
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNew members of the family of highly luminescent 1,3-bis(4-phenylpyridin-2-yl)-4,6-difluorobenzene platinum(II) complexes: exploring the effect of substituents on the 4-phenylpyridine unit†
Giulia
De Soricellis
ab,
Bertrand
Carboni
c,
Véronique
Guerchais
c,
J. A. Gareth
Williams
 d,
Daniele
Marinotto
e,
Alessia
Colombo
a,
Claudia
Dragonetti
a,
Francesco
Fagnani
*a,
Simona
Fantacci
f and
Dominique
Roberto
a
d,
Daniele
Marinotto
e,
Alessia
Colombo
a,
Claudia
Dragonetti
a,
Francesco
Fagnani
*a,
Simona
Fantacci
f and
Dominique
Roberto
a
aDipartimento di Chimica, Università degli Studi di Milano and UdR-INSTM di Milano, Via C. Golgi 19, I-20133 Milan, Italy. E-mail: francesco.fagnani@unimi.it
bDipartimento di Chimica, Università di Pavia, Via Taramelli 12, I-27100 Pavia, Italy
cUniversité de Rennes 1, CNRS, ISCR (Institut des Sciences Chimiques de Rennes) - UMR 6226, F-35000 Rennes, France
dDepartment of Chemistry, University of Durham, South Road, Durham, DH1 3LE, UK
eIstituto di Scienze e Tecnologie Chimiche (SCITEC) “Giulio Natta”, Consiglio Nazionale delle Ricerche (CNR), via C. Golgi 19, I-20133 Milan, Italy
fIstituto di Scienze e Tecnologie Chimiche “Giulio Natta” SCITEC, Consiglio Nazionale delle Ricerche (CNR), Computational Laboratory for Hybrid/Organic Photovoltaics (CLHYO), via Elce di Sotto 8, 06213, Perugia, Italy
First published on 5th June 2025
Abstract
The synthesis and characterization of Pt{1,3-bis(4-phenylpyridin-2-yl)-4,6-difluorobenzene}Cl and Pt{1,3-bis(4-(4-methoxy-2,6-dimethylphenyl)-pyridin-2-yl)-4,6-difluorobenzene}Cl are reported, along with their molecular structure optimized by Density Functional Theory. These new NCN-coordinated Pt(II) complexes are both very highly luminescent in deoxygenated solution in the blue region of the spectrum: λmax = 471–480 nm and Φlum = 0.89–0.98 at room temperature. Compared to the Pt{1,3-bis(pyridin-2-yl)-4,6-difluorobenzene}Cl analogue, the complex with a simple, unsubstituted phenyl ring at position 4 of the pyridinyl rings shows an improved luminescence quantum yield. However, a further enhancement is achieved with the 2,6-dimethyl-4-methoxyphenyl substituent, the steric hindrance of which inhibits the formation of bimolecular species, allowing high quantum yields to be maintained even in concentrated solutions (2 × 10−4 M).
Introduction
Luminescent platinum(II) complexes are extensively researched owing to their potential for application in nonlinear optics,1–8 bioimaging,9–25 sensing,26–28 photocatalysis,29–31 and electro-luminescent devices.32–46 The strong spin–orbit coupling (SOC) associated with the platinum atom promotes intersystem crossing to triplet excited states and their subsequent radiative decay by relaxing the spin selection rule. The phosphorescence is typically favoured by the introduction of platinum–carbon bonds within cyclometallated units, as the resulting strong ligand fields ensure that otherwise deactivating d–d excited states are destabilised, and non-radiative decay through them is suppressed.47,48 Unlike complexes of d6 metal ions such as Ir(III), the square planar geometry favours the formation of bimolecular states – either in the ground state (dimers or higher aggregates) or in the excited state (excimers) – thanks to Pt⋯Pt or ligand⋯ligand intermolecular interactions. These species may emit at lower energies than the isolated molecules, offering an intriguing route to deep-red/NIR-emitting materials, colour modulation according to the local concentration, and even white light emission.42,43,49–51Pt(II) complexes of the NCN-coordinating ligand 1,3-bis(pyridin-2-yl)benzene (dpyb) are among the brightest and most efficient Pt-based emitters, apparently owing to the combination of a particularly short Pt–C bond and high rigidity, minimising non-radiative decay.52–55 Recently, some of us studied the effect of the introduction of a polarizable π-delocalized bulky substituent on the phosphorescence properties of Pt(F2dpyb)Cl {F2dpybH = 1,3-bis(pyridin-2-yl)-4,6-difluorobenzene},56 demonstrating particularly high quantum yields for PtL1Cl and PtL2Cl {Scheme 1; HL1 = 1,3-bis(4-triphenylamine-pyridin-2-yl)-4,6-difluoro-benzene and HL2 = 1,3-bis(4-mesityl-pyridin-2-yl)-4,6-difluoro-benzene}.57,58 In this context, we were curious to see the effect of the introduction of other π-delocalized groups on the 4-position of the pyridine rings. Here we describe the synthesis and properties of the complex with a simple unsubstituted phenyl group on each pyridine ring (PtL3Cl, Scheme 1) and the related complex with a 2,6-dimethyl-4-methoxyphenyl group (PtL4Cl). Both are highly emissive, enlarging this fascinating, brightly luminescent family of complexes based on NCN ligands that feature 4-phenylpyridin-2-yl units.
 | ||
| Scheme 1 Chemical structures of the investigated complexes. | ||
Results and discussion
Preparation of the new pro-ligands HLn and of the related Pt(II) complexes
The new proligand 1,3-bis(4-phenylpyridin-2-yl)-4,6-difluoro-benzene (HL3) was prepared by Pd-catalysed cross-coupling of the pinacol ester of 1,3-difluoro-4,6-diboronic acid58 with 2-chloro-4-phenylpyridine. 1,3-Bis(4-(4-methoxy-2,6-dimethyl-phenyl)-pyridin-2-yl)-4,6-difluorobenzene (HL4) was prepared similarly using 2-chloro-4-(2,6-dimethyl-4-methoxyphenyl)-pyridine (Scheme 2). The two chlorinated reagents were likewise prepared by cross-couplings, as shown in Scheme 2. Reaction of the proligands with K2PtCl4 led to PtL3Cl and PtL4Cl in 93% and 81% yield, respectively. Full details of synthesis and characterisation are provided in the Experimental Section and in the ESI.† | ||
| Scheme 2 Synthesis of PtL3Cl and PtL4Cl. | ||
Photophysical properties
The absorption spectra of PtL3Cl and PtL4Cl in dichloromethane at selected concentrations between 1 × 10−6 and 1 × 10−4 M are shown in Fig. 1. There are intense bands in the range 260–320 nm, due to intraligand 1π–π* transitions of the NCN ligand, and less intense bands at 340–420 nm, due to charge-transfer transitions involving the cyclometalated ligand and the metal, as for related NCN-coordinated platinum(II) complexes.55–59 A weaker band at 471 and 467 nm (ε = 262 and 214 M−1 cm−1) for PtL3Cl and PtL4Cl, respectively (Fig. 1, insets), is attributed to the weak transition from the singlet ground state to the lowest triplet state. This formally spin forbidden S0 → T1 transition, facilitated by the high SOC associated with the Pt centre, is close to that of the parent Pt(F2dpyb)Cl (467 nm, 140 M−1 cm−1). The higher extinction coefficient for this transition in PtL3Cl and PtL4Cl may be an indication of more efficient SOC pathways. | ||
| Fig. 1 Absorption spectra of PtL3Cl (a) and PtL4Cl (b) at the concentrations indicated, in CH2Cl2 at 298 K. The weak bands at longer wavelengths are shown on an expanded scale in the insets, for clarity. | ||
The normalized emission spectra of PtL3Cl and PtL4Cl at various concentrations in degassed CH2Cl2 solution at room temperature are shown in Fig. 2 (see also Fig. S4–S8 and S18–S22 in the ESI†). Upon excitation at 380 nm in dilute solution (1 × 10−6 M), PtL3Cl displays a structured spectrum, λ0,0 = 480 nm, with a vibrational progression of around 1400 cm−1, attributed to emission from the T1 state.56 The corresponding band of PtL4Cl is a little blue-shifted to 471 nm, very similar to that reported for the mesityl analogue PtL2Cl58 and for Pt(F2dpyb)Cl.56 In contrast, the previously investigated triphenylamine-substituted complex PtL1Cl displayed much lower-energy emission, with λmax = 562 nm.57
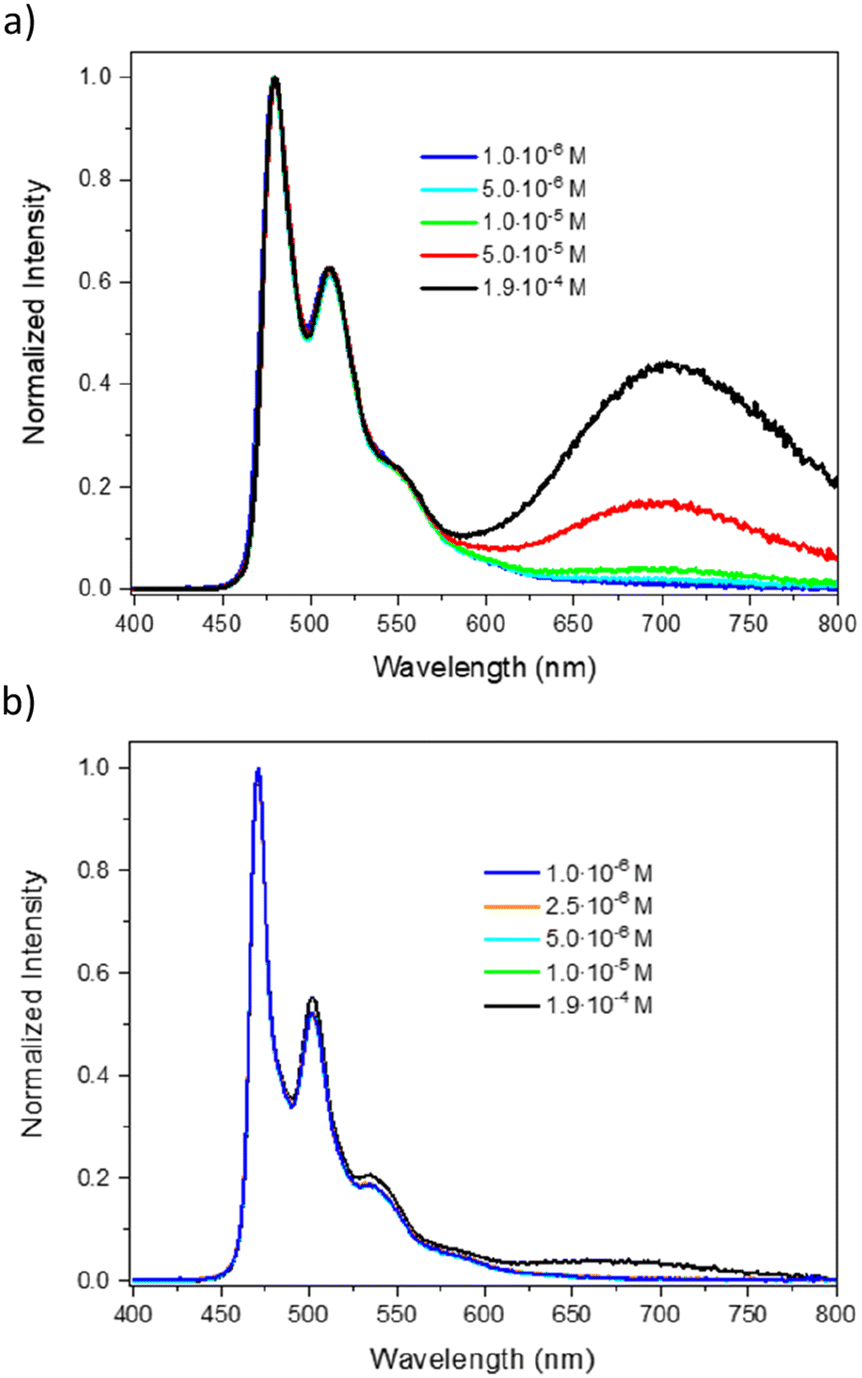 | ||
| Fig. 2 Normalized emission spectra of PtL3Cl (a; λex = 380 nm) and PtL4Cl (b; λex = 377 nm) in degassed dichloromethane solution at the concentrations indicated, at 298 K. | ||
For PtL3Cl, as the concentration is increased, a broad, structureless band progressively grows in, with λmax around 704 nm (Fig. 2a), attributed to the formation of emissive, bimolecular excited states (excimers and/or aggregates).59 For PtL4Cl, the corresponding band – centered at 680 nm in this case – is much less intense at a given concentration (compare Fig. 2a and b), probably due to the steric hindrance of the 4-methoxy-2,6-dimethyl phenyl substituent on the pyridyl rings. The presence of the two methyl groups ortho to the interannular C–C bond will inhibit the attainment of the planar conformation that favours face-to-face intermolecular interactions. The corresponding band was similarly weak in the mesityl-substituted PtL2Cl.58
Under the same conditions, PtL1Cl, PtL2Cl, and Pt(F2dpyb)Cl are bright Pt(II) emitters with reported luminescence quantum yields (Φlum) of 0.90,57 0.97,58 and 0.80,56 respectively. PtL3Cl and PtL4Cl are similarly here to be characterized by excellent quantum yields of 0.89 and 0.98, respectively, as determined with an integrating sphere (Table 1). Under air-equilibrated conditions, the Φlum values decrease to 0.28 and 0.16, respectively, due to oxygen quenching; given the efficacy of this quenching, efficient production of singlet oxygen can be anticipated, an interesting aspect for photodynamic therapy.19
| Parametera | PtL1Cl | PtL2Cl | PtL3Cl | PtL4Cl |
|---|---|---|---|---|
| a At a concentration of 5 × 10−6 M at 298 K in degassed CH2Cl2, unless otherwise indicated. b λ ex = 422, 334, 380, and 377 nm for PtL1Cl, PtL2Cl, PtL3Cl, and PtL4Cl, respectively. c λ max of the excimer at 1.9 × 10−4 M. d φ lum measured with an integrating sphere. e Radiative and non-radiative rate constants are calculated from the quantum yields and emission decay times according to kr = Φlum/τ and knr = (1 − Φlum)/τ. | ||||
| λ max,abs S0 → T1/nm [ε/M−1 cm−1] | Not detected | 467 [120] | 471 [277] | 467 [214] |
| λ max,em/nmb monomer [excimer]c | 562 [696] | 471 [680] | 480 [704] | 471 [680] |
| Φ lum [aerated] | 0.90 [0.059] | 0.97 [0.18] | 0.89 [0.28] | 0.98 [0.16] |
| Φ lum (1.9 × 10−4 M)d [aerated] | 0.66 [0.026] | 0.62 [0.12] | 0.53 [0.22] | 0.85 [0.13] |
| τ/μs | 104 | 4.8 | 3.5 | 4.5 |
| τ/μs (1.9 × 10−4 M) | 4.3 | 2.4 | 0.93 | 4.0 |
k
r![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) /s−1
/s−1 |
8.7 × 103 | 2.0 × 105 | 2.5 × 105 | 2.2 × 105 |
|
k
r
e
/s−1 (1.9 × 10−4 M) |
1.5 × 105 | 2.6 × 105 | 5.7 × 105 | 2.1 × 105 |
|
k
nr
/s−1 |
96 × 102 | 6.3 × 103 | 3.1 × 104 | 4.4 × 103 |
|
k
nr
e
/ s−1 (1.9 × 10−4 M) |
7.7 × 104 | 1.6 × 105 | 5.1 × 105 | 3.7 × 104 |
The formation of excimers and aggregates of Pt(NCN)Cl complexes typically comes at the expense of the quantum yields.55 For PtL3Cl and PtL4Cl, increasing the concentration does compromise the quantum yield, but only to a very modest extent: for instance, the values at 1.9 × 10−4 M remain remarkably high (0.53 and 0.85, respectively). Inspection of the values in Table 1, in the context also of PtL1,2Cl, indicates that Φlum for PtL4Cl is substantially less affected by concentration than the other three complexes. Apparently, the steric hindrance of the 4-methoxy-2,6-dimethyl phenyl substituents to the formation of bi-molecular emissive species – as noted above based on the low intensity of the low-energy excimer band – also ensures that excellent quantum yields are maintained, even at elevated concentrations.
In the literature, methods to determine the luminescence quantum yields can be divided into two main approaches: the use of an integrating sphere, or the use of standards relative to which the intensity of emission is compared.60 We were curious to compare the Φlum values measured by means of an integrating sphere (Table 1) with those obtained relative to different standards. By using Pt(dpyb)Cl as the standard (Φlum = 0.60 in degassed CH2Cl2, λemmax = 491 nm),52 we determined Φlum values of 0.83 and 0.93 for PtL3Cl and PtL4Cl, respectively, in good agreement with the values measured with the integrating sphere. By using [Ru(bpy)3]Cl2 as standard (Φlum = 0.04 in aerated H2O; λemmax = 628 nm),60 values of 0.84 and 0.71, respectively, were determined. We consider Pt(dpyb)Cl to be the more reliable standard, owing to the better match of the emissive regions and to a more comparable quantum yield.
Excited state decay measurements of PtL3Cl in degassed CH2Cl2 solutions at different concentrations were performed, exciting at 374 nm and monitoring the emission at 480 nm (ESI, Table S2 and Fig. S9–S13†). The longest lifetime, 3.8 μs, is observed at the lowest concentration (1 × 10−6 M). An increase of the concentration leads to a decrease of the lifetime (3.5 and 3.3 μs at 5 × 10−6 and 1 × 10−5 M, respectively), a much lower value (0.93 μs) being obtained at 1.9 × 10−4 M. A linear Stern–Volmer relationship is observed, with a self-quenching constant kQ of 4.3 × 109 M−1 s−1 determined from the gradient, consistent with excimer formation (i.e., diffusion-controlled excited-state quenching as opposed to ground-state aggregation). Moreover, the temporal evolution of the emission intensity of the most concentrated solution at 704 nm clearly shows an initial rise at short time intervals, associated with the formation of excimers, with a τ of 0.7 μs (Fig. S14 in the ESI†). Corresponding measurements were made for PtL4Cl, monitoring the emission intensity at 471 nm (ESI, Table S4 and Fig. S23–S27†). Again, the longest lifetime, 4.6 μs, is observed at the lowest concentration (1 × 10−6 M). In this case, the lifetime is only marginally reduced over the entire concentration range investigated (τ = 4.6, 4.5, 4.2 and 4.0 μs at 2.5 × 10−6, 5 × 10−6, 1 × 10−5, and 1.9 × 10−4 M, respectively), consistent with high quantum yields being maintained at high concentrations, and with the excimer band having almost negligible intensity. In this instance, the change in lifetime is simply too small to determine the self-quenching constant reliably. However, for the most concentrated solution, the emission intensity again shows an initial rise at short time intervals after excitation (Fig. S28†), from which a time constant for excimer formation of 1.5 μs was estimated. The process is thus evidently slower than for PtL3Cl. The effect of the 4-methoxy-2,6-dimethyl groups in hindering self-quenching and excimer formation is apparently rather larger than that associated with the mesityl groups in PtL2Cl, wherein the lifetime declined to a greater extent with concentration (Table 1).58 As expected, the presence of substituents on the phenyl rings of the pyridine rings reduces the propensity to form bimolecular species (see Fig. S29 in the ESI† for the plot of intensity of the lowest energy band vs. concentration).
The very long lifetime observed in the case of PtL1Cl, with a triphenylamine group on the pyridines, remains unique amongst the four complexes.57
Radiative kr and non-radiative knr decay constants can be estimated from the Φlum and τ values, assuming that the emitting state is formed with unit efficiency. It is clear from the values so calculated (Table 1) that the superior quantum yield of PtL4Cl compared to PtL3Cl originates from suppressed non-radiative processes in the former, both in dilute and concentrated solution. Meanwhile, the kr values of PtL2–4Cl are ≥2 × 105 s−1, around 3× that of the parent unsubstituted complex Pt(dpyb)Cl, and approaching that of the “gold standard” fac-Ir(ppy)3, for which kr is around 4 × 105 s−1.38 It is likely that the higher energy of the emissive triplet state in PtL2–4Cl leads to a smaller S1–T1 energy gap ΔES–T, and hence to more efficient promotion of the formally forbidden T1 → S0 transition (the efficiency of which is inversely related to ΔES–T47). The ΔES–T values estimated from the maxima of the lowest-energy absorption bands and the 0,0 component of the emission are approximately 5900 cm−1 for PtL1Cl, compared to lower values of 5100 for PtL2Cl, and 5300 cm−1 for PtL3Cl and PtL4Cl. Of course, relaxation of the spin selection rule also requires efficient mixing of metal orbitals into the excited state, to benefit from the high spin–orbit coupling associated with the metal. TD-DFT calculations on related complexes reveal heavily-mixed dPt|πL → πL* states.61,62 On the other hand, the electron-rich anilino pendants in PtL1Cl likely render the lowest-energy excited states of intraligand character (i.e., πAr → πpy*, where Ar = Ph2N–C6H4–), suppressing the degree of metal character and leading to an abnormally low kr and long observed lifetime τ.61
Computational modelling
To model the molecular structure of PtL3Cl and PtL4Cl, we turned to geometry optimization by Density Functional Theory (DFT). DFT calculations were performed both on the isolated complexes and on the related dimers. The dimers can be considered the simplest representative species of the aggregation phenomenon that may provide information on the packing of PtL3Cl and PtL4Cl in the crystal and the aggregation in solution. The monomer and the dimer geometry optimizations were performed with the Gaussian09 program package (G09)63 by using the B3LYP exchange–correlation functional64 integrated with the D3-BJ model65 to include the dispersion effects in the geometry optimizations. The dichloromethane solvation effects were included through the conductor-like polarizable continuum model as implemented in G09.66–69 All the atoms, except Pt, were described by 6-31G** basis set,70–72 while Pt was described with the LANL2DZ basis set along with the corresponding pseudopotentials.73 Such computational methodology has been demonstrated to be accurate for reproducing the geometry of the PtL2Cl complex and its aggregation phenomena in solution; moreover the optimized dimer structure calculated in solution mimics the main orientation of the PtL2Cl molecules in the crystal.58 In Fig. 3 and 4 the optimized molecular geometries of both PtL3Cl and PtL4Cl monomer and dimer, respectively, are reported, showing both the front and side views for the dimers. As already observed for (PtL2Cl)2,58 in (PtL3Cl)2 and (PtL4Cl)2 the two monomers are arranged in a head-to-tail configuration with respect to the Pt centers being staggered one from the other. In (PtL3Cl)2 and (PtL4Cl)2 the Pt⋯Pt distance is computed to be 4.88 Å and 6.54 Å, respectively, compared to a value of 6.31 Å for (PtL2Cl)2. In (PtL3Cl)2 the presence of unsubstituted phenyls allows for a closer approach of the two monomeric units; on the contrary, in (PtL4Cl)2, the methoxy substituent in place of methyl in para position slightly increases the Pt⋯Pt distance. | ||
| Fig. 3 Optimized molecular geometries of the monomer and dimer of PtL3Cl in dichloromethane. Both the front and side view have been shown for the dimer. | ||
 | ||
| Fig. 4 Optimized molecular geometries of the monomer and dimer of PtL4Cl in dichloromethane. Both the front and side view have been shown for the dimer. | ||
The dimerization energy for the formation of (PtL3Cl)2 in dichloromethane is computed to be slightly smaller than for (PtL2Cl)2 −43.34 kcal mol−1vs. −47.4 kcal mol−1, while that computed for (PtL4Cl)2 in solution is slightly larger, at −50.5 kcal mol−1. The dimerization energy provides an estimate of π⋯π and Pt⋯Pt interactions that allow the dimer to form.
Conclusion
In conclusion, two novel members of the family of 1,3-bis(4-phenylpyridin-2-yl)-4,6-difluoro-benzene Pt(II) complexes were easily prepared and well characterized. Both are highly luminescent (Φlum = 0.89–0.98) in the blue region (471–480 nm) with lifetimes of a magnitude typical for Pt(dpyb)Cl derivatives (ca. 4 μs). It appears that the introduction of a simple phenyl group on the position 4 of the pyridine rings is a useful way to improve the luminescence quantum yield of the parent complex Pt(F2dpyb)Cl. A further enhancement can be achieved with a suitable functionalization of the 4-phenylpyridine moiety. Thus, this work shows that the steric hindrance of the 4-methoxy-2,6-dimethyl phenyl moiety on the pyridinyl rings is particularly appropriate, and even better than that of a triphenylamine or mesityl group in hampering the formation of bimolecular species. It allows excellent quantum yields to be maintained even in concentrated solutions, an aspect of particular interest in future applications.Experimental section
General comments
All reagents and solvents were used as received from the supplier. The purifications were performed through column chromatography on silica gel (Merck Geduran 60, 0.063–0.200 mm).NMR characterizations were obtained recording on a Bruker AV III 300 MHz or AV III 400 MHz spectrometers. Chemical shifts of 1H, 19F and 13C NMR spectra are reported in parts per million (ppm) and the coupling constants are measured in Hertz (Hz). The multiplicities of signals are listed as singlet (s), d (doublet), t (triplet), quartet (q), multiplet (m).
UV-Visible spectra were collected by a Shimadzu UV3600 spectrophotometer. Luminescence measurements were carried out in CH2Cl2 solution after the Freeze–Pump–Thaw (FPT) procedure necessary to remove dissolved oxygen. Photoluminescence quantum yields, Φlum, were measured using a C11347 Quantaurus Hamamatsu Photonics K.K spectrometer (see ESI† for details) or relative to the standards indicated earlier.
Steady state and time-resolved fluorescence data were obtained using a FLS980 spectrofluorimeter (Edinburg Instrument Ltd). Emission spectra were corrected for background intensity and quantum efficiency of the photomultiplier tube. Excitation spectra were corrected for the intensity fluctuation of a 450 W Xenon arc lamp. Time-resolved fluorescence measurements were performed through the time-correlated single photon counting technique with an Edinburgh Picosecond Pulsed Diode Laser (emitted wavelength 374 nm).
Synthesis of PtL3Cl
HL3 (0.10 g, 0.24 mmol) was dissolved in acetonitrile (9 mL) and a solution of K2PtCl4 (0.16 g, 0.38 mmol) in water (1 mL) was added. Before sealing the flask, the reaction mixture was degassed by bubbling argon through the solution for 30 minutes. The mixture was heated at 110 °C for 72 h. The obtained suspension was allowed to cool to room temperature and was filtered through a 0.45 μm Nylon membrane. The collected solid was then washed with water, MeOH and diethyl ether, and dried under vacuum to obtain the desired Pt complex as a yellow powder (121 mg, 93% yield). 1H-NMR (300 MHz, CDCl3, δ): 9.32 (d, J = 6.1 Hz, 3J(195Pt) = 41 Hz, 2H), 8.11 (s, 2H), 7.69–7.78 (m, 4H), 7.58 (dd, J = 8.9, 5.0 Hz, 6H), 7.49 (dd, J = 6.0, 1.9 Hz, 2H), 6.77 (t, J = 11.3 Hz, 1H). 19F {1H} NMR (282.36 MHz, CDCl3, δ): −108.30 (s, 3J(195Pt) = 43 Hz, 2F). 13C {1H} NMR (75.48 MHz, CDCl3, δ): 151.8, 136.7, 130.3, 129.4, 127.2, 120.7, 120.4. Elemental anal. calcd for C28H17 F2N2195Pt35Cl: C, 51.74; H, 2.64; N, 4.31; Cl, 5.45; F, 5.85, Pt, 30.01; found: C, 51.83; H, 2.67; N, 4.33.Synthesis of PtL4Cl
HL4 (0.14 g, 0.27 mmol) was dissolved in acetonitrile (11.7 mL) and a solution of K2PtCl4 (0.22 g, 0.53 mmol) in water (1.3 mL) was added. Argon was bubbled through the mixture for 40 minutes before sealing the reaction vessel. The reaction mixture was heated to 110 °C for 72 h. The obtained suspension was allowed to cool to room temperature and then filtered through a 0.45 μm Nylon membrane. The collected solid waswashed with water, MeOH and diethyl ether, and dried under vacuum to obtain the desired Pt complex as a yellow powder (124 mg, 81%). 1H-NMR (300 MHz, CDCl3, δ): 9.40 (d, J = 5.9 Hz, 3J(195Pt) = 40 Hz, 2H), 7.76 (s, 2H), 7.17 (d, J = 5.8, 2H), 6.68–6.80 (m, 5H), 3.87 (s, 6H), 2.13 (s, 12H). 13C{1H}-NMR (75.48 MHz, CDCl3, δ): 164.2, 159.4, 153.8, 151.9, 136.3, 130.7, 124.9, 124.1, 113.3, 55.2, 21.2. 19F{1H}-NMR (282 MHz, CDCl3, δ): −108.1 (s, 2F). HRMS (ESI+): (M + Na)+ calcd for C34H29N2O2F235Cl Na195Pt, 788.1426; found: 788.1436. Elemental anal. calcd for C34H29ClF2N2O2Pt: C, 53.30; H, 3.82; N, 3.66; found: C, 53.55; H, 3.83; N, 3.64.Author contributions
Conceptualization, D.R. and V.G.; methodology, B.C., D.M., A.C., C.D., F.F., S.F.; investigation, G.D., D.M., F.F., J.A.G.W., S.F.; resources, A.C. and C.D.; data curation, G.D., D.M., F.F., J.A.G.W., S.F.; supervision, D.R. and V.G.; writing—original draft, F.F.; writing—review and editing, G.D., B.C., V.G., J.A.G.W., D.M., A.C., C.D., F.F., S.F., D.R. All authors have read and agreed to the published version of the manuscript.Data availability
The data supporting this article, including details on the synthesis and full characterization of the two complexes (1H, 13C, and 19F NMR spectra, HRMS, elemental analysis, photophysical characterization), computational modelling have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
Fondazione Cariplo and Regione Lombardia are acknowledged for the instrumentation bought during the SmartMatLab Centre project (2014). The work was supported by the National Interuniversity Consortium of Materials Science and Technology (Project TRI_25_073 Dragonetti and TRI_25_173 Colombo) and Università di Milano (Project PSR2023_DIP_005_PI_FTESS).References
- A. Colombo, C. Dragonetti, D. Marinotto, S. Righetto, D. Roberto, S. Tavazzi, M. Escadeillas, V. Guerchais, H. Le Bozec, A. Boucekkine and C. Latouche, Organometallics, 2013, 32, 3890–3894 CrossRef CAS.
- D. Espa, L. Pilia, C. Makedonas, L. Marchiò, M. L. Mercuri, A. Serpe, A. Barsella, A. Fort, C. A. Mitsopoulou and P. Deplano, Inorg. Chem., 2014, 53, 1170–1183 CrossRef CAS PubMed.
- D. Espa, L. Pilia, S. Attar, A. Serpe and P. Deplano, Inorg. Chim. Acta, 2018, 470, 295–302 CrossRef CAS.
- H. Zhao, E. Garoni, T. Roisnel, A. Colombo, C. Dragonetti, D. Marinotto, S. Righetto, D. Roberto, D. Jacquemin, J. Boixel and V. Guerchais, Inorg. Chem., 2018, 57, 7051–7063 CrossRef CAS PubMed.
- S. Attar, F. Artizzu, L. Marchik, D. Espa, L. Pilia, M. F. Casula, A. Serpe, M. Pizzotti, A. Orbelli-Biroli and P. Deplano, Chem. – Eur. J., 2018, 24, 10503–10512 CrossRef CAS PubMed.
- A. Colombo, C. Dragonetti, V. Guerchais and D. Roberto, Coord. Chem. Rev., 2021, 446, 214113–213133 CrossRef CAS.
- J. Boixel, A. Colombo, F. Fagnani, P. Matozzo and C. Dragonetti, Eur. J. Inorg. Chem., 2022, 22, e202200034 CrossRef.
- F. Fagnani, A. Colombo, C. Dragonetti, D. Roberto, V. Guerchais, T. Roisnel, D. Marinotto and S. Fantacci, Eur. J. Inorg. Chem., 2024, 27, e202400478 CrossRef.
- C.-K. Koo, K.-L. Wong, C. W.-Y. Man, Y.-W. Lam, L. K.-Y. So, H.-L. Tam, S.-W. Tsao, K.-W. Cheah, K.-C. Lau, Y.-Y. Yang, J.-C. Chen and M. H.-W. Lam, Inorg. Chem., 2009, 48, 872–878 CrossRef CAS PubMed.
- P. Wu, E. L. Wong, D. Ma, G. S. Tong, K. Ng and C. Che, Chem. – Eur. J., 2009, 15, 3652–3656 CrossRef CAS PubMed.
- V. Fernandez-Moreira, F. L. Thorp-Greenwood and M. P. Coogan, Chem. Commun., 2010, 46, 186–202 RSC.
- Q. Zhao, C. Huang and F. Li, Chem. Soc. Rev., 2011, 40, 2508–2524 RSC.
- E. Baggaley, J. A. Weinstein and J. A. G. Williams, Coord. Chem. Rev., 2012, 256, 1762–1785 CrossRef CAS.
- E. Baggaley, S. W. Botchway, J. W. Haycock, H. Morris, I. V. Sazanovich, J. A. G. Williams and J. A. Weinstein, Chem. Sci., 2014, 5, 879–886 RSC.
- M. Mauro, A. Aliprandi, D. Septiadi, N. S. Kehr and L. De Cola, Chem. Soc. Rev., 2014, 43, 4144–4166 RSC.
- K. Mitra, C. E. Lyons and M. C. T. Hartman, Angew. Chem., Int. Ed., 2018, 57, 10263–10267 CrossRef CAS PubMed.
- V. W.-W. Yam and A. S.-Y. Law, Coord. Chem. Rev., 2020, 414, 213298 CrossRef CAS.
- A. S.-Y. Law, L. C.-C. Lee, K. K.-W. Lo and V. W.-W. Yam, J. Am. Chem. Soc., 2021, 143, 5396–5405 CrossRef CAS PubMed.
- G. De Soricellis, F. Fagnani, A. Colombo, C. Dragonetti and D. Roberto, Inorg. Chim. Acta, 2022, 541, 121082 CrossRef CAS.
- J. Berrones Reyes, P. S. Sherin, A. Sarkar, M. K. Kuimova and R. Vilar, Angew. Chem., Int. Ed., 2023, e202310402 CAS.
- J. Wu, B. Xu, Y. Xu, L. Yue, J. Chen, G. Xie and J. Zhao, Inorg. Chem., 2023, 62, 19142–19152 CrossRef CAS PubMed.
- E. Clancy, S. Ramadurai, S. R. Needham, K. Baker, T. A. Eastwood, J. A. Weinstein, D. P. Mulvihill and S. W. Botchway, Sci. Rep., 2023, 13, 422 CrossRef CAS PubMed.
- A. Upadhyay, A. Nepalia, A. Bera, D. K. Saini and A. R. Chakravarty, Chem. – Asian J., 2023, 18, 1–11 Search PubMed.
- F. Fagnani, G. De Soricellis, A. Colombo, C. Dragonetti, D. Roberto, A. di Biase, S. Fantacci and D. Marinotto, Dyes Pigm., 2024, 225, 112064 CrossRef CAS.
- R. M. Dell'Acqua, F. Fagnani, M. Wojciechowska, D. Marinotto, G. Colombo, I. Dalle-Donne, J. Trylska, S. Cauteruccio and A. Colombo, Dalton Trans., 2025, 54, 3314–3322 RSC.
- A. Haque, H. El Moll, K. M. Alenezi, M. S. Khan and W. Y. Wong, Materials, 2021, 14, 4236–4261 CrossRef CAS PubMed.
- A. S.-Y. Law, M. C.-L. Yeung and V. W.-W. Yam, ACS Appl. Mater. Interfaces, 2017, 9, 41143–41150 CrossRef CAS PubMed.
- Y. Y. Ning, G. Q. Jin, M. X. Wang, S. Gao and J. L. Zhang, Curr. Opin. Chem. Biol., 2022, 66, 102097–102107 CrossRef CAS PubMed.
- M. Yoshida, K. Saito, H. Matsukawa, S. Yanagida, M. Ebina, Y. Maegawa, S. Inagaki, A. Kobayashi and M. Kato, J. Photochem. Photobiol., A, 2018, 358, 334–344 CrossRef CAS.
- D. Gómez de Segura, A. Corral-Zorzano, E. Alcolea, M. T. Moreno and E. Lalinde, Inorg. Chem., 2024, 63, 1589–1606 CrossRef PubMed.
- P. Domingo-Legarda, A. Casado-Sánchez, L. Marzo, J. Alemán and S. Cabrera, Inorg. Chem., 2020, 59, 13845–13857 CrossRef CAS PubMed.
- W. Sotoyama, T. Satoh, N. Sawatari and H. Inoue, Appl. Phys. Lett., 2005, 86, 153505–153507 CrossRef.
- M. Cocchi, D. Virgili, V. Fattori, D. L. Rochester and J. A. G. Williams, Adv. Funct. Mater., 2007, 17, 285–289 CrossRef CAS.
- X. Yang, Z. Wang, S. Madakuni, J. Li and G. E. Jabbour, Adv. Mater., 2008, 20, 2405–2409 CrossRef CAS.
- W. Y. Wong and C. L. Ho, Chem. Soc. Rev., 2009, 253, 1709–1758 CAS.
- C. M. Che, C. C. Kwok, S. W. Lai, A. F. Rausch, W. J. Finkenzeller, N. Y. Zhu and H. Yersin, Chem. – Eur. J., 2010, 16, 233–247 CrossRef CAS PubMed.
- J. Kalinowski, V. Fattori, M. Cocchi and J. A. G. Williams, Coord. Chem. Rev., 2011, 255, 2401–2425 CrossRef CAS.
- L. F. Gildea and J. A. G. Williams, Iridium and platinum complexes for OLEDs, in Organic Light-Emitting Diodes: Materials, Devices and Applications, ed. A. Buckley, Woodhead, Cambridge, 2013 Search PubMed.
- G. Cheng, P.-K. Chow, S. C. F. Kui, C.-C. Kwok and C.-M. Che, Adv. Mater., 2013, 25, 6765–6770 CrossRef CAS PubMed.
- C. Cebrian and M. Mauro, Beilstein J. Org. Chem., 2018, 14, 1459–1481 CrossRef CAS PubMed.
- X. Yang, H. Guo, X. Xu, Y. Sun, G. Zhou, W. Ma and Z. Wu, Adv. Sci., 2019, 6, 1801930 CrossRef PubMed.
- W.-C. Chen, C. Sukpattanacharoen, W.-H. Chan, C.-C. Huang, H.-F. Hsu, D. Shen, W.-Y. Hung, N. Kungwan, D. Escudero, C.-S. Lee and Y. Chi, Adv. Funct. Mater., 2020, 30, 2002494 CrossRef CAS.
- Y.-C. Wei, S. F. Wang, Y. Hu, L.-S. Liao, D.-G. Chen, K.-H. Chang, C.-W. Wang, S.-H. Liu, W.-H. Chan, J.-L. Liao, W.-Y. Hung, T.-H. Wang, P.-T. Chen, H.-F. Hsu, Y. Chi and P.-T. Chou, Nat. Photonics, 2020, 14, 570–577 CrossRef CAS.
- S.-F. Wang, B.-K. Su, X.-Q. Wang, Y.-C. Wei, K.-H. Kuo, C.-H. Wang, S.-H. Liu, L.-S. Liao, W.-Y. Hung, L.-W. Fu, W.-T. Chuang, M. Qin, X. Lu, C. You, Y. Chi and P.-T. Chou, Nat. Photonics, 2022, 16, 843–850 CrossRef CAS.
- F. Fagnani, A. Colombo, C. Dragonetti, D. Roberto and D. Marinotto, Inorg. Chim. Acta, 2022, 532, 120744 CrossRef CAS.
- D. Roberto, A. Colombo, C. Dragonetti, F. Fagnani, M. Cocchi and D. Marinotto, Molecules, 2022, 27, 5171 CrossRef CAS PubMed.
- H. Yersin, A. F. Rausch, R. Czerwieniec, T. Hofbeck and T. Fischer, Coord. Chem. Rev., 2011, 255, 2622–2652 CrossRef CAS.
- P. T. Chou, Y. Chi, M. W. Chung and C. C. Lin, Coord. Chem. Rev., 2011, 255, 2653–2665 CrossRef CAS.
- V. Adamovich, J. Brooks, A. Tamayo, A. M. Alexander, P. I. Djurovich, B. W. D'Andrade, C. Adachi, S. R. Forrest and M. E. Thompson, New J. Chem., 2002, 26, 1171–1178 RSC.
- M. Cocchi, J. Kalinowski, V. Fattori, J. A. G. Williams and L. Murphy, Appl. Phys. Lett., 2009, 94, 073309–073311 CrossRef.
- P. Brulatti, V. Fattori, S. Muzzioli, S. Stagni, P. P. Mazzeo, D. Braga, L. Maini, S. Milita and M. Cocchi, J. Mater. Chem. C, 2013, 1, 1823–1831 RSC.
- J. A. G. Williams, A. Beeby, S. Davies, J. A. Weinstein and C. Wilson, Inorg. Chem., 2003, 42, 8609–8611 CrossRef CAS PubMed.
- S. J. Farley, D. L. Rochester, A. L. Thompson, J. A. K. Howard and J. A. G. Williams, Inorg. Chem., 2005, 44, 9690–9703 CrossRef CAS PubMed.
- J. A. G. Williams, Chem. Soc. Rev., 2009, 38, 1783–1801 RSC.
- C. Dragonetti, F. Fagnani, D. Marinotto, A. Di Biase, D. Roberto, M. Cocchi, S. Fantacci and A. Colombo, J. Mater. Chem. C, 2020, 8, 7873–7881 RSC.
- A. F. Rausch, L. Murphy, J. A. G. Williams and H. Yersin, Inorg. Chem., 2012, 51, 312–319 CrossRef CAS PubMed.
- A. Colombo, G. De Soricellis, F. Fagnani, C. Dragonetti, M. Cocchi, B. Carboni, V. Guerchais and D. Marinotto, Dalton Trans., 2022, 51, 12161–12169 RSC.
- A. Colombo, G. De Soricellis, C. Dragonetti, F. Fagnani, D. Roberto, B. Carboni, V. Guerchais, T. Roisnel, M. Cocchi, S. Fantacci, E. Radicchi and D. Marinotto, J. Mater. Chem. C, 2024, 12, 9702–9715 RSC.
- J. Kalinowski, M. Cocchi, L. Murphy, J. A. G. Williams and V. Fattori, Chem. Phys., 2010, 378, 47–57 CrossRef CAS.
- H. Ishida, S. Tobita, Y. Hasegawa, R. Katoh and K. Nozaki, Coord. Chem. Rev., 2010, 254, 2449–2458 CrossRef CAS.
- Aniline-based electron-rich pendants on the central aryl ring have similarly been shown to lead to enhanced intraligand charge-transfer character, at the expense of the metal, and thus to longer lifetimes: see, for example, ref. 53 and: D. L. Rochester, S. Develay, S. Zalis and J. A. G. Williams, Dalton Trans., 2009, 1728–1741 RSC.
- P. Pander, A. Sil, R. J. Salthouse, C. W. Harris, M. T. Walden, D. S. Yufit, J. A. G. Williams and F. B. Dias, J. Mater. Chem. C, 2022, 10, 15084–15095 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- S. Miertus, E. Scrocco and J. Tomasi, Chem. Phys., 1981, 55, 117–129 CrossRef CAS.
- M. Cossi, V. Barone, R. Cammi and J. Tomasi, Chem. Phys. Lett., 1996, 255, 327–335 CrossRef CAS.
- V. Barone and M. Cossi, J. Phys. Chem. A, 1998, 102, 1995–2001 CrossRef CAS.
- M. Cossi, N. Rega, G. Scalmani and V. Barone, J. Comput. Chem., 2003, 24, 669–681 CrossRef CAS PubMed.
- A. D. McLean and G. S. Chandler, J. Chem. Phys., 1980, 72, 5639–5648 CrossRef CAS.
- A. J. H. Wachters, J. Chem. Phys., 1970, 52, 1033 CrossRef CAS.
- A. Petersson and M. A. Al-Laham, J. Chem. Phys., 1991, 94, 6081–6090 CrossRef.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 299–310 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5dt00682a |
| This journal is © The Royal Society of Chemistry 2025 |