
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Oxidative addition of Si–H bonds to metal-decorated Zintl clusters [Hyp3Ge9Ir(CO)PR3] (R = Ph, ptolyl, Me)†
Nicole S.
Willeit
ab,
Tim
Kratky
 c,
Viktor
Hlukhyy
a,
Sebastian
Günther
c and
Thomas F.
Fässler
*ab
c,
Viktor
Hlukhyy
a,
Sebastian
Günther
c and
Thomas F.
Fässler
*ab
aDepartment of Chemistry, TUM School of Natural Sciences, Technical University of Munich (TUM), Lichtenbergstraße 4, D-85748 Garching, Germany. E-mail: thomas.faessler@lrz.tum.de; Web: https://www.ch.nat.tum.de/acnm
bWacker Institute of Silicon Chemistry, Technical University of Munich (TUM), Lichtenbergstraße 4, D-85748 Garching, Germany
cDepartment of Chemistry, TUM School of Natural Sciences, Technical University of Munich (TUM), Lichtenbergstraße 4, D-85748 Garching, Germany. E-mail: sebastian.guenther@tum.de; Web: https://www.ch.nat.tum.de/pc5
First published on 18th June 2025
Abstract
Catalytic reactions with metalated Zintl clusters as catalysts represent a growing research field, whereby the concept of heterogeneous single-site catalysis is transferred towards homogeneous reactions, leading to so-called single-site homogeneous catalysts (SSHoCs). A synthetic protocol for three cluster compounds [Hyp3Ge9Ir(CO)PR3] (Hyp = Si{SiMe3}3; R = Ph, ptolyl, Me; 1–3) is presented, in which the iridium atom is embedded in the polyhedral cluster surface. The products are characterized by NMR, IR and LIFDI/MS and also structurally characterized for R = Ph by single crystal X-ray structure determination, revealing a closo-[Ge9Ir] cluster. The exchange of the phosphine ligand of 1 in solution, which is regarded as an important step to create the active site, is investigated for various phosphines. In subsequent reactions, oxidative addition of Si–H bonds of primary and secondary silanes SiHR2R′ (R/R′ = H/Ph, H/pMePh, H/p{OMe}Ph, H/p{NMe2}Ph, and Ph/H) to the Ir atom is investigated. The addition reaction is directly monitored by NMR spectroscopy. Additionally, LIFDI/MS, IR spectroscopy, and single crystal structure determination of the addition products confirm the reaction. X-ray photoelectron spectroscopy (XPS) reveals that the transition metal atom and the Ge atoms of the supporting cluster have a low oxidation state.
Introduction
There is increasing research interest in heterogeneous single-site catalysis and designing supported single-atom active sites to boost selectivity in catalytic transformations due to the uniformity of active sites.1–3 The synthesis of such catalysts requires highly sophisticated techniques. The single-atom active sites are however less stable if compared to conventional heterogeneous catalysts. Since the atoms are bonded mostly to an oxide-based support a strong influence of the support on the property of the transition metal (TM) and also a sterically restricted access of the substrate to the metal is anticipated. More effective than single-site catalysts, where the active site can consist of more than one atom, are heterogeneous single-atom catalysts (SACs) with single metal atoms individually anchored or incorporated onto the surface of a solid support. However, here the atoms tend to agglomerate leading to the formation of metal atom clusters and a limited metal loading to prevent agglomeration lowers the catalytic activity. Also, characterization of the dispersed nature of the active sites is a significant challenge.1–8 In contrast, homogeneous catalysts, in which typically a metal center is coordinated by ligands, are more difficult to separate from the product and might show a lower stability due to ligand loss during the reaction. However, they offer a large variety of characterization methods.9–11 Recently the so-called single-site homogeneous catalysts (SSHoCs) have been introduced in which the concepts of heterogeneous SACs and homogeneous catalysts are combined. Here, a TM is strongly incorporated into the surface of a soluble molecular main group atom cluster in oxidation states close to zero by forming mixed atom clusters.12 These compounds can also be considered as intermetalloids, which consist of at least two different metal elements with mainly metal–metal interactions, further confirming that the cluster has a low oxidation state.13 Therefore, homoatomic main-group element clusters as they occur as so-called Zintl clusters are especially suitable starting materials. Thus, catalytic reactions with Zintl clusters as catalysts have become an increasing research field. The advantage of these compounds is that all active sites are identical, the metal atom is strongly bound to the cluster core, the TM atom can retain a low oxidation state on the “support”, and the unit remains stable even after the dissociation of ligands that might be bound to the TM atom. Such clusters can be functionalized through the surface atoms to enhance the solubility allowing monitoring of the catalytic activity via in situ analytical methods.12 Zintl clusters simply form by reactions for example between alkali metal atoms and p-block (semi)metals at higher temperatures. They can easily be transferred in solution and functionalization of germanium clusters, which are best studied, is well developed. The binary phase K4Ge9 comprising [Ge9]4− ions readily forms [Hyp3Ge9]− or [Hyp2Ge9]2− (Hyp = Si{SiMe3}3) in acetonitrile with chlorotris(trimethylsilyl)silane (HypCl) depending on the stoichiometry.14,15 Besides these species, other silylated germanium clusters are also known, such as [(SiiBu3)3Ge9]−, [(SiiPr3)3Ge9]−, [(SiEt3)3Ge9]−, [(SiHtBu2)3Ge9]−, [Hyp2(SiPh2R)Ge9]−, [Hyp(Si{SiMe3}2{SiPh3})2Ge9]−, [(Si{SiMe3}2{SiPh3})2Ge9]2−, [(Si{SiMe3}2{SiiPr3})3Ge9]− and [(SiPh2R)3Ge9]− (R = –CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2, –(CH2)3CHCH2).16–18 Neutral cluster compounds [Hyp3R′Ge9] form by introducing a fourth ligand by the reaction of [Hyp3Ge9]− with organotin halides, acid chlorides, or halogenated hydrocarbons.19–22 Besides the organic functionalization of Zintl clusters, many metalation reactions are known.23,24 Through metalation, Zintl clusters can simply act as a ligand to the TM atom by η1- and η3-coordination, or the clusters can also incorporate TM atoms resulting in various mixed atom cluster isomers. Based on the pristine [E9]4− (E = Si–Pb) clusters, the incorporation of a TM can form two spherical ten-atom cluster isomers. Based on a bicapped square antiprism, the TM can either take the position of the cap of the square or be located in the square, resulting in coordination numbers 4 and 5, respectively, with surrounding tetrel atoms (Fig. 1).23,24
CH2, –(CH2)3CHCH2).16–18 Neutral cluster compounds [Hyp3R′Ge9] form by introducing a fourth ligand by the reaction of [Hyp3Ge9]− with organotin halides, acid chlorides, or halogenated hydrocarbons.19–22 Besides the organic functionalization of Zintl clusters, many metalation reactions are known.23,24 Through metalation, Zintl clusters can simply act as a ligand to the TM atom by η1- and η3-coordination, or the clusters can also incorporate TM atoms resulting in various mixed atom cluster isomers. Based on the pristine [E9]4− (E = Si–Pb) clusters, the incorporation of a TM can form two spherical ten-atom cluster isomers. Based on a bicapped square antiprism, the TM can either take the position of the cap of the square or be located in the square, resulting in coordination numbers 4 and 5, respectively, with surrounding tetrel atoms (Fig. 1).23,24
 | ||
| Fig. 1 Incorporation of transition metal atoms into [E9]4− (E = Si–Pb) cluster cores forming ten atomic cluster species: (a) η4-(capping the open square of the antiprism) and (b) η5-coordination (as part of the square). Green spheres indicate tetrel element atoms, blue spheres indicate either a tetrel element atom or a Ge-silyl group, and pink spheres represent transition metal atoms with ligands. | ||
The incorporation of transition metals to form ten atomic cluster species can be realized either for the naked Zintl anions [E9]4− or, especially in the case of germanium, for already functionalized clusters, such as [Hyp3Ge9]− and [Hyp3EtGe9]. Capping the open square of a mono-capped antiprism arises in η4-coordination to the metal atom. This binding mode is very common for ligand-free cluster species and a broad variety of compounds exist, such as [Ge9Ni(CO)]3−,25 [Ge9CuPR3]3− (R = iPr, Cy),26 [E9ZnR]3− (E = Si–Pb; R = Ph, iPr, Mes),27,28 [Ge9PdPPh3]3−,29 [E9M(CO)3]4− (E = Sn, Pb; M = Cr, Mo, W),30–33 and [Sn9IrCOD]3−.34 In contrast, concerning silylated germanium clusters bearing the same number of skeletal electrons of the cluster framework, this type of coordination is only observed for [Hyp3Ge9RhCOD] (COD = 1,5-cyclooctadiene).35,36 More frequently η5-coordination – through incorporation of the TM as a vertex in the square – is observed in clusters such as [Hyp3Ge9Rhdppe] (dppe = 1,2-bis(diphenylphosphino)ethane),35 [Hyp3Ge9M(CO)3]− (M = Cr, Mo, W),37,38 and [Hyp3EtGe9MPR3] (M/R = Ni/Ph, ptolyl, iPr, Me; Pd/PPh3; Pt/PPh3).12,39,40 This bonding mode also occurs but less frequently for naked cluster species of the heavier tetrel elements, as seen in [Pb9Mo(CO)3]4−33 and [Sn9M(CO)3]4− (M = Cr, Mo, W).32 The latter even pronounces a dynamic rearrangement between η5 and η4 coordination of the transition metal atom.32 In addition, it has been shown that the polyhedral Zintl clusters [Hyp3Ge9RhCOD],35,36 [Hyp3Ge9Rh(PPh3)],36,41 and [Hyp3EtGe9Ni(PR3)] (R = Ph, ptolyl, iPr, Me)12 – the latter comprising abundant Ni as the active transition metal – are capable of hydrogenation and isomerization reactions, as well as H/D exchange and H2/D2 scrambling, respectively. Since the catalysts significantly differ from transition metal complexes that are generally used in homogeneous catalysis, the expression single site homogeneous catalyst (SSHoC) has been introduced.12
Herein, we report the synthesis of three uncharged, molecular ten-atom closo-[Ge9Ir] clusters with different phosphine ligands, PPh3, Pptolyl3, and PMe3, bound to the iridium atom. [Hyp3Ge9Ir(CO)(PPh3)] (1) and [Hyp3Ge9Ir(CO)(Pptolyl3)] (2) were obtained by reacting K[Hyp3Ge9] with the Vaska complexes Ir(CO)(PR3)2Cl (R = Ph, ptolyl) (Scheme 1). [Hyp3Ge9Ir(CO)(PMe3)] (3) was obtained via a ligand exchange reaction from 1 (Scheme 1). In subsequent reactions, Si–H bond activation of primary and secondary SiHR2R′ (R/R′ = H/Ph, H/pMePh, H/p{OMe}Ph, H/p{NMe2}Ph, and Ph/H) is investigated (Scheme 2). The compounds 1, 2, and 3 were characterized by NMR and IR spectroscopy, as well as LIFDI/MS. Compound 1 could be obtained in good yields as a single crystalline material; thus the catalytic potential of [Hyp3Ge9Ir(CO)(PPh3)] was investigated. The products of the oxidative addition were attained in situ and therefore characterized by NMR spectroscopy, except for [Hyp3Ge9Ir(CO)(H)(SiH2p{OMe}Ph)], which was additionally investigated via single crystal structure determination.
 | ||
| Scheme 1 Reaction of K[Hyp3Ge9] with Ir(CO)(PR3)2Cl in toluene, yielding [Hyp3Ge9Ir(CO)(PR3)], R = Ph (1), ptolyl (2), and follow-up reaction of 1 with PMe3, yielding [Hyp3Ge9Ir(CO)(PMe3)] (3). | ||
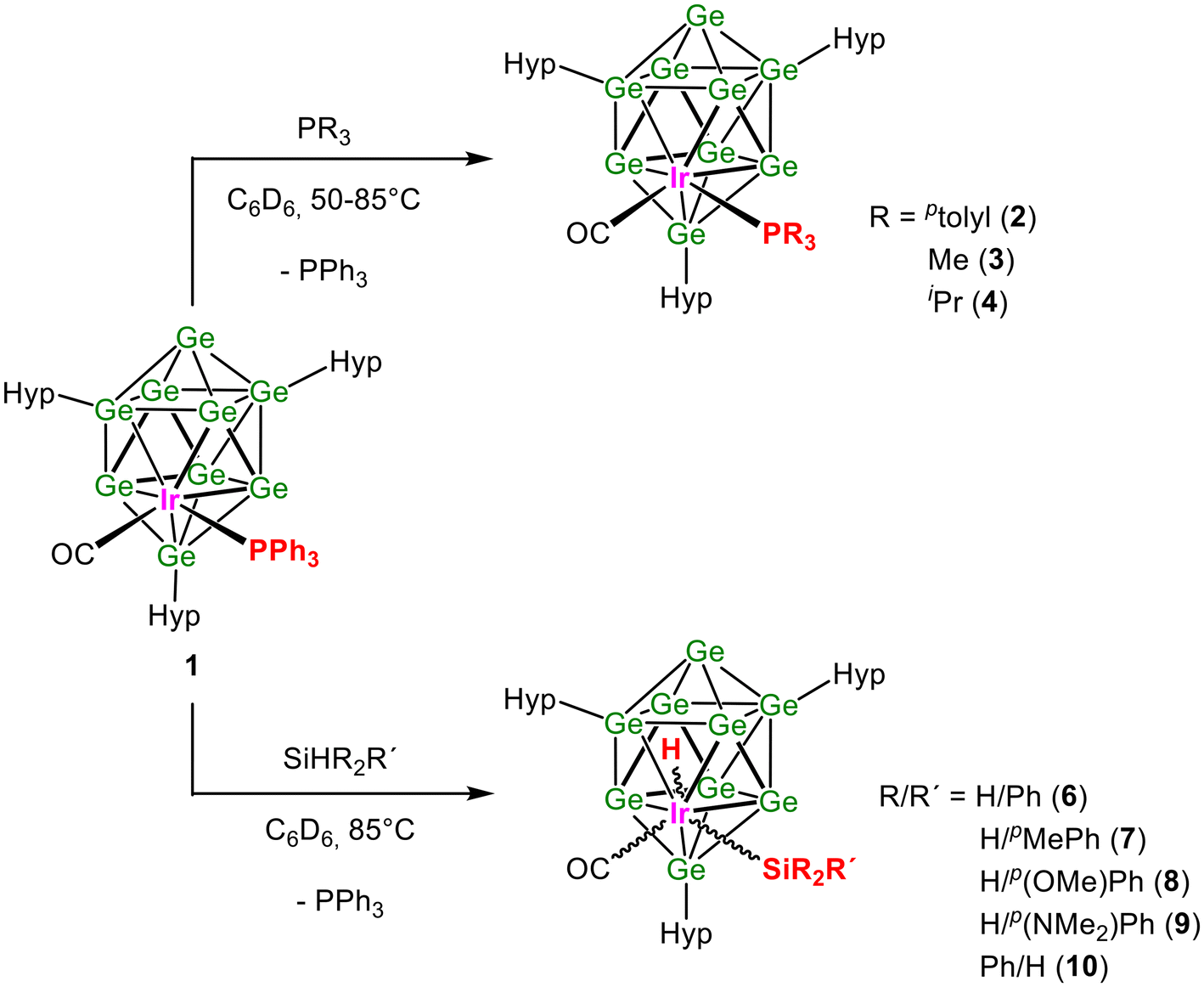 | ||
| Scheme 2 Reaction schemes for (top) the phosphine exchange reactions between 1 and PR3 (R = ptolyl, Me, iPr) and (bottom) the Si–H bond activation of SiHR2R′ [R/R′ = H/Ph, H/pMePh, H/p(OMe)Ph, H/p(NMe2)Ph, Ph/H] with 1. | ||
Results and discussion
Synthesis and structure
The three compounds with polyhedral ten-atom [Ge9Ir] cores [Hyp3Ge9Ir(CO)(PR3)] with R = Ph (1), ptolyl (2·Pptolyl3), and Me (3·[PPh3]xx ≈ 0.33) were synthesized either through the reaction of K[Hyp3Ge9] with Ir(CO)(PR3)2Cl (R = Ph, ptolyl) in toluene at room temperature or through a ligand exchange reaction, whereby the phosphine ligand of [Hyp3Ge9Ir(CO)(PPh3)] (1) was replaced by PMe3 resulting in 3·(PPh3)x. As shown below, cluster 2 can only be isolated in the presence of one equivalent of Pptolyl3, 2·Pptolyl3, and for 3 after ligand exchange some PPh3 remains, 3·(PPh3)x.Products 1, 2·Pptolyl3, and 3·(PPh3)x were analyzed by NMR, LIFDI mass, and IR spectroscopy. The LIFDI/MS measurements of toluene solutions of 1, 2·Pptolyl3, and 3·(PPh3)x show the mass peaks for [Hyp3Ge9Ir(CO)(PR3)] with R = Ph, ptolyl, and Me, respectively (Fig. 2a, Fig. S18, S25, and S32, ESI†).
 | ||
| Fig. 2 LIFDI mass spectra of (a) [Hyp3Ge9Ir(CO)(PPh3)] (1) and (b) [Hyp3Ge9Ir(CO)(H)(SiH2Ph)] (6). Experimental spectra and simulated isotope pattern are shown in black and red, respectively. | ||
The 1H NMR spectra of 1–3 each display three sharp signals indicating three different hypersilyl substituents for each compound, and the signals for the respective phosphine ligands (Fig. S12, S19, and S26, ESI†). The 1H NMR signals of the phosphine ligands are thereby only slightly shifted compared to the used Vaska complexes, but strongly shifted compared to unattached, free phosphine ligands. The 29Si NMR spectra of each compound show six different signals for the silyl groups: three for the silicon atoms directly bound to Ge atoms of the cluster and three for the TMS groups attached to these (Fig. S15, S22, and S29, ESI†), in agreement with the 1H NMR spectra. Also, the 13C NMR spectra show three individual signals for the Hyp groups (Fig. S14, S21, and S28, ESI†) next to the signals for the phosphines and the CO ligand. Three inequivalent silyl groups have also been observed in multimetallic cluster species, such as [(CODRh)2Ge9Hyp3Cl], [(CODRh)(CODIr)Ge9Hyp3Cl], and [(NBDRh)3Ge9Hyp3Cl2] (NBD = norbornadiene),42 and they hint at a C1 symmetric cluster. The signals for the phosphine ligands in the 31P NMR spectra (Fig. S13, S20, and S27, ESI†) are only slightly shifted compared to the used Vaska complexes Ir(CO)(PR3)2Cl (R = Ph, ptolyl) by 1 to 2 ppm but are strongly low field shifted compared to the unattached phosphine ligands by 20 to 30 ppm. This implies that upon Ir coordination to the cluster, a similar electronic environment is present to that existing for the Vaska complex itself. In all ligand exchange reactions at the Ir center of the used Vaska complexes, non-coordinating phosphine ligands are formed. In the case of 1, free PPh3 could be removed by vacuum sublimation. In the case of cluster 2, one equivalent of unattached Pptolyl3, which is generated during the reaction, is found in the product 2·Pptolyl3 and is also observed in the expected ratio in the NMR spectra. Attempts to remove the byproduct by fractionating crystallization or sublimation were not successful. For 3, which is formed by a ligand exchange reaction from 1, the product could be purified by vacuum sublimation of the byproduct PPh3; however, 0.33 equivalents remain in the product 3·(PPh3)x according to the NMR spectra. Additionally, the IR spectra of compounds 1, 2·Pptolyl3, and 3·(PPh3)x (Fig. S35–S37, ESI†) reveal the CO stretching vibration region bands at 1953 cm−1 (1, 2) and 1950 cm−1 (3), respectively, and thus differ only slightly from one another and are also close to the values of the Vaska complexes [Ir(CO)(PPh3)2Cl: 1950 cm−1 and Ir(CO)(Pptolyl3)2Cl 1954 cm−1] (Table S6, ESI†). The small deviations of the carbonyl (CO) streching frequencies indicate as well that the cluster imposes a similar electronic environment as it is present in average by the other ligands in the Vaska complexes.
Single crystals of [Hyp3Ge9Ir(CO)(PPh3)] (1) were obtained through crystallization from a diethyl ether solution at −32 °C (Fig. 3). The structural determination reveals that the [Ge9Ir] cluster core hereby adopts a distorted bicapped square antiprism with the iridium atom located at a vertex of one of the two squares and thus is five-fold connected to Ge atoms (Ge1, Ge3, Ge7, Ge8, and Ge9). Overall, the cluster bears five substituents: three hypersilyl groups bound to Ge1, Ge2, and Ge3 as well as the phosphine and carbon monoxide ligands coordinating to Ir. The overall symmetry of the cluster is reduced to point group C1 in agreement with the NMR spectra. Besides this, a transition metal atom, bearing different donor ligands, incorporated into the core of a Zintl cluster is to the best of our knowledge not known so far. The Ge–Ge bond lengths within the cluster core in 1 lie in the range of 2.485(1)–3.025(2) Å, with Ge6–Ge9 being the longest and Ge2–Ge4 being the shortest distance. The bond lengths in the [Ge9Ir] species are in good accordance with other known metalated cluster species in the literature, where the metal atom occupies the vertex of a square. See for example [Hyp3EtGe9M(PR3)] (M/R = Ni/PPh3, Pptolyl3, PiPr3, PMe3; Pd/PPh3; Pt/PPh3),12,39,40 [Hyp3Ge9Rhdppe],35 and [Hyp3Ge9M(CO)3]− (M = Mo, W, Cr)37,38 with Ge–Ge distances in the ranges 2.455–2.885 Å, 2.498–3.152 Å, and 2.502(5)–2.824(3) Å, respectively. Remarkably, the bond angles around Ir are all almost 90° each: ∡(Ge7–Ir–CCO) = 88.4(3)°, ∡(Ge7–Ir–Ge9) = 89.95(3)°, ∡(Ge9–Ir–P) = 89.80(6)°, and ∡(P–Ir–CCO) = 87.8(3)°, and thus CO is trans to Ge9. However, both ligands (CO and PPh3) are slightly tilted by of 9.11(3)° towards the Ge3 atom with respect to the plane built by Ir, Ge7, Ge6, and Ge9 (Fig. 3). To quantitatively evaluate the atomic ratio of Si, Ge, and Ir of compounds 1, 2·Pptolyl3, and 3·(PPh3)x as well as the oxidation states of the elements, X-ray photoelectron spectroscopy (XPS) was performed. The survey spectra (Fig. S38, ESI†) reveal all expected core level and Auger emissions from the elements present in 1, 2, and 3. The quantification of the composition based on Ge 3d, Si 2p, and Ir 4f confirms the stoichiometry of the expected compounds (Table S7, ESI†). Since the oxidation states of the cluster atoms, Ge and Ir, are of importance for the description of the SSHoC concept, implying a low valent transition metal atom embedded in an elemental germanium surface, a thorough analysis of the so-called chemical shift is given in the ESI.† In order to extract the oxidation states of the present elements, the binding energy of a certain core level is typically required to determine the chemical shift. As compounds 1–3 are electrical insulators, their XP spectra suffer from sample charging and, hence, charging correction needs to be applied first to obtain true binding energy values (see the ESI†). After this correction, a binding energy of around 61 eV for the Ir 4f7/2 core level suggests either metallic Ir or a low oxidation state as values of ∼62 eV are reported for Ir(IV) species.43 These results are in good accordance with the expected values of germanium near the oxidation state zero and iridium in oxidation state 1. So, the concept of building a single-site catalyst via the incorporation of a metal atom in low oxidation state into a near metallic cluster core seems appropriate.
 | ||
| Fig. 3 (a and b) Two orientations of the molecular structure of [Hyp3Ge9Ir(CO)(PPh3)] (1). All displacement ellipsoids are shown at a probability level of 50%. The [Ge9Ir] cluster cores are depicted as green polyhedra. Ge, Si, Ir, P, O, and C atoms are displayed in green, blue, pink, orange, red, and gray colors, respectively. All hydrogen atoms are omitted, and the ligands, despite CO, are shown in wire and frame modes for clarity. In (b), the phenyl rings of the phosphine are additionally omitted for clarity. | ||
Phosphine exchange reactions in 1
We have observed for Ni clusters that catalytically active sites can be created by ligand dissociation in solution.12 In order to create analogue sites here, we studied as a first step the thermally induced ligand dissociation in 1. According to the 31P NMR spectra (Fig. S39, ESI†), we observe an additional signal at −5.42 ppm indicative of the formation of free PPh3 with a maximum of 37% (324 hours at 70–85 °C). On addition of one and 20 equivalents of the phosphines Pptolyl3, PMe3 and PiPr3 to a solution of 1 at 70–85 °C (Scheme 2 top), we observe for a mixture of 1 and Pptolyl3 or PMe3 the appearance of new 31P NMR signals that indicate the formation of 2 and 3, respectively. This is in line with the formation of free PPh3 in solution. In more detail, for one equivalent of Pptolyl3 and PMe3 the new species are visible at approximately 60% and 40%, respectively, in the NMR spectra at 70–85 °C (Fig. S40 and 41 to S44 and 45 respectively, ESI,† and entries 1−3 in Table 1). At an excess of 20 phosphine equivalents, for both Pptolyl3 and PMe3, a full exchange in solution at 85 °C is observed (Fig. S42 and 43 to S46 and 47 respectively, ESI,† and entries 2−4 in Table 1). We notice that no significant cluster decomposition occurs. In the case of PiPr3, we observe for a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio only 20% of a desired new species after 253 hours of tempering (Fig. S48 and 49, ESI† and entry 5 in Table 1), and at 20 equivalents we reach 70% of exchange (Fig. S50 and 51, ESI† and entry 6 Table 1). This observation hints at an influence of the steric demand of PiPr3. The ligand cone angle of PiPr3 of 160° is considerably larger than for PPh3, Pptolyl3, and PMe3 with cone angles of 145°, 145°, and 115°, respectively.44 However, the exchange with PiPr3 also shows the appearance of new signals in the 1H-NMR in the region of the silyl groups, which cannot be assigned to specific compounds but indicate cluster decomposition.
:1 ratio only 20% of a desired new species after 253 hours of tempering (Fig. S48 and 49, ESI† and entry 5 in Table 1), and at 20 equivalents we reach 70% of exchange (Fig. S50 and 51, ESI† and entry 6 Table 1). This observation hints at an influence of the steric demand of PiPr3. The ligand cone angle of PiPr3 of 160° is considerably larger than for PPh3, Pptolyl3, and PMe3 with cone angles of 145°, 145°, and 115°, respectively.44 However, the exchange with PiPr3 also shows the appearance of new signals in the 1H-NMR in the region of the silyl groups, which cannot be assigned to specific compounds but indicate cluster decomposition.
| Entry | Phosphine | Ratio mixture | T [°C] | t [h] | Ratio of compound 1:new speciesb [%] |
|---|---|---|---|---|---|
| a Overall tempering time is independent of temperature. b According to 1H and 31P NMR. Decomposition products are not considered here. | |||||
| 1 | Pptolyl3 | 1:1 |
70 → 85 | 253 | 40:60 |
| 2 | Pptolyl3 | 1:20 |
85 | 172 | 0:100 |
| 3 | PMe3 | 1:1 |
85 | 5 | 60:40 |
| 4 | PMe3 | 1:20 |
85 | 119 | 0:100 |
| 5 | PiPr3 | 1:1 |
70 → 85 | 253 | 80:20 |
| 6 | PiPr3 | 1:20 |
85 | 172 | 30:70 |
In addition to monodentate phosphines, we investigated the phosphine replacement through bidentate dppe. Using a 1:1 dppe:1 ratio, small amounts of a new species 5 appear in the 31P NMR spectrum; however, this completely decomposes within 20 hours at 85 °C, and no reaction occurs at lower temperatures (Fig. S52 and 53, ESI†). If an excess of dppe is used instead, immediate decomposition to [Hyp3Ge9]− occurs at 85 °C and the formation of the new species 5 is never observable (Fig. S54 and 55, ESI†). Thus, most interestingly, this allows the extraction of the Ir atom from the cluster surface and a full recovery of the starting material [Hyp3Ge9]−. This is in accordance with the observation that the ligand strength of [Hyp3Ge9]− is between aryl and alkyl phosphines,45 and thus dppe as a stronger ligand leads to the observed extraction of Ir.
Oxidative addition of silanes to 1
Since we showed that ligand exchange reactions are possible, the formation of a catalytically active site seemed reasonable. Therefore, we investigated reactions of 1 with 1-hexene, hydrogen, and 1-hexene in the presence of hydrogen, which however according to the NMR spectra showed no reaction. However, reactions with phenylsilanes revealed the activation of Si–H bonds and the formation of oxidative addition products [Hyp3Ge9Ir(CO)(H)(SiR2R′)] [R/R′ = H/Ph (6) H/pMePh (7), H/p(OMe)Ph (8), H/p(NMe2)Ph (9), and Ph/H (10)] under substitution of the phosphine ligand (Scheme 2 bottom). As shown below in detail, all products were characterized in solution. Compound 6 was additionally characterized using LIFDI/MS as shown in Fig. 2b, which shows a mass peak corresponding to [Hyp3Ge9Ir(CO)(H)(SiH2Ph)] and its fragmentation products (Fig. S61, ESI†). A few single crystals of [Hyp3Ge9Ir(CO)(H)(SiH2p{OMe}Ph)] (8) suitable for single crystal X-ray structure determination were obtained from a diethyl ether solution at −32 °C (Fig. 4). The structure of 8 reveals a [Ge9Ir] polyhedron that is almost identical to the cluster core of [Hyp3Ge9Ir(CO)(PPh3)], 1, with Ge–Ge bond lengths of 2.516(2)–3.130(2) Å lying in the same range as in 1; as observed previously, Ge2–Ge4 is the shortest and Ge6–Ge9 the longest distance. The position of the hydrogen atom could be approximately localized using a Fourier difference plot and refined without constraints, resulting in an Ir–H bond length of 1.60(9) Å. This is in good agreement with known experimental and calculated Ir–H distances in the literature that are observed in the range of 1.49(5) to 2.214 Å.46–50 The Si–H bonds within the structure were fixed according to the literature to distances of about 1.5 Å.51 Another remarkable fact is that the CO-ligand is arranged like in 1 in the trans-position with respect to Ge9, but with a larger deviation from 90° bond angles around Ir formed by the connected ligand atoms: ∡(CCO–Ir–Sisilane) = 89.5(5)°, ∡(Ge7–Ir–Ge9) = 92.01(4)°, ∡(Ge9–Ir–Sisilane) = 77.6(1)°, and ∡(Ge7–Ir–CCO) = 85.4(4)°. However, both ligands – CO and SiH2p{OMe}Ph – are tilted by 18.2(6)° towards Ge3 with respect to the plane built by Ir, Ge7, Ge6, and Ge9 (Fig. 4), which is significantly higher than the angle in 1. Since only a few crystals of 8 could be obtained, no NMR of purified 8 could be measured. | ||
| Fig. 4 (a and b) Two orientations of the molecular structure of [Hyp3Ge9Ir(CO)(H)(SiH2p{OMe}Ph)], 8. All displacement ellipsoids are shown at a probability level of 50%. The [Ge9Ir] cluster cores are depicted as green polyhedra. Ge, Si, Ir, O, C, and H atoms are displayed in green, blue, pink, red, gray, and white colors, respectively. All hydrogen atoms at ligands and the TMS groups at the silyl substituents are omitted for clarity. The residue at the silane is shown in wire and frame modes for simplicity. | ||
Even though only one isomer in the crystalline product of 8 from the reaction of 1 with SiH3p{OMe}Ph was isolated, two new sets of signals with approximate ratios of 1:4 besides the signals for 1 and residual silane were observed in the 1H NMR spectra of the reaction solution. Details of the 1H NMR spectrum of 8 are shown in Fig. 5a and the reaction process is demonstrated in Fig. 5b in detail for 6. The spectra are shown as representatives and the corresponding spectra of 7, 8, 9, and 10 are shown in the ESI.† For the reaction solution of compound 8 (Fig. 5a), two signals are observed in the negative ppm range at −14.50 ppm and −9.11 ppm, which are assigned to hydrogen atoms connected to the Ir atoms labeled as 8a and 8b. Two new signal sets occur also for the hypersilyl ligands, which are shifted with respect to 1 to slightly lower and higher fields and are denoted in Fig. 5a as 8a and 8b, respectively. For the respective primary silane residue SiH2p(OMe)Ph, two new signal sets for the H atoms attached to the Si atom of the silane appear in the range 5.3–6.5 ppm, also denoted as 8a and 8b. Besides these signals, a singlet originating from the H atoms directly bound to Si from SiH3p{OMe}Ph, which was used in excess, can be detected at 4.31 ppm, and thus the new signal groups of 8 are low field shifted compared to the unattached silane. Considering the integral ratios for both species 8a and 8b, the expected values of 81:1:2 for the hypersilyl:hydride:Si–H groups can be found; however, 8a is clearly favored over 8b and even storing the sample at room temperature does not change the ratio between both species.
 | ||
| Fig. 5 Relevant excerpts of the 1H NMR spectra (400 MHz) of the reaction of 1 with (a) (4-methoxyphenyl)silane and (b) phenylsilane recorded in C6D6. The three sections of the spectra of Ir–H units, hypersilyl groups, and Si–H units are marked with a blue, green, and red box, respectively. In each section, the two different products formed are labeled as 6a (8a) and 6b (8b). The excerpts shown in the black boxes are enlarged areas of the corresponding sections of the NMR spectra. | ||
We assume that the more intensive signals arise from the compound that forms through crystallization – at least the interpretation of the 1H NMR-spectra are in agreement with the solid state structure. The second set of signals which arise with lower intensity but identical relative signal intensities could originate from a product, in which the hydride ligand and SiH2p{OMe}Ph at the Ir atom have a different orientation with respect to the Ge9 unit. Considering the structures of the two species, one can assume that for 8 the product with the majority occurrence in solution adopts the structure obtained from the single crystal X-ray structure determination. The silane being the most bulky ligand when compared to CO and H seems sterically less hindered in the trans-position to the Ge1–Hyp moiety. The formation of a second species 8b can be rationalized through the silane addition with reversed Si and H positions with respect to the CO ligand resulting in an exchange of the silane and H ligand at the Ir atom (Fig. 6).
 | ||
| Fig. 6 Both possible silane addition pathways leading to (a) the arrangement as observed in the crystal structure of 8 and (b) the exchanged orientation of hydride and silane ligands at the Ir center. | ||
Correspondingly for the reaction solution of compound 6 (Fig. 5b), two hydride species at −14.57 ppm and −9.16 ppm, two new signal sets for the hypersilyl ligands, and two sets for the H atoms attached to the Si atom of the silane in the range of 5.3–6.5 ppm can be detected in the 1H NMR spectrum denoted as 6a and 6b, respectively. Here again, the new Si–H groups are low field shifted compared to the unattached SiH3Ph, which shows a singlet at 4.23 ppm for the Si–H protons. The integral ratios of 6a and 6b are hereby also consistent with the expected values of 81:1:2 for the hypersilyl:hydride:Si–H groups. Accordingly, the 29Si NMR and 1H29Si HMBC NMR spectra of the reaction with phenylsilane to form 6 (Fig. S58 and S59,† ESI) show two new signals at −51.82 ppm and −61.09 ppm, which can be correlated to the new Si–H signals in the corresponding 1H NMR in the range of 5.3–6.5 ppm, further confirming the formation of two products. Analogously, similar results occur for the other reactions forming 7, 9 and 10, and thus for each two new signal sets can be observed in the 1H NMR spectra, with two signals in the negative range indicative of the iridium hydride units, two new signal sets for the hypersilyl ligands and two new signal sets for the protons directly bound to the Si atom of the silane residues, indicated with na and nb with n = 7, 9, and 10 (Fig. S65, S69, and S71, ESI†). In summary, all 1H NMR spectra of the reaction solutions forming 6–10 hint at the appearance of two similar products, while one is clearly favored. For the reaction with diphenylsilane to form 10, the 1H29Si HMBC NMR spectrum only shows the main species hinting at a strong preference for one species (Fig. S73, ESI†). Additionally, in the 31P NMR spectra of each reaction, a signal at about −5 ppm arises, indicating the formation of free PPh3 (Fig. S57, S66, S68, S70, and S72, ESI†). For 6, the formation was also observed in the IR spectrum. Thereby, a new band is observed at 2082 cm−1, which appears in the typical range for Ir–H bonds such as in Ir(H)(PPh3)2(CO)(H)(SiPh3), Ir(H)(PPh3)2(CO)(H)(SiPhMe2), or Ir(H)(Cl)(SiCl3)(CO)(PPh3)2 with bands at 2075 cm−1, 2080 cm−1, and 2128 cm−1, respectively.52,53 In addition, a shift of the CO stretching mode at 1953 cm−1 for 1 to higher wavenumbers at 1996 cm−1 appears in the IR spectrum of 6, which hints as well at the presence of an Ir-CO group after the oxidative addition of the silane (Fig. S63, ESI†).
Since products 6–10 were not isolated, the yields of the reactions were estimated from the ratio of 1 to the corresponding product (concerning both species for 6–10) that are in the range of 0 to 100 and 40 to 60 (entries 1–5, Table 2) according to the 1H NMR spectra. Considering the quantities of these two products, one species denoted as “a” is always clearly preferably formed with ratios of about na:nb = 80:20 for R/R′ = H/Ph, H/pMePh, H/p(OMe)Ph, H/p(NMe2)Ph (6–9, entries 1–4, Table 2), while it is even more preferred for R/R′ = Ph/H (10) with a ratio of 90:10 (entries 1–4, Table 2).
| Entry | Silane | Product-Nr. | T [°C] | t [h] | Ratio of compound 1:new species (a:b)a [%] |
|---|---|---|---|---|---|
| a According to 1H NMR. Decomposition products are not considered here. | |||||
| 1 | Phenylsilane | 6 | 85 | 41 | 0:100 (82:18) |
| 2 | (4-Methylphenyl)silane | 7 | 85 | 19 | 11:89 (81:19) |
| 3 | (4-Methoxyphenyl)silane | 8 | 85 | 17 | 18:82 (80:20) |
| 4 | (4-Dimethylaminophenyl)silane | 9 | 85 | 11.5 | 22:78 (81:19) |
| 5 | Diphenylsilane | 10 | 85 | 126 | 39:61 (89:11) |
| 6 | Triethylsilane | — | 85 | 41 | 100:0 (—:—) |
In contrast to previously reported reactions with metalated Zintl clusters, we observe a color change of the reaction solutions from yellowish-brown (1) to reddish-brown (6–10). Exemplary, for all silane reactions, UV/vis spectra of 1 and 6 are shown in the ESI (Fig. S62–S64†). We observe a slight shift of 5 nm of the absorption maxima of 6 (270 nm) compared to 1 (265 nm) in accordance with the visible red shift. Since the silane addition causes a change in the oxidation state of the Ir atom, the color change might originate from this reaction type. This change in the oxidation state from +I to +III is already known for other homogeneous iridium metal complexes, like the Vaska complex itself.52 Other bond activations, like B–H and N–H through oxidative addition to the Ir atom, have not been successful up to now, nor did we observe reactions between triethylsilane and the cluster (Fig. S74, ESI† and entry 6 in Table 2).
Conclusion
Through a straightforward synthetic protocol, three different iridium metalated clusters [Hyp3Ge9Ir(CO)(PR3)] (R = Ph, ptolyl, Me, 1–3) could be synthesized in good yields. The products were fully characterized using NMR, LIFDI/MS, and IR methods. For 1, single crystal structure determination could also be performed revealing a distorted bicapped square antiprism with C1-symmetry for the [Ge9Ir] polyhedron. The exchangeability of the phosphine ligand of 1 in solution was tested, while sterically less demanding ones, like PMe3 and Pptolyl3, can be introduced easily in contrast to PiPr3 with a big ligand cone angle. Additionally catalytic reactions with 1 were investigated, while oxidative addition of silanes was achieved, in which two different products were built. During this process, the CO-ligand remains at the iridium center while the phosphine ligand is exchanged by the silane. The reaction products [Hyp3Ge9Ir(CO)(H)(SiR2R′)] [R/R′ = H/Ph, H/pMePh, H/p(OMe)Ph, H/p(NMe2)Ph, Ph/H, 6–10] were characterized using NMR, IR, and LIFDI/MS. For 8, a single crystal of the major species was obtained, revealing an almost identical cluster to 1. Given the similarity between our cluster and the Vaska complex, the formation of a stable product upon oxidative addition of silanes can be expected. However, since educt activation is a crucial step in catalysis, our results nevertheless prove that metalated Zintl clusters are potent candidates for catalysis.Author contributions
Investigation, writing – review and editing and visualization, N. W. and T. K.; writing – original draft, N. W.; validation, V. H.; supervision, S. G. and T. F. F.; conceptualization, funding acquisition, project administration, and writing – review and editing, T. F. F. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Data availability
The experimental procedures, spectral data, and selected crystallographic data supporting this article have been included as part of the ESI.† Crystallographic data for 1 and 8 have been deposited at the CCDC under deposition numbers 2328043 and 2372675.†Acknowledgements
This work was supported by financial funding from Wacker Chemie AG. The authors thank B.Sc. Yasmina Boulesnam and B.Sc. Carolin Lauerburg for the synthesis and preparations of different reaction solutions. Furthermore, we thank M.Sc. Ivan Antsiburov and M.Sc. Jonas Gilch for performing the LIFDI/MS measurements and Manuel Seiler for the execution of the elemental analysis.References
- X. Liang, N. Fu, S. Yao, Z. Li and Y. Li, The Progress and Outlook of Metal Single-Atom-Site Catalysis, J. Am. Chem. Soc., 2022, 144, 18155–18174 CrossRef.
- Y. Shi, Y. Zhou, Y. Lou, Z. Chen, H. Xiong and Y. Zhu, Homogeneity of Supported Single-Atom Active Sites Boosting the Selective Catalytic Transformations, Adv. Sci., 2022, 9, e2201520 CrossRef PubMed.
- Q. Zhang and J. Guan, Applications of single-atom catalysts, Nano Res., 2021, 15, 38–70 CrossRef.
- G. G. Hlatky, Heterogeneous Single-Site Catalysts for Olefin Polymerization, Chem. Rev., 2000, 100, 1347–1376 CrossRef PubMed.
- Z. Li, D. Wang, Y. Wu and Y. Li, Recent advances in the precise control of isolated single-site catalysts by chemical methods, Natl. Sci. Rev., 2018, 5, 673–689 CrossRef.
- J. D. Pelletier and J. M. Basset, Catalysis by Design: Well-Defined Single-Site Heterogeneous Catalysts, Acc. Chem. Res., 2016, 49, 664–677 CrossRef CAS PubMed.
- B. Qiao, A. Wang, X. Yang, L. F. Allard, Z. Jiang, Y. Cui, J. Liu, J. Li and T. Zhang, Single-atom catalysis of CO oxidation using Pt1/FeOx, Nat. Chem., 2011, 3, 634–641 CrossRef CAS PubMed.
- J. M. Thomas, R. Raja and D. W. Lewis, Single-site heterogeneous catalysts, Angew. Chem., Int. Ed., 2005, 44, 6456–6482 CrossRef CAS PubMed.
- B. Cornils and W. A. Herrmann, Concepts in homogeneous catalysis: the industrial view, J. Catal., 2003, 216, 23–31 CrossRef CAS.
- C. Weetman and S. Inoue, The road travelled: after main–group elements as transition metals, ChemCatChem, 2018, 10, 4213–4228 Search PubMed.
- P. P. Power, Main-group elements as transition metals, Nature, 2010, 463, 171–177 CrossRef CAS PubMed.
- N. S. Willeit, W. Klein, P. Coburger, E. Fritz-Langhals and T. F. Fässler, Functionalised NiGe9 Clusters as Homogeneous Single-Site Catalysts for Olefin Isomerisation Reactions, ChemCatChem, 2024, 16, e202301200 CrossRef CAS.
- T. F. Fässler and S. D. Hoffmann, Endohedral Zintl ions: intermetalloid clusters, Angew. Chem., Int. Ed., 2004, 43, 6242–6247 CrossRef.
- F. Li and S. C. Sevov, Rational synthesis of [Ge9{Si(SiMe3)3}3]− from its parent Zintl ion Ge9(4−), Inorg. Chem., 2012, 51, 2706–2708 CrossRef CAS.
- O. Kysliak and A. Schnepf, {Ge9[Si(SiMe3)3]2}2−: a starting point for mixed substituted metalloid germanium clusters, Dalton Trans., 2016, 45, 2404–2408 RSC.
- L. J. Schiegerl, F. S. Geitner, C. Fischer, W. Klein and T. F. Fässler, Functionalization of [Ge9] with Small Silanes:[Ge9(SiR3)3]− (R = iBu, iPr, Et) and the Structures of (CuNHCDipp)[Ge9{Si(iBu)3}3], (K-18c6)Au[Ge9{Si(iBu)3}3]2, and (K-18c6)2[Ge9{Si(iBu)3}2], Z. Anorg. Allg. Chem., 2016, 642, 1419–1426 CrossRef.
- K. Mayer, L. J. Schiegerl, T. Kratky, S. Günther and T. F. Fässler, Targeted attachment of functional groups at Ge9 clusters via silylation reactions, Chem. Commun., 2017, 53, 11798–11801 RSC.
- O. Kysliak, T. Kunz and A. Schnepf, Metalloid Ge9R3−Clusters with Various Silyl Substituents: From Shielded to Open Cluster Cores, Eur. J. Inorg. Chem., 2017, 805–810, DOI:10.1002/ejic.201601134.
- S. Frischhut and T. F. Fässler, Synthesis of low-oxidation-state germanium clusters comprising a functional anchor group - synthesis and characterization of [(Ge(0))5(Ge-R)3(Ge-(CH2)n-CH=CH2)] with R = Si(SiMe3)3, Dalton Trans., 2018, 47, 3223–3226 RSC.
- S. Frischhut, W. Klein, M. Drees and T. F. Fässler, Acylation of Homoatomic Ge9 Cages and Subsequent Decarbonylation, Chem. – Eur. J., 2018, 24, 9009–9014 CrossRef CAS.
- F. Li, A. Munoz-Castro and S. C. Sevov, [Ge9{Si(SiMe3)3}3{SnPh3}]: a tetrasubstituted and neutral deltahedral nine-atom cluster, Angew. Chem., Int. Ed., 2012, 51, 8581–8584 CrossRef CAS PubMed.
- F. Li and S. C. Sevov, Synthesis, structures, and solution dynamics of tetrasubstituted nine-atom germanium deltahedral clusters, J. Am. Chem. Soc., 2014, 136, 12056–12063 CrossRef CAS PubMed.
- C. Liu and Z.-M. Sun, Recent advances in structural chemistry of Group 14 Zintl ions, Coord. Chem. Rev., 2019, 382, 32–56 CrossRef.
- R. J. Wilson, N. Lichtenberger, B. Weinert and S. Dehnen, Intermetalloid and Heterometallic Clusters Combining p-Block (Semi)Metals with d- or f-Block Metals, Chem. Rev., 2019, 119, 8506–8554 CrossRef.
- J. M. Goicoechea and S. C. Sevov, Deltahedral Germanium Clusters: Insertion of Transition-Metal Atoms and Addition of Organometallic Fragments, J. Am. Chem. Soc., 2006, 128, 4155–4161 CrossRef PubMed.
- S. Scharfe and T. F. Fässler, Varying Bonding Modes of the Zintl Ion [Ge9]4− in CuI Complexes: Syntheses and Structures of [Cu(η4−Ge9)(PR3)]3− (R = iPr, Cy) and [Cu(η4−Ge9)(η1−Ge9)]7−, Eur. J. Inorg. Chem., 2010, 2010, 1207–1213 CrossRef.
- J. M. Goicoechea and S. C. Sevov, Organozinc Derivatives of Deltahedral Zintl Ions: Synthesis and Characterization of closo-[E9Zn(C6H5)]3− (E = Si, Ge, Sn, Pb), Organometallics, 2006, 25, 4530–4536 CrossRef.
- B. Zhou, M. S. Denning, C. Jones and J. M. Goicoechea, Reductive cleavage of Zn-C bonds by group 14 Zintl anions: synthesis and characterisation of [E9ZnR]3− (E = Ge, Sn, Pb; R = Mes, iPr), Dalton Trans., 2009, 1571–1578, 10.1039/b815122a.
- Z.-M. Sun, Y.-F. Zhao, J. Li and L.-S. Wang, Diversity of Functionalized Germanium Zintl Clusters: Syntheses and Theoretical Studies of [Ge9PdPPh3]3− and [Ni@(Ge9PdPPh3)]2−, J. Cluster Sci., 2009, 20, 601–609 CrossRef.
- J. Campbell, H. P. A. Mercier, H. Franke, D. P. Santry, D. A. Dixon and G. J. Schrobilgen, Syntheses, Crystal Structures, and Density Functional Theory Calculations of the closo-[1-M(CO)3(η4-E9)]4− (E = Sn, Pb; M = Mo, W) Cluster Anions and Solution NMR Spectroscopic Characterization of [1-M(CO)3(η4-Sn9)]4− (M = Cr, Mo, W), Inorg. Chem., 2002, 41, 86–107 CrossRef PubMed.
- B. W. Eichhorn and R. C. Haushalter, Synthesis and Structure of closo-Sn9Cr(CO)34− : The First Member in a New Class of Polyhedral Clusters, J. Am. Chem. Soc., 1988, 110, 8704–8706 CrossRef.
- B. Kesanli, J. Fettinger and B. Eichhorn, The closo-[Sn9M(CO)3]4− Zintl ion clusters where M = Cr, Mo, W: two structural isomers and their dynamic behavior, Chem. – Eur. J., 2001, 7, 5277–5285 CrossRef PubMed.
- L. Yong, S. D. Hoffmann and T. F. Fässler, Crystal Structures of [K(2.2.2−crypt)]4[Pb9Mo(CO)3]—Isolation of the Novel Isomers [(η5−Pb9)Mo(CO)3]4− beside [(η4−Pb9)Mo(CO)3]4−, Eur. J. Inorg. Chem., 2005, 3663–3669, DOI:10.1002/ejic.200500060.
- J. Q. Wang, S. Stegmaier, B. Wahl and T. F. Fassler, Step-by-step synthesis of the endohedral stannaspherene [Ir@Sn12]3− via the capped cluster anion [Sn9Ir(cod)]3−, Chem. – Eur. J., 2010, 16, 1793–1798 CrossRef.
- O. P. E. Townrow, C. Chung, S. A. Macgregor, A. S. Weller and J. M. Goicoechea, A Neutral Heteroatomic Zintl Cluster for the Catalytic Hydrogenation of Cyclic Alkenes, J. Am. Chem. Soc., 2020, 142, 18330–18335 CrossRef.
- O. P. E. Townrow, S. B. Duckett, A. S. Weller and J. M. Goicoechea, Zintl cluster supported low coordinate Rh(I) centers for catalytic H/D exchange between H2 and D2, Chem. Sci., 2022, 13, 7626–7633 RSC.
- F. Henke, C. Schenk and A. Schnepf, [Si(SiMe3)3]3Ge9M(CO)3(−) (M=Cr, Mo, W): coordination chemistry with metalloid clusters, Dalton Trans., 2011, 40, 6704–6710 RSC.
- C. Schenk and A. Schnepf, {Ge9R3Cr(CO)5}− and {Ge9R3Cr(CO)3}−: a metalloid cluster (Ge9R3−) as a flexible ligand in coordination chemistry [R=Si(SiMe3)3], Chem. Commun., 2009, 3208–3210, 10.1039/b901870k.
- S. Frischhut, F. Kaiser, W. Klein, M. Drees, F. E. Kühn and T. F. Fässler, Capping nido-Nonagermanide Clusters with M-PPh3 and Dynamics in Solution: Synthesis and Structure of closo-[(Me3Si)3Si]3Et[Ge9M](PPh3) (M = Ni, Pt), Organometallics, 2018, 37, 4560–4567 CrossRef.
- F. Li, A. Muñoz-Castro and S. C. Sevov, [(Me3Si)Si]3EtGe9Pd(PPh3), a Pentafunctionalized Deltahedral Zintl Cluster: Synthesis, Structure, and Solution Dynamics, Angew. Chem., Int. Ed., 2016, 55, 8630–8633 CrossRef.
- N. S. Willeit, V. Hlukhyy and T. F. Fässler, Synthesis, Structure and Catalytic Properties of Hyp3[Ge9Rh]PPh3, Z. Anorg. Allg. Chem., 2024, 650, e202400171 CrossRef.
- O. P. E. Townrow, A. S. Weller and J. M. Goicoechea, Controlled cluster expansion at a Zintl cluster surface, Angew. Chem., Int. Ed., 2024, 63, e202316120 CrossRef.
- M. Rubel, R. Haasch, P. Mrozek, A. Wieckowski, C. DePauli and S. Trasatti, Characterization of IrO2-SnO2 thin layers by electron and ion spectroscopies, Vacuum, 1994, 45, 423–427 CrossRef.
- C. A. Tolman, Steric effects of phosphorus ligands in organometallic chemistry and homogeneous catalysis, Chem. Rev., 1977, 77, 313–348 CrossRef.
- C. Gienger and A. Schnepf, Neutral R3PAuGe9(Hyp)3 (R=Et, nPr, iPr, nBu, tBu, Cy) (Hyp=Si(SiMe3) Clusters give new insights into the ligand strength of the metalloid [Ge9(Hyp)3]− cluster, Z. Anorg. Allg. Chem., 2021, 647, 1695–1701 CrossRef.
- M. D. Fryzuk, P. A. MacNeil and S. J. Rettig, Stereoselective formation of rhodium and iridium hydrides via intramolecular hydrogen bonding, J. Am. Chem. Soc., 1987, 109, 2803–2812 CrossRef.
- F. T. Ladipo and J. S. Merola, Oxidative addition of nitrogen-hydrogen bonds to iridium: synthesis and structure of (heterocyclic amine)iridium hydride complexes, Inorg. Chem., 1990, 29, 4172–4173 CrossRef.
- H. V. Nanishankar, S. Dutta, M. Nethaji and B. R. Jagirdar, Dynamics of a cis-Dihydrogen/Hydride Complex of Iridium, Inorg. Chem., 2005, 44, 6203–6210 CrossRef PubMed.
- B. S. Omar, J. Mallah, M. Ataya, B. Li, X. Zhou, S. Malakar, A. S. Goldman and F. Hasanayn, H(2) Addition to Pincer Iridium Complexes Yielding trans-Dihydride Products: Unexpected Correlations of Bond Strength with Bond Length and Vibrational Frequencies, Inorg. Chem., 2018, 57, 7516–7523 CrossRef PubMed.
- P. Zaleski-Ejgierd, High-pressure formation and stabilization of binary iridium hydrides, Phys. Chem. Chem. Phys., 2014, 16, 3220–3229 RSC.
- F. H. Allen and I. J. Bruno, Bond lengths in organic and metal-organic compounds revisited: X-H bond lengths from neutron diffraction data, Acta Crystallogr., Sect. B:Struct. Sci., 2010, 66, 380–386 CrossRef PubMed.
- M. A. Bennett, R. Charles and P. J. Fraser, Oxidative Addition of Monosilanes to Planar Iridium(1) Complexes and Carbonylation of the Resulting Adducts, Aust. J. Chem., 1977, 30, 1201–1211 CrossRef.
- J. F. Harrod, D. F. R. Gilson and R. Charles, Oxidative addition of silicon hydrides to hydridocarbonyltris(triphenylphosphine)iridium(I), Can. J. Chem., 1968, 47, 2205 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2328043 and 2372675. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5dt01147g |
| This journal is © The Royal Society of Chemistry 2025 |