
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Recent progress and perspectives on composite structural layered transition metal oxides for sodium-ion batteries
Zhiyang
Fan
 a,
Yichen
Li†
a,
Jiawei
Pan
a,
Zhiyou
Zhou
ac,
Weipeng
Li
a,
Taifan
Yang
a,
Haihan
Zhang
a,
Chengyong
Shu
*a,
Weibo
Hua
a,
Yuping
Wu
*d and
Wei
Tang
*ab
a,
Yichen
Li†
a,
Jiawei
Pan
a,
Zhiyou
Zhou
ac,
Weipeng
Li
a,
Taifan
Yang
a,
Haihan
Zhang
a,
Chengyong
Shu
*a,
Weibo
Hua
a,
Yuping
Wu
*d and
Wei
Tang
*ab
aSchool of Chemical Engineering and Technology, Xi'an Jiaotong University, Xi'an, 710049, China. E-mail: kowscy-n@mail.xjtu.edu.cn; tangw2018@xjtu.edu.cn
bNational Innovation Platform (Center) for Industry-Education Integration of Energy Storage Technology, Xi'an, 710049, China
cNa Mey New Energy Technology (Luoyang) Limited Company, Luo'yang, 471000, China
dSchool of Energy and Environment, Southeast University, Nanjing 210096, China. E-mail: wuyp@seu.edu.cn
First published on 14th January 2025
Abstract
Layered transition metal oxides (LTMOs) are regarded as a compelling candidate for the prospective commercialization of sodium-ion batteries (SIBs), attributed to their simplistic architectural design, elevated operating potential window, and the feasibility of synthesis and tailoring. Nevertheless, a prevalent issue with many of these materials is their susceptibility to degradation under ambient conditions and suboptimal electrochemical behaviors. In light of these considerations, the multiphasic and synergistic effects exhibited by composite structural LTMOs for SIBs have emerged as a potent strategy to mitigate the aforementioned challenges. Furthermore, at present, the mechanistic insights into the synergistic interactions among multiple phases remain fragmented. This review offers a comprehensive synopsis of the advancements in research pertaining to composite structural LTMOs in SIBs, encompassing layered heterogeneous phases, multiphase composites, core–shell structures, and concentration gradient structures. Notably, this extensive overview delves into the intricate structure–function–performance relationship of phase interfaces and offers insights into predictive methodologies, synthetic strategies, and comprehensive materials characterization techniques, in order to pave a new avenue for future research, endeavors focused on composite-structured LTMOs, and fostering the development of more robust and efficient sodium-ion battery materials.
Broader contextSodium-ion batteries (SIBs) are regarded as an important supplement to lithium-ion batteries (LIBs) due to their advantages of abundant resources and low cost, showing great potential, especially in large-scale energy storage. In the field of SIB research, the development of high-performance cathode materials represents a crucial objective. Layered transition metal oxide (LTMO) cathode materials have attracted considerable attention due to their high specific capacity, structural diversity and component tunability. However, conventional single-phase layered oxide cathodes typically present a number of challenges, including irreversible phase transition, poor air stability and interfacial side reactions. Nevertheless, recent studies have demonstrated that composite structural LTMO cathode materials can effectively stabilize the phase transition of the material, enhance structural stability and markedly improve sodium-ion diffusion kinetics. Currently, the mechanism of synergistic interactions between each phase remains poorly understood. This review provides a comprehensive summary of the research progress in composite-phase layered oxides in sodium-ion batteries, discusses the possible mechanisms of composite-phase materials, and provides insights into future research directions. |
1. Introduction
Lithium-ion batteries (LIBs), as traditional commercial batteries, have been widely used in portable electronic products and electric vehicles.1–6 However, the scarcity of resources and high production costs have gradually become significant obstacles to their further development.7,8 Sodium is a highly abundant element on Earth, with an abundance 1180 times that of lithium,9 and the physical and chemical characteristics of sodium, which is situated in close proximity to Li in the periodic table, are analogous to those of lithium and result in a comparable rocking-chair mechanism.10,11 Furthermore, the reactive resistance between Na and Al negates the necessity for costly Cu current collectors, thereby conferring a substantial cost benefit.12 Besides, a puncture experiment demonstrated that sodium-ion batteries (SIBs) exhibit high stability during the reaction process and are less prone to decomposition.13 Currently, the primary cathode materials for SIBs include layered oxides,14–16 polyanionic compounds,17,18 Prussian blue analogs,19,20 and organic materials.21,22 Layered oxides have attracted considerable research attention due to a number of favorable characteristics, including a high specific capacity, rapid sodium-ion diffusion rates, structural simplicity, and cost-effectiveness. Sodium-ion transition metal layered oxides are capable of forming layered oxides with a variety of elements (Ni, Co, Mn, Fe, Ti, V, Cr, and Cu),23 whereas lithium exhibits electrochemical activity only with a selected few elements (Ni, Co, and Mn).24 Nevertheless, despite their promising attributes, several impediments persist, hindering the commercialization of LTMOs. A notable challenge is the propensity of layered oxide cathode materials to frequently undergo irreversible phase transitions under high voltages,25,26 which are typically accompanied by significant volume fluctuations, further triggering the fracture of particles or their detachment from the current collector and impairing the diffusive kinetics of sodium ions. On the other hand, LTMOs exhibit heightened susceptibility to degradation with water (H2O) and carbon dioxide (CO2) molecules readily absorbed and intercalated into the layered structure when exposed to humid air; LTMOs are rapidly degraded, leading to a loss of active sodium and the formation of surface alkaline residues, which adversely affect electrode fabrication and battery performance.27,28To tackle these challenges, researchers have devised multifaceted strategies, including element doping,29–32 surface coating,33–35 and the design of structure and morphology36 to bolster performance. Nevertheless, non-uniform doping may lead to local phase structure instability, which in turn affects the consistency and stability of the material's performance during electrochemical cycling. Additionally, the thickness of the coating and imperfections at the interface between the coating layer and the substrate can undermine structural integrity and performance.37–39 Amidst this backdrop, composite structural layered cathode materials have emerged as a vibrant frontier in high-performance SIB research (Fig. 1).
 | ||
| Fig. 1 Schematic diagram of composite structural LTMO design in SIBs. | ||
Scientists are now exploring the development of composite structural materials that offer superior integrated electrochemical properties compared to single-phase materials. Reflecting on past studies, the development of composite structural materials has yielded numerous achievements, primarily in the areas of materials synthesis and performance optimization.40–42 Nevertheless, a notable gap persists in the comprehensive understanding of the fundamental mechanisms underpinning their functionality. Moreover, the sintering temperature, along with the precise stoichiometry of transition metals and sodium ions, as well as the strategic doping of other ions, profoundly impacts the formation of the ultimate structure of composite materials. These factors further complicate the characterization and design of their structure. In the regard, this review delves into the remarkable endeavors and advancements achieved in composite structural LTMOs in recent years, embarking from the spatial heterogeneity in their crystalline structures. It meticulously summarizes the diverse categories of composite structural LTMOs, encompassing layered heterostructures, multiphase composites, intricate core–shell configurations, and innovative concentration gradients. By offering an encompassing overview, it elucidates the mechanisms underpinning phase regulation at varying scales, highlighting how these intricate interactions influence the crucial properties of materials such as high-rate performance, cycling stability, and air stability. Furthermore, this review sheds light on promising avenues for future research, providing a roadmap for exploring the untapped potential of these innovative materials.
2. Limitations and structural prediction of LTMOs
In SIBs, sodium based LTMOs with the composition NaxTMO2 (TM = transition metal, 0 ≤ x ≤ 1) are a typical type of cathode material. Based on their morphology, transition metal oxides can be classified into two main categories: layered and tunnel structures.43,44 The layered structure is composed of transition metal oxide octahedra (TMO2) with shared edges, in which Na+ is located between TMO2 sheets. Delmas first categorized the LTMOs into O-type and P-type structures by delineating the distinctive chemical environments that surround the Na+ layers within the layers.45 In the O-type structure, sodium ions occupy the octahedral sites formed by oxygen atoms (2/3 < x < 1), while in the P-type structure, they occupy the prismatic sites formed by oxygen atoms (1/2 < x < 2/3). Conversely, at lower sodium contents (x < 1/2),46 the oxides predominantly adopt a tunnel-like structure.47 Although three-dimensional tunnel oxides possess a stable structure, the initial sodium content is insufficient, resulting in a low initial charge–discharge capacity ratio and, consequently, limiting their practical application.48,49 The number of unit layers within a single cell allows for further classification of O-type and P-type materials into O2, O3, and P2, P3. The majority of reported layered metal oxides are found in O350–53 and P254–56 forms, where the numerals 2 and 3 represent the minimal layer counts of the periodic sequences formed by oxygen stacking, ABBAABBA and ABCABC, respectively. In the O3 type, sodium ions are interconnected with transition metal ions in an edge-sharing manner, occupying octahedral sites, exhibiting a higher sodium content and increased initial capacity. As depicted in Fig. 2a, the diffusion of sodium ions occurs primarily through the edge-sharing tetrahedral sites positioned between these layers in the O3 structure. This process encounters substantial energy barriers, stemming from the unfavorable accommodation of sodium ions within the tetrahedral sites, a phenomenon attributed to inherent size mismatches. Conversely, in the P2 structure, sodium ions traverse a relatively roomy plane, facilitating a decrease in energy barriers and thereby accelerating the diffusion kinetics. Nevertheless, this configuration's limitation lies in its reduced sodium content, ultimately leading to a diminished specific capacity.57 Since these single-phase structures exhibit a range of distinctive strengths and weaknesses, the accurate prediction of the structure of synthesized materials can result in significant savings in terms of manpower and resources by enabling the targeted preparation of desired products and accelerates the development of new LTMOs. As illustrated in Fig. 2b, Zhao et al. discovered that a ratio of approximately 1.62 between the interlayer distances d(O–Na–O) in the alkali metal layer and d(O–M–O) in the transition metal layer can serve as an indicator to distinguish between P2 and O3 type structures.58 Subsequently, Zhao et al. put forth the concept of the “cationic potential” parameter to guide the design, synthesis, or optimization of layered oxide cathode materials, which is defined as follows: | (1) |
 | (2) |
 | (3) |
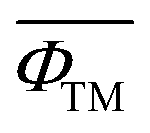 is the weighted ionic potential for transition metal ions. ωi, ni and Ri correspond to the stoichiometric coefficient, charge, and ionic radius of species, respectively.
is the weighted ionic potential for transition metal ions. ωi, ni and Ri correspond to the stoichiometric coefficient, charge, and ionic radius of species, respectively.  is the weighted ionic potential for the sodium ion site,
is the weighted ionic potential for the sodium ion site,  is considered to be constant at 28.571 nm−1
is considered to be constant at 28.571 nm−1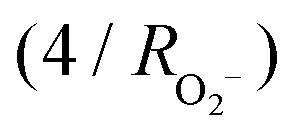 . Extensive experiments have revealed a preference for the formation of the O3 phase when
. Extensive experiments have revealed a preference for the formation of the O3 phase when  (Fig. 2c).59 Upon reversing this inequality, the P2 phase emerges as the preferred configuration. An increased ratio favors the formation of the O3 phase, whereas a decreased ratio promotes the genesis of the P2 phase. This underscores the viability of the design and prediction of layered structures as a strategic approach. Moreover, the relentless pursuit of novel sodium-ion materials, coupled with the optimization of the composition and structure of existing materials, holds paramount significance for advancing the practical application of SIBs.
(Fig. 2c).59 Upon reversing this inequality, the P2 phase emerges as the preferred configuration. An increased ratio favors the formation of the O3 phase, whereas a decreased ratio promotes the genesis of the P2 phase. This underscores the viability of the design and prediction of layered structures as a strategic approach. Moreover, the relentless pursuit of novel sodium-ion materials, coupled with the optimization of the composition and structure of existing materials, holds paramount significance for advancing the practical application of SIBs.
 | ||
| Fig. 2 Crystal structures of O3 and P2 phases and predictive methodologies for their identification. (a) Crystalline structure of layered oxide cathode materials and the Na+ migration path.57 Copyright 2024, Royal Society of Chemistry. (b) The ratio of the interlayer distance between the alkali metal layer d(O–Na–O) and the transition metal layer d(O–TM–O).58 Copyright 2018, Wiley-VCH GmbH. (c) Various sodium based LTMOs with the O3 and P2 structures, which show clear tendency toward each structure depending on the sodium ionic potential and the overall cationic potential.59 Copyright 2020, The American Association for the Advancement of Science. | ||
3. Layered heterogeneous phases
Layered heterogeneous materials refer to the materials composed of two or more layered phases with different properties, which are distributed unevenly on a macroscopic or microscopic scale. In the realm of battery electrode materials, these materials exhibit a special spatial arrangement of elements or crystal structures, endowing them with a diverse array of physicochemical attributes. For instance, layered heterogeneous materials may incorporate an active phase renowned for its high specific capacity, alongside an inactive phase that safeguards structural stability to mitigate the volume expansion, and bolster its high-rate charge–discharge capabilities simultaneously.In 2014, Lee et al. pioneeringly embarked on exploring the incorporation of Li into O3-NaNi0.5Mn0.5O2, culminating in the formation of a topotactic intergrowth O3 (primary phase)/P2-Na1−xLixNi0.5Mn0.5O2+d composite material (Fig. 3a).60 Particularly, upon initial cycling, this doped material evolved into a P2/P3 intergrowth structure, which electrochemically stabilized, circumventing the energy costs typically incurred during the repeated O3 to P3 phase transition (Fig. 3c). Consequently, this structurally robust layered material optimizes sodium-ion diffusion between the trigonal prismatic sites throughout the majority of the charge–discharge progression, unhindered by macroscopic lattice rearrangements. The corresponding Li-NMR spectroscopy result reveals that Li is predominantly located within the alkali layers of the phases, while a minor fraction of Li incorporated as a dopant within the TM layers induces the nanoscale intergrowth of phases. This intergrowth effect serves to stabilize the structural changes associated with phase transitions, thereby improving the electrochemical performance of the material.
 | ||
| Fig. 3 The influence of doping on the phase transformation of layered heterogeneous phase structures in SIBs. (a) XRD patterns of Na1−xLixNi0.5Mn0.5O2+d when the Li content varies from 0 to 1 and the major phase evolution as a function of Li content is summed up by color switches in the right. (b) High-resolution TEM images that show the P2 and O3 lattice intergrowth stacking of different magnitudes. (c) In situ synchrotron X-ray diffraction of Na0.7Li0.3Ni0.5Mn0.5O2 during initial charge–discharge.60 Copyright 2014, Wiley-VCH GmbH. (d) Schematic diagram of the phase proportion at different Ti contents. (e) HRTEM image of NMT3, (f) STEM image of ABF and (g) HAADF image of NMT3 at the [010] zone axis.62 Copyright 2022, Elsevier. (h) Schematic illustration of the synthesis of MCA cathodes by the dealloying–annealing strategy. (i) STEM-HAADF image of the P2/P3 MCA-3 sample.69 Copyright 2014, Wiley-VCH GmbH. | ||
Apart from the partial substitution of Li within the alkali metal layers, the introduction of dopants in the transition metal layers can also modulate the ratio of heterogeneous phases, such as with Mg,61 Ti,62–64 Sn65–67 and Cu.68 Yu et al. prepared a series of Na0.85Ni0.34Mn0.66−xTixO2 layered oxides by strategically adjusting the content of Ti (denoted as NM, NMT1, NMT2, NMT3, and NMT4 for x = 0, 0.11, 0.22, 0.33, and 0.44, respectively). As the content of Ti increases, the proportion of the O3 phase in the material correspondingly increases, and when x = 0.44, the material exclusively embodies the O3 phase (Fig. 3d). This phenomenon can be elucidated by the “cationic potential”, wherein Ti4+ possesses a lower potential compared to Mn4+, which favors the formation of the O3 phase. The high-resolution transmission electron microscopy (HRTEM) and high-angle annular dark field (HAADF) images clearly distinguish between interlayer distances of 5.4 Å and 5.6 Å, affirming the topological intergrowth structure of P2 and O3 phases at the atomic level in P2/O3-NMT3 (Fig. 3e–g). The P2/O3 heterogeneous phases undergo a more intricate phase transition sequence during cycling. During the initial charging process, the P2 phase serves as a stabilizing scaffold, alleviating the structural stress associated with the O3 to P3 phase transition. As the voltage increases, P2-NM and P3-NMT4 undergo complete transformations into the O2 and OP2 phases, respectively, a process that is accompanied by substantial volume fluctuations and structural stresses. The P2 phase in the P2/O3 heterogeneous phases remains inert, generating stress in the opposite direction of the gliding plane, thereby hindering the sliding motion of the P3 transition metal layer. The interlocking effect of the P2 and O3 phases mitigates lattice mismatch and minimizes the sliding of the transition metal layers, contributing to enhanced structural stability.
In another study,69 a series of heterogeneous phase materials was meticulously crafted through a dealloying and annealing process. As the Co content in the material increased, the proportion of the P3 phase within the composite phase gradually increased (Fig. 3h). In particular, upon achieving a Co addition of 30%, the P3 phase proportion peaked at 37.8%. The doping of Co in the transition metal layer delicately modulated the Na environment, triggering a cationic rearrangement that facilitated the emergence of the P3 phase structure with lower sodium ion diffusion barriers. The fast Fourier transform (FFT) pattern along the [110] direction revealed the optimized intergrowth of non-phasic domains in the Na0.67Mn0.64Co0.30Al0.06O2 (MCA-3) sample (Fig. 3i), underscoring its optimized microstructure. During the cycling process, the unique P2/P3 phase structure effectively suppressed the P2–P2′ phase transition, with no discernible formation of extra phases, according to the mitigation of the Jahn–Teller effect in the MCA-3 cathode. Contemporary research endeavors have convincingly demonstrated that the incorporation of dopants into a lattice can proficiently engender alternating heterostructured nanoarchitectures and incite localized lattice distortions, thereby fostering the stable coexistence of disparate phases.
Moreover, in the synthesis process, the sintering temperature also has a profound effect on the composite phase materials, intricately intertwined with their microstructural characteristics, phase composition, and ultimate electrochemical behaviors.70,71 Throughout the sintering process, the extent of temperature elevation influences the lattice energy, which dictates the tenacity of ionic bonding and the facility of ion diffusion, consequently affecting the crystallization and grain growth of the material. Guo et al. illuminated the synergistic advantages of the P2 and P3 phases in the hybrid cathode Na0.7Li0.06Mg0.06Ni0.22Mn0.67O2, utilizing high-temperature in situ XRD to track phase evolution across a temperature range from 450 °C to 900 °C.72 As shown in Fig. 4a, the P3 phase is produced at 670 °C, while the P2 phase emerges above 750 °C, and pure P3 and P2 structures are formed below 750 °C and above 830 °C, respectively. Upon increasing the temperature, the diffraction peaks of the P3 phase gradually diminish and eventually vanish, while the diffraction peaks of the P2 phase progressively emerge and intensify. What's more, with increasing calcination temperature, sodium layer gliding in the P-type layered cathode is observed, and it can be clearly observed from the HRTEM diagram that P2 (104) and P3 (012) have a symbiotic structure (Fig. 4b), indicating that the P3–P2 phase transition necessitates substantial energy to facilitate breaking/reforming the Me–O bonds.
 | ||
| Fig. 4 The impact of sintering temperature, sodium content, and transition metal elements on the phase transformation of layered heterogeneous phase structures in SIBs. (a) The roadmap of high-temperature in situ XRD patterns of the obtained precursor. (b) The P2/P3-Na0.7Li0.06Mg0.06Ni0.22Mn0.67O2 composite HRTEM images.72 Copyright 2019, Elsevier. (c) XRD patterns of NaxNi0.5Mn0.3Co0.2O2 composites (0.50 ≤ x ≤ 1.00). (d) Phase evolution derived from the XRD analysis. (e) Rate capability of the NaxNi0.5Mn0.3Co0.2O2 cathodes.78 Copyright 2018, American Chemical Society. (f) Synthesis phase diagram of NaxMnyNi1−yO2 with a sintering temperature of 900 °C. (g) Cationic potentials (black dots) of each Na0.85Mn0.5Ni0.4M0.1O2 (M = Ti, Co, Fe, Cu, Ni, and Mn) material and its corresponding P2 ratio (red dots). (h) Cycling performances of Na0.85Mn0.5Ni0.4M0.1O2 (M = Ti, Co, Fe, Cu, Ni, and Mn) and NaMn0.5Ni0.4Fe0.1O2 between 2 and 4 V at 1C with the first 3 cycles under 0.1C (1C = 120 mA g−1).79 Copyright 2021, Elsevier. | ||
Obviously, the sodium content and the ratio of transition metals play a crucial role in tailoring the material's phase structure. By adjusting these parameters, researchers can further optimize the performance of the material. Sodium-rich stoichiometric approaches tend to favor the promotion of the O3 phase, while sodium-deficient methods are more inclined to encourage the formation of the P2 phase.73,74 An increase in sodium content within a specific voltage range can result in an enhanced capacity but introduce more complex phase transitions under a high voltage, emphasizing the paramount importance of meticulously examining sodium concentration and the composition of transition metal elements during the material selection process75–77 For instance, a study showed fabrication of a class of nickel-based layered NaxNi0.5Mn0.3Co0.2O2 oxide composites by adjusting the sodium content and annealing temperature (Fig. 4d).78 When the sodium content is relatively low (x = 0.5 and 0.6), the P2/P′3 phase dominates. As the sodium content escalates, so does the quantity of the P′3 phase. Upon increasing the sodium content to 0.7, the presence of the P2 phase is further diminished, with the emergence of the O′3 phase leading to a coexistence of P′3/P2/O′3 phases. Upon elevating the sodium content to 0.8, a distinct tri-phase composite of P′3/O3/O′3 is observed, while the O3 and O′3 phases become dominant, with the concomitant disappearance of the NiO impurity phase at a sodium content of 0.9. Among these, the P2/P′3/O′3-tri-phase composite exhibited a high specific capacity of 126 mA h g−1 within a voltage range of 1.5–4.2 V and a high-rate capability of approximately 72% of its initial capacity at 5C (Fig. 4e). Analogously, Xiao et al. have systematically investigated NaxMnyNizTM1−y−zO2-based cathode materials (Fig. 4f), leveraging the cation potential to fine-tune phase proportions through elemental substitution, and further promoting the structural robustness and electrochemical performance of the materials.79 The ratio of O3 to P2 phases, taken as the characteristic of the O3/P2 structure, can be modulated by partially replacing other transition metals (TMs) with Mn substituents with various TMs, revealing that the P2 phase preferentially forms in Mn-rich environments following sequentially by Ni, Cu, Fe, Co, and Ti in descending order (Fig. 4g), whereas a pure O3 phase emerges in multiple element doping. Notably, the Na0.85Mn0.5Ni0.4Ti0.1O2 cathode material stands out, delivering an impressive specific capacity of approximately 130 mA h g−1 within the 2 to 4 V voltage window, accompanied by a remarkable capacity retention rate of 91% after 500 cycles (Fig. 4h). Extensive research has demonstrated that meticulously modulating the cation potential via the compositional fine-tuning of transition metals and sodium is instrumental in governing phase ratios and mitigating the growth of non-uniform intergrowth phases. The cornerstone of developing heterogeneous phase materials lies in understanding and controlling the interactions between different phases and their impact on the overall material properties. The formation of heterogeneous phases is influenced by a variety of factors. Doping can change lattice parameters and grain boundary properties to improve ion transport and electrochemical stability of materials. Moreover, temperature plays a critical role in affecting the diffusion of sodium and transition metals within the bulk phase, subsequently impacting the microstructural features and interfacial attributes of the material. By meticulously optimizing these pivotal factors, one can significantly bolster the cycling stability and energy density of the material, thereby catering to the stringent demands of advanced battery applications.
4. Multiphase composites
Beyond layered materials, tunnel-type80 and spinel-structured81–84 materials have also garnered significant interest due to their unique 3D crystallographic structures and expanded tunnels for sodium insertion/extraction. In pursuit of augmenting the electrochemical performance of materials, efforts have been directed towards integrating layered cathodes with tunnel-type and spinel structures.Xiao et al. introduced a novel layered-tunnel intergrowth structure, comprising the stoichiometric composition Na0.6MnO2, which has been effectively synthesized via a straightforward thermal polymerization technique in conjunction with a solid-state reaction process.87In situ X-ray diffraction (XRD) analysis reveals a phase transformation from P2–O2 to P2–OP4 within the originally monophasic material. The OP4 phase, distinguished by an alternating sequence of P–O–P–O, mitigates the substantial interlayer spacing variations associated with the P–O transition due to the presence of the P-type structure. Consequently, the layered-tunnel multiphasic structure of Na0.6MnO2 displays superior cycling stability, retaining 85.1% of its capacity after 100 cycles at a 1C rate. Furthermore, building upon the Na0.6MnO2 material, Gao et al. have conducted further investigations into the inducing effects of sodium content and cobalt substitution on the heterogeneity of the layered tunnel structure. A hybrid P2 + Tunnel phase, NaxCo0.1Mn0.9O2 (0.44 ≤ x ≤ 0.7), is distinguished by a multitude of interfacial features that facilitate high-performance sodium storage.85 HRTEM images clearly demonstrate the coexistence of the P2 and T phases with the presence of lattice fringes with the interspacing of 2.84 Å and 5.49 Å, respectively, which correspond to the (100) planes of the P2 structure and the (050) planes of the tunnel structure, respectively. Fig. 5a provides a clear delineation of the interface between the two phases within the material, confirming the formation of growth regions in the P2 + Tunnel composite material. The corresponding FFT patterns (Fig. 5a1′–a3′) demonstrate the FFT diffractions, which can be indexed along the [100] and [050] zone axes, respectively. The interface is observed to manifest as a loose polycrystalline structure, which can be attributed to the irregular arrangement of atoms, suggesting the presence of a coherent orientation relationship between the intergrowth of nanoscale domains. The tunnel phase effectively accelerates sodium-ion diffusion and exhibits remarkable structural stability, while the layered P2 phase significantly contributes to achieving a high specific capacity. Moreover, the P2 + Tunnel phase interface provides additional channels and active sites for charge transfer and surplus charge storage. As a consequence, the prepared NaxCo0.1Mn0.9O2 (0.44 ≤ x ≤ 0.7) exhibits an impressive discharge capacity of 219 mA h g−1 at 0.1C, retaining 117 mA h g−1 even at a high rate of 5C.
 | ||
| Fig. 5 Multiphase LTMO cathode materials in SIBs. (a) HRTEM images of the P2 + T material. The amplified figure of the rectangular region (a1–a3) and the corresponding fast Fourier transform (FFT) diffraction (a1′–a3′). (b) Two phase bonded interface model.85 Copyright 2018, Royal Society of Chemistry. (c) HRTEM images of the LS-NFM sample. The red and white dashed squares indicate the spinel and layered components, respectively, where the corresponding FFT images are shown as insets. The red and white circles indicate the spinel (111) plane with the zone axis [110] and layered (003) plane with the zone axis [100], respectively. (d) GITT profile and Na diffusivity as a function of the voltage of LS-NFM and NFM cathodes in (e) charge and (f) discharge processes. (g) Rate capability of LS-NFM and NFM cathodes.86 Copyright 2018, American Chemical Society. | ||
In addition to the composite of layered and tunnel structures, the hybridization of layered and spinel structures has emerged as a potent approach to bolster the electrochemical performance and stability of cathode materials. As reported by Xiong and colleagues, a Li doped O3/spinel composite material, Li0.25Na0.87Ni0.4Fe0.2Mn0.4O2+δ (LSNFM), was successfully fabricated. HRTEM images vividly depict the nanoscale integration of the layered O3 (003) and spinel structured (111) planes in intimate contact (Fig. 5c), while the phase interfaces are observed to be tightly packed together with excellent structural compatibility and connectivity.86 Following 50 cycles, the retention of both layered and spinel structures underscores the robust structural stability of LSNFM. The rate of charge transfer within the LS-NFM cathode is faster than that of O3-NFM. The results of the galvanostatic intermittent titration (GITT) measurement from the charge–discharge processes reveal that the LS-NFM cathode boasts a superior diffusion rate, initiating from the initial charge and persisting up to an electrode potential of 3.75 V (Fig. 5d–f). Specifically, the diffusion coefficient of LS-NFM was determined to be 2.4 × 10−13 cm2 s−1 at 2.67 V, significantly outperforming the 1.44 × 10−14 cm2 s−1 of NFM at 2.80 V. Furthermore, across the entire potential range, the LS-NFM electrode sustains a diffusion rate during discharge that is an order of magnitude higher than that of the NFM electrode. This result is consistent with the observed behavior of layered-spinel materials in LIBs88–91 and the integration of 3D spinel and 2D layered phase channels results in a reduction of the sodium-ion diffusion pathway, thereby enhancing the rate capability.
In conclusion, the fusion of stable tunnel-like or spinel structured materials with layered materials offers a promising avenue to amplify the high-capacity characteristics and superior rate performance advantages of both. However, the formidable challenge in crafting multiphasic cathode materials resides in the intricate integration of layered architectures with tunnel or spinel structures. The aim is to harness the synergistic strengths while mitigating the inherent limitations of each phase, thereby achieving unparalleled electrochemical performance. It is essential that the interfaces between phases are compatible and connected, providing additional conduits and active sites for accelerated sodium transport and charge storage. Moreover, the spatial distribution and ratio of the two phases must be subjected to meticulous consideration, as tunnel-type materials are inherently susceptible to sodium deficiency, and an insufficiency in sodium content may result in a reduction in the material's capacity.
5. Core–shell structure
Despite the high theoretical capacities exhibited by most O3-type LTMOs,92 their structural fragility and heightened reactivity in humid environments93,94 give rise to residual alkali formation,95,96 ultimately curtailing the cathode's longevity. In order to protect the layered matrix, certain coatings have been incorporated, including metal oxides (such as Al2O3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 97,98 and MgO99), metal phosphides,100 and other inert materials.101,102 However, these coatings are typically only around 10 nm thick and are electrochemically inert. To reconcile the need for material stability with high-capacity retention, researchers have devised core–shell structures as a solution to this issue. The design of core–shell structures has yielded several promising outcomes in LIBs,103–105 inspiring the current study's proposal for a heterostructured cathode. A rational design of a hetero-structured cathode is proposed, combining a high-capacity O3-type core with a structurally stable P2-type Na2/3MnO2 coating on the surface (Fig. 6a).106 The protective P2-Na2/3MnO2 layer not only bolsters the cathode's resilience against air and heat exposure but also enhances electrochemical kinetics and minimizes charge transfer resistance. A half-cell with this optimized heterostructure cathode retains 85.3% of its capacity after 150 cycles at 1C (Fig. 6b). HRTEM imaging coupled with FFT analysis demonstrates that the O3-type core particles are encapsulated within a P2-NMO protective shell with an approximate thickness of 15 nm. The lattice fringes within the O3 phase core were measured at 2.43 and 5.34 Å, and correspond to the rhombohedral symmetry of the R3m space group, a characteristic of the O3 phase. On the particle surface, the (110) and (004) planes attributed to the P2 phase are observable, with the interface between the two phases exhibiting a high degree of connectivity, thereby ensuring facile sodium ion transport. XRD results indicate that the lower sodium content in the P2 phase leads to an increase in the c-axis parameter of the heterostructure, enhancing the repulsive forces between adjacent layers and expanding the interlayer spacing, which improves the kinetics of ion transportation.
97,98 and MgO99), metal phosphides,100 and other inert materials.101,102 However, these coatings are typically only around 10 nm thick and are electrochemically inert. To reconcile the need for material stability with high-capacity retention, researchers have devised core–shell structures as a solution to this issue. The design of core–shell structures has yielded several promising outcomes in LIBs,103–105 inspiring the current study's proposal for a heterostructured cathode. A rational design of a hetero-structured cathode is proposed, combining a high-capacity O3-type core with a structurally stable P2-type Na2/3MnO2 coating on the surface (Fig. 6a).106 The protective P2-Na2/3MnO2 layer not only bolsters the cathode's resilience against air and heat exposure but also enhances electrochemical kinetics and minimizes charge transfer resistance. A half-cell with this optimized heterostructure cathode retains 85.3% of its capacity after 150 cycles at 1C (Fig. 6b). HRTEM imaging coupled with FFT analysis demonstrates that the O3-type core particles are encapsulated within a P2-NMO protective shell with an approximate thickness of 15 nm. The lattice fringes within the O3 phase core were measured at 2.43 and 5.34 Å, and correspond to the rhombohedral symmetry of the R3m space group, a characteristic of the O3 phase. On the particle surface, the (110) and (004) planes attributed to the P2 phase are observable, with the interface between the two phases exhibiting a high degree of connectivity, thereby ensuring facile sodium ion transport. XRD results indicate that the lower sodium content in the P2 phase leads to an increase in the c-axis parameter of the heterostructure, enhancing the repulsive forces between adjacent layers and expanding the interlayer spacing, which improves the kinetics of ion transportation.
 | ||
| Fig. 6 Electrochemical behavior of core–shell structured LTMO cathode materials in SIBs. (a) HRTEM image of the O3@5% P2. (b) Cycling stability of the O3-NNMO and O3@5% P2 at 1C.106 Copyright 2022, Elsevier. (c) Schematic illustration of the core–shell structured Na0.8[(Ni0.5Co0.2Mn0.3)0.6(Ni0.33Mn0.67)0.4]O2. (d) Cross-sectional SEM image of the O3/O′3 electrode after 50 cycles. (d1, d2) Enlarged SEM images corresponding to the rectangular regions in (d). (e) Cross-sectional SEM image of the O3/O′3–P2 electrode after 50 cycles. (e1, e2) Enlarged images corresponding to the rectangular regions in (e). CV profiles of the O3/O′3 (f) and O3/O′3–P2 (g) samples.107 Copyright 2020, American Chemical Society. | ||
In a parallel study, a groundbreaking O3/O′3–P2 core–shell composite material was successfully synthesized,107 characterized by a nanoscale coherent P2/O3 intergrowth architecture seamlessly bridging the microscale P2 shell and the O3-type core (Fig. 6c). This innovative design imparts exceptional reversible capacity, outstanding rate performance, and enhanced cycling stability to the material. The cross-sectional electron probe micro-analysis (EPMA) results of the O3/O′3–P2 core–shell composite material reveal a distinct gradient in transition metal concentrations, with Ni and Co enriched in the core and Mn predominantly concentrated on the surface. The distribution of transition metals alters the material's electronic structure and crystallographic properties, while X-ray photoelectron spectroscopy (XPS) identified a lower oxidation state of surface Ni, attributed to the Mn-rich P2 shell that partially reduces nickel cations to a lower oxidation state, further corroborating this specific transition metal ion concentration. A comparative postmortem examination of the O3/O′3 and O3/O′3–P2 electrodes after 50 cycles underscores the significant protective role of the P2 shell. SEM cross-sections of the cycled electrodes show significant microcracks in the O3/O′3 electrode without a P2 shell, while the O3/O′3–P2 electrode, with its uniform P2 phase shell, suppresses volume phase changes during cycling and prevents electrolyte penetration into microcracks, thus decelerating material degradation (Fig. 6d and e). Analysis of the electrodes’ CV curves highlights minimal shifts in peak positions and intensities within the 2.3–3.0 V range for the O3/O′3–P2 electrode, signifying reduced polarization and sustained structural integrity before and after cycling (Fig. 6f and g).
The segregation in the elemental composition of particle surfaces is a common phenomenon in metal oxides, driven by surface Gibbs free energy.108 This phenomenon can be intentionally exploited to modulate the electronic and chemical properties of the surface, thereby facilitating the preparation of composite materials. Yan et al. prepared a P2@P3 Na0.78Cu0.33−xZnxMn0.67O2 composite without Ni and Co to further reduce the price, which sheds light on the intricate redox behavior of Cu2+/3+.109 Zn doping not only triggers the formation of a P3 phase, but also fosters the emergence of a P3 nanometer-scale thickness and a phase as a protective coating layer, characterized by a distinct discernible organization of crystals and atoms. This composite structure provides a high and flat voltage profile, demonstrating excellent electrochemical performance, improved structural and electrochemical stability, and moisture resistance.
In essence, the role of the core–shell structure in layered cathode materials for batteries is to encapsulate high-capacity core materials within a shell that possesses high electrical conductivity and chemical stability, thereby protecting the active core and providing expedited pathways for ion and electron transport. This architecture not only bolsters the structural integrity of the material and mitigates volume expansion during charge–discharge cycles but also enhances the electrochemical performance by elevating charge–discharge rates, augmenting cycling stability, and ameliorating safety.
6. Concentration gradient structure
Currently, layered oxide materials featuring a concentration gradient have showcased remarkable electrochemical performance, as evidenced in various studies.113–116 The introduction of a spatial variation in chemical composition within the material allows for the modulation of electrochemical properties, which then leads to enhanced structural stability and the promotion of uniform strain distribution during charge–discharge cycles. Furthermore, the concentration gradient serves as a regulatory mechanism, enhancing electronic conductivity and optimizing ion diffusion pathways, which in turn bolsters rate capability and cycling stability, contributing to an overall improvement in battery performance.For instance, Hwang et al. have ingeniously crafted high-density spherical particles that exhibit a unique structural transition from the inner end (Na[Ni0.75Co0.02Mn0.23]O2) to the outer end (Na[Ni0.58Co0.06Mn0.36]O2) (Fig. 7a).110 This material features a radially aligned layered prismatic structure, exhibiting a chemical compositional gradient that progressively varies from the core towards the periphery. The heightened nickel content within the particle core facilitates the multi-electron reactions of Ni4+/Ni3+ and Ni3+/Ni2+ in layered oxide cathodes for SIBs, thereby ensuring a high energy capacity. Meanwhile, the enriched manganese concentration at the exterior reinforces the material's structural stability. The narrow interstices between these prismatic structures not only augment tap density but also minimize undesired side reactions. This singular architecture harmoniously combines high specific capacity, extended cycling stability, and enhanced tap density. As depicted in Fig. 7b, the precursor's XRD pattern reveals a distinct layered hydroxide structure, which, upon thermal treatment, transforms into an O3-type layered structure as seen in Fig. 7d. Notably, the spherical secondary particles maintain a uniform size of approximately 10 μm, both prior to and following the sintering process. The radially aligned prismatic structure remains intact, with no discernible migration of transition metals observed, as evidenced in Fig. 7c–e. The rationally designed concentration gradient cathode material exhibits favorable temperature performance, with these results attributed to the distinctive chemical properties that facilitate Ni redox reactions and minimize the surface area in contact with corrosive electrolytes. Consequently, following 300 cycles at 0.5C and assembly in a full cell with hard carbon, the capacity retention of the RAHC cell remains at 80%, which is significantly higher than that of the bulk material NaNi0.60Co0.05Mn0.35O2 with a capacity retention of 49.2% under the same test conditions. Subsequently, their team further explored such spoke-like nanorods of Na[Ni0.61Co0.12Mn0.27]O2 with a concentration gradient.117 Mechanical strength assessment revealed that the particle strength of these secondary nanostructured assemblies (SNAs) was superior to their constant concentration (CC) counterparts. In addition, to further validate the high stress tolerance of the radially aligned hierarchical prismatic structures, both types of cathodes were immersed in the electrolyte and subjected to increasing discharge currents. Optical microscopy showed that the spherical particles of the CC control group fragmented, while the SNAs remained intact. This difference demonstrates the excellent electrochemical and mechanical properties of SNAs, which result from their radially arranged prismatic structure with a concentration gradient. Concentration gradients can typically enhance the bonding performance at material interfaces, especially in composite materials. This enhanced interfacial bonding helps to improve the overall mechanical properties of the material, particularly in terms of tensile strength and impact resistance.
 | ||
| Fig. 7 The strategic implementation of concentration gradients within layered oxide cathodes to enhance structural stability. (a) Design scheme for a RAHC particle compared with a bulk particle (b) and (d) XRD patterns of RAHC [Ni0.60Co0.05Mn0.35](OH)2 and sodiated Na[Ni0.60Co0.05Mn0.35]O2 (inset: scanning electron microscopy (SEM) image), (c) and (e) cross-sectional TEM image of RAHC [Ni0.60Co0.05Mn0.35](OH)2 and sodiated Na[Ni0.60Co0.05Mn0.35]O2.110 Copyright 2015, Springer Nature. (f) Schematic diagram of a single CG particle and the P2 structure. (g) In situ XRD pattern of the (002) peak of the CG sample during the first charge/discharge process. (h) The corresponding intensity contour map.111 Copyright 2024, Elsevier. (i) A schematic of the P3-type manganese-based CG-Na0.5[Ni0.216Mn0.784]O2(NaNMO) cathode, displaying varying atomic ratios of manganese and nickel, along with a gradual increase in low-spin Mn4+ concentration that prevents Jahn–Teller distortions. (j) Cycling stability for CG-NaNMO and bulk-NaNMO.112 Copyright 2023, American Chemical Society. | ||
What's more, a novel concentration gradient oxide (CG) Na0.67Ni0.17Mn0.66O2 with a P2 structure was designed and prepared using the co-precipitation approach (Fig. 7f).111 This unique composition boasts a gradation of Mn concentration, progressively intensifying from the core to the outer surface of each individual spherical primary particle, whereas the Ni concentration follows an inverse trend. To demonstrate the concentration gradient structure, the elemental cross-sectional distribution of individual precursor particles of CG was examined through energy dispersive spectroscopy (EDS) measurement to reveal a linear escalation of Mn concentration toward the particle surface, accompanied by a marginal decrease in Ni and Co content. In situ XRD tests were conducted to further elucidate the impact of the concentration gradient structure on the structural evolution of the cathode during cycling (Fig. 7h). During the charging and discharging processes, the 002 peak initially shifts to a lower degree due to the intercalation of sodium ions, subsequently reverting to its original position upon ion extraction. Significantly, the absence of O2 phase-related characteristic peaks in the high-voltage region underscores the efficacy of the Mn-rich surface concentration gradient in stymieing the P2–O2 phase transition, thereby mitigating the capacity fading.
P3-type LTMOs have garnered significant attention due to their high theoretical specific capacity and cost-effective synthesis methods. These materials demonstrate favorable sodium ion diffusion kinetics, attributed to their large open channels in prismatic sodium sites, and can be synthesized at lower solid-state reaction temperatures (below 750 °C) compared to P2-type and O3-type materials, which require temperatures exceeding 850 °C, enhancing energy efficiency during production.118 However, the presence of both Mn3+ and Mn4+ oxidation states within P3 materials can lead to Jahn–Teller (J–T) distortions, negatively impacting discharge capacity and promoting disproportionation reactions that cause the dissolution of active materials into the electrolyte (Fig. 7i). To address these challenges, compositionally graded (CG) structures in P3-type manganese-based Na0.5[Ni0.216Mn0.784]O2 (NaNMO) particles have been developed,112 where the atomic ratio of nickel increases linearly from the core to the surface while the atomic ratio of manganese decreases, enhancing the concentration of stable Mn4+ ions at the surface. This concentration gradient structure effectively mitigates J–T distortions and improves overall structural stability, resulting in CG-NaNMO cathodes that exhibit remarkable cycling performance with a high reversible capacity of approximately 170 mA h g−1. The CG-NaNMO electrode demonstrates enhanced cycling stability at 50 °C compared to bulk-NaNMO (Fig. 7j). These advancements underscore the potential of CG structures in enhancing the performance of sodium-ion batteries.
In conclusion, the pursuit of novel layered oxide cathodes featuring concentration gradient structures presents a highly promising avenue for advancing battery technology. The concentration gradient acts as a buffer, alleviating the detrimental consequences of cation mixing and phase transitions commonly encountered in cathode materials, thereby extending the service life and reliability of the battery. Compared to core–shell structures, where a concentration gradient outer layer replaces the shell, the intrinsic gradient acts as an internal stabilizer, optimizing the material's resilience against the mechanical and chemical stresses inherent to electrochemical energy storage. This addresses the fundamental issue of the tens-of-nanometer gap and structural disparity often encountered between core and shell particles in core–shell architectures, which can impede ion and electron transfer pathways within the core.119,120 Nevertheless, the precise synthesis of concentration gradient structures poses a formidable challenge, necessitating the development of more intricate and sophisticated synthetic methodologies to meticulously control elemental distribution and gradient formation.
7 Summary and outlook
To conclude, SIBs hold substantial potential for large-scale energy storage systems and small electric vehicles owing to the low cost and broad temperature operating range (Fig. 8). In the research of sodium-ion batteries, layered oxide cathodes have garnered significant attention due to their high energy density. However, with the advancement of technology, the limitations of single-phase layered structures have gradually become apparent. Specifically, while these materials theoretically show superior electrochemical performance, they often face challenges in practical applications, such as insufficient stability and slow ion diffusion rates. Therefore, designing novel composite structures that integrate the advantages of layered oxides with the properties of other materials has become an effective approach to enhancing battery performance (Fig. 8). Recent research endeavors have illuminated that composite-structured LTMOs can stabilize phase transitions, bolster structural stability, and improve both capacity and rate performance. Specifically, heterogeneous phase materials can refine electrochemical performance and mitigate phase transitions by meticulously regulating phase interfaces within secondary particles, without the need for direct modification of individual phase characteristics. Furthermore, nanoscale intergrowth of different phases can stabilize a specific phase or heterogeneous phases through a locking mechanism during the cycling process, minimizing strain energy at phase interfaces and inhibiting interlayer slipping of transition metals, thereby fostering cycling stability and rate performance. Moreover, the incorporation of additional phases, such as tunnel and spinel phases, can create three-dimensional pathways that enhance the diffusion rate of sodium ions, thereby facilitating the rapid migration of these ions. The core–shell structure exhibits reduced layer expansion during charging, attributed to shell layer compression and crack suppression. The robust shell safeguards the high-capacity core, enhancing both material and air stability. In addition, robust core–shell interfacial adhesion is crucial for preventing phase delamination during cycling, highlighting the importance of controllable shell thickness and distribution in future developments. In this regard, the unique concentration gradient architecture augments the electrochemical kinetics of the material and mitigates side reactions, as well as suppressing the dissolution of transition metals at the interface during cycling. The interfacial stress within heteroepitaxial structures further inhibits phase transition phenomena during charge–discharge cycles. Additionally, the accumulation of inactive phase components on the material's surface safeguards against atmospheric erosion by water and carbon dioxide, reducing transportation and storage costs. This review comprehensively summarizes the current advancements in composite-structured LTMOs, categorizing the types and mechanisms of composite phase structures, and emphasizing the impact of interfacial and spatial heterogeneity distribution in multiphase materials on their performance as summarized in Table 1. | ||
| Fig. 8 Summary and outlook of composite structural LTMO cathodes for SIBs. | ||
| Materials | Phases | Discharge capacity (mA h g−1) | Voltage range (V, vs. Na/Na+) | Capacity retention | Comment | Ref. |
|---|---|---|---|---|---|---|
| Na0.7Li0.3Ni0.5Mn0.5O2+d | P2/O3 | ∼130 at 15 mA g−1 | 2–4.05 | No long-term cycling | ∼120 at 150 mA g−1 | 47 |
| Na0.66Li0.18Mn0.71Ni0.21Co0.08O2+δ | P2/O3 (94.2%:5.8%) |
200 at 10 mA g−1 | 1.5–4.5 | 75% after 150 cycles at 0.5C | High energy density (640 mA h g−1) | 127 |
| Na0.67Li0.11Fe0.36Mn0.36Ti0.17O2 | P2/O3 (63%:37%) |
235 at 0.1C | 1.5–4.2 | 85.4% after 100 cycles at 1C | Triggering reversible anionic redox activity | 128 |
| Na0.67Mn0.55Ni0.25Li0.2O2 | P2/O3 (85.1%:14.9%) |
158 at 12 mA g−1 | 1.5–4.2 | 84% after 50 cycles at 24 mA g−1 | Raising average discharge voltage | 129 |
| Na0.8Li0.2Fe0.2Mn0.6O2 | P2/O3 (34%:60%) |
174 at 0.1C | 2.0–4.6 | 82% after 100 cycles at 0.1C | Li acting as the structure stabilizer | 130 |
| Na0.85Ni0.34Mn0.33Ti0.33O2 | P2/O3 (24.8%:75.2%) |
126.6 at 0.1C | 2.2–4.4 | 80.6% after 200 cycles at 1C | 82.4 mA h g−1 at 10C | 49 |
| Na0.67Li0.2Fe0.2Mn0.6O2 | P2/O3 (89.1%:6.6%) |
227 at 10 mA g−1 | 1.5–4.5 | 83.5% after 60 cycles at 10 mA g−1 | Suppressing P2-Z phase transition and Jahn–Teller distortion | 131 |
| Na2/3Li0.18Mn0.8Fe0.2O2 | P2/O3 (mainly P2) | 125 at 0.1C | 1.5–4.2 | 69.6% after 100 cycles at 0.1C | The O3 phase appears to be electrochemically inactive | 132 |
| Na0.78Ni0.2Fe0.38Mn0.42O2 | P2/O3 (31.7%:68.3%) |
86 at 0.1C | 2.5–4.0 | 92% after 100 cycles at 0.1C | 56.8 at 10C, and 90% after 1500 cycles | 133 |
| Na0.73Ni0.4Mn0.4Ti0.2O2 | P2/O3 (33%:67%) |
140 at 20 mA g−1 | 1.5–4.4 | ∼73% after 50 cycles at 200 mA g−1 | Almost 100% initial coulombic efficiency | 50 |
| Na0.85MnyNizM1−y−zO2 (M = Ti, Fe, Cu, Mn) | O3/P2 | 2.0–4.0 | Constructing phase diagram | 79 | ||
| M = Ti | O3 | 131 at 12 mA g−1 | 2.0–4.0 | 91% after 500 cycles at 120 mA g−1 | ||
| M = Fe | P2/O3 | 130 at 12 mA g−1 | 2.0–4.0 | 86.2% after 500 cycles at 120 mA g−1 | ||
| M = Cu | P2/O3 | 117 at 12 mA g−1 | 2.0–4.0 | 90.6% after 500 cycles at 120 mA g−1 | ||
| M = Mn | P2/O3 | ∼112 at 12 mA g−1 | 2.0–4.0 | 81.6% after 500 cycles at 120 mA g−1 | ||
| Na0.9Cu0.2Fe0.28Mn0.52O2 | P3/O3 (mainly O3) | 125 at 10 mA g−1 | 2.0–4.0 | 99.3% after 100 cycles at 10 mA g−1 | Revealing the charge compensation mechanism | 68 |
| Na0.75Co0.125Cu0.125Fe0.125Ni0.125Mn0.5O2 | P2/P3 (36.9%:59.6%) |
90 at 0.1C | 2.0–4.0 | 96% after 500 cycles at 1C | 84.2% in a full cell after 1000 cycles at 1C | 134 |
| Na0.67Mn0.64Co0.30Al0.06O2 | P2/P3 (62.2%:37.8%) |
160 at 20 mA g−1 | 1.5–4.0 | 73% after 100 cycles at 200 mA g−1 | 83 mA h g−1 at 1700 mA g−1 | 56 |
| Na0.7Li0.06Mg0.06Ni0.22Mn0.67O2 | P2/P3 (46.7%:53.3%) |
119 at 0.2C | 2.0–4.4 | 97.2% after 150 cycles at 0.1C | High average voltage (3.36 V) and energy density (218 W h kg−1) | 59 |
| Na0.7Ni0.5Mn0.3Co0.2O2 | P2/P′3/O′3 | 126 at 0.1C | 1.5–4.2 | 68.4% after 100 cycles at 0.5C | Layered heterogeneous phase regulation achieves equilibrium | 135 |
| Na0.48Co0.1Mn0.9O2 | P2/T (32.4%:63.6%) |
219 at 0.1C | 1.5–4.4 | 80% after 50 cycles at 0.1C | 1C = 176 mA g−1 and retains 117 mA h g−1 at 5C | 70 |
| Na0.6MnO2 | P2/T | 198.2 at 0.2C | 1.5–4.3 | 85.1% after 100 cycles at 1C | Highly reversible structural evolution | 69 |
| Na0.6Mn0.9Cu0.1O2 | P2/T (86.8%:13.2%) |
∼168 at 0.1C | 2.0–4.1 | 80% after 100 cycles at 1C | 85 mA h g−1 at 8C | 136 |
| Na0.50Ni1/6Co1/6Mn2/3O2 | P2/spinel | 145 at 75 mA g−1 | 2.0–4.5 | 60% after 100 cycles at 0.2C | 85 mA h g−1 at 1500 mA g−1 | 137 |
| Na0.87Li0.25Ni0.4Fe0.2Mn0.4O2+δ | O3/spinel | 131 at 12 mA g−1 | 2.0–4.2 | 86% after 100 cycles at 100 mA g−1 | 112 mA g−1 at 100 mA g−1 | 71 |
| Na0.5[Ni0.2Co0.15Mn0.65]O2 | P3/P2/spinel | ∼180 at 0.1C | 1.5–4.0 | 87.6% after 100 cycles at 0.1C | 105 mA h g−1 at 10C | 138 |
| O3-NaNi0.5Mn0.5O2@P2-Na2/3MnO2 | O3@5%P2 | 141.4 at 0.1C | 1.5–3.9 | 85.3% capacity retention after 150 cycles at 1C | 103.7 mA h g−1 at 15C crack suppression | 78 |
| Na0.8[Ni0.5Co0.2Mn0.3]O2@Na0.8[Ni0.33Mn0.67]O2 | O3/O′3@P2 | 146 at 0.1C | 1.5–4.0 | 75% after 200 cycles at 0.1C | Crack suppression | 76 |
| Na0.78Cu0.27Zn0.06Mn0.67O2 | P2@P3 | 84 at 1C | 2.5–4.1 | 85% after 200 cycles at 1C | Revealing a possible Cu2+/3+ redox reaction mechanism | 88 |
| Na[Ni0.75Co0.02Mn0.23]O2 (center) Na[Ni0.58Co0.06Mn0.36]O2 (surface) | Concentration gradient | 157 at 15 mA g−1 | 1.5–4.1 | 80% after 100 cycles at 0.1C | Good temperature performance | 89 |
| Na[Ni0.75Co0.08Mn0.17]O2 (center) Na[Ni0.58Co0.14Mn0.28]O2 (surface) | Concentration gradient | 160 at 15 mA g−1 | 1.5–4.1 | ∼81.6% after 100 cycles at 0.5C | 130 mA h g−1 at 1500 mA g−1 | 95 |
| Na0.67Ni0.17Co0.17Mn0.66O2 | Concentration gradient | 143.2 at 20 mA g−1 | 2.0–4.5 | 91.5 after 100 cycles at 20 mA g−1 | Restraining phase transition | 96 |
While composite structures hold immense potential in augmenting the materials properties significantly, the inherent heterogeneity of these materials poses additional layers of complexity in the research endeavors. Presently, the understanding of single-phase materials has progressed considerably, yet the exploration into the intricate mechanisms that dictate multiphase interactions stands as a relatively uncharted territory. To further extend the utilization of composite phase LTMOs in SIBs and cutting-edge energy storage applications, elucidating the mechanisms of composite phases is not only necessary but also of paramount importance; thus future research endeavors should consider the following aspects:
(1) Design: The conventional approach to materials development, often a synthesis-followed-by-verification methodology, is significantly influenced by a myriad of factors such as composition, synthesis routes, and environmental conditions, thereby consuming substantial labor and resources. Artificial intelligence (AI) and machine learning (ML) merge as powerful allies in the pursuit of novel materials development.121 In the future, AI-assisted development, coupled with a profound understanding of the intricate interactions between their constituent phases such as the concept of “cation potential” and the principles of solid-state reaction thermodynamics, could systematically unravel the intricacies of the formation mechanisms of coexisting composite phase LTMOs. The establishment of universal design and synthesis principles would not only provide a robust theoretical framework but also pave the way for the innovative development and design of oxide-based cathode materials.
(2) Synthesis: The current synthetic approaches for layered oxide materials primarily encompass high-temperature solid-state reactions, co-precipitation, sol–gel processes, spray pyrolysis, and ion exchange, among others. Particularly, LTMOs exhibit heightened sensitivity to the distribution of transition metal elements, where the solid-state high-temperature sintering process can expedite the diffusion of transition metal ions, potentially disrupting the delicate heterogeneous material structure.122 The co-precipitation method allows for the regulation of secondary particle composition and spatial heterogeneity by controlling the concentration of precursors, droplet acceleration, and stirring velocity. The precursors generated via co-precipitation exhibit an advantageous distribution of transition metals, and enable a decrease in sintering temperature and a reduction in the duration of the heat treatment due to the reduced reliance on solid-state diffusion. In addition, the optimal particle size and morphology exploration remains an underexplored frontier, necessitating further probing. It is also paramount to acknowledge that the final morphology and grain size of the resultant material are intricately influenced by temperature, composition, and synthetic conditions, rendering the reproducibility of materials with intricate nanoscale interfaces challenging.123 This, in turn, can obfuscate the accurate portrayal of the material's inherent performance. Furthermore, the production of materials with composite structures incurs higher costs and lower efficiencies compared to simpler materials, hindering their progression towards industrial-scale production. Consequently, the exploration of cost-effective, safe, and reliable synthetic methods is of significant importance for the realization of high-performance composite-structured cathode materials.
(3) Characterization: The inherent heterogeneity of composite structures poses a formidable challenge in the characterization process, necessitating the utilization of high spatial resolution techniques for an accurate understanding of the structure–performance relationships.124–126 For instance, advanced microscopy techniques such as STEM and TXM can uncover detailed information at the atomic or molecular level, making them applicable for characterizing nanoscale phase interfaces. Crystallographic techniques like XRD and SXRD are utilized to investigate phase transition phenomena during the cycling processes. Furthermore, delineating the length scales of heterogeneous structures is crucial, as the nanoscale phase intergrowth and mechanical mixing of secondary particles significantly impact phase transitions, charge transfer, and interfacial side reactions. A comprehensive application of multiple characterization techniques can provide a holistic view of electrode materials, including the interface structure of coexisting regions, as well as the chemical composition and valency. Additionally, there is a pressing need to explore more in situ characterization methods and establish systematic characterization protocols tailored to diverse composite structures, fostering the development of mixed-structure materials. The judicious application of cutting-edge characterization techniques will facilitate a more profound understanding of these materials, thereby expanding the spectrum of hybrid-structure compounds and capitalizing on the synergistic merits of composite architectures.
In conclusion, numerous studies have convincingly demonstrated the substantial potential of composite structures in achieving remarkable improvements in integrated electrochemical performance. Nonetheless, the development of layered composite structural materials is still confronted with challenges, chief among them being the rational prediction of the structures of materials, the comprehensive optimization of synthetic approaches, and the efficient application of characterization techniques to understand the phase evolution during synthesis and electrochemical processes. As future research endeavors delve deeper into the systematic exploration of composite structures, it is anticipated that multiphase composite layered metal oxide materials will emerge as promising contenders for SIBs. These materials hold promise for delivering high energy density and unparalleled cycling stability, thereby addressing the pressing demands of modern energy storage systems. By addressing the aforementioned challenges and leveraging the latest advancements in materials science and engineering, we can pave the way for the widespread adoption of these advanced composites, revolutionizing the landscape of energy storage technologies.
Author contributions
Zhiyang Fan: conceptualization, formal analysis, investigation, and writing – original draft. Yichen Li: formal analysis and investigation. Jiawei Pan: writing – review & editing and supervision. Zhiyou Zhou: methodology and software. Weipeng Li: supervision. Taifan Yang: provision of study materials. Haihan Zhang: investigation. Chengyong Shu: conceptualization, supervision, and writing – review & editing. Weibo Hua: conceptualization. Yuping Wu: conceptualization and supervision. Wei Tang: conceptualization, funding acquisition, and resources.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This project was supported by the National Key R&D Program of China (2022YFB2404600), the National Natural Science Foundation of China (Grant No. 22379120), the Key Research and Development Plan of Shanxi Province (China, Grant No. 2018ZDXM-GY-135, 2021JLM-36), the Higher Education Institution Academic Discipline Innovation and Talent Introduction Plan (“111 Plan”) (No. B23025), “Young Talent Support Plan” of Xi'an Jiaotong University (71211201010723).References
- J. Jiang, Y. Y. Li, J. P. Liu, X. T. Huang, C. Z. Yuan and X. W. Lou, Adv. Mater., 2012, 24, 5166–5180 CrossRef PubMed
.
- H. S. Chen, T. N. Cong, W. Yang, C. Q. Tan, Y. L. Li and Y. L. Ding, Prog. Nat. Sci.: Mater. Int., 2009, 19, 291–312 CrossRef
- B. Dunn, H. Kamath and J. M. Tarascon, Science, 2011, 334, 928–935 CrossRef PubMed
- M. Z. Chen, J. Zhang, J. L. Zhang, B. K. Yu, L. M. Zhou, Y. Xiao, X. Gao, J. Xiao, C. S. Li, Y. Sun, H. K. Liu, S. X. Dou and S. L. Chou, Nanoscale, 2023, 15, 13076–13085 RSC
- J. W. Tang, Y. J. Zhou, X. Y. Li, X. Huang, W. Tang and B. B. Tian, Energy Mater., 2024, 4, 400022 Search PubMed
- X. Ao, Y. Kong, S. Zhao, Z. Chen, Y. Li, X. Liao and B. Tian, Angew. Chem., Int. Ed., 2024, e202415036, DOI:10.1002/anie.202415036
- J. P. Mensing, T. Lomas and A. Tuantranont, Carbon, 2022, 197, 264–281 CrossRef CAS
- Y. X. Lu, C. L. Zhao, X. H. Rong, L. Q. Chen and Y. S. Hu, Acta Phys. Sin., 2018, 67, 108801 CrossRef
- J. Q. Deng, W. B. Luo, S. L. Chou, H. K. Liu and S. X. Dou, Adv. Energy Mater., 2018, 8, 1701428 CrossRef
- H. L. Pan, Y. S. Hu and L. Q. Chen, Energy Environ. Sci., 2013, 6, 2338–2360 RSC
- S. H. Yuan, J. X. Li, W. Xia, Z. P. Li, W. Y. Tian, Y. Yang, X. B. Ji and P. Ge, Adv. Funct. Mater., 2024 DOI:10.1002/adfm.202409295
- C. Vaalma, D. Buchholz, M. Weil and S. Passerini, Nat. Rev. Mater., 2018, 3, 18013 CrossRef
- Q. Li, G. Liu, H. R. Cheng, Q. J. Sun, J. L. Zhang and J. Ming, Chem. – Eur. J., 2021, 27, 15842–15865 CrossRef CAS
- X. Z. Wang, Y. T. Zuo, Y. B. Qin, X. Zhu, S. W. Xu, Y. J. Guo, T. R. Yan, L. Zhang, Z. B. Gao, L. Z. Yu, M. T. Liu, Y. X. Yin, Y. H. Cheng, P. F. Wang and Y. G. Guo, Adv. Mater., 2024, 36, 2312300 CrossRef CAS PubMed
- Y. H. Liu, Y. H. Zhang, J. Ma, J. W. Zhao, X. Li and G. L. Cui, Chem. Mater., 2023, 36, 54–73 CrossRef
- Y. F. Liu, K. Han, D. Peng, L. Y. Kong, Y. Su and H. W. Li, InfoMat, 2023, 5, e12422 CrossRef CAS
- C. L. Xu, J. M. Zhao, C. Yang and Y. S. Hu, ACS Cent. Sci., 2023, 9, 1721–1736 CrossRef
- Z. Ahsan, Z. F. Cai, S. Wang, M. Moin, H. C. Wang, D. M. Liu, Y. Z. Ma, G. S. Song and C. E. Wen, Adv. Energy Mater., 2024, 14, 2400373 CrossRef
- J. Peng, W. Zhang, Q. N. Liu, J. Z. Wang, S. L. Chou, H. K. Liu and S. X. Dou, Adv. Mater., 2022, 34, 2108384 CrossRef PubMed
- Y. J. Yang, J. B. Zhou, L. L. Wang, Z. Jiao, M. Y. Xiao, Q. A. Huang, M. M. Liu, Q. S. Shao, X. L. Sun and J. J. Zhang, Nano Energy, 2022, 99, 107424 CrossRef
- L. M. Zhu, Y. F. Shen, M. Y. Sun, J. F. Qian, Y. L. Cao, X. P. Ai and H. X. Yang, Chem. Commun., 2013, 49, 11370–11372 RSC
- S. W. Wang, L. J. Wang, Z. Q. Zhu, Z. Hu, Q. Zhao and J. Chen, Angew. Chem., Int. Ed., 2014, 53, 5892–5896 CrossRef
- S. K. Jiang, H. L. Wang, T. Wang, L. M. Zhou, H. Xia, H. K. Liu, S. X. Dou and M. Z. Chen, Battery Energy, 2024, 3, 20230071 CrossRef
- N. Yabuuchi, K. Kubota, M. Dahbi and S. Komaba, Chem. Rev., 2014, 114, 11636–11682 CrossRef
- F. X. Ding, X. H. Rong, H. B. Wang, Y. Yang, Z. L. Hu, R. B. Dang, Y. X. Lu and Y. S. Hu, Acta Phys. Sin., 2022, 71, 108801 CrossRef
- Q. Huang, Y. M. Feng, L. Wang, S. Qi, P. G. He, X. B. Ji, C. P. Liang, S. Q. Chen, L. J. Zhou and W. F. Wei, Chem. Eng. J., 2022, 431, 133454 CrossRef
- H. Y. Yang, Q. Zhang, M. H. Chen, Y. Yang and J. B. Zhao, Adv. Funct. Mater., 2024, 34, 2308257 CrossRef
- T. Chen, B. Ouyang, X. Fan, W. Zhou, W. Liu and K. Liu, Carbon Energy, 2022, 4, 170–199 CrossRef
- B. Peng, G. L. Wan, N. Ahmad, L. Yu, X. Y. Ma and G. Q. Zhang, Adv. Energy Mater., 2023, 13, 2300334 CrossRef CAS
- Q. Ni, Y. J. Zhao, X. Y. Yuan, J. B. Li and H. B. Jin, Small, 2022, 18, 2200289 CrossRef CAS PubMed
- H. R. Yao, P. F. Wang, Y. Gong, J. N. Zhang, X. Q. Yu, L. Gu, C. Y. OuYang, Y. X. Yin, E. Y. Hu, X. Q. Yang, E. Stavitski, Y. G. Guo and L. J. Wan, J. Am. Chem. Soc., 2017, 139, 8440–8443 CrossRef CAS
- H. Lei, X. Cui, Z. Zeng, C. Zhu, W. Sun, Y. Yang and P. Ge, Angew. Chem., Int. Ed., 2024, e202414918, DOI:10.1002/anie.202414918
- U. Nisar, N. Muralidharan, R. Essehli, R. Amin and I. Belharouak, Energy Storage Mater., 2021, 38, 309–328 CrossRef
- Y. You, A. Dolocan, W. D. Li and A. Manthiram, Nano Lett., 2019, 19, 182–188 CrossRef CAS
- S. X. Deng, B. W. Xiao, B. Q. Wang, X. Li, K. Kaliyappan, Y. Zhao, A. Lushington, R. Y. Li, T. K. Sham, H. Wang and X. L. Sun, Nano Energy, 2017, 38, 19–27 CrossRef CAS
- S. Y. Chu, S. H. Guo and H. S. Zhou, Chem. Soc. Rev., 2021, 50, 13189–13235 RSC
- W. Chang, J. W. Choi, J. C. Im and J. K. Lee, J. Power Sources, 2010, 195, 320–326 CrossRef CAS
- T. T. Gao, P. Tian, Q. J. Xu, H. C. Pang, J. W. Ye and G. L. Ning, ACS Appl. Energy Mater., 2024, 7, 3904–3915 CrossRef CAS
- S. Kalluri, M. Yoon, M. Jo, H. K. Liu, S. X. Dou, J. Cho and Z. P. Guo, Adv. Mater., 2017, 29, 1605807 CrossRef PubMed
- H. Su and H. J. Yu, Small Methods, 2019, 3, 1800205 CrossRef CAS
- C. Hakim, N. Sabi and I. Saadoune, J. Energy Chem., 2021, 61, 47–60 CrossRef
- Q. Wang, S. Y. Chu and S. H. Guo, Chin. Chem. Lett., 2020, 31, 2167–2176 CrossRef
- J. F. Whitacre, A. Tevar and S. Sharma, Electrochem. Commun., 2010, 12, 463–466 CrossRef
- X. Zhou, A. L. Zhao, Z. X. Chen and Y. L. Cao, Electrochem. Commun., 2021, 122, 106897 CrossRef
- A. Mendiboure, C. Delmas and P. Hagenmuller, J. Solid State Chem., 1985, 57, 323–331 CrossRef
- D. Zhang, W. J. Shi, Y. W. Yan, S. D. Xu, L. Chen, X. M. Wang and S. B. Liu, Electrochim. Acta, 2017, 258, 1035–1043 CrossRef
- S. Heng, S. Jaffer and Y. Haijun, Energy Storage Mater., 2016, 5, 116–131 CrossRef
- K. H. Dai, J. Mao, X. Y. Song, V. Battaglia and G. Liu, J. Power Sources, 2015, 285, 161–168 CrossRef
- X. He, J. Wang, B. Qiu, E. Paillard, C. Z. Ma, X. Cao, H. D. Liu, M. C. Stan, H. D. Liu, T. Gallash, Y. S. Meng and J. Li, Nano Energy, 2016, 27, 602–610 CrossRef
- N. Yabuuchi, H. Yoshida and S. Komaba, Electrochemistry, 2012, 80, 716–719 CrossRef
- H. Yoshida, N. Yabuuchi and S. Komaba, Electrochem. Commun., 2013, 34, 60–63 CrossRef
- H. J. Yu, S. H. Guo, Y. B. Zhu, M. Ishida and H. S. Zhou, Chem. Commun., 2014, 50, 457–459 RSC
- P. Vassilaras, A. J. Toumar and G. Ceder, Electrochem. Commun., 2014, 38, 79–81 CrossRef
- R. Berthelot, D. Carlier and C. Delmas, Nat. Mater., 2011, 10, 74–U73 CrossRef
- M. Guignard, C. Didier, J. Darriet, P. Bordet, E. Elkaïm and C. Delmas, Nat. Mater., 2013, 12, 74–80 CrossRef PubMed
- N. Yabuuchi, M. Kajiyama, J. Iwatate, H. Nishikawa, S. Hitomi, R. Okuyama, R. Usui, Y. Yamada and S. Komaba, Nat. Mater., 2012, 11, 512–517 CrossRef PubMed
- Y. J. Guo, R. X. Jin, M. Fan, W. P. Wang, S. Xin, L. J. Wan and Y. G. Guo, Chem. Soc. Rev., 2024, 53, 7828–7874 RSC
- C. L. Zhao, M. Avdeev, L. Q. Chen and Y. S. Hu, Angew. Chem., Int. Ed., 2018, 57, 7056–7060 CrossRef PubMed
- C. L. Zhao, Q. D. Wang, Z. P. Yao, J. L. Wang, B. Sánchez-Lengeling, F. X. Ding, X. G. Qi, Y. X. Lu, X. D. Bai, B. H. Li, H. Li, A. Aspuru-Guzik, X. J. Huang, C. Delmas, M. Wagemaker, L. Q. Chen and Y. S. Hu, Science, 2020, 370, 708–711 CrossRef PubMed
- E. Lee, J. Lu, Y. Ren, X. Y. Luo, X. Y. Zhang, J. G. Wen, D. Miller, A. DeWahl, S. Hackney, B. Key, D. Kim, M. D. Slater and C. S. Johnson, Adv. Energy Mater., 2014, 4, 1400458 CrossRef
- Y. F. Zhu, Y. Xiao, W. B. Hua, S. Indris, S. X. Dou, Y. G. Guo and S. L. Chou, Angew. Chem., Int. Ed., 2020, 59, 9299–9304 CrossRef
- L. Z. Yu, Z. W. Cheng, K. Xu, Y. X. Chang, Y. H. Feng, D. Si, M. T. Liu, P. F. Wang and S. L. Xu, Energy Storage Mater., 2022, 50, 730–739 CrossRef
- J. H. Hong, S. Xiao, L. Deng, T. X. Lan and G. He, Ionics, 2020, 26, 3911–3917 CrossRef
- K. Wang, Z. G. Wu, G. Melinte, Z. G. Yang, A. Sarkar, W. B. Hua, X. K. Mu, Z. W. Yin, J. T. Li, X. D. Guo, B. H. Zhong and C. Kübel, J. Mater. Chem. A, 2021, 9, 13151–13160 RSC
- R. R. Li, X. Qin, X. L. Li, J. X. Zhu, L. R. Zheng, Z. T. Li and W. D. Zhou, Adv. Energy Mater., 2024, 14, 2400127 CrossRef
- Y. Xiao, H. R. Wang, H. Y. Hu, Y. F. Zhu, S. Li, J. Y. Li, X. W. Wu and S. L. Chou, Adv. Mater., 2022, 34, 2202695 CrossRef
- R. R. Li, Y. Y. Liu, Z. Wang and J. L. Li, Electrochim. Acta, 2019, 318, 14–22 CrossRef
- M. M. Rahman, Y. H. Xu, H. Cheng, Q. L. Shi, R. H. Kou, L. Q. Mu, Q. Liu, S. H. Xia, X. H. Xiao, C. J. Sun, D. Sokaras, D. Nordlund, J. C. Zheng, Y. J. Liu and F. Lin, Energy Environ. Sci., 2018, 11, 2496–2508 RSC
- N. Jiang, Q. N. Liu, J. W. Wang, W. F. Yang, W. S. Ma, L. Q. Zhang, Z. Q. Peng and Z. H. Zhang, Small, 2021, 17, 2007103 CrossRef PubMed
- L. G. Chagas, D. Buchholz, C. Vaalma, L. Wu and S. Passerini, J. Mater. Chem. A, 2014, 2, 20263–20270 RSC
- Y. C. Lei, X. Li, L. Liu and G. Ceder, Chem. Mater., 2014, 26, 5288–5296 CrossRef
- Y. N. Zhou, P. F. Wang, Y. B. Niu, Q. H. Li, X. Q. Yu, Y. X. Yin, S. L. Xu and Y. G. Guo, Nano Energy, 2019, 55, 143–150 CrossRef
- X. Gao, H. Q. Liu, H. Y. Chen, Y. Mei, B. W. Wang, L. Fang, M. Z. Chen, J. Chen, J. Q. Gao, L. S. Ni, L. Yang, Y. Tian, W. T. Deng, R. Momen, W. F. Wei, L. B. Chen, G. Q. Zou, H. S. Hou, Y. M. Kang and X. B. Ji, Sci. Bull., 2022, 67, 1589–1602 CrossRef PubMed
- X. Liang and Y. K. Sun, Adv. Funct. Mater., 2022, 32, 2206154 CrossRef
- G. L. Xu, R. Amine, Y. F. Xu, J. Z. Liu, J. Gim, T. Y. Ma, Y. Ren, C. J. Sun, Y. Z. Liu, X. Y. Zhang, S. M. Heald, A. Solhy, I. Saadoune, W. L. Mattis, S. G. Sun, Z. H. Chen and K. Amine, Energy Environ. Sci., 2017, 10, 1677–1693 RSC
- X. Q. Chen, X. L. Zhou, M. Hu, J. Liang, D. H. Wu, J. P. Wei and Z. Zhou, J. Mater. Chem. A, 2015, 3, 20708–20714 RSC
- T. Y. Yu, J. Kim, G. Oh, M. H. Alfaruqi, J. Y. Hwang and Y. K. Sun, Energy Storage Mater., 2023, 61, 102908 CrossRef
- C. Chen, B. Han, G. X. Lin, Q. Huang, S. Zhao, D. T. Zhang, C. Ma, D. G. Ivey and W. F. Wei, ACS Appl. Mater. Interfaces, 2018, 10, 28719–28725 CrossRef PubMed
- B. W. Xiao, X. Liu, M. Song, X. Yang, F. Omenya, S. Feng, V. Sprenkle, K. Amine, G. L. Xu, X. L. Li and D. Reed, Nano Energy, 2021, 89, 106371 CrossRef
- Z. X. Chen, T. C. Yuan, X. J. Pu, H. X. Yang, X. P. Ai, Y. Y. Xia and Y. L. Cao, ACS Appl. Mater. Interfaces, 2018, 10, 11689–11698 CrossRef
- X. Q. Xie, D. W. Su, B. Sun, J. Q. Zhang, C. Y. Wang and G. X. Wang, Chem. – Eur. J., 2014, 20, 17125–17131 CrossRef PubMed
- Z. H. Chen, K. L. Huang, S. Q. Liu and H. Y. Wang, Trans. Nonferrous Met. Soc. China, 2010, 20, 2309–2313 CrossRef
- O. K. Park, Y. Cho, S. Lee, H. C. Yoo, H. K. Song and J. Cho, Energy Environ. Sci., 2011, 4, 1621–1633 RSC
- Y. F. Zhu, Y. Xiao, S. X. Dou, Y. M. Kang and S. L. Chou, eScience, 2021, 1, 13–27 CrossRef
- G. F. Gao, D. Tie, H. Ma, H. J. Yu, S. S. Shi, B. Wang, S. M. Xu, L. L. Wang and Y. F. Zhao, J. Mater. Chem. A, 2018, 6, 6675–6684 RSC
- C. J. Deng, P. Skinner, Y. Z. Liu, M. L. Sun, W. Tong, C. R. Ma, M. L. Lau, R. Hunt, P. Barnes, J. Xu and H. Xiong, Chem. Mater., 2018, 30, 8145–8154 CrossRef
- Y. Xiao, P. F. Wang, Y. X. Yin, Y. F. Zhu, X. N. Yang, X. D. Zhang, Y. S. Wang, X. D. Guo, B. H. Zhong and Y. G. Guo, Adv. Energy Mater., 2018, 8, 1800492 CrossRef
- A. Bhaskar, S. Krueger, V. Siozios, J. Li, S. Nowak and M. Winter, Adv. Energy Mater., 2015, 5, 1401156 CrossRef
- D. Luo, G. S. Li, C. C. Fu, J. Zheng, J. M. Fan, Q. Li and L. P. Li, Adv. Energy Mater., 2014, 4, 1400062 CrossRef
- P. K. Nayak, J. Grinblat, M. D. Levi, O. Haik, E. Levi, M. Talianker, B. Markovsky, Y. K. Sun and D. Aurbach, Chem. Mater., 2015, 27, 2600–2611 CrossRef
- P. K. Nayak, E. Levi, J. Grinblat, M. Levi, B. Markovsky, N. Munichandraiah, Y. K. Sun and D. Aurbach, ChemSusChem, 2016, 9, 2404–2413 CrossRef PubMed
- X. H. Liang, J. Y. Hwang and Y. K. Sun, Adv. Energy Mater., 2023, 13, 2301975 CrossRef
- P. F. Wang, Y. You, Y. X. Yin and Y. G. Guo, Adv. Energy Mater., 2018, 8, 1701912 CrossRef
- H. R. Yao, X. G. Yuan, X. D. Zhang, Y. J. Guo, L. T. Zheng, H. Ye, Y. X. Yin, J. X. Li, Y. M. Chen, Y. Y. Huang, Z. G. Huang and Y. G. Guo, Energy Storage Mater., 2023, 54, 661–667 CrossRef
- S. P. Jia, S. Kumakura and E. McCalla, Energy Environ. Sci., 2024, 17, 4343–4389 RSC
- J. Y. Hwang, T. Y. Yu and Y. K. Sun, J. Mater. Chem. A, 2018, 6, 16854–16862 RSC
- J. Alvarado, C. Z. Ma, S. Wang, K. Nguyen, M. Kodur and Y. S. Meng, ACS Appl. Mater. Interfaces, 2017, 9, 26518–26530 CrossRef
- K. Kaliyappan, J. Liu, A. Lushington, R. Y. Li and X. L. Sun, ChemSusChem, 2015, 8, 2537–2543 CrossRef PubMed
- M. Z. Leng, J. Q. Bi, W. L. Wang, Z. Xing, W. K. Yan, X. C. Gao, J. Y. Wang and R. Liu, J. Alloys Compd., 2021, 855, 157533 CrossRef
- Y. Zhang, Y. Pei, W. Liu, S. Zhang, J. J. Xie, J. Xia, S. Nie, L. Liu and X. Y. Wang, Chem. Eng. J., 2020, 382, 122697 CrossRef
- N. Li, J. Ren, R. B. Dang, K. Wu, Y. L. Lee, Z. B. Hu and X. L. Xiao, J. Power Sources, 2019, 429, 38–45 CrossRef
- H. B. Wang, F. X. Ding, Y. Q. Wang, Z. Han, R. B. Dang, H. Yu, Y. Yang, Z. Chen, Y. Q. Li, F. Xie, S. G. Zhang, H. Z. Zhang, D. W. Song, X. H. Rong, L. Q. Zhang, J. P. Xu, W. Yin, Y. X. Lu, R. J. Xiao, D. Su, L. Q. Chen and Y. S. Hu, ACS Energy Lett., 2023, 8, 1434–1444 CrossRef
- W. J. Lu, X. T. Guo, Y. Q. Luo, Q. Li, R. M. Zhu and H. Pang, Chem. Eng. J., 2019, 355, 208–237 CrossRef
- S. W. Kim, J. Ryu, C. B. Park and K. Kang, Chem. Commun., 2010, 46, 7409–7411 RSC
- J. W. Shin and J. T. Son, J. Nanosci. Nanotechnol., 2019, 19, 1335–1339 CrossRef PubMed
- X. Liang, T. Y. Yu, H. H. Ryu and Y. K. Sun, Energy Storage Mater., 2022, 47, 515–525 CrossRef
- C. Chen, Z. Han, S. Q. Chen, S. Qi, X. Y. Lan, C. C. Zhang, L. Chen, P. Wang and W. F. Wei, ACS Appl. Mater. Interfaces, 2020, 12, 7144–7152 CrossRef PubMed
- P. Y. Hou, M. H. Dong, F. Li, Z. Z. Lin and X. J. Xu, Adv. Funct. Mater., 2024 DOI:10.1002/adfm.202409518
- Z. C. Yan, L. Tang, Y. Y. Huang, W. B. Hua, Y. Wang, R. Liu, Q. F. Gu, S. Indris, S. L. Chou, Y. H. Huang, M. H. Wu and S. X. Dou, Angew. Chem., Int. Ed., 2019, 58, 1412–1416 CrossRef PubMed
- J. Y. Hwang, S. M. Oh, S. T. Myung, K. Y. Chung, I. Belharouak and Y. K. Sun, Nat. Commun., 2015, 6, 6865 CrossRef CAS PubMed
- Y. Duan, Z. H. Ma, Q. X. Wan, M. M. Li, Y. Y. Huang, L. L. Li, X. H. Han, S. Bao and J. L. Lu, J. Colloid Interface Sci., 2024, 662, 69–75 CrossRef CAS PubMed
- P. Y. Hou, M. H. Dong, Z. Z. Lin, Z. B. Sun, M. S. Gong, F. Li and X. J. Xu, ACS Sustainable Chem. Eng., 2023, 11, 10785–10794 CrossRef CAS
- S. Bao, S. H. Luo, Z. Y. Wang, S. X. Yan, Q. Wang and J. Y. Li, J. Power Sources, 2018, 396, 404–411 CrossRef
- K. Wu, N. Li, K. R. Hao, W. Yin, M. Wang, G. F. Jia, Y. L. Lee, R. B. Dang, X. Deng, X. L. Xiao, E. Y. Zhao and Z. J. Wu, J. Phys. Chem. C, 2021, 125, 20171–20183 CrossRef
- P. Y. Hou, F. Li, Y. Y. Wang, J. M. Yin and X. J. Xu, J. Mater. Chem. A, 2019, 7, 4705–4713 RSC
- Q. Q. Zhao, F. K. Butt, Z. F. Guo, L. Q. Wang, Y. Q. Zhu, X. Y. Xu, X. L. Ma and C. B. Cao, Chem. Eng. J., 2021, 403, 126308 CrossRef
- J. Y. Hwang, S. T. Myung, C. S. Yoon, S. S. Kim, D. Aurbach and Y. K. Sun, Adv. Funct. Mater., 2016, 26, 8083–8093 CrossRef
- Z. G. Liu, S. Y. Chu, J. H. Wu, C. Cheng, L. Zhang, S. H. Guo and H. S. Zhou, Chem. Eng. J., 2022, 435, 134944 CrossRef
- Y. K. Sun, S. T. Myung, B. C. Park, J. Prakash, I. Belharouak and K. Amine, Nat. Mater., 2009, 8, 320–324 CrossRef PubMed
- Y. K. Sun, S. T. Myung, B. C. Park and K. Amine, Chem. Mater., 2006, 18, 5159–5163 CrossRef
- Z. H. Wang, L. Wang, H. Zhang, H. Xu and X. M. He, Nano Convergence, 2024, 11, 8 CrossRef
- E. Gabriel, D. W. Hou, E. Lee and H. Xiong, Energy Sci. Eng., 2022, 10, 1672–1705 CrossRef
- K. X. Gu, Z. P. Shi, X. Li, B. Qiu and Z. P. Liu, J. Mater. Chem. A, 2024, 12, 24727–24745 RSC
- L. Yu, M. Li, J. G. Wen, K. Amine and J. Lu, Mater. Chem. Front., 2021, 5, 5186–5193 RSC
- Z. Y. Zhang, S. Said, K. Smith, R. Jervis, C. A. Howard, P. R. Shearing, D. J. L. Brett and T. S. Miller, Adv. Energy Mater., 2021, 11, 2101518 CrossRef
- R. Huang and Y. Ikuhara, Curr. Opin. Solid State Mater. Sci., 2012, 16, 31–38 CrossRef
- S. H. Guo, P. Liu, H. J. Yu, Y. B. Zhu, M. W. Chen, M. Ishida and H. S. Zhou, Angew. Chem., Int. Ed., 2015, 54, 5894–5899 CrossRef PubMed
- C. Chen, W. Y. Huang, Y. W. Li, M. J. Zhang, K. Q. Nie, J. O. Wang, W. G. Zhao, R. Qi, C. J. Zuo, Z. B. Li, H. C. Yi and F. Pan, Nano Energy, 2021, 90, 106504 CrossRef
- Z. Y. Li, J. C. Zhang, R. Gao, H. Zhang, L. R. Zheng, Z. B. Hu and X. F. Liu, J. Phys. Chem. C, 2016, 120, 9007–9016 CrossRef
- L. T. Yang, J. M. L. del Amo, Z. Shadike, S. M. Bak, F. Bonilla, M. Galceran, P. K. Nayak, J. R. Buchheim, X. Q. Yang, T. Rojo and P. Adelhelm, Adv. Funct. Mater., 2020, 30, 2003364 CrossRef
- J. E. Wang, H. Kim, Y. H. Jung, D. K. Kim and D. J. Kim, Small, 2021, 17, 2100146 CrossRef
- M. Bianchini, E. Gonzalo, N. E. Drewett, N. Ortiz-Vitoriano, J. M. L. del Amo, F. J. Bonilla, B. Acebedo and T. Rojo, J. Mater. Chem. A, 2018, 6, 3552–3559 RSC
- X. G. Qi, L. L. Liu, N. N. Song, F. Gao, K. Yang, Y. X. Lu, H. T. Yang, Y. S. Hu, Z. H. Cheng and L. Q. Chen, ACS Appl. Mater. Interfaces, 2017, 9, 40215–40223 CrossRef PubMed
- M. M. Rahman, J. Mao, W. H. Kan, C. J. Sun, L. X. Li, Y. Zhang, M. Avdee, X. W. Du and F. Lin, ACS Mater. Lett., 2019, 1, 573–581 CrossRef
- M. Keller, T. Eisenmann, D. Meira, G. Aquilanti, D. Buchholz, D. Bresser and S. Passerini, Small Methods, 2019, 3, 1900239 CrossRef
- T. R. Chen, T. Sheng, Z. G. Wu, J. T. Li, E. H. Wang, C. J. Wu, H. T. Li, X. D. Guo, B. H. Zhong, L. Huang and S. G. Sung, ACS Appl. Mater. Interfaces, 2018, 10, 10147–10156 CrossRef PubMed
- J. M. Zheng, P. F. Yan, W. H. Kan, C. M. Wang and A. Manthiram, J. Electrochem. Soc., 2016, 163, A584–A591 CrossRef
- P. Y. Hou, J. M. Yin, X. H. Lu, J. M. Li, Y. Zhao and X. J. Xu, Nanoscale, 2018, 10, 6671–6677 RSC
Footnote |
| † Current address: Heze No. 1 Middle School, HeZe 274002, Shandong, China. |
| This journal is © The Royal Society of Chemistry 2025 |