in the context of a carbon-neutral future and the evolving high-performance electric vehicle industry, we summarize and provide insights into the application prospects and developmental directions of layered oxide cathode materials from perspectives of cost, performance, safety, and environmental friendliness.
 Ziqi Wu | Ziqi Wu received his Bachelor's degree in 2022 at Chang'an University. He is currently pursuing his master's degree in engineering at Beijing Institute of Technology, under the supervision of Prof. Ruiwen Shao. His main research interests lie in oxide cathode materials and the precise characterization of electron microscopy. |
 Wen Yang | Wen Yang obtained his Ph.D. from the Changchun Institute of Applied Chemistry, Chinese Academy of Sciences in 2009. From 2010 to 2011, he served as a postdoctoral fellow at the Max Planck Institute for Colloids and Interfaces in Germany. His main research directions are solid-state electrochemistry and lithium-ion batteries. |
 Ruiwen Shao | Ruiwen Shao is an associate professor at the School of Medical Technology, Beijing Institute of Technology. He obtained his Ph.D. from Beijing University of Technology in 2016. From 2016 to 2018, he served as a postdoctoral fellow at the Electron Microscope Laboratory of Peking University. His primary research interests include electron microscopy and modification research on new energy batteries. |
Broader contextThe development of lithium-ion battery technology has profoundly transformed human life and is expected to revolutionize the electric vehicle industry. The cathode is the highest-cost component of the battery pack, and the battery's overall performance, particularly energy density and cycling stability, largely depends on it. However, due to the inherent structural instability of conventional cathode materials, key performance parameters like voltage and capacity deteriorate rapidly under extreme conditions such as high voltage and fast charging, hampering the growth of the electric vehicle market and contributing to issues like “range anxiety”. The accumulation of nanoscale strain and lattice displacement within cathode particles is a key driving force for structural degradation, yet this may be among the most enigmatic characteristics of battery materials. In this review, we elucidate the electrochemical–mechanical failure mechanisms of cathode materials from atomic to macroscopic scales, emphasizing the interrelations between various failure mechanisms, particularly lattice defects, nanostrain and microscopic mechanical anisotropy. We also provide an outlook on the application prospects and development directions of layered oxide cathode materials concerning cost, performance, safety, and environmental friendliness. |
1. Introduction
Over the past 30 years, LIBs have revolutionized human living standards,1 and led to the award of the Nobel Prize in Chemistry in 2019. The cathode constitutes the highest-cost component within the battery pack,2 and the overall performance of the battery, particularly its energy density and cycling stability, is largely dependent on the cathode. Over the past 25 years, the energy density of LIBs has increased at an approximate rate of 3% per year.3 However, in recent years, the rate of improvement in energy density has slowed, with the most advanced commercial LIBs currently achieving energy densities of only 300–330 W h kg−1 and 750–830 W h L−1, which falls short of meeting societal demands.4,5 Various governments have set ambitious targets for rechargeable batteries. According to “Made in China 2025”, the energy density of power batteries is expected to reach 500 W h kg−1 by 2030. Thus, developing the next-generation LIBs with superior electrochemical properties has become a top priority, with the crux of the matter lying in the pursuit of suitable cathode materials.In the context of power systems in new energy vehicles, the ideal next-generation lithium battery cathode material should exhibit the following performance characteristics: (1) high specific capacity and energy density (>500 W h kg−1 and 1000 W h L−1),6 thereby enhancing driving range while reducing the size and weight of the battery pack; (2) excellent ionic conductivity and rate capability to ensure fast-charging performance and reliable operation in cold conditions;7,8 and (3) robust cycling stability, which extends the battery's operational lifespan.9 However, current cathode materials face significant challenges in increasing the operating voltage to enhance capacity, enabling fast charging, and performing under extreme environmental conditions.10–14
During a battery's charge–discharge process, the insertion and extraction of Li+ ions within the layered structure induce a lithium concentration gradient that varies from the surface to the interior. This process simultaneously generates non-uniform elastic strain and volume changes. The extent of these volume changes, whether the lattice expands or contracts, and whether such expansion or contraction is symmetric are closely related to the electrochemical properties and microstructure of the cathode material.15 Generally, charging reduces the lattice parameters due to the extraction of Li+ from the structure, while the re-insertion of Li+ during discharging increases lattice size. The expansion rates of secondary cathode particles range from 3.3% in NCM111 to 7.8% in NCM811. In the confined space within a battery, the volume cannot change, leading to substantial stress generated by the expansion or contraction of microscopic particles.16,17 This phenomenon is particularly pronounced in solid-state batteries. If the tensile component of this interfacial stress exceeds the material's strength, it may result in brittle fracture.
Most cathode materials exhibit brittleness, leading to fracture when subjected to excessive stress, “a phenomenon” particularly observed in polycrystalline materials.18 Data analysis on the micromechanical behavior of cathode materials reveals that the fracture toughness of single crystals of LiNi0.5Mn0.3Co0.2O2 is 0.3 MPa m1/2, while that of polycrystalline agglomerates is 0.1 MPa m1/2.19 During prolonged charging and discharging, the active cathode material responds to the electrochemical loads induced by lithium insertion and extraction within the host structure, similar to the fatigue effects observed in mechanical structures subjected to cyclic external loads. For cathodes, these changes result in the accumulation of irreversible damage at both the macroscopic and microscopic levels. At the macroscopic level, this manifests as stress-induced particle fracture, traditionally considered a primary cause of cathode degradation.20–23 At the microscopic level, nanoscale strains develop, yet the destructive impact on structural stability remains unclear due to the extreme difficulty in detecting atomic-scale changes. In summary, one of the most promising strategies to mitigate mechanical instability is to eliminate the gradient of elastic strain at the microscopic level, which is inevitably generated by localized and non-uniform Li+ transport kinetics.24 Although this task may appear formidable, with its atomic-scale mechanisms still under debate, a significant amount of current research is centered around this concept.
Recent studies have demonstrated that microstrains caused by structural defects, such as dislocations and stacking faults, play a critical role in the degradation of layered transition metal oxides.25 On the one hand, localized and non-uniform Li+ transport kinetics lead to the concentration of local stresses and the generation of crystalline defects. These lattice distortions directly affect the material's electrical conductivity and ionic mobility, thereby influencing the battery's overall performance and cycle life. At the nanoscale, the impact of lattice distortions is particularly pronounced, where even minor distortions, such as crystal plane bending, lattice rotation, stacking faults, and tensile/compressive stresses, can trigger significant performance degradation.26,27 On the other hand, lattice distortions can also lead to phase transitions and crack formation, further accelerating material degradation and failure. Therefore, an in-depth investigation of the failure mechanisms driven by electrochemical–mechanical coupling at the microscopic level is crucial for understanding the root causes of cathode material failure. This understanding not only provides a theoretical foundation for developing advanced electrode materials with enhanced stability and longer cycle life but also offers guidance for optimizing battery design and improving overall battery performance.
This paper reviews recent advances in the study of failure mechanisms of layered oxide cathode materials under extreme conditions, such as high voltage and fast charging, with a particular focus on the relationship between electrochemical failure and anisotropic mechanical strain. Firstly, the electrochemical–mechanical failure mechanisms of cathode materials are explained from the microscopic to the macroscopic scale, highlighting the interrelations between various failure mechanisms, especially lattice defects and microscopic mechanical anisotropy. Secondly, representative strategies and recent advancements in modifying layered oxide cathode materials are discussed. Finally, layered oxide cathode materials’ application prospects and future development directions are summarized and projected from cost, performance, safety, and environmental friendliness perspectives.
2. Structure and kinetics fundamentals
Layered oxides have the most widespread application and promising development prospects among the various cathode materials. Therefore, they can be primarily categorized into three main types: lithium cobalt oxides (LiCoO2, LCO), lithium ternary oxides (LiNi1−x–yCoxMnyO2, NCM, LiNi1−x−yCoxAlyO2, NCA), and lithium-rich manganese-based oxides (xLi2MnO3·(1 − x)LiMO2, LRMO, where M mainly represents Ni, Co, and Mn). LCO possesses a high energy density, making it dominant in the consumer electronics (3C) sector, where high volumetric and energy density are critical.28 In NCM, a large amount of Ni is used to replace the expensive Co,29 significantly reducing costs, leading to its widespread adoption in electric vehicles.30–32 LRMO cathode materials exhibit an extremely high specific capacity (exceeding 250 mA h g−1); however, they face several technical challenges, including low initial coulombic efficiency and poor cycling stability. If these issues can be effectively addressed, LRMO has the potential for broad application, especially in the EV field.33,34LCO and layered ternary oxides exhibit similar crystal structures, adopting the α-NaFeO2-type layered structure with the R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m space group; the closely packed hexagonal oxygen layers form the structural framework, where Li and transition metal (TM) ions are arranged in an alternating manner to occupy the octahedral sites of the oxygen framework. The oxygen layers exhibit an ABCABC-stacking sequence; hence, this structure is referred to as the O3 structure.11,34,35 In contrast, LRMO can be viewed as composites of the LiMO2 (the same as LCO) and Li2MnO3 phases. The heterogeneity of this structure determines the electrochemical instability of LRMO materials.36
m space group; the closely packed hexagonal oxygen layers form the structural framework, where Li and transition metal (TM) ions are arranged in an alternating manner to occupy the octahedral sites of the oxygen framework. The oxygen layers exhibit an ABCABC-stacking sequence; hence, this structure is referred to as the O3 structure.11,34,35 In contrast, LRMO can be viewed as composites of the LiMO2 (the same as LCO) and Li2MnO3 phases. The heterogeneity of this structure determines the electrochemical instability of LRMO materials.36
According to the bipolar diffusion theory, the effective diffusion coefficient DLi of Li0 is given by DLi = 2DLi+De/(DLi+ + De). Here, DLi+ is the diffusion coefficient of Li+, and De is the diffusion coefficient of electrons. In the cathode, De is generally much greater than DLi+. Therefore, the diffusion rate of Li+ limits the diffusion kinetics within each cathode particle. In an ideal layered structure, transition metal (TM) and oxygen ions are immobile, and Li+ diffuses through a vacancy mechanism. However, when the level of delithiation is relatively high, the layered structure of the cathode material becomes unstable, and the cathode particles tend to decompose to eliminate these lithium vacancies (this will be discussed in detail in the phase transition section of the next chapter). Conversely, lithium vacancy concentration is extremely low at high lithium insertion levels, causing a sharp decline in DLi. This is one of the reasons for the low initial coulombic efficiency of layered cathodes.37
3. Mechanisms of electrochemical–mechanical failure
For LCO and NCM cathodes, the reaction equations during the charging process can be expressed as follows (where M mainly represents Ni, Co, and Mn):| LiMO2 → Li1−xMO2 + xLi+ + xe− | (1) |
In contrast, the situation becomes more complex for LRMO. According to results of previous research, when the voltage is below 4.4 V, the LiMnO2 domain undergoes preferential oxidation, accompanied by the extraction of Li+:
| xLi2MnO3·(1 − x)LiMO2 → xLi2MnO3·(1 − x)MO2 + (1 − x)Li | (2) |
But when the charging voltage exceeds 4.5 V, the reaction equation is as follows:
 | (3) |
As the charging capacity increases, the Li2MnO3 domain becomes activated in the second stage. In this process, Li+ and oxygen are released in the form of Li2O, forming a MnO2 rock-salt structure and a significant number of oxygen vacancies. Simultaneously, an uneven electrochemical–mechanical coupling spans both micro- and macro-scales.
In layered cathode materials, the electrochemical–mechanical coupling processes span from the atomic to the macroscopic scale, forming a complex and interconnected framework of failure mechanisms. Fig. 1 systematically illustrates the multiscale characteristics of these mechanisms and their interactions. At the atomic and nanoscale, Li+ extraction induces irreversible lattice instabilities, phase transitions, and oxygen loss, which lay the foundation for structural degradation. Progressing to the microscale, uneven lithium extraction and anisotropic volume changes generate heterogeneous stress distributions within particles, leading to particle fracture and planar gliding. At the further macroscale, due to the constrained volumetric changes within the battery, these particulate-level damages amplify through cumulative effects among particles, ultimately compromising the overall mechanical integrity and electrochemical performance of the electrode.
 | ||
| Fig. 1 The failure mechanism of the cathode. All figures are reproduced with permission.20,27,38–43 | ||
These multiscale failure processes are closely coupled and mutually reinforcing, collectively determining the degradation behavior of layered cathodes during cycling. Therefore, understanding the interconnections among these failure mechanisms is crucial for revealing the nature of electrochemical–mechanical coupling and provides valuable insights for improving the performance of cathode materials.
3.1 Atomic-scale microstructural evolution
 | ||
| Fig. 2 Anisotropic lattice strain. (A to C) Lattice parameters along (A) the a-axis and (B) c-axis of the four NCMs on charge as a function of Li content. (C) The c/a ratio is based on panels (A) and (B), showing the degree of “anisotropy” of lattice changes. Reproduced with permission from ref. 38. Copyright 2019, American Chemical Society. (D) Schematic of the correlation of strain generation and O release and transition metal migration in LRMO. Reproduced with permission from ref. 44. Copyright 2022, Springer Nature. | ||
LRMO consists of two structurally coherent nanoscale domains (LiTMO2 and Li2MnO3), electrochemically activated through different reactions within distinct voltage ranges. This nanoscale heterogeneity contributes to more pronounced mechanical anisotropy in lithium-rich cathodes.46,47 As shown in Fig. 2D,44 in the initial state, both nanoscale domains exhibit a randomly distributed and coherent lattice structure. During charging, the LiTMO2 domain is activated first, leading to increased local electrostatic repulsion and a tendency for lattice expansion. At this stage, the Li2MnO3 domain remains inactive, and its lattice expansion is constrained, resulting in significant nanoscale strain at the boundary between the two domains. Lattice strain begins at the particle surface and gradually extends into the bulk, peaking when the LiTMO2 domain is nearly fully delithiated. This strain substantially disrupts the structural stability, causing decomposition of Li2MnO3 and oxygen release. This strain will gradually accumulate in long-term cycles, inevitably leading to structural degradation and rapid electrochemical decay.
Researchers employed a three-dimensional continuous rotation electron diffraction method (cRED), and auxiliary high-resolution transmission electron microscopy (HRTEM), to investigate the structural differences at the atomic level between two commercial LiCoO2 materials: a normal LCO (N-LCO) and a high-voltage LCO (H-LCO). The most prominent structural difference between the two LCOs is the degree of curvature in their cobalt oxide layers, particularly in the near-surface region, where the occurrence of curvature is significantly more frequent in N-LCO (Fig. 3A and B). The curvature of the cobalt oxide layers near the surface plays a crucial role in determining the material's structural stability at high potentials, which, in turn, influences its electrochemical performance.40
 | ||
| Fig. 3 (A and B) Schematic illustration of the LCO structural evolutions during charge. (A) N-LCO. (B) H-LCO. The flatness of the cobalt oxide layers, especially in the near-surface region, clearly shows the LCO structural stability at high potentials. Reproduced with permission from ref. 40. Copyright 2021, The Author(s), under exclusive license to Springer Nature Limited. N-LCO, normal LCO. H-LCO, high-voltage LCO. (C) The high-angle annular dark-field (HAADF) STEM image of the LRMO after 50 cycles. (D) In the enlarged STEM image of (C), the yellow dashed arrow represents the degree of bending. (E) The enlarged STEM image of the region is marked by the dashed green box in (D), indicating severe O release at the point of maximum curvature. (F) The atomic-resolution STEM image in the region of maximum curvature. Reproduced with permission from ref. 48. Copyright 2023, Wiley-VCH. | ||
Surface layer bending is a ubiquitous phenomenon in layered oxide cathode materials, and layer bending has also been observed in LRMO. Fig. 3C presents a HAADF image of LRMO after 50 cycles, with Fig. 3D and E displaying magnified images. Nano-voids induced by O release are concentrated at locations with the highest curvature in the bent layers and propagate inward from the exterior (indicated by the red dashed box) to the interior (indicated by the blue dashed box). Detailed Electron Energy Loss Spectroscopy (EELS) analysis reveals the presence of a significant number of oxygen vacancies at the location of maximum curvature, accompanied by a slight reduction of Co (Fig. 3F). Layer bending not only accumulates stress, causing the layered structure to become unstable but also induces the release of oxygen, ultimately leading to rapid capacity fade.48
Rotation is another form of lattice instability. Fig. 4A–F illustrate the issue of lattice rotation in high-nickel cathodes.27 In high-nickel materials, interlayer expansion/contraction is reversible during charge–discharge cycles; however, lattice rotation is difficult to reverse and exhibits a significantly greater magnitude of change than interlayer spacing. During the continuous cycling process, irreversible lattice rotations lead to the accumulation of plastic deformation and mechanical degradation, disrupting Li+ diffusion and the reversible deintercalation/intercalation of Li+, ultimately leading to severe capacity decay of the cathode particles.
 | ||
| Fig. 4 (A to F) 3D lattice evolution of SC-NCM individual particle during charged and discharged process. (A to C) SDXM images of the (003) peak at OCV (A), charged to 4.1 V (B), and charged to 4.5 V (C). Blue and red colors express the d-spacing variation. The length and orientation of the quiver represent the magnitude and direction of lattice rotation, respectively. (D) Schematic illustration of two types of lattice distortions: lattice expansion and contraction (top) and lattice rotation (bottom). (E and F) Comparison of d-spacing variation (E) and lattice rotation magnitude (F) of the selected particles in (A) to (C). Reproduced with permission from ref. 27. Copyright 2024, The American Association for the Advancement of Science. SDXM, scanning diffraction X-ray microscopy. (G and H) STEM/GPA analyses of pristine (G) and tenth-cycled O2–Lix(Li0.25Ni0.05Mn0.7)O2 (H) electrodes. The colored trapezoids in the STEM images represent the respective stacking sequence of the transition-metal layers. The white and red dotted boxes indicate the regions where intragranular cracks and dislocation defects occurred. The yellow circles in the figures indicate singularity points, and the white boxes show areas where the singularities are linearly distributed. Reproduced with permission from ref. 49. Copyright 2024, The Author(s), under exclusive license to Springer Nature Limited. | ||
The rotational stacking faults (RSFs) in lithium-rich cathodes arise from specific stacking sequences at various angles.49 STEM and GPA analyses demonstrate that the pristine cathode containing RSFs undergoes severe deformation after 10 cycles, exhibiting numerous mechanical failures and dislocations (Fig. 4G and H). Adjusting the annealing temperature can reduce RSFs in the pristine material and maintain fewer cracks and mechanical failures after cycling.
A distinct lattice degradation pathway is observed in solid-state batteries compared to traditional liquid batteries. The surface structural frustration caused by oxygen loss, combined with the interlayer shear induced by delithiation, destabilizes the layered oxides and forms a rock salt phase layer with fragmented lattice structures on the particle surface.50Fig. 5A and B display EDS and EELS line scan images at the interface between the sulfide solid electrolyte LPSCl and NCM811 particles, respectively, revealing the distribution of sulfur (S) and phosphorus (P) as well as evident Ni reduction at the surface. Atomic resolution imaging indicates the formation of a ∼10 nm disrupted lattice layer on the particle's exterior surface, composed of extremely small nanocrystalline domains (denoted by dashed circles). FFT analysis confirms that these nanocrystals are electrochemically inactive NiO phases (Fig. 5C). This finding suggests significant oxygen loss at the cathode/solid electrolyte interface, with primary particles directly interfacing with the solid electrolyte being more prone to surface instability compared to particles located within secondary particles.
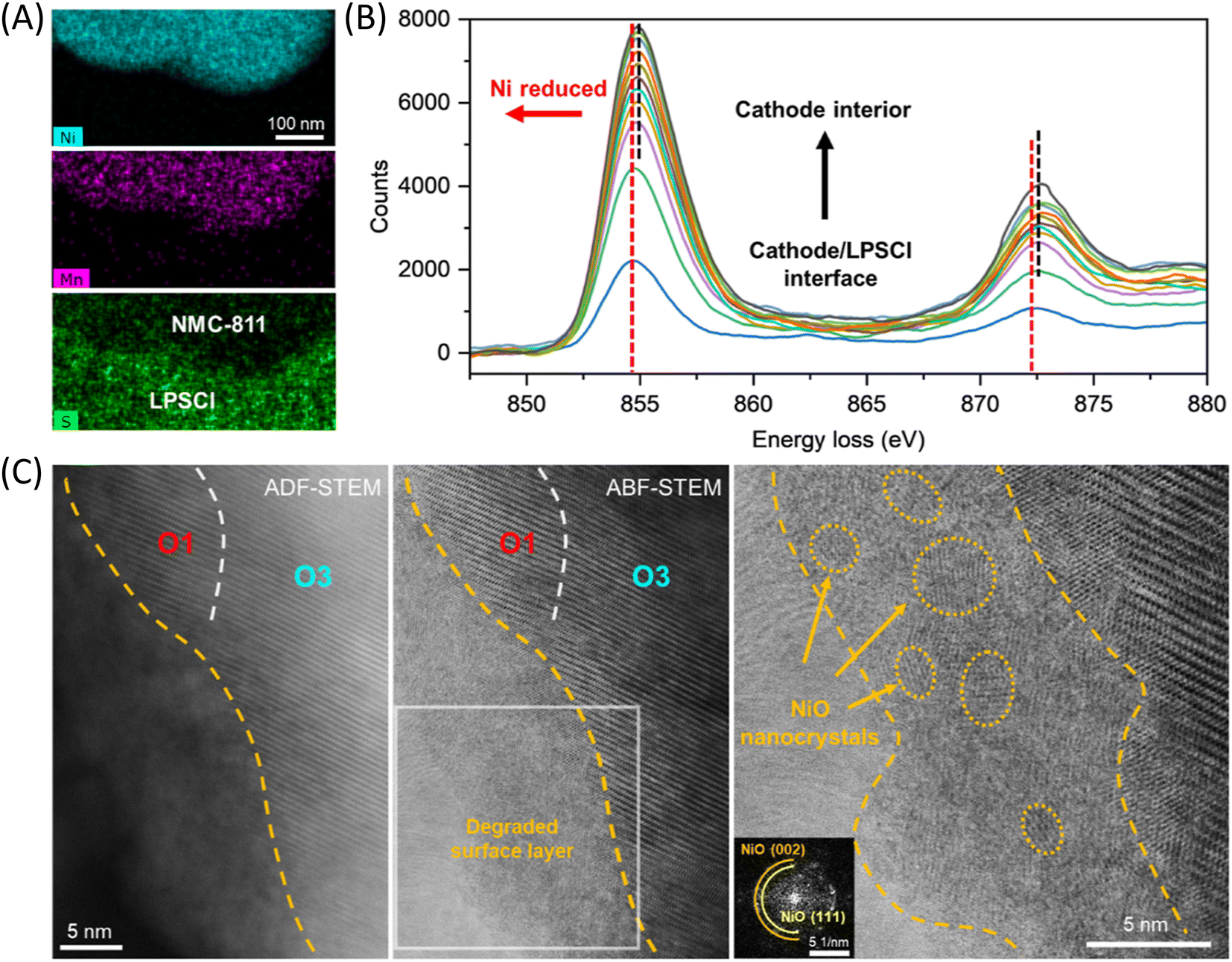 | ||
| Fig. 5 Surface nanocrystallization and deactivation driven by the electrochemically triggered cathode–electrolyte reaction. (A) EDS maps of pristine NCM-811 cathode particles in contact with sulfide solid electrolyte LPSCl after electrochemical cycling. (B) EELS profiles of charged (second cycle) NCM-811 from the cathode–electrolyte interface to the cathode particle's interior. (C) Representative ADF-STEM (left panel) and BF-STEM (middle panel) images showing the electrochemically driven oxygen loss and structural degradation at the cathode surface. The right panel shows a zoomed-in image corresponding to the boxed region in the middle panel. The atomic structure of the nanodomains (highlighted by dashed circles) can be assigned to the rock salt phase, which also agrees well with the FFT of the degraded surface layer (inset). Reproduced with permission from ref. 50. Copyright 2024, American Chemical Society. | ||
Meanwhile, in solid-state batteries, the crystal orientation at the cathode/electrolyte interface is also critically important. In traditional composite cathodes, due to the crystallographically diverse cathode/electrolyte interfaces formed by randomly oriented particles, introduce complexities arising from varying (electro)chemical compatibilities. To investigate this issue, researchers have utilized an epitaxial model system to precisely control the crystal orientation of the cathode and solid electrolyte while employing in situ electron microscopy to monitor the interface in real-time during the co-sintering process.51 Experimental results demonstrate that interfacial reactions during sintering are highly dependent on crystal orientation/alignment, particularly on the availability of open ion channels. NCM interfaces with open ion channels exhibit greater mutual diffusion but tend to stabilize by forming an early-stage passivation layer. In contrast, interfaces with closed ion channels exhibit stability at intermediate temperatures but deteriorate rapidly at higher temperatures due to oxygen release, increasing interfacial resistance. The elucidation of these distinct interfacial behaviors highlights the necessity of decoupling collective interfacial properties to enable rational design in solid-state batteries.
Due to the direct contact of the cathode particles’ surface with the binder, conductive carbon black, and electrolyte, and the shorter diffusion distances for oxygen atoms and Li+ in the surface region, the release of lattice oxygen induced by an elevated state of charge (SOC) starts at the surface and gradually extends towards the interior of the particles. As shown in Fig. 6A and B, after 40 cycles at 4.6 V, nano-voids associated with oxygen loss appear on the LCO surface. When heated to 400 degrees Celsius, these voids expand dramatically, causing severe degradation of the cathode particles.58 EELS line scans display a gradual decrease in the O/Co ratio from the interior to the surface, which becomes more pronounced after heating. This indicates that oxygen release begins at the surface and progressively diffuses inward. The consumption of oxygen atoms forms oxygen vacancies, which accumulate on specific crystal planes, generating cracks and creating new surfaces in contact with the electrolyte. Simultaneously, oxygen vacancies accelerate the migration of TM ions and, together with the accumulation of strain, lead to irreversible phase transitions from the layered phase to the spinel phase and eventually to the rock salt phase. This series of transformations is considered a primary cause of battery performance degradation.
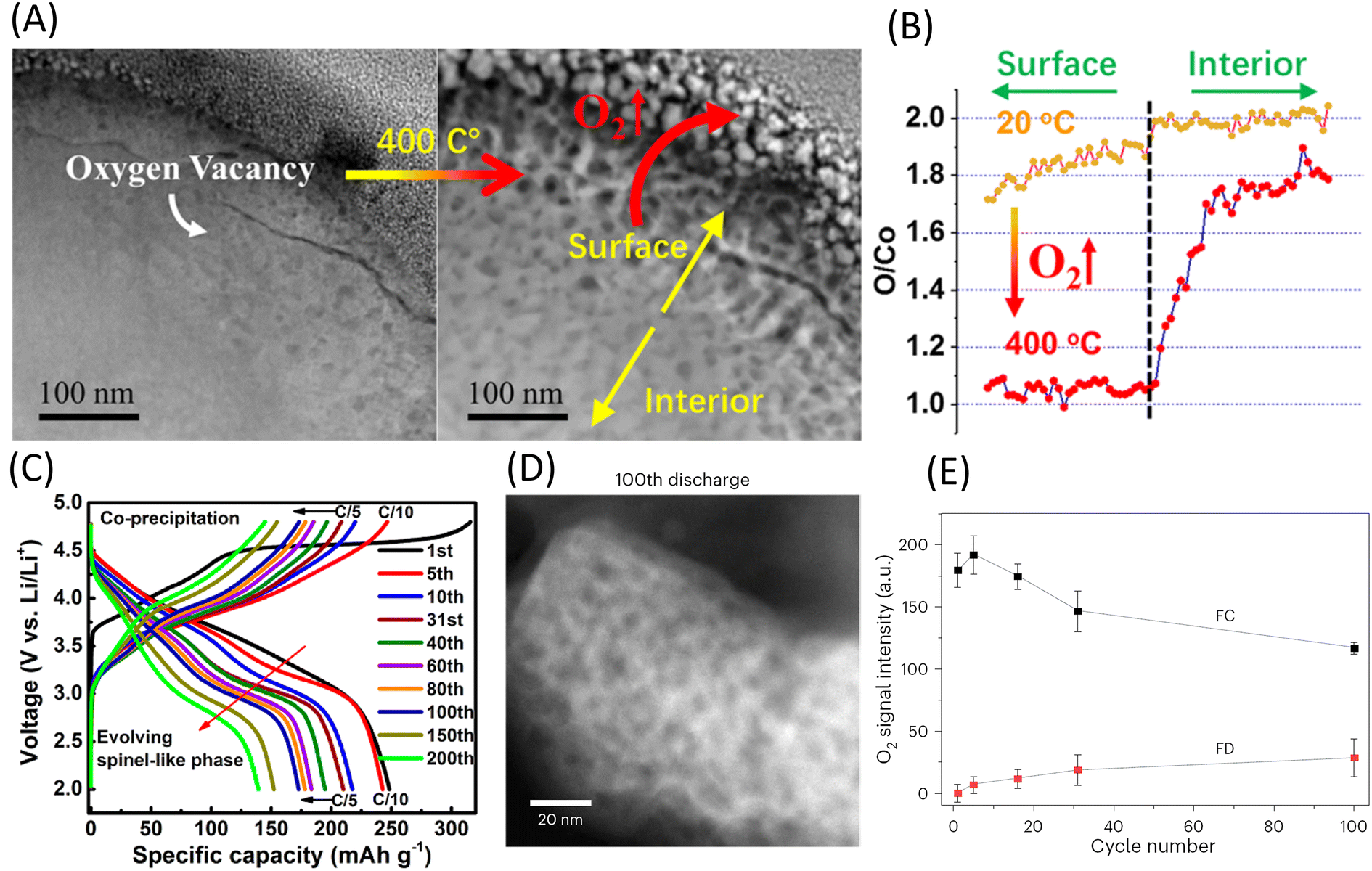 | ||
| Fig. 6 (A and B) Thermal behavior of the high-voltage-cycled LiCoO2 cathode. (A) Time-lapse HAADF STEM images. (B) Comparison of the EELS quantification of the O/Co ratio from surface to interior. Reproduced with permission from ref. 58. Copyright 2020, American Chemical Society. (C) Voltage plateau of LRMO during the initial charge. Reproduced with permission from ref. 41. Copyright 2014, American Chemical Society. (D and E) Oxygen release in LRMO. (D) ADF-STEM images of the 100th discharge show the formation of voids about 4–12 nm in diameter over extended cycling. (E) Evolution in the amount of trapped O2 over cycling. Reproduced under terms of the CC-BY license.59 Copyright 2024, The Authors, published by Springer Nature. | ||
The issue of oxygen release is particularly pronounced in LRMO cathode materials.60,61 As shown in Fig. 6C, during the initial charge of the LRMO cathode, a distinctive voltage plateau appears, corresponding to oxygen loss during the first charge, a phenomenon referred to as “cathode activation”.41 Moreover, the voltage of LRMO rapidly decays over cycling, severely impeding its commercialization. Recent studies indicate that in lithium-rich cathode materials, O2− is continuously oxidized to O2 during charging and reduced back to O2− during discharging. The produced O2 is stored in nanoscale voids within the cathode material (Fig. 6D).59 Marie et al. observed these oxygen-storing nanoscale voids in LRMO. It noted that the decline in voltage and capacity during cycling is due to the reduced reversibility of the O2−/O2 redox process. As shown in Fig. 6E, the O2 stored in the voids gradually loses its reactivity, resulting in an increasing amount of inactive O2. This expansion of voids hinders the transport of electrons and Li+, ultimately leading to particle fracture and oxygen release.
3.2 Nanoscale phase transition processes
In recent years, the development of cathode materials has leaned towards adopting high-voltage strategies to enhance capacity. High voltage implies a greater degree of delithiation, which is thermodynamically unstable and can lead to the decomposition of the cathode to eliminate lithium vacancies, especially when heated, and this process is accompanied by a series of side reactions triggered by the release of lattice oxygen. The greater the oxygen loss, the closer the TM to O ratio in the cathode particles approaches 1, resulting in more severe phase transitions. For example, in lithium cobalt oxide, as delithiation and oxygen loss progress, the initial layered LiCoO2 gradually transforms into the spinel phase Co3O4 (Fdm) and eventually into the rock salt phase CoO (RS, Fmm).63 The atomic configurations and corresponding HAADF images of these three structures are shown in Fig. 7A–F.62
 | ||
| Fig. 7 (A to F) The atomic structural models (A to C) and corresponding high-resolution HAADF-STEM images (D to F) of the layered, spinel, and rock-salt phases, respectively. The black rhomboids indicate similar unit cells with cubic close-packed oxygen arrays. Reproduced with permission from ref. 62. Copyright 2020, Wiley–VCH GmbH. The subscript: L: layered, S: spinel, R: rock-salt. (G and H) Surfaces in direct contact with the liquid electrolyte (G) and surfaces that did not have contact with the electrolyte (H). Reproduced with permission from ref. 42. Copyright 2020, American Chemical Society. | ||
The layered phase LCO and the spinel phase LiCo2O4 possess structures that allow Li+ diffusion, while the Co3O4-type TM-rich spinel and rock salt phases theoretically do not provide Li+ diffusion channels. The electrochemically inert shell formed on the surface slows down the lithium intercalation kinetics of the underlying layered phase, thereby reducing the overall electrochemical performance of the cathode particles. Furthermore, the brittle rock salt phase is more prone to fracture and crack under stress, severely affecting the performance of the cathode material. Fig. 7G and H show the reconstructed layer on the surface of the cathode particles after cycling. Due to more severe oxygen loss at the particle surface, the surface in contact with the electrolyte exhibits a thicker inert rock salt phase.42
Furthermore, phase transitions have a significant impact on the fast-charging performance of batteries. The rate performance of the cathode is determined by the Li+ insertion kinetics at the atomic level. After the surface of the cathode material undergoes oxygen loss and transitions to an inert rock-salt phase structure, the Li+ insertion kinetics of the particles are reduced. Consequently, Li+ cannot be inserted and extracted quickly and efficiently, thereby impacting the rate performance of the particles.
However, the phase transition from the layered phase to the rock-salt phase can also occur through Li/TM ion disorder without changes in chemical composition. Unlike the pure rock-salt phase that contains only TM ions, the cation-disordered rock-salt phase can provide Li+ diffusion pathways.64,65 Due to the pillar effect, an appropriate amount of cation mixing is beneficial for enhancing the stability of the crystal structure and suppressing harmful phase transitions.
As delithiation progresses, the cathode material undergoes a series of O3 (H1)–O1 phase transitions. As shown in Fig. 8A–C, the oxygen ions in the O3 phase (containing three TM-O6 octahedra per unit cell) are stacked in an ABCABC sequence, while in the O1 phase (containing one TM-O6 octahedron per unit cell), the oxygen ions are stacked in an ABAB sequence.66 This phase transition process can be observed at the microscopic level by examining the atomic columns in HAADF images: the O1 structural units exhibit a rectangular shape, while the O3 structural units exhibit a parallelogram shape. This phase transition process is typically characterized using in situ XRD and dQ/dV curves at the macroscopic level. As shown in Fig. 8D, the pristine LCO sample exhibits a pure H1 phase (O3 type) with a (003)H1 peak at 18.92°.40 As the degree of delithiation increases, continuous phase transitions to H2, M1, and H3 occur, and the H1–3 and O1 phases appear upon deep charging to 4.8V, accompanied by changes in interlayer spacing. It is generally believed that the irreversible H1–3 phase transition is one of the reasons for the poor reversibility of LCO at high voltage. As shown in Fig. 8E, the dQ/dV peak intensity of single-crystal NCM material significantly decreases and shifts after long cycling, indicating that single-crystal NCM particles suffer from irreversible structural evolution and electrochemical degradation under high voltage.27
 | ||
| Fig. 8 (A to C) Atomic models of O3 phase (space group Rm), O3 phase with partially transited O1 phase, and pure O1 phase. Li, Ni, and O are denoted by green, gray, and red symbols, respectively. Reproduced under terms of the CC-BY license.66 Copyright 2021, The Authors, Published by Elsevier Inc. (D) The voltage profile and corresponding in situ PXRD evolution of LCO. Reproduced with permission from ref. 40. Copyright 2021, The Author(s), under exclusive license to Springer Nature Limited. (E) The differential capacity curves (dQ/dV) of SC-NCM between 2.8 and 4.5 V. PXRD, in situ powder X-ray diffraction. Reproduced with permission from ref. 27. Copyright 2024, The American Association for the Advancement of Science. (F) Atomic-resolution HAADF-STEM and corresponding super-resolution images of a charged (second cycle) NCM-811 particle showing local O1 formation driven by local lattice bending. The O3 phase and O1 phase are denoted by red and orange symbols, respectively. (G) Atomistic schematic illustration showing the interlayer-shear-induced O3/O1 phase transformation triggered by bending deformation. Reproduced with permission from ref. 67. Copyright 2023, Elsevier Inc. | ||
It is generally believed that the O3–O1 phase transition is achieved through layer shearing induced by delithiation, although mechanical bending during electrochemical operation may also lead to the O3–O1 phase transition. Fig. 8F shows atomic-resolution and super-resolution images of a locally deformed region near the surface of a charged (second cycle) NCM-811 primary particle, with the bent region indicated by pink dashed lines. The unbent area on the left remains in the O3 phase (highlighted with red symbols), while the O1 phase appears at the right end of the bent region (highlighted with orange symbols). The absence of the O1 phase in the adjacent unbent area indicates a positive correlation between lattice bending and the presence of the O1 phase. Fig. 8G presents an atomic-scale schematic of the O3/O1 phase transition under mechanical bending deformation. The O3/O1 transformation is accomplished via layer-by-layer interlayer shear under a resolved shear stress introduced by the bending deformation. This stress-driven phase transition pathway is widely observed in bending bands and kinks.67
Recent research has revealed that the layered-to-rock salt phase transition and the O3–O1 phase transition occur sequentially, with mechanical shear displacement linking these two transitions. As shown in Fig. 9, atomic-level in situ transmission electron microscopy has captured the two-step transformation process from the O1 phase (denoted by yellow symbols) to the rock-salt phase (denoted by orange symbols), which involves cation mixing followed by shear displacement along the (003) plane (Fig. 9A and B). The nudged elastic band (NEB) calculations are consistent with the experimental observations, indicating that the energy barrier for the migration of transition metals to the lithium layer is lower in the O1 phase compared to the O3 phase (Fig. 9C). Consequently, the O1 phase provides preferential nucleation sites for the transition to the rock salt phase. This novel phase transition mechanism bridges the gap between the two traditional phase transition pathways.66
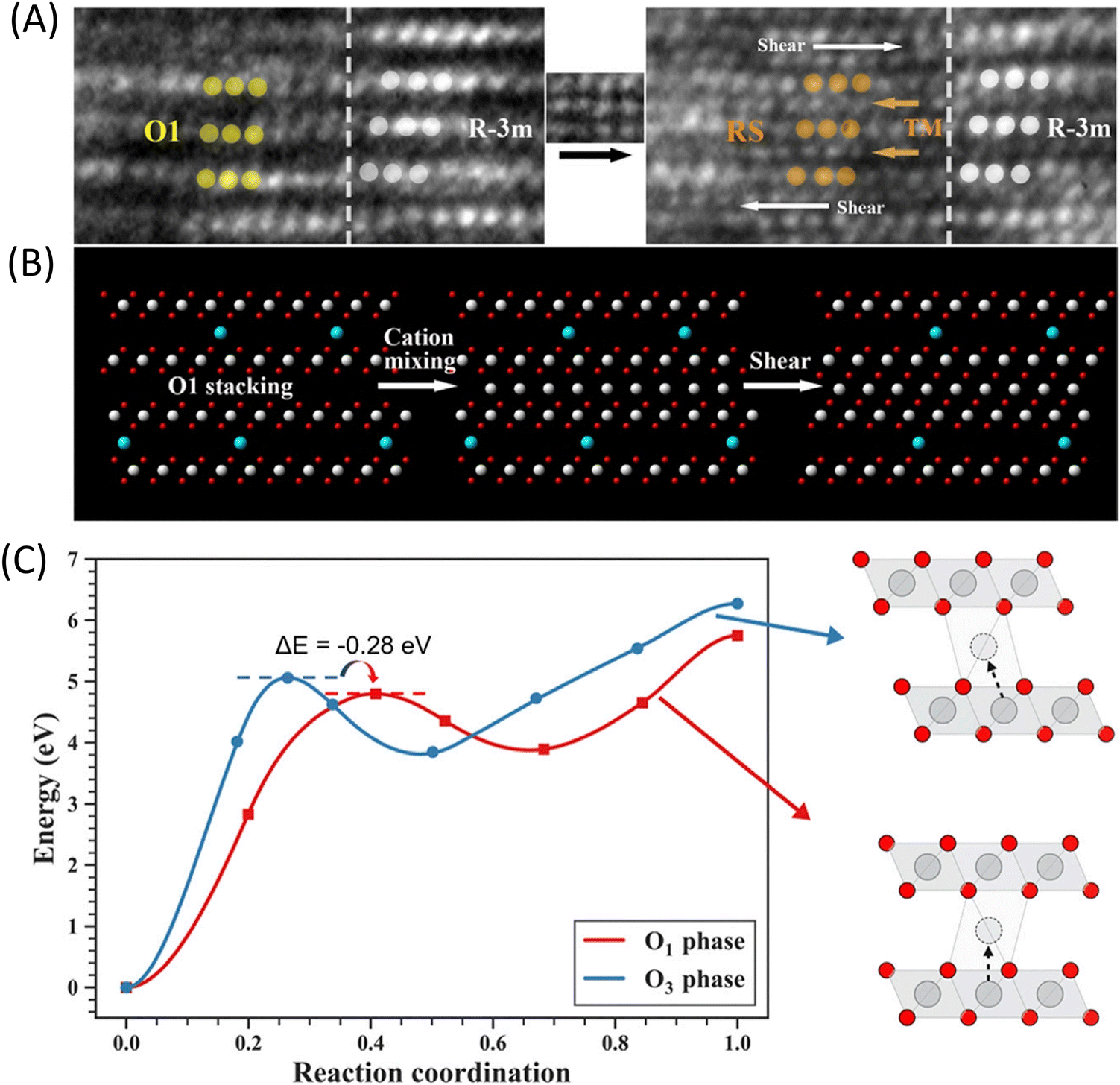 | ||
| Fig. 9 Preferential transformation from O1 phase to RS (rock-salt). (A) In situ HRTEM images showing the transformation from O1 phase to RS. O1 phase and RS are denoted by yellow and orange symbols, respectively. During the transformation, the region on the right side with perfect layered structure (white symbols) remains stable. (B) Schematic illustration of the two-step transformation pathway. (C) The minimum energy path calculation of moving Ni from the TM layer to the Li site in the O3 and O1 phases, respectively. Reproduced under terms of the CC-BY license.66 Copyright 2021, The Authors, Published by Elsevier Inc. | ||
Phase transition is also an issue in LRMO.68 LRMO consists of a coexisting phase of hexagonal LiMO2 (space group Rm) and monoclinic Li2MnO3 (space group C2/m). There are two main perspectives in the academic community regarding its structure. Thackeray et al. proposed that lithium-rich manganese-based materials are a single-phase solid solution formed by LiMO2 and Li2MnO3.69 In contrast, Lu et al. argued that lithium-rich manganese-based materials are composed of a two-phase composite of LiMO2 and Li2MnO3.70
The phase transitions occurring in LRMO cathode materials typically progress from a layered structure to a spinel-like structure and eventually to a disordered rock-salt structure.71 It is worth noting that the layered structure consists of the Rm phase and the C2/m phase, and phase transitions can also occur between these two similar structures. Furthermore, in addition to the formation of the spinel phase driven by conventional electrochemical processes, other factors such as composition and calcination temperature can also induce phase transitions. Theoretical calculations and experimental results have shown that oxygen vacancies promote TM (transition metal) ion migration and structural transformation in LMR cathode materials. However, there are differing perspectives on the pathways of these phase transitions. Gu et al. reported that the phase transition behavior differs among the various layered phases within the LRMO cathode material.72 The transition from the LiTMO2Rm phase to the spinel phase occurs through the migration of TM ions to Li sites without disrupting the lattice, leading to the formation of spinel grains with an intergrown structure within the parent particles. In contrast, the transition from the Li2MnO3C2/m phase to the spinel phase involves the removal of Li+ and O2−, generating significant lattice strain, which destroys the parent lattice and leads to randomly oriented spinel grains within the same particle. Zheng et al. proposed an alternative phase transition pathway for LMR cathode materials.73 According to their study, the electrode material undergoes a transformation from a layered structure (initially transitioning from the activated C2/m phase to the Rm phase) to a low-temperature LiCoO2 (LT-LiCoO2)-type defective spinel structure (with the Fd3m space group), and finally to a disordered rock-salt structure (with the Fmm space group).
3.3 Microscale mechanical behavior abnormality
During the electrochemical cycling of the cathode, the accumulation of anisotropic stress leads to the gradual formation of cracks in the cathode particles.74 These cracks impede the transport of Li+, resulting in the degradation of electrochemical performance. Studies have shown that cracks in layered cathodes preferentially propagate along the 003 plane of the layered phase. The kinetics of crack growth remain controversial. One viewpoint suggests that the rock-salt phase, formed from oxygen loss starting at the particle surface, gradually infiltrates the particle's interior. Due to its brittleness, the rock-salt layer lattice is disrupted by the stress induced by electrochemical cycling, leading to the formation of (003) cracks. Cracks near the particle surface expose unprotected active material surfaces, causing more severe oxygen loss, phase transitions, and further crack propagation.Insertion and extraction rates of Li+ accelerate during fast charging, leading to a larger lithium concentration gradient and a more pronounced stress mismatch among various components. When the energy release rate or stress exceeds a certain threshold, cracks can form in the cathode particles, accompanied by the fracture of the CEI film and the loss of contact between the particles, conductive carbon, and binders. The phenomenon of mechanical pulverization induced by fast charging has been confirmed in materials such as NCM and LCO.75–78
The failure mechanisms induced by fast charging are deeply interconnected: first, cracks result in poor electrical contact; second, cracks expose more fresh surfaces to react with the electrolyte; and the high temperatures generated during fast charging accelerate these side reactions. These reactions, in turn, promote the growth of the SEI, exacerbating impedance increase, loss of active material, and loss of lithium inventory. Finally, the electrolyte consumption reduces the electrode surface's wettability, hindering ion transport. This interconnected positive feedback loop of failure mechanisms accelerates the degradation of the cathode under fast-charging conditions.79
Due to differences in microstructure, the impact of charging speed on LRMO differs from that on other materials.80 Slow charging primarily induces oxygen vacancies in LRMO, which subsequently leads to progressive phase transitions and the formation of nanoscale voids. In contrast, fast charging results in lattice displacement dominated by oxygen distortion, dissolution of transition metal ions, and changes in lithium sites, causing more significant damage to the mechanical structure of the layered material.
In addition to the formation of cracks during Li+ (de)intercalation, NCM and LCO cathode materials also face the issue of planar gliding. Researchers observed planar gliding and microcracks perpendicular to the c-axis on the surface of single-crystal NCM particles charged to 4.8V (Fig. 10A). After discharging to 2.7V (Fig. 10B), although some “traces” (indicated by red boxes) were visible, the glided layers nearly returned to their original positions.81 High-resolution HAADF images (Fig. 10C) confirmed that the interlayer spacing of the (003) planes remained constant at 0.48 nm on both sides of the slip plane (yellow dashed line), and the layered structure was well-maintained after the slip traces appeared (Fig. 10D). EELS analysis showed that Ni, Mn, Co, and O were still uniformly distributed near the slip plane (Fig. 10E). The reversible formation of microstructural defects is related to local stress caused by the gradient of lithium atom concentration in the lattice. Similarly, planar gliding was also observed in LCO.39Fig. 10F and G show SEM images of LCO before cycling and after 300 cycles, respectively. Before cycling, LCO mostly existed as uniform individual particles and aggregates. However, after 300 cycles at 0.1 C, widespread particle fracture and shear were observed, with many cycled particles exhibiting a step-like morphology. This is due to planar gliding along the layered direction driven by electrochemically induced mechanical stress during cycling. Severe planar gliding exposes fresh surfaces to the electrolyte, leading to significant surface degradation and side reactions.
 | ||
| Fig. 10 The planar gliding in layered cathode materials. (A to E) Planar gliding in single-crystalline NCM76. (A) SEM image of single-crystalline NCM76 initially charged to 4.8 V (versus Li+ /Li). (B) SEM image of single-crystalline NCM76 discharged to 2.7 V (after being charged to 4.8 V versus Li+/Li). (C) HAADF-STEM image from around the slicing area. The upper inset is a magnified image of the gliding area enclosed by the red square. (D) SAED of the gliding area. (E) EELS mapping of the gliding area. Reproduced with permission from ref. 81. Copyright 2020, The American Association for the Advancement of Science. SAED, selected area electron diffraction. (F and G) planar gliding in LCO. (F) SEM images of pristine coated –LiCoO2 particles. (G) Coated –LiCoO2 particles extracted from cells after 300 cycles. Reproduced with permission from ref. 39. Copyright 2018, American Chemical Society. | ||
The microscale stress induced by lithium extraction ultimately leads to mechanical deformation, which in turn affects the transport pathways of Li+. Stress or strain within cathode particles profoundly impacts the diffusion kinetics of Li+ by altering the defect formation energy and migration energy.
First-principles calculations have shown that external stress or strain can significantly alter the Li+ migration energy barrier in LiCoO2 cathode materials, thereby affecting the diffusion coefficient and electrical conductivity. Tensile strain (3%–5%) applied along the c-axis can enhance the Li+ diffusion coefficient by 3 to 5 orders of magnitude, whereas compressive strain increases the migration energy barrier, weakening diffusion kinetics.82 Similarly, in-plane strain (within the ab plane), although having a weaker effect on diffusion, is still non-negligible. For instance, even 1% tensile strain in the plane can more than double the ionic conductivity of the material, while out-of-plane strain may have an even more pronounced impact, leading to conductivity changes exceeding an order of magnitude.83 This stress-regulation effect suggests that by designing appropriate mechanical strain, the charge–discharge performance of LIBs cathodes can be effectively optimized.
3.4 Macroscopic inhomogeneities
 | ||
| Fig. 11 The heterogeneity among particles at the macroscopic scale. (A to C) Finite element analysis of the heterogeneous electrochemistry and mechanics in an NCM composite electrode. (A) A 3D rendering of the nanotomography data of an arbitrarily selected region in the NCM622 cathode. (B) The normalized Li concentration within the NCM particles and the local Li flux (arrows) within the electrolyte in the composite electrode. (C) The cross-sectional view of Li concentration (left) and the internal stress σeq (right) in the NCM particles near the separator (upper) versus near the current collector (lower). Reproduced with permission from ref. 84. Copyright 2019, WILEY–VCH Verlag GmbH & Co. KGaA, Weinheim. (D) The reconstructed 3D volume and quantified cracking volume percentage of NCM811 thick electrode after cycling between 4.4 and 2.8 V (left), between 4.7 and 2.5 V (right) (green, cracks; red, NCM particles). (E) The reconstructed and quantified contact surface areas between NCM particles and carbon-binder domain components after cycling between 4.4 and 2.8 V (left) and 4.7 and 2.5 V (right). Reproduced with permission from ref. 74. Copyright 2022, Elsevier Inc. (F and G) The spatial distributions of the severely damaged particles in the (F) 10-cycled and (G) 50-cycled electrodes. (H) Probability distributions of the distance between two neighboring severely damaged particles in 10-cycled and 50-cycled electrodes. Reproduced with permission from ref. 85. Copyright 2022, The American Association for the Advancement of Science. | ||
Liquid electrolytes naturally provide complete coverage of active materials, whereas in solid-state batteries, the electrolyte forms point contact with the active materials. This insufficient contact limits both stability and electrochemical performance. At the atomic scale, the (electro)chemical instability between solid electrolytes and high-nickel cathode active materials leads to resistive by-products at the interface, resulting in increased impedance, reduced ion mobility, and ultimately suppressed rate performance. Furthermore, chemical reactivity at the interface can induce structural degradation of the active materials, reducing capacity retention and long-term reliability. In addition, the solid contact issue is exacerbated during charge–discharge cycling due to the volumetric expansion and contraction (approximately 5.7%) of layered cathodes, which promotes localized stress concentration and mechanical strain, ultimately causing premature electrode degradation and capacity fading.86
| LiCoO2 ⇌ Li2O + CoO | (4) |
Thermodynamically, this reaction is negligible, but the Li2O produced by the reaction will be gradually consumed by the following reactions, which will continuously drive the equation to proceed further to the right, resulting in a greater extent of LiCoO2 decomposition:
| LiPF6 + H2O + Li2O → 2HF + 2LiF↓ + LiPOF | (5) |
The Li2O produced by the decomposition of lithium cobalt oxide is consumed in side reactions, while cobalt oxide remains. Since these side reactions are more concentrated on the surface, a cobalt oxide layer forms on the surface of the lithium cobalt oxide particles. According to recent studies, the curvature of the cobalt oxide layer significantly determines the high-voltage stability of the cathode material (as discussed in previous sections).
Additionally, the O2 released from the cathode can react with carbon in conductive carbon black or organic carbon-containing electrolytes to produce CO2 and CO. CO2 can then react with Li2O to form Li2CO3. This is a reversible reaction; residual Li2CO3 from the cathode preparation process can decompose into gases such as CO2 and CO as the temperature rises and the battery cycles, ultimately leading to cell swelling.87 It is important to note that Li2CO3 and LiF are primary components of the cathode-electrolyte interphase (CEI).
Side reactions occurring on the cathode surface reduce the valence state of TM ions, making them more prone to detachment from the lattice and dissolution into the electrolyte.88 This causes cathode degradation and results in the migration of dissolved TM ions to the anode side, leading to excessive growth of the solid electrolyte interphase (SEI) and adversely affecting battery performance.89
When a battery is charged at a high rate, the overall heat generation significantly increases, which not only accelerates degradation but also poses potential safety risks.90,91 Moreover, the thermal runaway behavior of the battery changes after fast charging.92,93 For instance, ARC (Accelerating Rate Calorimetry) tests on high-energy soft-pack batteries after fast charging showed that the thermal runaway temperature of fast-charged batteries is noticeably lower than regular batteries. However, these effects appear reversible if sufficient rest time is provided. The re-intercalation of lithium can explain this phenomenon in the anode during the resting period and the reaction of plated lithium with the electrolyte to form a new SEI layer.
A series of chain reactions trigger thermal runaway. Typically, thermal runaway in batteries is initiated by a short circuit, followed by reactions involving the electrolyte, leading to the battery reaching its peak temperature. The thermal runaway process can be divided into three stages for batteries after fast charging.94 In the first stage, the reaction between plated lithium and the electrolyte causes the battery to heat up, but the temperature remains relatively low. In the second stage, a large amount of lithium metal is consumed during its reaction with the electrolyte, resulting in a rapid temperature rise. In the third stage, reactions occur between the cathode, anode, and electrolyte and between the cathode and anode themselves. Eventually, the battery reaches its peak temperature and undergoes thermal runaway.
4. Structural stabilization strategy
4.1 Surface coating and bulk doping
Surface coating and bulk doping are the most commonly used methods for cathode modification. Surface coating can regulate Li+ transport on the particle surface and suppress surface-side reactions, enabling normal ion transport in cathode materials even at high voltages. Bulk doping, on the other hand, can alter the concentration and distribution of elements, thereby modifying the material's intrinsic properties. As a result, this allows cathode particles to maintain good mechanical stability during fast charging.The traditional method for surface coating is the wet chemical approach. However, this method has the drawback of being difficult to form uniform coatings and accurately control the coating thickness. Recently, with advancements in technology, new vapor-phase methods have emerged, such as atomic layer deposition (ALD) and chemical vapor deposition (CVD).95,96 These methods enable the construction of uniformly controlled coatings with precise thickness, and they have been widely adopted in modifying cathode materials.
An ideal surface coating should exhibit the following characteristics: firstly, the coating must be sufficiently thin and provide channels for electron and ion transport, otherwise, it may hinder the insertion/extraction of Li+. Secondly, the coating layer should uniformly cover the entire cathode particle, ensuring no exposed surfaces are in contact with the electrolyte. Lastly, the coating should be inactive towards delithiation reactions and effectively suppress oxygen evolution.97
Currently, various materials are used for surface coating, including oxides, fluorides, phosphates, and polymers.100–104Fig. 12A–E illustrates examples of perovskite phases used as coating layers to enhance the stability of cathode materials.98 Researchers have exchanged Li+ with La3+ and Ca2+ on the surface of LCO, reconstructing the surface lattice into a lithium-deficient perovskite structure protective shell. The surface of the modified material is divided into a three-layer structure, with the outermost Region I confirmed as La1−wCawCoO3−δ. A buffer layer with a gradient strain structure, namely Region II, appears between Region I and the layered Region III. Subsequent experiments confirmed that the constructed surface structure physically separates the organic electrolyte from the cathode particles. High-voltage cycling stability has been greatly enhanced, up to 4.8 V versus Li+/Li, including in practical pouch-type full cells. The strong Co–O bonds in the perovskite phase and its capacity to store oxygen species stabilize the lattice oxygen in the surface layer, thereby mitigating oxygen release and reducing side reactions.
 | ||
| Fig. 12 (A) HAADF-STEM of La-LCO surface (after heat treatment) showing an architecture consisting of Region I, II, and III, separated by the dashed lines. (B) HAADF-STEM of Region I. (C) FFT pattern of (B) matching perovskite La1−wCawCoO3−δ. (D) HAADF-STEM of Region III. (E), FFT pattern of (D). Reproduced with permission from ref. 98. Copyright 2023, The Author(s), under exclusive license to Springer Nature Limited. (F) The in situ delithiated HE-LNMO and LNO strain state was obtained by geometrical phase analysis (GPA). Reproduced with permission from ref. 99. Copyright 2022, The Author(s), under exclusive license to Springer Nature Limited. HE-LNMO, LiNi0.8Mn0.13Ti0.02Mg0.02Nb0.01Mo0.02O2. LNO, LiNiO2. | ||
Another widely adopted method for modifying cathode materials is bulk doping.105 The layered structure of these materials naturally facilitates the incorporation of doping elements, resulting in high solubility for many elements, especially at elevated temperatures. Therefore, doping is typically carried out via high-temperature annealing. Mg,106 Al,107 and Ti108 are among the most commonly used doping elements,109 as they can expand the interlayer spacing, enhance electrical conductivity, and enhance structural stability (also known as the pillar effect).110 Tang et al. employed Zr doping to enhance nickel-rich cathode's Li+ diffusion and mechanical properties. Due to the stronger Zr–O bonds, the modified cathode exhibited a high capacity retention rate of 97.6%, even during rapid charging at a rate of 5C.111 In addition to single-element doping, co-doping can be employed to leverage the synergistic effects of various elements, thereby collectively enhancing the performance of the cathode material.112Fig. 12F illustrates a high-entropy-doped, high-nickel, cobalt-free layered cathode material, specifically LiNi0.8Mn0.13Ti0.02Mg0.02Nb0.01Mo0.02O2 (HE-LNMO).99 Characterization techniques such as X-ray diffraction, transmission electron microscopy, and nano-tomography reveal that HE-LNMO exhibits nearly zero volumetric change over a wide electrochemical window, significantly reducing lattice defects and locally strain-induced cracks. In situ, TEM delithiation experiments were conducted on HE-LNMO and LiNiO2. Strain analysis indicates that HE-LNMO maintains an almost defect-free and strain-free state during delithiation, whereas LiNiO2 forms numerous dislocations. The enhanced thermal stability and zero volumetric change of HE-LNMO lead to a substantially improved capacity retention rate.
However, it must be noted that dopants tend to cause a lower initial discharge capacity and higher capacity loss.113 Therefore, careful consideration should be given to the selection of dopant dosage to avoid increased costs and decreased capacity resulting from excessive doping levels.
4.2 Stabilizing the lattice
As discussed in previous sections, cathode materials’ fragile, layered structure is highly susceptible to damage during high-voltage cycling. Lattice instability can trigger a series of side reactions, such as oxygen loss and phase transformations, ultimately leading to a decline in battery performance. Therefore, the core issue in modifying cathode materials is stabilizing the lattice.In recent years, numerous approaches have been developed to stabilize the lattice, including introducing a more stable structure to act as a scaffold.116,117 As shown in Fig. 13A, researchers have incorporated the perovskite variant phase La4[LiTM]O8 (LLMO) as a “rivet” into the layered structure, forming a mechanically stable crystal structure.114 HAADF-STEM images display the coherent growth of the LLMO phase along the [100] direction within the layered NCM (Fig. 13B). This robust structure exhibits high stability during subsequent electrochemical cycling, attributed to the rivet phase's high phase transformation barrier (or pinning effect). This strategy significantly suppresses structural distortion and planar gliding within the layered structure, thereby achieving a pseudo-zero-strain cathode. Compared to traditional materials, the modified material demonstrates nearly a 70% reduction in lattice strain per cycle, demonstrating long-term cycling stability even at a high cutoff voltage of 4.6 V, with a capacity retention rate of up to 94.7% after 200 cycles.
 | ||
| Fig. 13 (A and B) Introducing perovskite phase as a ‘rivet’ to stabilize the lattice (A) HAADF-STEM images of D-NCM. Scale bars, 2 nm. Inset: the corresponding fast Fourier transform of the whole HAADF-STEM image. (B) Schematic illustrations of material structural design. D-NCM designed strain-retardant NCM. Reproduced with permission from ref. 114. Copyright 2022, The Author(s), under exclusive license to Springer Nature Limited. (C to G) CSRD configuration of LiCoO2. (C) Atomic-resolution STEM-HAADF image and the corresponding FFT pattern (inset) of the as-prepared CSRD-LiCoO2 material. (D and F) The magnified bulk and edge regions from (C). (E) Line profiles along the horizontal direction of the bulk and edge regions, in which the green arrows denote the local short-range disorder distribution of Co. (G) NPD pattern of the as-prepared CSRD–LiCoO2 material recorded at a temperature of 300 K. Reproduced with permission from ref. 115. Copyright 2024, The Author(s), under exclusive license to Springer Nature Limited. CSRD, chemical short-range disorder. NPD, neutron powder diffraction. | ||
Another method of stabilizing the lattice involves adjusting the annealing temperature, duration, and other process parameters to enable certain elements to occupy Li+ sites.118 These elements exhibit strong interactions with the neighboring oxygen atoms, making the layers less prone to sliding, thus stabilizing the lattice and inhibiting harmful phase transitions.119–121 Researchers have developed chemical short-range disorder (CSRD) LCO, as shown in Fig. 13C–F.115 In the HAADF images of both the bulk and edge, several positions within the Li layer display varying contrasts, indicating the presence of Co ions at these sites. Neutron powder diffraction (NPD) refinement results reveal that approximately 2.6% of Co occupies Li sites, which is the cause of the CSRD structure (Fig. 13G). In addition to the “rivet effect”, the introduction of local disorder reduced local strain in several adjacent coordination layers due to the weaker Li–O interactions. Additionally, it alters the electronic structure of the cathode, effectively closing the bandgap and thereby enhancing the electronic conductivity of the cathode material. Compared to traditional cathodes, this CSRD cathode material exhibits exceptional cycling stability even under high voltage conditions (4.6 V) and fast charging rates (5C). CSRD holds significant promise for tuning advanced oxide materials’ electronic and structural properties and other related compounds.
4.3 Constructing zero-strain cathodes
As previously discussed, layered oxide cathode materials undergo anisotropic volume changes during lithium (de)intercalation, which is a fundamental cause of battery failure. Zero-strain materials can address this issue, thus offering significant prospects in high-voltage and fast-charging environments.122 In these materials, the lattice parameters do not change or are restricted to a very narrow range during lithium insertion and extraction, resulting in a net strain of zero.123 Li4Ti5O12 is a well-known zero-strain material, but its low voltage limits its large-scale commercial application.124 Extensive doping of conventional cathode materials can impart zero-strain characteristics; however, this approach often significantly reduces specific capacity. In contrast, high-entropy doping is a promising solution to achieve near-zero strain with minimal dopants without compromising capacity.99 In addition to modifying existing materials, exploring new zero-strain materials is a promising direction for developing cathode materials. Well-calibrated first-principles calculations have established general principles for designing low-strain cathodes based on materials with FCC anion frameworks. Transition-metal redox centers with non-bonding electronic configurations, isotropic structures, cation disorder, inactive elements, and octahedral-to-tetrahedral migration of lithium during charging can reduce volume changes associated with delithiation. Experimentally determined that Li1.3V0.4Nb0.3O2 and Li1.25V0.55Nb0.2O1.9F0.1 are representative examples of nearly strain-free cathodes.1254.4 Single-crystallization
Polycrystalline NCM particles contain numerous spherical primary particles of various sizes and orientations. These particles contain substantial internal voids and experience stresses of differing magnitudes and directions during lithiation/delithiation processes. The development of these stresses leads to an increasing number of intergranular cracks, which expose fresh layered structures to the surface, triggering intense side reactions. Converting cathode particles into single crystals can resolve these issues.128 Single-crystal NCM, consisting of numerous single-crystal particles, not only eliminates the problem of intergranular cracks during cycling but also allows for higher pressures during calendaring. This significantly enhances the tap density and energy density of the cathode material while reducing the occurrence of harmful phase transitions and cracks,57 as illustrated in Fig. 14A.126 | ||
| Fig. 14 (A) 3D tomographic reconstruction and multislicing analysis of a representative PC9622 and SC9622 secondary particle after long-term cycling. Reproduced with permission from ref. 126. Copyright 2021, American Chemical Society. SC9622, a novel single-crystalline ultrahigh-Ni layered cathode with a designed composition of LiNi0.96Mn0.02Co0.02O2. PC9622, a baseline polycrystalline cathode with the same designed composition. (B) B-doped micron-sized single-crystal Co-free LRMO were obtained by molten-salt (LiNO3 and H3BO3)-assisted sintering. Reproduced with permission from ref. 127. Copyright 2024, American Chemical Society. | ||
Single-crystal cathodes pose new requirements for the synthesis process.129,130 It is necessary to synthesize the cathode material at a relatively low temperature to reduce the degree of cation mixing. Additionally, selecting high-quality precursors is crucial to ensure rapid crystal growth after nucleation. As shown in Fig. 14B, B-doped micron-sized single-crystal cobalt-free LRMO was prepared using a molten salt (LiNO3 and H3BO3) assisted sintering method.127 The results indicate that low-melting-point molten salts can serve as a liquid-phase medium to improve the efficiency of atomic mass transfer, crystal nucleation, and growth. The modified single-crystal LRMO cathode can resist anisotropic stress and strain accumulation during cycling, reducing interfacial side reactions and achieving excellent high-voltage stability and kinetic performance.
However, several challenges remain despite the enhanced cycling stability of single-crystal NCM particles. Firstly, due to the absence of diffusion pathways along grain boundaries, the Li+ migration path is relatively long, particularly in larger single-crystal particles. This can lead to a significant decline in performance during high-rate charging. Secondly, single-crystal particles experience more severe planar gliding during high-voltage cycling, forming additional cracks compared to polycrystalline particles. The long-range layered structure within single-crystal particles is more susceptible to structural stress induced by variations in Li+ concentration during lithium extraction/insertion, which promotes planar gliding and compromises the mechanical stability of the particles. Therefore, selecting an appropriate particle size, constructing Li-ion channels along grain boundaries, and optimizing internal vacancies are promising strategies for modification.
4.5 Macroscopic regulation strategies
To enhance battery performance under high-voltage and fast-charging conditions, it is essential to modify cathode particles at the microscopic level, optimize the overall performance of the cathode electrode at the macroscopic level, and improve charging strategies, which also play a pivotal role.Macroscopic modification strategies significantly enhance the electrochemical performance and mechanical stability of LIBs by optimizing the overall design of the battery. Compared to microscopic modification methods, such as surface coating, elemental doping, and lattice stabilization, macroscopic strategies focus more on the structural engineering of the electrodes and the battery. Specifically, these strategies address issues caused by mechanical stress, chemical degradation, and interfacial failure during cycling by regulating the geometric structure of the electrode, optimizing the surface properties of current collectors, designing functional electrolytes, and strengthening interfacial interactions.
The optimization of electrode structures and current collectors is one of the core strategies for macroscopic modifications. Adjusting the electrode thickness, porosity, and coating uniformity can enhance ion/electron transport rates while reducing local current density and alleviating polarization effects. For instance, appropriately increasing the electrode porosity can provide Li+ with more accessible transport pathways, thereby reducing ionic migration resistance within the material.131 Additionally, surface modifications of current collectors, such as the introduction of conductive carbon coatings or chemically inert protective layers, can significantly improve the stability of the electrode–current collector interface while enhancing electrical conductivity.132,133 In recent years, the adoption of porous metal current collectors and multilayer structured current collectors has further improved the mechanical stability and electrochemical uniformity of electrodes.134,135
Flexible electrode design offers an effective approach to address stress accumulation caused by material volume expansion. These designs typically integrate highly elastic binders with conductive materials to form a three-dimensional flexible conductive network, effectively mitigating the volume changes of cathode materials during charge and discharge cycles.133,136 For example, crosslinked polymer-based binders, benefiting from meticulously engineered hydrogen-bonding segments and exceptional adhesion properties, provide outstanding flexibility. Even under significant electrode deformation, they maintain structural integrity and enable stable cycling performance and voltage output during dynamic deformation tests.
The modification of electrolytes and interfaces plays a crucial role at the macroscopic level, particularly in enhancing the interfacial stability between electrodes and electrolytes. By introducing specific functional additives, such as lithium salts or interfacial stabilizers, stable solid electrolyte interphase (SEI) or cathode electrolyte interphase (CEI) can be formed on the electrode surface, thereby reducing the occurrence of side reactions.137 Meanwhile, optimizing the wettability of the electrolyte can significantly improve the contact performance between the electrode and electrolyte, further lowering interfacial impedance.138 Additionally, electrolyte strategies incorporating solvent-free or all-solid-state designs have been demonstrated to effectively minimize side reactions and mitigate the risks of thermal runaway associated with conventional liquid electrolytes.139
Although many material-level modification strategies show promising application prospects, they remain some distance away from large-scale commercialization. In contrast, optimizing charging strategies currently represents the simple and most efficient approach to fast charging.79 The CC–CV (constant current–constant voltage) charging strategy is the most commonly used protocol due to its simplicity and wide applicability across various charging scenarios. This strategy consists of two stages: first, charging under constant current (CC stage) until the set cutoff voltage is reached, followed by constant voltage (CV stage) charging until the current decreases to nearly zero. However, a significant drawback of the CC–CV method is the extended duration of the CV stage, which limits the overall charging speed.140,141 To address this limitation, the MCC (multi-constant current) charging strategy has been proposed, which introduces two or more constant current steps before the constant voltage stage.142 For example, a higher charging current can be applied in the initial charging stage since the anode potential is less likely to drop to the lithium plating potential, thereby shortening the total charging time. Another common fast-charging method is the pulse charging strategy, in which the charging current varies periodically.143 This approach effectively reduces concentration polarization, prevents the local potential from becoming negative and mitigates the mechanical stress caused by localized Li+ extraction. One of the advantages of pulse charging is that it does not exhibit significant differences from traditional CC–CV charging in terms of capacity degradation over the same charging duration. Studies have shown that CC–CV is more suitable for fast charging in high-power batteries. In contrast, MCC is typically employed when lithium plating risks must be avoided. Researchers have proposed variable current charging strategies for more complex fast-charging requirements, such as VCD (Varying Current Decay) and UVP (Universal Voltage Protocol).144,145 These strategies dynamically adjust the charging current profile based on the battery's aging state and internal resistance changes. Typically, the internal resistance is relatively high during the initial charging phase (low SOC), so the charging current remains low. As SOC increases, the internal resistance decreases, allowing the current to rise rapidly and peak in the low SOC range. Subsequently, as the Li+ intercalation level increases and Li+ diffusion becomes more limited, the charging current gradually decreases.
Most fast-charging strategies are only effective under standard temperature conditions and for specific battery configurations. Since high current induces greater mechanical stress within electrode particles and leads to significant non-uniformities in current and temperature distribution, caution is required when applying fast charging to different types of batteries. The general applicability of many current charging strategies still needs further experimental validation. With the expansion of electric vehicles in low-temperature regions, more research is needed to develop fast-charging strategies suitable for low-temperature conditions. Furthermore, the battery's internal temperature, rather than the ambient temperature, determines its performance. Therefore, changes in battery temperature during the charging process must also be considered.
In solid-state batteries, to overcome the challenge of poor contact between the electrolyte and active materials and to fully utilize the capacity of all-solid-state batteries (ASSBs), novel electrode fabrication techniques have been explored. The dry electrode process has been actively investigated for LIBs cathodes as an alternative to conventional slurry-based wet processes. This technique enables the fabrication of high-loading cathodes (>5 mA h cm−2), thereby increasing energy density. In recent years, this process has also been applied to the cathodes of ASSBs. In wet processes, the solvent-drying step often leads to carbon black agglomeration and cracking due to binder migration under high-loading conditions. In contrast, the dry process, through solvent-free processing, achieves uniform mixing and intimate contact between the active materials and solid electrolytes, resulting in superior rate capability and cycling stability. Lee et al. systematically studied the shear force effects of the dry process by comparing binder-free hand-mixed particles, wet-processed electrodes, and dry-processed electrodes.86 Based on microscopic image analysis, they proposed the concept of coverage, a critical parameter defined as the area ratio of active material in contact with the solid electrolyte. In dry-processed electrodes, the coverage was significantly improved to 67.2% due to the ductility of sulfide-based solid electrolytes. Physicochemical modeling revealed that coverage is a key determinant of electrode performance, as it influences rate capability by limiting Li+ solid-state diffusion within the active material.
5. Summary and prospects
Over the past 30 years, LIB technology has significantly contributed to advancing human society and protecting the Earth's environment, with cathode materials playing a crucial role. However, due to the limited reversible capacity, charging speed, and thermal stability of existing cathode materials, the electric vehicle industry faces growing concerns over range anxiety and safety. Operating under high voltage, rapid charge–discharge rates, and a wide range of temperatures accelerates the degradation of cathode materials. Therefore, research on cathode materials’ failure mechanisms and modification strategies is critically important. In this paper, we provide a comprehensive review of recent advances in the study of key failure mechanisms, representative stabilization strategies, and advanced characterization methods for cathode materials from the microscopic to the macroscopic level. Based on this understanding, special attention is given to the relationship between electrochemical failure and anisotropic mechanical strain and modification strategies. These strategies hold the potential to drive further progress in the renewable energy sector; however, achieving large-scale commercialization will require continued efforts from both academia and industry.Firstly, in layered cathode materials, electrochemical–mechanical coupling extends from atomic to macroscopic scales, forming a complex and interconnected framework of failure mechanisms. At atomic and nanoscales, Li+ extraction induces irreversible lattice instability, phase transitions, and oxygen loss, which lay the foundation for structural degradation. Progressing to the microscale, uneven lithium extraction and anisotropic volume changes generate heterogeneous stress distributions within particles, leading to particle fracture and planar gliding. At the macroscopic scale, these particle-level damages are amplified through cumulative effects between particles due to constrained volume changes within the battery, ultimately compromising the electrode's overall mechanical integrity and electrochemical performance. Although numerous studies in recent years have revealed the complexity of failure mechanisms and material behaviors induced by electrochemical–mechanical coupling, particularly the connection between nanoscale lattice mechanical behavior and high-pressure/fast-charging conditions, these mechanisms remain incompletely elucidated. To ensure the long-term stability of batteries under ultra-fast charging and ultra-high voltage conditions, it is essential to clarify the material failure and capacity degradation issues induced by electrochemical–mechanical coupling across various scales.
Secondly, ensuring the utmost safety of batteries under complex electrochemical–mechanical coupling conditions is crucial, especially in scenarios where rapid charging could induce thermal runaway. In recent years, the modification strategies applied to cathode systems have continuously evolved, providing new insights for the large-scale commercial application of high-voltage, fast-charging automotive batteries. Beyond traditional methods such as coating and doping, both academia and industry have gradually recognized that microscopic lattice stability is a more fundamental cause of cathode failure. Based on this understanding, novel structural modification techniques have been developed, including chemical short-range disorder, high-entropy doping, zero-strain material, gradient strain-buffering layers, and perovskite “rivet” structures, with preliminary successes. Moreover, many new electrode materials have been proposed that may improve the fast-charging capabilities of LIBs, though their stability, degradation mechanisms, and ease of manufacturing still require further exploration. LRMO offers lower cost and higher capacity; however, its heterogeneous two-phase structure is highly susceptible to severe electrochemical–mechanical coupling failures. To become a mainstream cathode material for LIBs, it is essential to address these challenges related to performance degradation and stability.
Thirdly, modifications at the macroscopic level and optimization of charging strategies are also crucial. Regulating electrode structures, optimizing the surface properties of current collectors, and improving the CEI layer can significantly enhance the electrochemical performance and mechanical stability of batteries. Meanwhile, the introduction of flexible electrode designs and porous metal current collectors effectively mitigates issues such as volumetric expansion and interfacial failure during cycling. In terms of charging strategies, the traditional CC–CV mode is straightforward but time-consuming. In contrast, strategies such as MCC, multi-step constant current, pulse charging, and variable current charging adjust the current profile to reduce charging time while minimizing the risks of lithium deposition and mechanical stress. However, studies on fast-charging protocols remain largely empirical or experimental, meaning their performance has only been evaluated under limited battery chemistries, physical dimensions, and operating conditions. These results cannot be easily generalized to other battery types or environmental temperatures, as findings from different researchers often contradict each other. On the other hand, many model-based charging optimization studies rely on models such as the single particle model or the equivalent circuit model, which may become inaccurate under high-current conditions. Moreover, their results are typically validated only against other models. Accurate and validated cell and pack models and improved understanding of limiting phenomena are needed to enable the design of charging protocols without the need for extensive laboratory testing.
Fourthly, to date, there is a lack of reliable methods for comprehensively detecting the stress–strain distribution and its corresponding structural evolution mechanisms at the microscale, especially under practical operating conditions (e.g., fast charging, high voltage, and high temperature). The stress–strain behavior within cathode materials under such complex environments often exhibits highly dynamic and localized characteristics, making real-time and precise characterization a highly challenging task. Existing experimental techniques, while capable of partially revealing structural evolution, are limited by resolution, time scale, and testing environment, making it difficult to simultaneously capture the distribution patterns of microscale stress and strain and their coupling mechanisms with structural changes in the material. Therefore, advancing more sophisticated characterization techniques and multi-scale simulation methods to investigate stress–strain behavior and structural evolution mechanisms of cathode materials under real-world operating conditions with high spatiotemporal resolution has become critical to address this scientific challenge. For instance, X-ray absorption near-edge structure spectroscopy (XANES), as part of synchrotron radiation techniques, can be employed to study the valence state changes of elements within the material and the dynamic evolution of the local chemical environment. Similarly, three-dimensional X-ray fluorescence (3D-XRF) can reveal materials’ internal elemental distribution and migration patterns during dynamic charge–discharge processes, providing crucial insights into the electrochemical heterogeneities induced by microscale stress–strain. Characterization methods based on neutron scattering are also gaining widespread attention. Neutron diffraction, which is more sensitive to light elements such as lithium, can directly analyze the distribution of Li+ and their insertion/extraction behavior across different crystal lattices, offering a novel perspective for studying stress-induced Li+ transport barriers. Furthermore, in situ and ex situ 4D-STEM, which combines spatial and momentum resolution, provides detailed information on the local electronic structure and chemical bonding within materials. These advanced techniques can capture dynamic material evolution across different temporal, spatial, and energy scales, providing multidimensional analytical tools to unveil complex scientific phenomena. Consequently, they offer critical theoretical support and experimental evidence for designing and optimizing battery materials.
Cathode materials are entering a golden era of development, with numerous scientific discoveries being rapidly translated into productivity. Ongoing interdisciplinary research and collaboration between industry, academia, and research institutions are expected to overcome the aforementioned challenges and advance the practical application of high-performance cathode materials. This provides potential solutions for safer and more energy-dense energy storage, paving the way for sustainable and efficient societal development.
Author Contributions
Ziqi Wu: conceptualization, investigation, writing – original draft, writing – review and editing, visualization. Ruiwen Shao: conceptualization, investigation, writing – review and editing, supervision; Wen Yang: conceptualization, investigation, supervision.Data availability
Data sharing is not applicable to this article as no new data were created or analyzed in this study.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (no. 12274025, 21975025), Hainan Province Science and Technology Special Fund (ZDYF2023GXJS022). These funding agencies are acknowledged. The authors acknowledge the Analysis and Testing Center at the Beijing Institute of Technology.References
- A. Yoshino, Angew. Chem., Int. Ed., 2012, 51, 5798–5800 CrossRef PubMed.
- C. Yang and X.-Y. Mu, Front. Energy, 2023, 17, 822–832 CrossRef.
- H. Li, Joule, 2019, 3, 911–914 CrossRef.
- J. Wen, D. Zhao and C. Zhang, Renewable Energy, 2020, 162, 1629–1648 CrossRef.
- W. Cao, J. Zhang and H. Li, Energy Storage Mater., 2020, 26, 46–55 CrossRef.
- D. Wang, J. Qiu, N. Inui, R. Hagiwara, J. Hwang and K. Matsumoto, ACS Energy Lett., 2023, 8, 5248–5252 CrossRef CAS.
- C.-Y. Wang, T. Liu and X.-G. Yang, Nature, 2022, 611, 485–490 CrossRef CAS.
- Z. Li, Y. Yao, S. Sun, C. Jin, N. Yao, C. Yan and Q. Zhang, Angew. Chem., Int. Ed., 2023, 62, e202303888 CrossRef CAS PubMed.
- L. Luo, Z. Sun, Y. You, X. Han, C. Lan, S. Pei, P. Su, Z. Zhang, Y. Li, S. Xu, S. Guo, D. Lin, G. Lin, C. Li, W. Huang, S. Wu, M.-S. Wang and S. Chen, ACS Nano, 2024, 18, 2917–2927 CrossRef CAS PubMed.
- K. Wang, J. Wan, Y. Xiang, J. Zhu, Q. Leng, M. Wang, L. Xu and Y. Yang, J. Power Sources, 2020, 460, 228062 CrossRef CAS.
- J. Xiang, Y. Wei, Y. Zhong, Y. Yang, H. Cheng, L. Yuan, H. Xu and Y. Huang, Adv. Mater., 2022, 34, 2200912 CrossRef CAS.
- C. Wang, J. Weng, S. Wu, X. Zhang, Q. Tong and M. Zhu, Mater. Chem. Front., 2022, 6, 2319–2337 RSC.
- A. Tomaszewska, Z. Chu, X. Feng, S. O'Kane, X. Liu, J. Chen, C. Ji, E. Endler, R. Li, L. Liu, Y. Li, S. Zheng, S. Vetterlein, M. Gao, J. Du, M. Parkes, M. Ouyang, M. Marinescu, G. Offer and B. Wu, eTransportation, 2019, 1, 100011 CrossRef.
- N. Piao, X. Gao, H. Yang, Z. Guo, G. Hu, H.-M. Cheng and F. Li, eTransportation, 2022, 11, 100145 CrossRef.
- K. Märker, P. J. Reeves, C. Xu, K. J. Griffith and C. P. Grey, Chem. Mater., 2019, 31, 2545–2554 CrossRef.
- A. O. Kondrakov, A. Schmidt, J. Xu, H. Geßwein, R. Mönig, P. Hartmann, H. Sommer, T. Brezesinski and J. Janek, J. Phys. Chem. C, 2017, 121, 3286–3294 CrossRef.
- R. Koerver, W. Zhang, L. De Biasi, S. Schweidler, A. O. Kondrakov, S. Kolling, T. Brezesinski, P. Hartmann, W. G. Zeier and J. Janek, Energy Environ. Sci., 2018, 11, 2142–2158 RSC.
- S. Kalnaus, N. J. Dudney, A. S. Westover, E. Herbert and S. Hackney, Science, 2023, 381, eabg5998 CrossRef PubMed.
- L. S. de Vasconcelos, N. Sharma, R. Xu and K. Zhao, Exp. Mech., 2019, 59, 337–347 CrossRef.
- H. Dong, M. Xie, M. Cai, H. Liu, Z. Zhang, B. Ye, P. Zhao, W. Dong and F. Huang, Chem. Eng. J., 2022, 436, 135227 CrossRef.
- Y. Zhu, D. Wu, X. Yang, L. Zeng, J. Zhang, D. Chen, B. Wang and M. Gu, Energy Storage Mater., 2023, 60, 102828 CrossRef.
- K.-J. Lee, Y.-W. Byeon, H.-J. Lee, Y. Lee, S. Park, H.-R. Kim, H.-K. Kim, S. J. Oh and J.-P. Ahn, Energy Storage Mater., 2023, 57, 326–333 CrossRef.
- S. Lee, L. Su, A. Mesnier, Z. Cui and A. Manthiram, Joule, 2023, 7, 2430–2444 CrossRef.
- A. Mistry, T. Heenan, K. Smith, P. Shearing and P. P. Mukherjee, ACS Energy Lett., 2022, 7, 1871–1879 CrossRef.
- C. Xu, K. Märker, J. Lee, A. Mahadevegowda, P. J. Reeves, S. J. Day, M. F. Groh, S. P. Emge, C. Ducati, B. Layla Mehdi, C. C. Tang and C. P. Grey, Nat. Mater., 2021, 20, 84–92 CrossRef CAS.
- Y. Li, X. Li, C. Du, H. Sun, Y. Zhang, Q. Liu, T. Yang, J. Zhao, C. Delmas, S. J. Harris, H. Chen, Q. Huang, Y. Tang, L. Zhang, T. Zhu and J. Huang, ACS Energy Lett., 2021, 6, 3960–3969 CrossRef CAS.
- W. Huang, T. Liu, L. Yu, J. Wang, T. Zhou, J. Liu, T. Li, R. Amine, X. Xiao, M. Ge, L. Ma, S. N. Ehrlich, M. V. Holt, J. Wen and K. Amine, Science, 2024, 384, 912–919 CrossRef CAS.
- C. Lin, J. Li, Z. Yin, W. Huang, Q. Zhao, Q. Weng, Q. Liu, J. Sun, G. Chen and F. Pan, Adv. Mater., 2023, 36, 2307404 CrossRef PubMed.
- Q. Xie, Z. Cui and A. Manthiram, Adv. Mater., 2021, 33, 2100804 CrossRef CAS.
- J. Lu, C. Xu, W. Dose, S. Dey, X. Wang, Y. Wu, D. Li and L. Ci, Chem. Soc. Rev., 2024, 53, 4707–4740 RSC.
- J. Yang, X. Liang, H.-H. Ryu, C. S. Yoon and Y.-K. Sun, Energy Storage Mater., 2023, 63, 102969 CrossRef.
- Z. Cui, X. Li, X. Bai, X. Ren and X. Ou, Energy Storage Mater., 2023, 57, 14–43 CrossRef.
- S. Jiao, J. Wang, Y.-S. Hu, X. Yu and H. Li, ACS Energy Lett., 2023, 8, 3025–3037 CrossRef CAS.
- W.-J. Kong, C.-Z. Zhao, S. Sun, L. Shen, X.-Y. Huang, P. Xu, Y. Lu, W.-Z. Huang, J.-Q. Huang and Q. Zhang, Adv. Mater., 2024, 36, 2310738 CrossRef CAS.
- Y. Lyu, X. Wu, K. Wang, Z. Feng, T. Cheng, Y. Liu, M. Wang, R. Chen, L. Xu, J. Zhou, Y. Lu and B. Guo, Adv. Energy Mater., 2021, 11, 2000982 CrossRef.
- C. Shen, L. Hu, Q. Duan, X. Liu, S. Huang, Y. Jiang, W. Li, B. Zhao, X. Sun and J. Zhang, Adv. Energy Mater., 2023, 13, 2302957 CrossRef.
- H. Zhou, F. Xin, B. Pei and M. S. Whittingham, ACS Energy Lett., 2019, 4, 1902–1906 CrossRef.
- W. Li, H. Y. Asl, Q. Xie and A. Manthiram, J. Am. Chem. Soc., 2019, 141, 5097–5101 CrossRef PubMed.
- W. Zhang, F. H. Richter, S. P. Culver, T. Leichtweiss, J. G. Lozano, C. Dietrich, P. G. Bruce, W. G. Zeier and J. Janek, ACS Appl. Mater. Interfaces, 2018, 10, 22226–22236 CrossRef PubMed.
- J. Li, C. Lin, M. Weng, Y. Qiu, P. Chen, K. Yang, W. Huang, Y. Hong, J. Li, M. Zhang, C. Dong, W. Zhao, Z. Xu, X. Wang, K. Xu, J. Sun and F. Pan, Nat. Nanotechnol., 2021, 16, 599–605 CrossRef PubMed.
- J. Zheng, M. Gu, A. Genc, J. Xiao, P. Xu, X. Chen, Z. Zhu, W. Zhao, L. Pullan, C. Wang and J.-G. Zhang, Nano Lett., 2014, 14, 2628–2635 CrossRef PubMed.
- L. Zou, W. Zhao, H. Jia, J. Zheng, L. Li, D. P. Abraham, G. Chen, J. R. Croy, J.-G. Zhang and C. Wang, Chem. Mater., 2020, 32, 2884–2892 CrossRef.
- Z. Zhang, C. Qin, X. Cheng, J. Li, Y. Zhang, W. Zhao, L. Wang, Y. Du, M. Sui and P. Yan, Electron, 2023, 1, e9 CrossRef.
- T. Liu, J. Liu, L. Li, L. Yu, J. Diao, T. Zhou, S. Li, A. Dai, W. Zhao, S. Xu, Y. Ren, L. Wang, T. Wu, R. Qi, Y. Xiao, J. Zheng, W. Cha, R. Harder, I. Robinson, J. Wen, J. Lu, F. Pan and K. Amine, Nature, 2022, 606, 305–312 CrossRef PubMed.
- G. Amatucci, Solid State Ionics, 1996, 83, 167–173 CrossRef.
- N. Leifer, T. Penki, R. Nanda, J. Grinblat, S. Luski, D. Aurbach and G. Goobes, Phys. Chem. Chem. Phys., 2020, 22, 9098–9109 RSC.
- H. Yu, R. Ishikawa, Y. So, N. Shibata, T. Kudo, H. Zhou and Y. Ikuhara, Angew. Chem., 2013, 125, 6085–6089 CrossRef.
- H. Peng, H. Zhuo, F. Xia, W. Zeng, C. Sun and J. Wu, Adv. Funct. Mater., 2023, 33, 2306804 CrossRef.
- D. Eum, S.-O. Park, H.-Y. Jang, Y. Jeon, J.-H. Song, S. Han, K. Kim and K. Kang, Nat. Mater., 2024, 23, 1093–1099 CrossRef PubMed.
- C. Wang, Y. Jing, D. Zhu and H. L. Xin, J. Am. Chem. Soc., 2024, 146, 17712–17718 CrossRef PubMed.
- S. Lee, H. Park, J. Y. Kim, J. Kim, M.-J. Choi, S. Han, S. Kim, W. Kim, H. W. Jang, J. Park and K. Kang, Nat. Commun., 2024, 15, 7947 CrossRef PubMed.
- D. Eum, B. Kim, J.-H. Song, H. Park, H.-Y. Jang, S. J. Kim, S.-P. Cho, M. H. Lee, J. H. Heo, J. Park, Y. Ko, S. K. Park, J. Kim, K. Oh, D.-H. Kim, S. J. Kang and K. Kang, Nat. Mater., 2022, 21, 664–672 CrossRef PubMed.
- R. Sim, Z. Cui and A. Manthiram, ACS Energy Lett., 2023, 8, 5143–5148 CrossRef.
- R. Jung, M. Metzger, F. Maglia, C. Stinner and H. A. Gasteiger, J. Electrochem. Soc., 2017, 164, A1361–A1377 CrossRef.
- X.-Y. Hou, Y. Kimura, Y. Tamenori, K. Nitta, H. Yamagishi, K. Amezawa and T. Nakamura, ACS Energy Lett., 2022, 7, 1687–1693 CrossRef.
- P. Liu, L. Yang, B. Xiao, H. Wang, L. Li, S. Ye, Y. Li, X. Ren, X. Ouyang, J. Hu, F. Pan, Q. Zhang and J. Liu, Adv. Funct. Mater., 2022, 32, 2208586 CrossRef.
- Y. Song, Y. Cui, B. Li, L. Geng, J. Yan, D. Zhu, P. Zhou, J. Zhou, Z. Yan, Q. Xue, Y. Tang and W. Xing, Nano Energy, 2023, 116, 108846 CrossRef.
- C. Sun, X. Liao, F. Xia, Y. Zhao, L. Zhang, S. Mu, S. Shi, Y. Li, H. Peng, G. Van Tendeloo, K. Zhao and J. Wu, ACS Nano, 2020, 14, 6181–6190 CrossRef PubMed.
- J.-J. Marie, R. A. House, G. J. Rees, A. W. Robertson, M. Jenkins, J. Chen, S. Agrestini, M. Garcia-Fernandez, K.-J. Zhou and P. G. Bruce, Nat. Mater., 2024, 23, 818–825 CrossRef.
- P. Yan, J. Zheng, Z.-K. Tang, A. Devaraj, G. Chen, K. Amine, J.-G. Zhang, L.-M. Liu and C. Wang, Nat. Nanotechnol., 2019, 14, 602–608 CrossRef.
- Z. Hao, H. Sun, Y. Ni, G. Yang, Z. Yang, Z. Hao, R. Wang, P. Yang, Y. Lu, Q. Zhao, W. Xie, Z. Yan, W. Zhang and J. Chen, Adv. Mater., 2024, 36, 2307617 CrossRef PubMed.
- S. Li, Z. Yao, J. Zheng, M. Fu, J. Cen, S. Hwang, H. Jin, A. Orlov, L. Gu, S. Wang, Z. Chen and D. Su, Angew. Chem., Int. Ed., 2020, 59, 22092–22099 CrossRef PubMed.
- Z. Wu, G. Zeng, J. Yin, C.-L. Chiang, Q. Zhang, B. Zhang, J. Chen, Y. Yan, Y. Tang, H. Zhang, S. Zhou, Q. Wang, X. Kuai, Y.-G. Lin, L. Gu, Y. Qiao and S.-G. Sun, ACS Energy Lett., 2023, 8, 4806–4817 CrossRef.
- W. Ding, H. Ren, Z. Li, M. Shang, Y. Song, W. Zhao, L. Chang, T. Pang, S. Xu, H. Yi, L. Zhou, H. Lin, Q. Zhao and F. Pan, Adv. Energy Mater., 2024, 14, 2303926 CrossRef.
- W. Wang, Y. Shi, P. Li, R. Wang, F. Ye, X. Zhang, W. Li, Z. Wang, C. Xu, D. Xu, Q. Xu and X. Cui, Energy Environ. Sci., 2024, 17, 4283–4294 RSC.
- C. Wang, L. Han, R. Zhang, H. Cheng, L. Mu, K. Kisslinger, P. Zou, Y. Ren, P. Cao, F. Lin and H. L. Xin, Matter, 2021, 4, 2013–2026 CrossRef.
- C. Wang, X. Wang, P. Zou, R. Zhang, S. Wang, B. Song, K.-B. Low and H. L. Xin, Matter, 2023, 6, 1265–1277 CrossRef.
- W. He, Q.-S. Xie, J. Lin, B.-H. Qu, L.-S. Wang and D.-L. Peng, Rare Met., 2022, 41, 1456–1476 CrossRef.
- M. M. Thackeray, S.-H. Kang, C. S. Johnson, J. T. Vaughey, R. Benedek and S. A. Hackney, J. Mater. Chem., 2007, 17, 3112 RSC.
- Z. Lu and J. R. Dahn, J. Electrochem. Soc., 2002, 149, A815–A822 CrossRef.
- Z. Ding, C. Zhang, S. Xu, J. Liu, C. Liang, L. Chen, P. Wang, D. G. Ivey, Y. Deng and W. Wei, Energy Storage Mater., 2019, 21, 69–76 CrossRef.
- M. Gu, I. Belharouak, J. Zheng, H. Wu, J. Xiao, A. Genc, K. Amine, S. Thevuthasan, D. R. Baer, J.-G. Zhang, N. D. Browning, J. Liu and C. Wang, ACS Nano, 2013, 7, 760–767 CrossRef PubMed.
- J. Zheng, P. Xu, M. Gu, J. Xiao, N. D. Browning, P. Yan, C. Wang and J.-G. Zhang, Chem. Mater., 2015, 27, 1381–1390 CrossRef.
- M. Zhang, M. Chouchane, S. A. Shojaee, B. Winiarski, Z. Liu, L. Li, R. Pelapur, A. Shodiev, W. Yao, J.-M. Doux, S. Wang, Y. Li, C. Liu, H. Lemmens, A. A. Franco and Y. S. Meng, Joule, 2023, 7, 201–220 CrossRef.
- S. Xia, L. Mu, Z. Xu, J. Wang, C. Wei, L. Liu, P. Pianetta, K. Zhao, X. Yu, F. Lin and Y. Liu, Nano Energy, 2018, 53, 753–762 CrossRef.
- R. Xu, L. S. De Vasconcelos, J. Shi, J. Li and K. Zhao, Exp. Mech., 2018, 58, 549–559 CrossRef.
- P. Yan, J. Zheng, M. Gu, J. Xiao, J.-G. Zhang and C.-M. Wang, Nat. Commun., 2017, 8, 14101 CrossRef.
- K. Zhao, M. Pharr, J. J. Vlassak and Z. Suo, J. Appl. Phys., 2010, 108, 073517 CrossRef.
- A. Tomaszewska, Z. Chu, X. Feng, S. O'Kane, X. Liu, J. Chen, C. Ji, E. Endler, R. Li, L. Liu, Y. Li, S. Zheng, S. Vetterlein, M. Gao, J. Du, M. Parkes, M. Ouyang, M. Marinescu, G. Offer and B. Wu, eTransportation, 2019, 1, 100011 CrossRef.
- Z. Liu, Y. Zeng, J. Tan, H. Wang, Y. Zhu, X. Geng, P. Guttmann, X. Hou, Y. Yang, Y. Xu, P. Cloetens, D. Zhou, Y. Wei, J. Lu, J. Li, B. Liu, M. Winter, R. Kostecki, Y. Lin and X. He, Nat. Nanotechnol., 2024, 19, 1821–1830 CrossRef PubMed.
- Y. Bi, J. Tao, Y. Wu, L. Li, Y. Xu, E. Hu, B. Wu, J. Hu, C. Wang, J.-G. Zhang, Y. Qi and J. Xiao, Science, 2020, 370, 1313–1317 CrossRef.
- F. Ning, S. Li, B. Xu and C. Ouyang, Solid State Ionics, 2014, 263, 46–48 CrossRef.
- A. Moradabadi, P. Kaghazchi, J. Rohrer and K. Albe, Phys. Rev. Mater., 2018, 2, 015402 CrossRef.
- Y. Yang, R. Xu, K. Zhang, S.-J. Lee, L. Mu, P. Liu, S. Spence, Z. Xu, C. Wei, D. J. Kautz, Q. Yuan, Y. Dong, Y.-S. Yu, X. Xiao, H.-K. Lee, P. Pianetta, P. Cloetens, J.-S. Lee, K. Zhao, F. Lin and Y. Liu, Adv. Energy Mater., 2019, 9, 1900674 CrossRef.
- J. Li, N. Sharma, Z. Jiang, Y. Yang, F. Monaco, Z. Xu, D. Hou, D. Ratner, P. Pianetta, P. Cloetens, F. Lin, K. Zhao and Y. Liu, Science, 2022, 376, 517–521 CrossRef PubMed.
- D. Lee, Y. Shim, Y. Kim, G. Kwon, S. H. Choi, K. Kim and D.-J. Yoo, Nat. Commun., 2024, 15, 4763 CrossRef PubMed.
- N. Mahne, S. E. Renfrew, B. D. McCloskey and S. A. Freunberger, Angew. Chem., Int. Ed., 2018, 57, 5529–5533 CrossRef.
- L. Rynearson, C. Antolini, C. Jayawardana, M. Yeddala, D. Hayes and B. L. Lucht, Angew. Chem., Int. Ed., 2024, 63, e202317109 CrossRef.
- H. Xu, Z. Li, T. Liu, C. Han, C. Guo, H. Zhao, Q. Li, J. Lu, K. Amine and X. Qiu, Angew. Chem. Int. Ed., 2022, 61, e202202894 CrossRef.
- T. Grandjean, A. Barai, E. Hosseinzadeh, Y. Guo, A. McGordon and J. Marco, J. Power Sources, 2017, 359, 215–225 CrossRef.
- U. S. Kim, J. Yi, C. B. Shin, T. Han and S. Park, J. Electrochem. Soc., 2013, 160, A990–A995 CrossRef.
- Y. Li, X. Feng, D. Ren, M. Ouyang and L. Lu, Meet. Abstr., 2019, MA2019-01, 585 CrossRef.
- T. Waldmann, J. B. Quinn, K. Richter, M. Kasper, A. Tost, A. Klein and M. Wohlfahrt-Mehrens, J. Electrochem. Soc., 2017, 164, A3154–A3162 CrossRef.
- X. Feng, M. Ouyang, X. Liu, L. Lu, Y. Xia and X. He, Energy Storage Mater., 2018, 10, 246–267 CrossRef.
- H.-J. Guo, Y. Sun, Y. Zhao, G.-X. Liu, Y.-X. Song, J. Wan, K.-C. Jiang, Y.-G. Guo, X. Sun and R. Wen, Angew. Chem., Int. Ed., 2022, 61, e202211626 CrossRef PubMed.
- G.-L. Xu, Q. Liu, K. K. S. Lau, Y. Liu, X. Liu, H. Gao, X. Zhou, M. Zhuang, Y. Ren, J. Li, M. Shao, M. Ouyang, F. Pan, Z. Chen, K. Amine and G. Chen, Nat. Energy, 2019, 4, 484–494 CrossRef.
- U. Nisar, N. Muralidharan, R. Essehli, R. Amin and I. Belharouak, Energy Storage Mater., 2021, 38, 309–328 CrossRef.
- M. Cai, Y. Dong, M. Xie, W. Dong, C. Dong, P. Dai, H. Zhang, X. Wang, X. Sun, S. Zhang, M. Yoon, H. Xu, Y. Ge, J. Li and F. Huang, Nat. Energy, 2023, 8, 159–168 CrossRef.
- R. Zhang, C. Wang, P. Zou, R. Lin, L. Ma, L. Yin, T. Li, W. Xu, H. Jia, Q. Li, S. Sainio, K. Kisslinger, S. E. Trask, S. N. Ehrlich, Y. Yang, A. M. Kiss, M. Ge, B. J. Polzin, S. J. Lee, W. Xu, Y. Ren and H. L. Xin, Nature, 2022, 610, 67–73 CrossRef PubMed.
- Y. Sun, W. Huang, G. Zhao, Q. Liu, L. Duan, S. Wang, Q. An, H. Wang, Y. Yang, C. Zhang and H. Guo, ACS Energy Lett., 2023, 8, 1629–1638 CrossRef.
- Z. Tan, X. Chen, Y. Li, X. Xi, S. Hao, X. Li, X. Shen, Z. He, W. Zhao and Y. Yang, Adv. Funct. Mater., 2023, 33, 2215123 CrossRef.
- K. Wu, P. Ran, B. Wang, F. Wang, J. Zhao and E. Zhao, Adv. Sci., 2024, 11, 2308258 CrossRef.
- F. Xin, H. Zhou, Y. Zong, M. Zuba, Y. Chen, N. A. Chernova, J. Bai, B. Pei, A. Goel, J. Rana, F. Wang, K. An, L. F. J. Piper, G. Zhou and M. S. Whittingham, ACS Energy Lett., 2021, 6, 1377–1382 CrossRef CAS.
- H. Yu, H. Zhu, H. Jiang, X. Su, Y. Hu, H. Jiang and C. Li, Natl. Sci. Rev., 2023, 10, nwac166 CrossRef CAS.
- G. Ko, S. Jeong, S. Park, J. Lee, S. Kim, Y. Shin, W. Kim and K. Kwon, Energy Storage Mater., 2023, 60, 102840 CrossRef.
- J. Chen, H. Chen, S. Zhang, A. Dai, T. Li, Y. Mei, L. Ni, X. Gao, W. Deng, L. Yu, G. Zou, H. Hou, M. Dahbi, W. Xu, J. Wen, J. Alami, T. Liu, K. Amine and X. Ji, Adv. Mater., 2022, 34, 2204845 CrossRef.
- X. Ou, T. Liu, W. Zhong, X. Fan, X. Guo, X. Huang, L. Cao, J. Hu, B. Zhang, Y. S. Chu, G. Hu, Z. Lin, M. Dahbi, J. Alami, K. Amine, C. Yang and J. Lu, Nat. Commun., 2022, 13, 2319 CrossRef.
- Z. Liu, S. Liu, L. Yang, C. Zhang, X. Shen, Q. Zhang, H.-J. Lin, C.-T. Chen, Z. Hu, Y. Yang, J. Ma, R. Yu, X. Wang, Z. Wang and L. Chen, ACS Appl. Mater. Interfaces, 2022, 14, 18353–18359 CrossRef.
- J.-N. Zhang, Q. Li, C. Ouyang, X. Yu, M. Ge, X. Huang, E. Hu, C. Ma, S. Li, R. Xiao, W. Yang, Y. Chu, Y. Liu, H. Yu, X.-Q. Yang, X. Huang, L. Chen and H. Li, Nat. Energy, 2019, 4, 594–603 CrossRef.
- E. Wang, X. Ye, B. Zhang, B. Qu, J. Guo and S. Zheng, Nanomaterials, 2024, 14, 147 CrossRef.
- Y. Tang, Z. Huang, W. Wang, Y. Wen, S. Zhang, X. Chen, Z. Zhang, Z. Yin, T. Yang, T. Li, L. C. Gallington, H. Zhu, S. Lan, S. Wang, Y. Ren, Z. Wu and Q. Liu, Nano Energy, 2024, 128, 109908 CrossRef.
- X. Tan, Y. Zhang, S. Xu, P. Yang, T. Liu, D. Mao, J. Qiu, Z. Chen, Z. Lu, F. Pan and W. Chu, Adv. Energy Mater., 2023, 13, 2300147 CrossRef.
- F. Zhou, W. Luo, X. Zhao and J. R. Dahn, J. Electrochem. Soc., 2009, 156, A917 CrossRef.
- L. Wang, T. Liu, T. Wu and J. Lu, Nature, 2022, 611, 61–67 CrossRef PubMed.
- Q. Wang, Z. Yao, J. Wang, H. Guo, C. Li, D. Zhou, X. Bai, H. Li, B. Li, M. Wagemaker and C. Zhao, Nature, 2024, 629, 341–347 CrossRef PubMed.
- Y. Yan, Q. Fang, X. Kuai, S. Zhou, J. Chen, H. Zhang, X. Wu, G. Zeng, Z. Wu, B. Zhang, Y. Tang, Q. Zheng, H. Liao, K. Dong, I. Manke, X. Wang, Y. Qiao and S. Sun, Adv. Mater., 2023, 36, 2308656 CrossRef PubMed.
- R. Wang, J. Chen, Y. Zhang, Z. Li, S. Cao, X. Liu, H. Hu, L. Wu, Y. Shen and X. Wang, Chem. Eng. J., 2024, 485, 149827 CrossRef.
- Y. Song, Y. Cui, L. Geng, B. Li, L. Ge, L. Zhou, Z. Qiu, J. Nan, W. Wu, H. Xu, X. Li, Z. Yan, Q. Xue, Y. Tang and W. Xing, Adv. Energy Mater., 2024, 14, 2303207 CrossRef.
- R. J. Clément, Z. Lun and G. Ceder, Energy Environ. Sci., 2020, 13, 345–373 RSC.
- J. Li, G. Liang, W. Zheng, S. Zhang, K. Davey, W. K. Pang and Z. Guo, Nano Mater. Sci., 2023, 5, 404–420 CrossRef.
- Y. Wang, A. Outka, W. M. Takele, M. Avdeev, S. Sainio, R. Liu, V. Kee, W. Choe, B. Raji-Adefila, D. Nordlund, S. Zhou, W. H. Kan, T. G. Habteyes and D. Chen, Adv. Mater., 2023, 35, 2306396 CrossRef PubMed.
- N. Li, M. Sun, W. H. Kan, Z. Zhuo, S. Hwang, S. E. Renfrew, M. Avdeev, A. Huq, B. D. McCloskey, D. Su, W. Yang and W. Tong, Nat. Commun., 2021, 12, 2348 CrossRef PubMed.
- E. Lee, B. J. Kwon, F. Dogan, Y. Ren, J. R. Croy and M. M. Thackeray, ACS Appl. Energy Mater., 2019, 2, 6170–6175 CrossRef.
- R. Koerver, W. Zhang, L. De Biasi, S. Schweidler, A. O. Kondrakov, S. Kolling, T. Brezesinski, P. Hartmann, W. G. Zeier and J. Janek, Energy Environ. Sci., 2018, 11, 2142–2158 RSC.
- X. Zhao, Y. Tian, Z. Lun, Z. Cai, T. Chen, B. Ouyang and G. Ceder, Joule, 2022, 6, 1654–1671 CrossRef.
- C. Wang, R. Zhang, C. Siu, M. Ge, K. Kisslinger, Y. Shin and H. L. Xin, Nano Lett., 2021, 21, 9797–9804 CrossRef.
- K. Liu, Q. Zhang, Z. Lu, H. Zhu, M. Song, L. Chen, C. Zhang and W. Wei, ACS Appl. Mater. Interfaces, 2024, 16, 14902–14911 CrossRef.
- J. Langdon and A. Manthiram, Energy Storage Mater., 2021, 37, 143–160 CrossRef.
- M. Yoon, Y. Dong, Y. Huang, B. Wang, J. Kim, J.-S. Park, J. Hwang, J. Park, S. J. Kang, J. Cho and J. Li, Nat. Energy, 2023, 8, 482–491 CrossRef.
- F. Li, K. Fan, Y. Tian, P. Hou, H. Zhang, Y. Sun, J. Huang, X. Xu and H. Huang, J. Mater. Chem. A, 2022, 10, 16420–16429 RSC.
- H. Zhao, Q. Yang, N. Yuca, M. Ling, K. Higa, V. S. Battaglia, D. Y. Parkinson, V. Srinivasan and G. Liu, Nano Lett., 2016, 16, 4686–4690 CrossRef.
- H. Hao, R. Tan, C. Ye and C. T. J. Low, Carbon Energy, 2024, e604 CrossRef.
- W. Shen, K. Li, Y. Lv, T. Xu, D. Wei and Z. Liu, Adv. Energy Mater., 2020, 10, 1904281 CrossRef.
- Z. Zhang, Y. Song, B. Zhang, L. Wang and X. He, Adv. Energy Mater., 2023, 13, 2302134 CrossRef.
- Y. Ye, R. Xu, W. Huang, H. Ai, W. Zhang, J. O. Affeld, A. Cui, F. Liu, X. Gao, Z. Chen, T. Li, X. Xiao, Z. Zhang, Y. Peng, R. A. Vila, Y. Wu, S. T. Oyakhire, H. Kuwajima, Y. Suzuki, R. Matsumoto, Y. Masuda, T. Yuuki, Y. Nakayama and Y. Cui, Nat. Energy, 2024, 9, 643–653 CrossRef.
- Y. Wang, J. Zhu, A. Chen, X. Guo, H. Cui, Z. Chen, Y. Hou, Z. Huang, D. Wang, G. Liang, S. C. Cao and C. Zhi, Adv. Mater., 2023, 35, 2303165 CrossRef PubMed.
- H. Ren, J. Hu, H. Ji, Y. Huang, W. Zhao, W. Huang, X. Wang, H. Yi, Y. Song, J. Liu, T. Liu, M. Liu, Q. Zhao and F. Pan, Adv. Mater., 2024, 36, 2408875 CrossRef PubMed.
- L. Zhao, Y. Peng, P. Dou, Y. Li, T. He and F. Ran, InfoMat, 2024, 6, e12597 CrossRef CAS.
- S. Kalnaus, N. J. Dudney, A. S. Westover, E. Herbert and S. Hackney, Science, 2023, 381, eabg5998 CrossRef CAS.
- Z. Li, H. Yi, W. Ding, H. Ren, Y. Du, M. Shang, W. Zhao, H. Chen, L. Zhou, H. Lin, Q. Zhao and F. Pan, Adv. Funct. Mater., 2023, 34, 2312837 CrossRef.
- S. S. Zhang, K. Xu and T. R. Jow, J. Power Sources, 2006, 160, 1349–1354 CrossRef CAS.
- K. Yu, A. M. Adeyinka, S.-Y. Choe and W. Lee, J. Power Sources, 2025, 626, 235759 CrossRef CAS.
- J. Guo, Y. Xu, M. Exner, X. Huang, Y. Li, Y. Liu, H. Wang, J. Kowal, Q. Zhang, P. K. Kristensen, D. Wang, K. Pedersen, L. Gurevich, D. Stroe and P. Adelhelm, Adv. Energy Mater., 2024, 14, 2400190 CrossRef CAS.
- G. Sikha, P. Ramadass, B. S. Haran, R. E. White and B. N. Popov, J. Power Sources, 2003, 122, 67–76 CrossRef CAS.
- Z. Guo, B. Y. Liaw, X. Qiu, L. Gao and C. Zhang, J. Power Sources, 2015, 274, 957–964 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2025 |