
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Design of new thorium nuclear clock materials based on polyatomic ions†
Harry W. T.
Morgan
 *ab,
Hoang Bao Tran
Tan
cd,
Andrei
Derevianko
c,
Ricky
Elwell
e,
James E. S.
Terhune
e,
Eric R.
Hudson
e and
Anastassia N.
Alexandrova
*a
*ab,
Hoang Bao Tran
Tan
cd,
Andrei
Derevianko
c,
Ricky
Elwell
e,
James E. S.
Terhune
e,
Eric R.
Hudson
e and
Anastassia N.
Alexandrova
*a
aDepartment of Chemistry and Biochemistry, University of California, Los Angeles, 607 Charles E Young Drive, Los Angeles, California 90095, USA. E-mail: harry.morgan@manchester.ac.uk; ana@chem.ucla.edu
bDepartment of Chemistry, University of Manchester, Oxford Road, Manchester M13 9PL, UK
cDepartment of Physics, University of Nevada, Reno, Nevada 89557, USA
dLos Alamos National Laboratory, P.O. Box 1663, Los Alamos, New Mexico 87545, USA
eDepartment of Physics and Astronomy, University of California, Los Angeles, California 90095, USA
First published on 17th June 2025
Abstract
Compounds of polyatomic anions are investigated theoretically as hosts for thorium in nuclear clock devices. The 229Th nucleus has an excited state at 8.355 eV which can be reached using a VUV laser as demonstrated in recent experiments. Incorporating 229Th into a host crystal is an essential step towards developing an ultra-stable nuclear clock. To be a suitable host, a material must have a band gap larger than the nuclear transition frequency. Thus, most research to date has focused on fluorides like LiSrAlF6 and CaF2, for they feature ionic bonding and large band gaps. Ionicity of chemical bonding can be pushed to its limits by superhalogens – polyatomic anions whose electron affinity can be greater than that of the halogens. An additional concern for fluorides is the presence of 19F nuclear spins in the material that can couple to the spin of the 229Th, detrimental to clock performance. In this work we investigate salts containing superhalogen anions, with the goal of identifying promising new hosts for 229Th clocks. Specifically, we investigate compounds of Ca, Sr, and Ba with three anions – BF4−, ClO4−, and SO42−. By computing the electronic properties of the pure and thorium-doped materials, we predict that M(BF4)2 have the widest band gaps, making them a promising material class. On the other hand, MSO4 could produce the most accurate clocks by eliminating inhomogeneous broadening due to nuclear spin interactions. These results may guide experimental searches for new 229Th clock materials and offer new opportunities to study this unique nuclear transition in the solid state.
1 Introduction
Accurate chronometry is essential for fundamental science, precise physical measurements, and communication and navigation technology. Clocks more precise than the current state-of-the-art could even open up the field of relativistic geodesy, using gravitational redshift of the clock frequency to predict earthquakes and volcanic eruptions.1,2 One such clock could be the thorium nuclear clock. The 229Th isotope has a nuclear excited state at 8.4 eV, similar to the states probed in Mössbauer spectroscopy but at a fraction of the energy, with an extremely long lifetime and narrow transition linewidth.3 In a solid-state clock, a large number of Th nuclei are interrogated simultaneously, so a given level of accuracy can be attained quickly, allowing rapid measurements. In the parlance of clock technology, solid-state clocks are exceptionally stable. Since the excitation primarily involves the nucleus, it is expected to be less susceptible to environmental perturbations than traditional atomic clocks based on electronic transitions, such as the hyperfine transition in atomic caesium.4 Furthermore, a solid-state clock could be more practical “in the field” than systems involving atomic vapour or trapped ions. Laser excitation of the 229Th nucleus has recently been demonstrated in thorium-doped single crystals of LiSrAlF6 and CaF2,5–7 and in ThF4 thin films.8One essential requirement for a candidate host material is that it must have a band gap larger than the nuclear excitation energy of 8.4 eV; if it does not then the incident laser radiation will be absorbed by the electrons of the material rather than the 229Th nuclei. Research so far has focused largely on s-block metal fluorides such as MgF2, CaF2, LiCaAlF6, and LiSrAlF6,5,6,9–14 and stoichiometric thorium fluorides.8,15 Because those metals have low ionization energies and fluorine has a large electron affinity, fluorides have large band gaps, reflecting the amount of energy required to move an electron from F − to a Mq+ cation. In searching for ‘simple’ ionic compounds for clock applications, we are almost completely restricted to fluorides because no other element is electronegative enough to produce such a large band gap.16
Chemistry can reveal new directions in the search for clock materials. “Superhalogens” are molecular species with electron affinities greater than halogen atoms, so we expect compounds of their anions with s-block metals to have the required band gaps.17,18 Commonly encountered superhalogen anions include BF4−, PF6−, and ClO4−, which are often used as non-coordinating anions in solution or in ionic liquids.19 The large ionization energies stem from delocalization of the negative charge over multiple electronegative atoms, and strong covalent bonding. We anticipate that the large ionization energies of the anion will correspond to large band gaps in corresponding salts.
Here we investigate three groups of compounds by studying the electronic properties of the pure phases and thorium defects: metal sulfates MSO4, perchlorates M(ClO4)2, and tetrafluoroborates M(BF4)2 (M = Ca, Sr, Ba). All three ions have strong covalent bonding and charge delocalization, so we expect them to produce large-gap salts. Strictly, SO42− is not a superhalogen because its double negative charge gives it a negative first ionization energy in the gas phase, but in an ionic crystal it is similar to ClO4−.20 Most of the compounds have been synthesized and characterized in anhydrous and hydrated forms.21–32 We expect the hydrates to have smaller band gaps due to the additional electronic states introduced by water, so we have chosen to focus on the anhydrous materials. For each group we will begin by computing the bulk band gaps to determine whether the host is transparent to photons at the nuclear excitation energy, and then perform thorium doping studies to find the thorium defect electronic properties, specifically 5f orbital energies and markers of Th-anion covalency. We will then determine the viability of polyatomic anions in thorium clocks and identify the best candidates for experimental study.
2 Methods
DFT calculations were performed with VASP,33 version 6.4.2, using the PAW34 method with a plane-wave cutoff of 500 eV and a spin-restricted formalism. The PBE35 functional was used for structural optimizations and defect formation energies, and the modified Becke–Johnson (MBJ)36,37 and HSE0638,39 functionals were used for electronic properties. Structural optimizations were performed using Monkhorst–Pack k-meshes with point spacings of 0.03 Å−1 for bulk materials and 0.04 Å−1 for the defect supercells. Structures were visualized using VESTA.40 MBJ calculations used point spacings of 0.02 Å−1 for the bulk materials and 0.025 Å−1 for the defect supercells. In our defect models the thorium atom replaces a metal cation and a neighbouring cation vacancy is created to balance the charge. It is also possible that the charge could be compensated by additional anions, but we have not explored that possibility here for consistency between materials and because we cannot identify what those anions might be due to uncertainties in the synthetic methods.3 Results
3.1 Undoped phases
 | ||
| Fig. 1 Unit cells of sulfates CaSO4 and SrSO4. | ||
| MBJ (eV) | HSE06 (eV) | |
|---|---|---|
| CaSO4 | 8.53 | 7.91 |
| SrSO4 | 8.71 | 8.07 |
| BaSO4 | 8.40 | 8.02 |
 nuclei. A perchlorate-based clock would therefore suffer from nuclear spin broadening, but possibly not as much as a fluoride-based clock because in a perchlorate only one fifth of the anion atoms are spin-active and because the closest nuclei to 229Th are I = 0. Furthermore, perchlorates are very hygroscopic, deliquescent,51 and oxidizing, making them comparatively difficult to handle experimentally. Based on the properties of the bulk materials, the sulfates appear to be better candidates.
nuclei. A perchlorate-based clock would therefore suffer from nuclear spin broadening, but possibly not as much as a fluoride-based clock because in a perchlorate only one fifth of the anion atoms are spin-active and because the closest nuclei to 229Th are I = 0. Furthermore, perchlorates are very hygroscopic, deliquescent,51 and oxidizing, making them comparatively difficult to handle experimentally. Based on the properties of the bulk materials, the sulfates appear to be better candidates.
 | ||
| Fig. 2 Optimized unit cells of Ca(ClO4)2, Ba(ClO4)2, Pcba-Sr(BF4)2 (high square antiprismatic SrF8), and C2/m-Ba(BF4) (showing pentagonal antiprismatic BaF10). | ||
| MBJ (eV) | HSE06 (eV) | |
|---|---|---|
| Ca(ClO4)2 | 7.82 | 7.86 |
| Sr(ClO4)2 | 8.39 | 7.84 |
| Ba(ClO4)2 | 7.72 | 7.63 |
The computed band gaps are all larger than 10 eV, significantly larger than the band gaps of the fluorides, perchlorates and sulfates, supporting our hypothesis that superhalogen salts are excellent candidates for nuclear clocks. Full results are given in Table 3. PDOS plots in the ESI† show that the band gap in these materials is a charge transfer from F − 2p to M2+ d.
| Material | MBJ (eV) | HSE06 (eV) |
|---|---|---|
| Ca(BF4)2 | 10.59 | 10.78 |
| Sr(BF4)2 | 12.01 | 10.59 |
| Ba(BF4)2Pcba | 11.05. | 10.45 |
| Ba(BF4)2C2/m | 10.45 | 10.53 |
3.2 Thorium-doped phases
We modelled thorium-doped phases by replacing an M2+ atom by Th4+ and removing a second M2+ to balance the charge. Note that these charge states are dictated by the typical chemistry of the elements and are not enforced in the calculations. Supercells of the hosts were used to reduce unphysical Th–Th interactions such that all Th–Th distances were 11 Å or more. Optimized structures and pictures of the thorium local coordination environments are in the ESI.† The thorium ions are 8- or 9-coordinate across all host material types, with unsymmetrical coordination geometries due to the electrostatic interaction between Th4+ and the anions. Most anions coordinate in a monodentate mode, though bidentate coordination can be found for all anions and is most common in the sulfates.For the tetrafluoroborates we computed defect formation energies by balancing the thorium doping reaction with binary metal fluorides, and for sulfates we computed defect formation energies by a reaction between metal sulfates. The results, in Tables S1 and S2,† show that the lowest defect formation energies in tetrafluoroborates are found in Sr(BF4)2 and Ba(BF4)2, and the lowest in sulfates is found in SrSO4. Strontium hosts therefore appear particularly good for thorium doping. f-element doping of sulfates is well-established because similar systems (e.g. Dy:CaSO4) are used for radiation dosimetry.52
The thorium PDOS plots for Th:SrSO4, Th:Sr(ClO4)2, and Th:Sr(BF4)2 are shown in Fig. 3. The electronic character of thorium is similar in all three materials. The 5f orbitals form narrow peaks around 5 eV and the 6d orbitals appear as sharp peaks at higher energies. The energies of the 5f peak are highest in Sr(BF4)2 and lowest in Sr(ClO4)2, weakly matching the trend in the band gaps of the host crystals. The 6d orbitals form a set of sharp peaks at higher energies, mixing into the conduction band in SrSO4 and Sr(ClO4)2. Thorium makes no contribution to the upper part of the valence band, appearing only in states below 1 eV, so the lowest thorium-centered electronic transitions are actually higher than the 5f energy relative to EF. This is a characteristic feature of cells containing cation vacancies because undercoordinated anions form the top of the valence band.
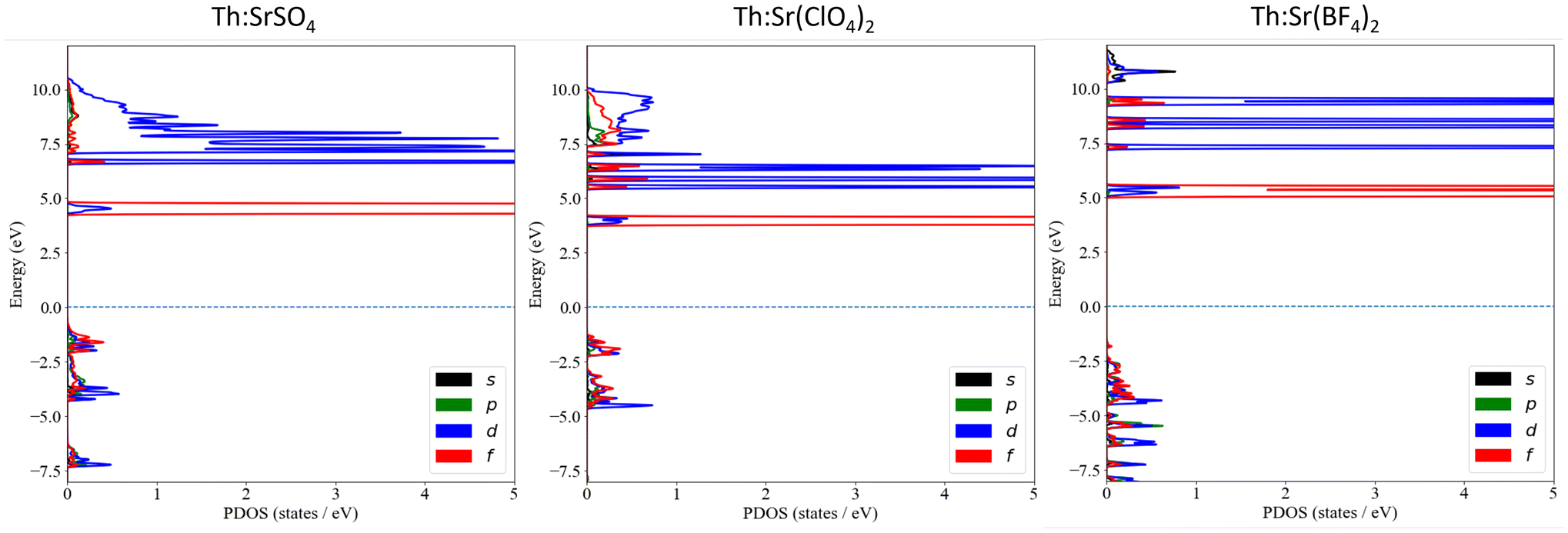 | ||
| Fig. 3 Thorium projected density of states plots for Th:SrSO4, Th:Sr(ClO4)2, and Th:Sr(BF4)2. | ||
The electronic part of IC is transfer of an electron from the anions to the vacant Th4+ 5f orbitals, so IC will be fast if there is strong orbital hybridization between Th 5f and the anions, and slow if there is weak hybridization. The thorium 5f PDOS in the valence band shows the extent of hybridization. For a quantitative comparison between compounds we integrate this PDOS over the valence band to give the “IPDOS” (integrated projected density of states), which is equivalent to the population of the Th 5f shell. Another benefit of the IPDOS is that it is not sensitive to the energies of the unoccupied 5f orbitals, which are subject to well-known DFT errors.
Th 5f IPDOSs for all compounds in this study can be found in Table S2.† The values are smallest for the sulfates and highest for the tetrafluoroborates. The IPDOS for Th:LiSrAlF6, for which radiative decay of the nuclear excited state has been observed, is in the same range as the polyatomic anion materials. A second descriptor of hybridization, the width of the 5f peak, is also discussed in the ESI.† The conclusion here is that the orbital hybridization calculations suggest slow IC in thorium-doped sulfates, supporting our conclusion that they are excellent candidates for experimental study.
 where ν0 is the transition frequency and Δν is the linewidth. We have estimated Q = 5 × 107 for disordered SrxBa1−x(BF4)2, which is comparable to the accuracy of quartz oscillators. The details of our estimation can be found in the ESI.† This result suggests that ordered crystalline materials will be required for accurate crystal clocks.
where ν0 is the transition frequency and Δν is the linewidth. We have estimated Q = 5 × 107 for disordered SrxBa1−x(BF4)2, which is comparable to the accuracy of quartz oscillators. The details of our estimation can be found in the ESI.† This result suggests that ordered crystalline materials will be required for accurate crystal clocks.
4 Conclusions
In conclusion, we predict alkaline earth tetrafluoroborates M(BF4)2 and sulfates MSO4 (M = Ca, Sr, Ba) to be excellent candidate materials for solid-state 229Th nuclear clocks. Inspired by non-coordinating anions in solution-phase chemistry and the concept of superhalogens, we identified the BF4−, ClO4−, and SO42− ions as having the right electronic properties for crystal clocks: they have large ionization potentials, giving a large band gaps in salts, and strong internal covalent bonds, minimizing Th–X covalency in crystals. Both of these effects combine to give a good opportunity for observing radiative decay of the isomeric state of 229Th after laser excitation.The largest host band gaps are possessed by M(BF4)2, so they should all be transparent to the VUV laser. All have similar Th 5f energies, but Ba(BF4)2 has the lowest markers of Th–F covalency, implying a desirably low IC rate. Sr(BF4)2 and Ba(BF4)2 have the lowest defect formation energies so we might expect a higher concentration of thorium than in Ca(BF4)2. Overall, Ba(BF4)2 is therefore the best candidate for laser excitation and radiative decay of the nucleus. The tetrafluoroborate series may offer exploration of the effects of small electronic changes on nuclear decay processes.
The sulfates have a major advantage for clock accuracy in being nuclear-spin-free. The absence of nuclear spins could make a sulfate clock more accurate than a fluoride clock by six orders of magnitude. We cannot be certain of which sulfates are transparent to the exciting radiation, based on computations of the band gaps, so experimental measurements of the optical band gaps of high-purity single crystals are needed. The perchlorates are inferior to the sulfates in several ways: they contain nuclear spins on chlorine; they have smaller band gaps and lower Th 5f energies; and they have undesirable chemical properties.
We have also found that ordered materials are likely to be a necessity for any of the cutting-edge applications proposed for nuclear clocks because the variation in electric field gradients created by cation disorder reduces the accuracy of the clock by many orders of magnitude. These results point to a broad class of materials, featuring polyatomic anions, that can provide us with new opportunities to study 229Th photophysics and that may outperform metal fluorides in crystal clocks.
Author contributions
H. W. T. Morgan: conceptualization; data curation; formal analysis; investigation; methodology; project administration; resources; visualization; writing – original draft; writing – review & editing. H. B. Tran Tan: formal analysis; investigation; methodology; writing – review & editing. A. Derevianko: data curation; formal analysis; funding acquisition; investigation; methodology; project administration; resources; supervision; writing – review & editing. R. Elwell: formal analysis; investigation; methodology; writing – review & editing. J. E. S. Terhune: formal analysis; investigation; methodology; writing – review & editing. E. R. Hudson: data curation; formal analysis; funding acquisition; investigation; methodology; project administration; resources; supervision; writing – review & editing. A. N. Alexandrova: conceptualization; data curation; formal analysis; funding acquisition; investigation; methodology; project administration; supervision; resources; writing – review & editing.Conflicts of interest
E.R.H. has US Patent No. 63/757422 pending.Data availability
The data supporting this article have been included as part of the ESI.†Acknowledgements
This work was supported by NSF awards PHYS-2013011, PHY-2207546, and PHYS-2412982, and ARO award W911NF-11-1-0369. ERH acknowledges institutional support by the NSF QLCI Award OMA-2016245. This work used Bridges-2 at Pittsburgh Supercomputing Center through allocation PHY230110 from the ACCESS program, which is supported by NSF grants #2138259, #2138286, #2138307, #2137603, and #2138296.References
- J. Müller, D. Dirkx, S. Kopeikin, G. Lion, I. Panet, G. Petit and P. Visser, Space Sci. Rev., 2018, 214, 5 CrossRef.
- T. Mehlstäubler, G. Grosche, C. Lisdat, P. Schmidt and H. Denker, Rep. Prog. Phys., 2018, 81, 064401 CrossRef PubMed.
- C. Campbell, A. Radnaev, A. Kuzmich, V. Dzuba, V. Flambaum and A. Derevianko, Phys. Rev. Lett., 2012, 108, 120802 CrossRef CAS PubMed.
- W. Rellergert, D. DeMille, R. Greco, M. Hehlen, J. Torgerson and E. Hudson, Phys. Rev. Lett., 2010, 104, 200802 CrossRef PubMed.
- R. Elwell, C. Schneider, J. Jeet, J. Terhune, H. Morgan, A. Alexandrova, H. Tan, A. Derevianko and E. Hudson, Phys. Rev. Lett., 2024, 133, 013201 CrossRef CAS PubMed.
- J. Tiedau, M. Okhapkin, K. Zhang, J. Thielking, G. Zitzer, E. Peik, F. Schaden, T. Pronebner, I. Morawetz, L. De Col, F. Schneider, A. Leitner, M. Pressler, G. Kazakov, K. Beeks, T. Sikorsky and T. Schumm, Phys. Rev. Lett., 2024, 132, 182501 CrossRef CAS PubMed.
- C. Zhang, T. Ooi, J. S. Higgins, J. F. Doyle, L. von der Wense, K. Beeks, A. Leitner, G. A. Kazakov, P. Li, P. G. Thirolf, T. Schumm and J. Ye, Nature, 2024, 633, 63–70 CrossRef CAS PubMed.
- C. K. Zhang, L. von der Wense, J. F. Doyle, J. S. Higgins, T. Ooi, H. U. Friebel, J. Ye, R. Elwell, J. E. S. Terhune, H. W. T. Morgan, A. N. Alexandrova, H. B. T. Tan, A. Derevianko and E. R. Hudson, Nature, 2024, 636, 603–608 CrossRef CAS PubMed.
- M. Pimon, J. Gugler, P. Mohn, G. A. Kazakov, N. Mauser and T. Schumm, J. Phys.:Condens. Matter, 2020, 32, 255503 CrossRef CAS PubMed.
- M. Pimon, P. Mohn and T. Schumm, Adv. Theory Simul., 2022, 5, 2200185 CrossRef CAS.
- B. S. Nickerson, M. Pimon, P. V. Bilous, J. Gugler, K. Beeks, T. Sikorsky, P. Mohn, T. Schumm and A. Pálffy, Phys. Rev. Lett., 2020, 125, 032501 CrossRef CAS PubMed.
- B. S. Nickerson, M. Pimon, P. V. Bilous, J. Gugler, G. A. Kazakov, T. Sikorsky, K. Beeks, A. Grüneis, T. Schumm and A. Pálffy, Phys. Rev. A, 2021, 103, 053120 CrossRef CAS.
- Q. Gong, S. Tao, S. Li, G. Deng, C. Zhao and Y. Hang, Phys. Rev. A, 2024, 109, 033109 CrossRef CAS.
- S. V. Pineda, P. Chhetri, S. Bara, Y. Elskens, S. Casci, A. N. Alexandrova, M. Au, M. Athanasakis-Kaklamanakis, M. Bartokos, K. Beeks, C. Bernerd, A. Claessens, K. Chrysalidis, T. E. Cocolios, J. G. Correia, H. De Witte, R. Elwell, R. Ferrer, R. Heinke, E. R. Hudson, F. Ivandikov, Y. Kudryavtsev, U. Köster, S. Kraemer, M. Laatiaoui, R. Lica, C. Merckling, I. Morawetz, H. W. T. Morgan, D. Moritz, L. M. C. Pereira, S. Raeder, S. Rothe, F. Schaden, K. Scharl, T. Schumm, S. Stegemann, J. Terhune, P. G. Thirolf, S. M. Tunhuma, P. Van Den Bergh, P. Van Duppen, A. Vantomme, U. Wahl and Z. Yue, Phys. Rev. Res., 2025, 7, 013052 CrossRef CAS.
- J. Ellis, X. Wen and R. Martin, Inorg. Chem., 2014, 53, 6769–6774 CrossRef CAS.
- A. L. Allred, J. Inorg. Nucl. Chem., 1961, 17, 215–221 CrossRef CAS.
- A. Srivastava, Chem. Commun., 2023, 59, 5943–5960 RSC.
- G. Gutsev and A. Boldyrev, Chem. Phys., 1981, 56, 277–283 CrossRef CAS.
- A. Srivastava, A. Kumar and N. Misra, J. Phys. Chem. A, 2021, 125, 2146–2153 CrossRef CAS PubMed.
- S. Nielsen and A. Ejsing, Chem. Phys. Lett., 2007, 441, 213–215 CrossRef CAS.
- J. Forero-Saboya, M. Lozinsek and A. Ponrouch, J. Power Sources Adv., 2020, 6, 100032 CrossRef.
- T. Bunic, G. Tavcar, E. Goreshnik and B. Zemva, Acta Crystallogr., Sect. C:Cryst. Struct. Commun., 2007, 63, I75–I76 CrossRef CAS.
- T. Jordan, B. Dickens, L. Schroeder and W. Brown, Acta Crystallogr., Sect. B, 1975, 31, 669–672 CrossRef.
- E. Mikuli, E. Szostak and B. Grad, Phase Transitions, 2007, 80, 539–546 CrossRef CAS.
- E. Goreshnik, A. Vakulka and G. Tavcar, IUCrData, 2023, 8, x230488 CrossRef CAS.
- D. O. Charkin, S. N. Volkov, L. S. Manelis, A. N. Gosteva, S. M. Aksenov and V. A. Dolgikh, J. Struct. Chem., 2023, 64, 253–261 CrossRef CAS.
- J. Hyoung, H. Lee, S. Kim, H. Shin and S. Hong, Acta Crystallogr., Sect. E:Crystallogr. Commun., 2019, 75, 447–450 CrossRef CAS PubMed.
- J. Lee, J. Kang, S. Lim and S. Hong, Acta Crystallogr., Sect. E:Crystallogr. Commun., 2015, 71, 588–591 CrossRef CAS PubMed.
- P. Murthy and C. Patel, J. Inorg. Nucl. Chem., 1963, 25, 310–312 CrossRef CAS.
- A. Christensen, M. Olesen, Y. Cerenius and T. Jensen, Chem. Mater., 2008, 20, 2124–2132 CrossRef CAS.
- A. Lauer, R. Hellmann, G. Montes-Hernandez, N. Findling, W. Ling, T. Epicier, A. Fernandez-Martinez and A. Van Driessche, J. Chem. Phys., 2023, 158, 054501 CrossRef CAS PubMed.
- M. Krystek, Phys. Status Solidi A, 1979, 54, K133–K135 CrossRef CAS.
- G. Kresse and J. Furthmuller, Phys. Rev. B, 1996, 54, 11169–11186 CrossRef CAS PubMed.
- P. E. Blochl, Phys. Rev. B, 1994, 50, 17953–17979 CrossRef PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- F. Tran and P. Blaha, Phys. Rev. Lett., 2009, 102, 226401 CrossRef PubMed.
- A. D. Becke and E. R. Johnson, J. Chem. Phys., 2006, 124, 221101 CrossRef PubMed.
- J. Heyd, G. E. Scuseria and M. Ernzerhof, J. Chem. Phys., 2003, 118, 8207–8215 CrossRef CAS.
- A. Krukau, O. Vydrov, A. Izmaylov and G. Scuseria, J. Chem. Phys., 2006, 125, 224106 CrossRef PubMed.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2008, 41, 653–658 CrossRef CAS.
- H. W. T. Morgan, J. E. S. Terhune, R. Elwell, H. B. T. Tan, U. C. Perera, A. Derevianko, E. R. Hudson and A. N. Alexandrova, A spinless crystal for a high-performance solid-state 229Th nuclear clock, arXiv, 2025, 2503.11374, DOI:10.48550/arXiv.2503.11374.
- U. Betke and M. S. Wickleder, Eur. J. Inorg. Chem., 2011, 2012, 306–317 CrossRef.
- A. Abrao, A. de Freitas and F. de Carvalho, J. Alloys Compd., 2001, 323, 53–56 CrossRef.
- A. Albrecht, G. Sigmon, L. Moore-Shay, R. Wei, C. Dawes, J. Szymanowski and P. Burns, J. Solid State Chem., 2011, 184, 1591–1597 CrossRef CAS.
- V. Wang, N. Xu, J. Liu, G. Tang and W. Geng, Comput. Phys. Commun., 2021, 267, 108033 CrossRef CAS.
- D. J. Singh, Phys. Rev. B, 2010, 82, 205102 CrossRef.
- W. Chen and A. Pasquarello, Phys. Rev. B, 2012, 86, 035134 CrossRef.
- S. H. Jagdale and S. H. Pawar, Bull. Mater. Sci., 1983, 5, 389–397 CrossRef CAS.
- A. Kadari, K. Mahi, R. Mostefa, M. Badaoui, A. Mameche and D. Kadri, J. Alloys Compd., 2016, 688, 32–36 CrossRef CAS.
- H. Nagabhushana, G. Nagaraju, B. Nagabhushana, C. Shivakumara and R. Chakradhar, Philos. Mag. Lett., 2010, 90, 289–298 CrossRef CAS.
- D. Nuding, E. Rivera-Valentin, R. Davis, R. Gough, V. Chevrier and M. Tolbert, Icarus, 2014, 243, 420–428 CrossRef CAS.
- N. Ingle, S. Omanwar, P. Muthal, S. Dhopte, V. Kondawar, T. Gundurao and S. Moharil, Radiat. Meas., 2008, 43, 1191–1197 CrossRef CAS.
- H. W. T. Morgan, H. B. T. Tan, R. Elwell, A. N. Alexandrova, E. R. Hudson and A. Derevianko, Theory of internal conversion of the thorium-229 nuclear isomer in solid-state hosts, arXiv, 2024, 2411.15641, DOI:10.48550/arXiv.2411.15641.
- X. Gui, B. Lv and W. Xie, Chem. Rev., 2021, 121, 2966–2991 CrossRef CAS PubMed.
- E. George, D. Raabe and R. Ritchie, Nat. Rev. Mater., 2019, 4, 515–534 CrossRef CAS.
- M. Gawande, R. Pandey and R. Jayaram, Catal.:Sci. Technol., 2012, 2, 1113–1125 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Details of methods and additional results and figures as described in the text, and DFT-optimized geometries in VASP POSCAR format. See DOI: https://doi.org/10.1039/d5dt00736d |
| This journal is © The Royal Society of Chemistry 2025 |